

Abstract
The maintenance of species boundaries despite interspecific gene flow has been a continuous source of interest in evolutionary biology. Many hybridizing species have porous genomes with regions impermeable to introgression, conferring reproductive barriers between species. We used ecological niche modelling to study the glacial and postglacial recolonization patterns between the widely hybridizing spruce species Picea glauca and P. engelmannii in western North America. Genome-wide estimates of admixture based on a panel of 311 candidate gene single nucleotide polymorphisms (SNP) from 290 genes were used to assess levels of admixture and introgression and to identify loci putatively involved in adaptive differences or reproductive barriers between species. Our palaeoclimatic modelling suggests that these two closely related species have a long history of hybridization and introgression, dating to at least 21 000 years ago, yet species integrity is maintained by a combination of strong environmental selection and reduced current interspecific gene flow. Twenty loci showed evidence of divergent selection, including six loci that were both Fst outliers and associated with climatic gradients, and fourteen loci that were either outliers or showed associations with climate. These included genes responsible for carbohydrate metabolism, signal transduction and transcription factors.
Keywords: admixture, ecological niche modelling, outlier loci, spruce
Introduction
The nature of genetic barriers that isolate species from interspecific gene flow is of great biological interest (Abbott et al. 2013; Feder et al. 2013). Understanding the maintenance of species boundaries requires addressing a fundamental question: are the genomes or genes the units of specific differentiation? Under the most widely recognized species concept, the biological species concept, the genomes of species are coadapted units that are separated from other units by reproductive barriers. This concept implies that species divergence only occurs through whole-genome isolation and therefore hybridizing species are not ‘true’ species. By contrast, the genic view of speciation proposes that the gene is the unit of species differentiation (Wu 2001), and reproductive isolation is a consequence of natural selection acting on individual genes. Species boundaries are ‘semi-permeable’; some genomic regions share introgressed genes between species, whereas other regions accumulate divergence between species in response to natural selection (Strasburg et al. 2012).
Despite recent advances in population genetics and genomics, little is known about how species boundaries are maintained between closely related species that hybridize. In hybridizing species showing adaptations to different environments, divergent selection acts on a subset of genes, counteracting the homogenizing effect of gene flow and preventing introgression in surrounding genomic regions (Chapman et al. 2013). As a result, species boundaries are maintained despite hybridization and introgression (Andrew & Rieseberg 2013). Species that hybridize can also coexist without merging or becoming swamped in environments where hybrids are favoured by selection over pure species (fitness higher than pure species), that is, in intermediate environments (Arnold 1997).
White spruce [Picea glauca (Moench) Voss] and Engelmann spruce (P. engelmannii Parry) are wind-dispersed, long-lived, closely related conifers that hybridize extensively in British Columbia and the western part of Alberta, Canada, as well as in some parts of the western United States. Their extensive hybrid zone is mainly composed of hybrids with a clinal intergradation of morphological and genetic characteristics along elevational gradients between parental species' habitats (Ledig et al. 2006).
Recent studies using neutral microsatellite markers have found that introgression is extensive in the hybrid zone and asymmetric towards Engelmann spruce (Haselhorst & Buerkle 2013; De La Torre & Aitken, unpublished). Despite extensive interspecific gene flow, natural selection acting along environmental gradients (exogenous selection) is responsible for maintenance of this hybrid zone occupying mountainous areas, with white spruce adapted to low-elevation boreal and subboreal environments, Engelmann spruce adapted to high-elevation environments and hybrid populations inhabiting intermediate elevations. This hybrid zone follows a bounded hybrid superiority model (Moore 1977) in which hybrids are fitter than pure species in intermediate environments (De La Torre et al. 2014).
Over the last century, studies of the evolutionary and genetic relationships between white and Engelmann spruce have focused on whether these two closely related species represent extreme phenotypes along a genetic continuum (Rajora & Dancik 2000) or whether they deserve species-level recognition. Although previous studies in the white x Engelmann spruce contact zone have provided a broad idea of the evolutionary relationships between these species, they have failed to identify the factors contributing to isolating barriers between species. Some limitations of previous studies included small numbers of loci that lacked species-specific diagnostic markers and a limited geographical scope of sampling within the hybrid zone.
