

Abstract
Patients with 13q deletion syndrome are characterized with different phenotypical features depending on the size and location of the deleted region on chromosome 13. These patients fall into three groups: In Group 1, deleted region is in the proximal and does not extend into q32; in Group 2, deleted region involves proximal to the q32 and in Group 3 q33-q34 is deleted. We present two cases with 13q syndrome with two different deleted region and different severity on clinical features: One case with interstitial deletion belongs to the Group 1 with mild mental retardation and minor malformations and the other case with terminal deletion belongs to Group 3 with moderate to severe mental retardation and major malformations.
Keywords: 13q deletion syndrome, mental retardation, multiple anomalies
Introduction
Phenotypical features of chromosome 13q syndrome have been described over the past 30 years. The patients have various clinical features depending on the size and deleted region on chromosome 13. This region was investigated as a critical region for these patients.[1,2] The 13q syndrome is characterized into 3 groups: Group 1 in which proximal region (q12.2-q31) is involved in deletion and patients tend to appear with minor abnormalities, mild to moderate mental retardation and susceptible to retinoblastoma.[3,4] Group 2 included those with proximal to q32 (q12.2-q32), these patients appear with major malfunctions, moderate to severe mental retardation and microcephaly[5] and growth retardation. Group 3 comprised those with q33-q34; these patients have severe mental retardation without major malfunctions and growth retardation.[1,2]
Case Reports
Case 1
A 5-year-old boy was referred for genetic counseling with different phenotypical features [Table 1]. The parents were unrelated with history of one abortion in the previous pregnancy. The patient was the product of a full-term pregnancy and normal vaginal delivery. Birth weight was 2.4 kg. He had seizure beginning from 2nd months; brain computed tomography (CT) scan displayed agenesis of vermis of the cerebellum [Figure 1], the clinical features in this case were:
Table 1.
Clinical features of the patients
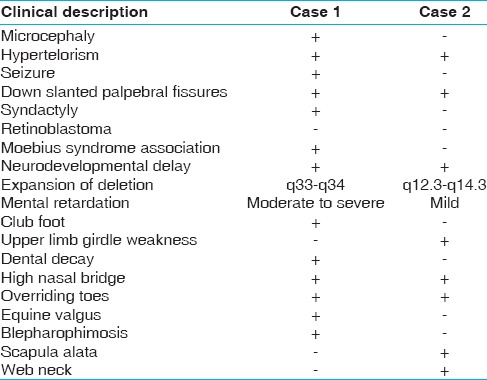
Figure 1.
(a) Note to the ptosis, down slanted palpebral fissures, strabismus, and high nasal bridge. (b) Over riding of fourth toes. (c) Hypospadias. (d) Agenesis of the vermis cerebellum
Cleft palate, microcephaly (head circumference of 45 cm [<-2 SD (2%)][6]), moderate to severe mental retardation, speech difficulty, asymmetric in left skulp, ptosis, club feet, down slanted palperbal fissures, hypospadias, neurodevelopmental delay, syndactyly in both hands fingers, short metacarp, overriding of the fourth toes, strabismus, left blepharophimosis in the left side, high nasal bridge, thin lips, micrognathia, no ovala, dental decay, small ears, and hypoplastic thumb.
Case 2
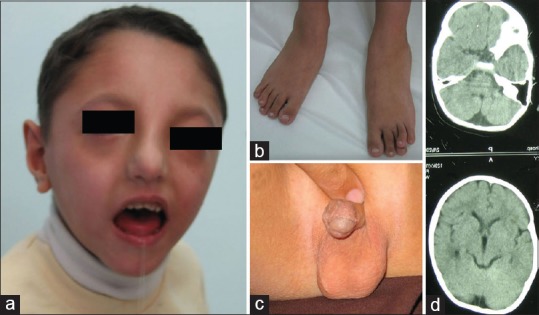
A 12-year-old girl was referred with dysmorphic features, mild mental retardation, and neurodevelopmental delay [Table 1]. The girl was the product of a normal pregnancy and birth weight and height were 3.5 kg and 49 cm, respectively. Head circumference was 53 cm normal for the age of 12 years, and she is studying in the elementary school. Parents were first cousins once removed with the history of one abortion in the mother. There are two other healthy male and female siblings in the family. She was evaluated by nerve conduction velocity test and exhibited some sort of neuropathy. Clinical characterizations included [Figure 2]: Generalized weakness of muscle bulk (hypotonia), scapula alata, upper limb girdle weakness, high nasal bridge, wide mandibular angle, high arched palate, thin and long narrow face, down slanted palpebral fissures, hypoplasia of alae nasi, low hairline, webbed neck, overriding toes, and sandal gap. There were small nevi on her posterior trunk.
