

Abstract
Constitutional mismatch repair deficiency syndrome is a rare autosomal recessive syndrome caused by homozygous mutations in mismatch repair genes. This is characterized by the childhood onset of brain tumors, colorectal cancers, cutaneous manifestations of neurofibromatosis-1 like café au lait spots, hematological malignancies, and occasionally other rare malignancies. Here, we would like to present a family in which the sibling had glioblastoma, and the present case had acute lymphoblastic lymphoma and colorectal cancer. We would like to present this case because of its rarity and would add to literature.
Keywords: Constitutional mismatch repair deficiency, hereditary nonpolyposis colorectal cancer, Lynch syndrome, mismatch repair genes
Introduction
Deoxyribonucleic acid (DNA) mismatch repair (MMR) mechanisms play a vital role in repairing the mutations that occur in it. If unrepaired they lead to development of errors in replication, defective transcription, and protein synthesis, which may lead to development of malignancies. They also play a role in response to agents causing DNA damage and postmeiotic segregation (PMS2) helps in immunologlobin class switch recombination.[1,2] Various monoallelic mutations that occur in the MMR genes, include mutL homolog1 (MLH1), mutS homolog 6 (MSH6), MSH2, PMS2 increased, which may lead to the development of malignancies described as Lynch syndrome.[3] Lynch syndrome commonly includes nonpolyposis colon cancer, stomach cancer, endometrial, ovary cancer, and rarely causes hematological malignancies like Leukemia's or lymphomas. But in presence of biallelic mutations of MMR genes, they lead to a severe form of the syndrome called as constitutional mismatch repair deficiency syndrome (CMMRD).[4,5]
The CMMRD syndrome is characterized by (a) early childhood onset of hematological, brain tumors and other rare malignancies e.g., infantile myofibromatosis (b) childhood onset of LS associated malignancies (e.g., Colorectal cancer) and (c) cutaneous features of neurofibromatosis 1 (NF-1) like café au lait spots.[6] This syndrome was first recognized in 1999. In a literature search for CMMRD 146 cases were reported. Here, we would like to report a case of CMMRD syndrome wherein the patient had non-Hodgkin's lymphoma and colon cancer, and his sibling had glioblastoma multiforme. It is the first case to be reported from India to the best of our knowledge.
Case Report
An 8-year-old male child presented with weakness and radicular pain of both limbs for 3 months. There was no history of trauma or any other constitutional symptoms. On examination, there was bilateral hypotonic, power of 4/5 in both the lower limbs with no sensory or autonomic deficits. A magnetic resonance imaging spine was performed, which showed multiple osteolytic lesions involving L1, L2, and L3 with suspicion of psoas abscess [Figure 1a-e]. Patient was started on empirical antituberculous therapy and planned for surgery. He underwent L2-L3 laminectomy and removal of tumor with spinal cord decompression. The histopathology of the excised tumor showed a round cell tumor and further immunohistochemistry confirmed Lymphoblastic lymphoma. On general examination, he had multiple café au lait macules involving the trunk and extremities [Figure 2a and b]. He was started on chemotherapy as per MCP-841 protocol for the acute lymphoblastic lymphoma. After completion of chemotherapy, he was disease-free from then and was on follow-up. In November 2010, he presented with bleeding per rectum and per abdominal, and per rectal examination was normal. With persistence of frank bleeding, patient was considered for colonoscopy, which showed a noduloulcerative circumferential growth involving rectosigmoid and upper third of the rectum. There were multiple sessile polyps involving sigmoid colon, transverse colon, and the Caecum. However, the number of polyps were <100 [Figure 2c]. A biopsy of the growth was performed, which showed adenocarcinoma Grade 2. With this, the patient was planned for total proctocolectomy with J-shaped ileal pouch and temporary ileostomy [Figure 2d]. Histopathology revealed adenocarcinoma Grade 2, T2 N1 M0 malignancy identified in recto-sigmoid colon and transverse colon. However, the overall number of polyps was <100. Patient was given adjuvant chemotherapy of FOLFOX4 regimen for 3 months. After completion of therapy, restoration of bowel continuity was performed. The child is under follow-up and is free of disease. He had a sibling who was diagnosed with glioblastoma multiforme at the age of 7 years in 2000 and underwent decompression craniotomy and excision of tumor. He was treated with external beam radiotherapy for the brain. Later, he developed recurrence in the year 2002 and was succumbed to death. There were no other family members with brain tumor or hematological malignancies or polyposis coli. The parents and child are given genetic counseling and are advised for molecular gene testing. However, the parents were not willing to pursue gene testing and hence the child was kept for follow-up.