This study combines population genomic approaches with ecological niche modelling to assess historical and contemporary evolutionary relationships between white and Engelmann spruce. Our primary objective is to understand how white and Engelmann spruce maintain their species integrity despite interspecific gene flow. To answer this question, we inferred the recent histories of white and Engelmann spruce by using palaeoclimatic analysis to study potential climatic niche-based range expansions and contractions and to infer past opportunities for secondary contact between species. We determined the extent and direction of introgression using a genome scan approach with markers from 290 candidate genes and identified a subset of genes that may be involved in genetic barriers between these species.
Materials and methods
Ecological niche modelling
We used ecological niche modelling to study past demographic processes during glacial and postglacial range contractions and expansions. While these models are based on assumptions such as environmental stability and niche conservation (Araujo & Guisan 2006), they have been used effectively, particularly at continental scales, for both past species range reconstructions and future range projections (Elith & Leathwick 2009). The ecological niche model was built using mapped ecoregion delineations for North America from various public sources. Model training and model projection were carried out using these ecoregion classes as a response variable, as this approach was shown to be effective at limiting range overprediction in model hindcasts (Roberts & Hamann 2012). Following model projection/hindcast of these ecoregion classes, species frequencies were attached to classes based on a summary of sample plot records, from the Forest Inventory and Analysis (FIA) programme data (http://www.fia.fs.fed.us) and from proprietary Canadian sample plot data, falling geographically within the boundaries of each original mapped ecoregion (Roberts & Hamann 2012). Environmental predictors comprised ten climate variables, interpolated at 1 km resolution: mean annual temperature, mean coldest and mean warmest month temperatures, continentality, mean annual and mean growing season precipitations, number of frost-free days, number of degree-days above 5 °C and estimates of annual and summer heat–moisture indices (Wang et al. 2012). Climate for the modern period was based on the 1961–1990 climate record. Palaeoclimate data were generated by overlaying the modern climate data with climatic anomaly data generated by two general circulation models (GCMs): the Community Climate Model (CCM1) (Kutzbach et al. 1998) and the Geophysical Fluid Dynamics Laboratory model (GFDL) (Bush & Philander 1999).
Models were trained with data from the present day and projected with the palaeoclimate data for past periods. Ecoregion model projections were made by a majority vote of three modelling strategies: a discriminant analysis, a randomized bootstrapped classification tree and a minimum multivariate distance (Roberts & Hamann 2012). Modelled reconstructions of modern ranges were validated against the 55 744 individual sample plots in the FIA data. Model projections for the past periods were validated against 931 fossil pollen and plant macrofossil records, largely from the Neotoma Paleoecology Database (http://www.neotomadb.org), using the area under the receiver-operating characteristic (AUC). Values for this statistic range from 0 to 1 where 1 represents a perfect model and 0.5 represents random chance.
Sample collection for genomic analyses
Newly flushed needle tissue of 745 samples of white spruce, Engelmann spruce and their hybrids were collected from common garden experiments, previously established by the British Columbia Ministry of Forests, Lands and Natural Resources Operations' spruce breeding programme (Table 1, Fig. 1). Seed planning zones (SPZs) are geographical units for genetic management based on ecosystem classification and adaptive traits of populations. In this study, we collected samples from 200 open-pollinated families (progeny of individual seed parents sampled from natural populations) in the West Kootenay, East Kootenay Mount Robson and Quesnel Lakes SPZs. Thirty-three putatively pure white spruce (22 from Fort Nelson and 11 from Prince George), and 40 putatively pure Engelmann spruce from southwestern United States were obtained from grafts of trees sampled from natural populations. After the population structure and admixture analyses, the Prince George population was reassigned as a hybrid population (Table 1). The same genetic materials were analysed in De La Torre et al. (2014).
Table 1.