Figure 2.
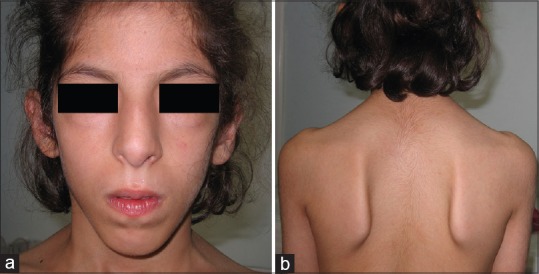
(a) Note to the high nasal bridge, wide mandibular angle, thin and long narrow face, down slanted palpebral fissures, web neck. (b) Scapula alata
Materials and Methods
Standard high-resolution GTG banding was carried out in order to investigate the patients’ chromosomes. Metaphase chromosomes prepared on slides using pancreatin enzyme for banding preparation.
Results and Discussion
We present here two cases with 13q deletion syndrome with different phenotypical features. Case 1, a boy with deletion of q33-q34 [Figure 3] who had microcephaly, moderate to severe mental retardation, neurodevelopmental delay, and primary diagnosis was Moebius syndrome. There was agenesis of vermis cerebellum after CT scan evaluation. Case 2 was a girl with deletion of q12.3-q14.3 [Figure 3], with dismorphic features, neurodevelopmental delay and neuropathy, but not microcephaly and seizure; this case did not have any retinoblastoma.
Figure 3.
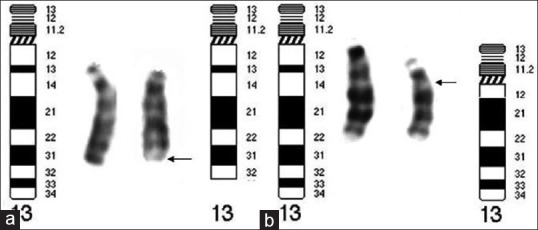
(a) The abnormal chromosome displays deleted region from q33 to q34 which, belongs to Group 3. (b) The abnormal chromosome, with deleted region of q12.3-q14.3 belongs to Group 1
Case 1 related to the Group 3 with more severe phenotypical and neurological features and Case 2 related to the Group 1 with less clinical and neurological characterizations. Overall Case 1 exhibited more dysmorphic characterizations and more severe mental retardation than Case 2. Our findings are consistent with previous cases reported. It is recommended that children with seizure, neuropathy, mental retardation, and dismorphic features can be the candidate for chromosomal investigation as well as other tests required for evaluation.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Brown S, Gersen S, Anyane-Yeboa K, Warburton D. Preliminary definition of a “critical region” of chromosome 13 in q32: report of 14 cases with 13q deletions and review of the literature. Am J Med Genet. 1993;45:52–9. doi: 10.1002/ajmg.1320450115. [DOI] [PubMed] [Google Scholar]
- 2.Brown S, Russo J, Chitayat D, Warburton D. The 13q-syndrome: The molecular definition of a critical deletion region in band 13q32. Am J Hum Genet. 1995;57:859–66. [PMC free article] [PubMed] [Google Scholar]
- 3.Lele KP, Penrose LS, Stallard HB. Chromosome deletion in a case of retinoblastoma. Ann Hum Genet. 1963;27:171–4. doi: 10.1111/j.1469-1809.1963.tb00209.x. [DOI] [PubMed] [Google Scholar]
- 4.Sparkes RS, Sparkes MC, Wilson MG, Towner JW, Benedict W, Murphree AL, et al. Regional assignment of genes for human esterase D and retinoblastoma to chromosome band 13q14. Science. 1980;208:1042–4. doi: 10.1126/science.7375916. [DOI] [PubMed] [Google Scholar]
- 5.Fryns JP. Microcephaly/lymphedema and terminal deletion of the long arm of chromosome 13. Am J Med Genet. 1995;57:504. doi: 10.1002/ajmg.1320570332. [DOI] [PubMed] [Google Scholar]
- 6.Jones KL. 6th ed. Philadelphia: Elsevier-Saunders; 2006. Smith's Recognizable Patterns of Human Malformation; p. 849. [Google Scholar]