Figure 1.
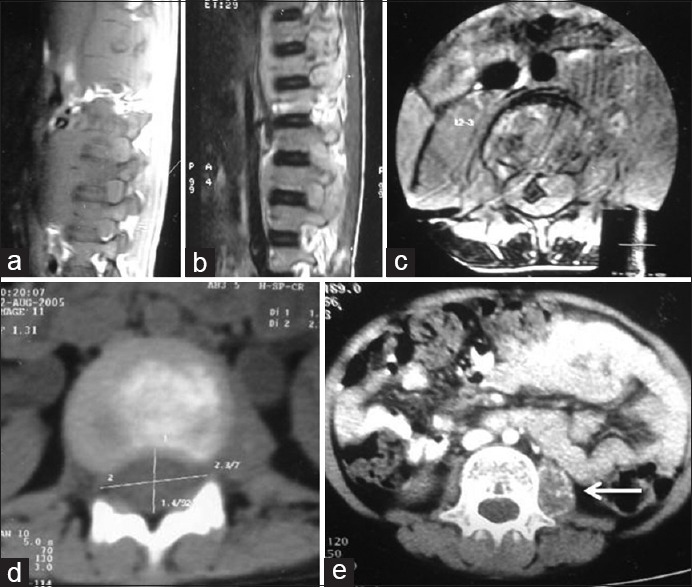
Magnetic resonance imaging images of dorsolumbar spine (axial and sagittal section) shows destruction of vertebrae L1, L2, and L3 with thecal compression (a-c); contrast-enhanced computer tomography scan showing osteolytic lesion involving the lumbar vertebra with cord compression (d), irregular calcification involving the left psoas muscle (arrow) (e)
Figure 2.
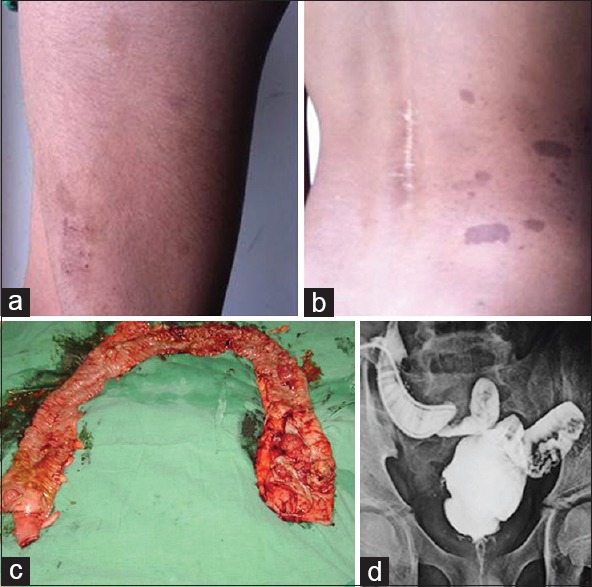
Image showing café au lait spots over the left thigh and back with a scar over the spine showing previous history of surgery (a and b); image of the total proctocolectomy specimen with growth seen at the recto-sigmoid and transverse colon with <100 polyps (c); distal loop ileogram showing the J-pouch ileo-anal anastomosis (d)
Discussion
Constitutional mismatch repair deficiency is an autosomal recessive condition, which is characterized by early presentation with brain tumors, colorectal malignancies, hematological malignancies, and skin manifestations.[7] Heterozygous mutations of MMR genes lead to the development of hereditary nonpolyposis colorectal cancer syndrome, which is an adult cancer syndrome, whereas biallelic gene mutation leads CMMRD syndrome in whom pediatric malignancies occur more commonly. The most common malignancies that occur are non-Hodgkin's lymphoma, colorectal cancer, and glioblastomas.[6] The genes involved in the development of CMMRD syndrome are MLH1, MSH2, MSH6, and PMS2. A recent review of 146 reported cases of CMMRD revealed PMS2 mutations identified in 60% of cases and 40% had MLH1, MSH2 or MSH6 mutations which is in contrary to LS were heterozygous MLH1 or MSH2 are more common.[8] There is a difference in presentation between MLH1 or MSH2 and MLH6 and PMS2. Patients with MLH1/MSH2 mutations have higher incidence of hematological malignancies, present at an earlier age and have poorer survival when compared to families with MSH6/PMS2 germ line mutations. The incidence of brain tumors and second malignancy is more in patients with MSH6/PMS2 mutations as they survive first malignancy.[6]
Hence, there is a need to identify this syndrome as most of them would have been missed. CMMR-D syndrome should be suspected in patients who develop childhood malignancies of CMMRD with skin features of NF-1, family history of LS-associated malignancies, consanguinity, sibling suffering from childhood cancer, development of second malignancy. They need to be differentiated from Turcots syndrome and Lynch syndrome that usually presents with adult cancers. Patients with PMS2 mutations may not fulfill the Amsterdam or the revised Bethesda criteria.[6] The families of these children who were suspected with CMMRD syndrome should undergo analysis for microsatellite instability or immunohistochemistry for all 4 genes (MLH1, MSH2, MSH6, and PMS2). As there is biallelic loss congenitally, the loss of MMR genes can be demonstrated both in normal and neoplastic tissues. Polymerase chain reaction can help in detecting MMR gene mutations in normal tissue.