Geographical coordinates and climatic variables of parent trees for Picea glauca, P. engelmannii and their hybrids analysed with 311 single nucleotide polymorphism loci. Two-letter codes are used to identify populations in subsequent tables and graphs
| Population | Province | Elevation range (m) | Latitude (degrees) | Longitude (degrees) | MAT (°C) | MAP (mm) | Sample size |
|---|---|---|---|---|---|---|---|
| Picea glauca | |||||||
| Fort Nelson (FN) | B.C | 350–600 | 58.4–59.4 | 120.5–126.3 | 0.3 | 509 | 22 |
| P. glauca x P. engelmannii | |||||||
| Prince George (PG) | B.C | 610–793 | 53.5–54 | 121.6–122 | 2.8 | 769 | 11 |
| Quesnel Lakes (QL) | B.C | 680–1555 | 51.8–53.2 | 119.4–122.1 | 2.3 | 914 | 220 |
| Mount Robson (MR) | B.C | 701–1525 | 52.2–53.8 | 118.4–121.5 | 1.7 | 1167 | 197 |
| East Kootenay (EK) | B.C | 1006–1677 | 49.4–50.8 | 115.1–116.6 | 1.9 | 944 | 204 |
| West Kootenay (WK) | B.C | 690–1966 | 49–50.5 | 114.9–118.4 | 2.5 | 1168 | 124 |
| Picea engelmannii | |||||||
| Salmon River (E1) | Idaho | 1859–2530 | 43.8–46.2 | 113.7–115.9 | 2.8 | 1028 | 9 |
| Teton-Wasatch (E2) | Wyo. | 2347–3048 | 40.4–43.8 | 109.5–111.6 | 2.6 | 885 | 13 |
| Fishlake-Lasal (E3) | Col. | 2606–3383 | 37.5–39.8 | 109.2–112.8 | 3.4 | 768 | 18 |
MAT, Mean annual temperature; MAP, mean annual precipitation; B.C, British Columbia; Wyo, Wyoming; Col, Colorado.
Fig 1.
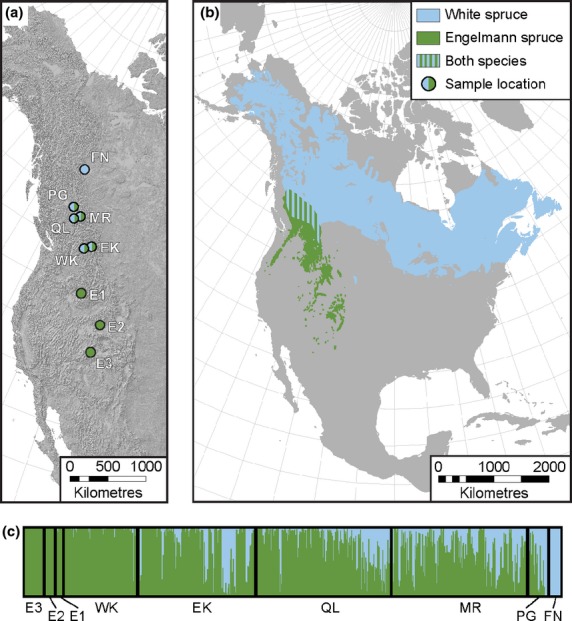
(a) Geographical locations of populations of pure Picea glauca (FN), pure P. engelmannii (E1, E2 and E3), and their hybrids (all other populations). Population names corresponding to two-letter codes in Table 1. (b) Map showing the location of the hybrid zone in North America; and (c) posterior estimates of cluster membership for the Picea glauca x P. engelmannii hybrid zone with TESS for K = 2. Populations are ordered by increasing latitude from left to right, finishing with the P. glauca reference population (FN).
DNA extraction and genotyping
Needles were stored at −80 °C prior to DNA isolation. Each sample was isolated using a modified CTAB protocol (Doyle & Doyle 1987). After the extractions, DNA quality and concentration of each sample was assessed using 0.8% agarose gels and quantified based on Nanodrop 2000C spectrophotometer readings (Thermo Fisher Inc., Waltham, MA, USA). DNA samples from all individuals were SNP-genotyped at the Genome Quebec/McGill Innovation Centre using an Illumina bead array chip (Illumina Inc., San Diego, CA, USA) with the GoldenGate allele-specific assay.
Samples from allopatric pure species populations and from hybrid populations were assayed in two different SNP arrays. White spruce, Engelmann spruce and Sitka spruce samples were tested with the first array comprising 1536 SNPs from a large panel of genes putatively involved in cold hardiness and insect herbivory resistance (Holliday et al. 2008). A total of 230 SNPs were selected based on their genotyping quality (GenTrain score >0.40). For the second SNP array, 154 additional SNPs previously tested in other studies (Namroud et al. 2008; Porth et al. 2011) were added to the analysis. In the second SNP array, SNPs selected from the first array and other studies (384) were used to genotype 745 samples from the hybrid zone. Due to the selection process, SNPs may be subject to ascertainment bias in terms of underrepresentation of rare alleles (Namroud et al. 2008).
Data files with florescence intensity for each SNP were loaded directly into GenomeStudio Genotyping Module v1.0 (Illumina 2010). The call rate cut-off for SNP selection was 90%; however, most of the SNPs (91%) had call rates >98%. Of 384 SNPs included in the second GoldenGate array, 311 SNPs corresponding to 290 widely distributed genes were successfully genotyped and met both genotyping quality and data normalization criteria. Annotation and position of these genes in the white spruce genome (Pavy et al. 2012; B. Pelgas, N. Isabel & J. Bousquet, unpublished data) can be found in Supporting Information Table S1 (Supporting information).