As this is relatively rare syndrome, genetic counseling to the parents should be given. If heterozygosity was detected in family members, they should be followed-up as LS-based guidelines.[9] Recently suggestions were made by european consortium ‘care for CMMRD’ in which a diagnostic criteria was developed and screening guidelines were described. According to this scoring system patients with ≥3 points are to considered for testing by microsatellite instability(MSI) and /or immunohistochemical staining of 4 MMR proteins (MLH1, MSH2, MSH6, and PMS2). In contrast to LS these defects can be detected even in non-neoplastic cells.[8].
Conclusion
Constitutional mismatch repair deficiency is a rare autosomal recessive syndrome associated with wide spectrum of malignancies. This syndrome is not studied well, and needs increased awareness among clinicians. Our case adds to the literature a case of CMMRD syndrome.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 2.Péron S, Metin A, Gardès P, Alyanakian MA, Sheridan E, Kratz CP, et al. Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. J Exp Med. 2008;205:2465–72. doi: 10.1084/jem.20080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;15(21):1174–9. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 4.Ricciardone MD, Ozçelik T, Cevher B, Ozdag H, Tuncer M, Gürgey A, et al. Human MLH1 deficiency predisposes to hematological malignancy and neurofibromatosis type 1. Cancer Res. 1999;59:290–3. [PubMed] [Google Scholar]
- 5.Wang Q, Lasset C, Desseigne F, Frappaz D, Bergeron C, Navarro C, et al. Neurofibromatosis and early onset of cancers in hMLH1-deficient children. Cancer Res. 1999;59:294–7. [PubMed] [Google Scholar]
- 6.Wimmer K, Kratz CP. Constitutional mismatch repair-deficiency syndrome. Haematologica. 2010;95:699–701. doi: 10.3324/haematol.2009.021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jasperson KW, Samowitz WS, Burt RW. Constitutional mismatch repair-deficiency syndrome presenting as colonic adenomatous polyposis: Cslues from the skin. Clin Genet. 2011;80:394–7. doi: 10.1111/j.1399-0004.2010.01543.x. [DOI] [PubMed] [Google Scholar]
- 8.Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, et al. Diagnostic Criteria for Constitutional Mismatch Repair Deficiency Syndrome: Suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD) J Med Genet. 2014;51:355–65. doi: 10.1136/jmedgenet-2014-102284. [DOI] [PubMed] [Google Scholar]
- 9.Vasen HF, Möslein G, Alonso A, Bernstein I, Bertario L, Blanco I, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer) J Med Genet. 2007;44:353–62. doi: 10.1136/jmg.2007.048991. [DOI] [PMC free article] [PubMed] [Google Scholar]