Genomic admixture
Samples of putatively pure species from allopatric populations were genotyped for a subset of the SNPs used to genotype individuals from the hybrid zone. These 86 SNPs were used to assess population structure and admixture levels using the program STRUCTURE 2.3.3 (Pritchard et al. 2000). Admixture models with a putative number of clusters (K) from one to 16 were tested using 50 000 iterations for the pre- and postburn periods. Each run was replicated twenty times to estimate K using the method developed by Evanno et al. (2005) with the program Structure Harvester version 0.6.7 (Earl & VonHoldt 2011). Based on STRUCTURE results for K = 2, individuals with Bayesian admixture proportions (Q) >0.9 from Fort Nelson (white spruce) and E1, E2, E3 (Engelmann spruce) were used as reference genotypes for ‘pure species’ to calculate hybrid index within the zone with 86 SNPs using the INTROGRESS 1.1 (Gompert & Buerkle 2010) package in R 2.13.1 (R Core Team 2013).
Population structure and admixture levels were also estimated using TESS version 2.3 (Chen et al. 2007). Unlike STRUCTURE, TESS uses a hierarchical Bayesian algorithm to include spatial prior distributions on the individual admixture proportions (Durand et al. 2009), giving a reliable estimation of admixture when admixture proportions are variable across space. The admixture model was performed for values of Kmax ranging from 2 to 14. Markov chain Monte Carlo (MCMC) algorithms were run for a length of 50 000 sweeps with burn-in periods of 30 000 sweeps. Each run was replicated twenty times.
Principal component analysis (PCA) was conducted using a correlation matrix of allele frequencies with SAS Enterprise Guide 4.2. The first twenty principal components were tested for correlation with elevation using PROC CORR. Fst values were calculated using GenAlEx version 6.4 (Peakall & Smouse 2006).
Linkage disequilibrium
Pairwise linkage disequilibrium (LD; r2) among all informative sites for 23 of the genes was calculated from inferred haplotypes using PHASE algorithms with the program DnaSP v.5.10.01 (Librado & Rozas 2009). Statistical significance of LD tests was determined by Fisher's exact tests with Bonferroni correction. Because LD can be affected by differences in sample sizes and allele frequencies, r2 was calculated from 22 randomly selected individuals in each of the populations. LD was compared between parental and hybrid populations to assess the likelihood of recent admixture in the hybrid zone. If admixture were recent in the hybrid zone, we would expect to find higher LD, on average, in hybridizing populations due to newly recombinant alleles (Barton & Hewitt 1985).
Detection of loci potentially affected by selection
To test for signatures of selection, we followed two different approaches. First, we used BayeScan v2.0 (Foll & Gaggiotti 2008) to identify loci that deviated significantly from neutrality. This Bayesian program decomposes Fst coefficients into a population-specific component (β) and a locus-specific component (α) using logistic regression. When the pattern of diversity cannot be explained by β alone (α significantly different from 0), the locus is considered to be under selection. Positive values of α suggest diversifying selection and negative values suggest balancing selection (Table 2). Several runs were performed to ensure consistency, with 5000 iterations and burn-in period of 50 000 iterations. False discovery rate (q), defined as the expected proportion of false positives among outlier markers, was set at 0.03.
Table 2.
Significant single nucleotide polymorphism (SNP) outliers detected using (A) 311 SNP loci within the contact zone (Fig. 4a); and (B) 86 SNP loci across the hybrid zone (Fig. 4b) with BayeScan. A positive value of α suggests diversifying selection, and a negative value, balancing selection. Cut-off for Bayesian posterior probability [prob(α≠0)] was 0.7. This probability cannot be compared with traditional P-values. SNP short ID identifies SNPs in Fig. 4
| SNP ID | SNP short ID | Prob (α≠0) | log (PO) | Alpha | Fst |
|---|---|---|---|---|---|
| (A) | |||||
| 13_496 | 17 | 0.96779 | 1.4778 | 1.29 | 0.10417 |
| 208pg12875c | 56 | 1 | 1 | 1.9987 | 0.1767 |
| 295_78 | 62 | 0.89898 | 0.94934 | 1.045 | 0.084892 |
| C2270-contig1.NC1-384 | 129 | 0.79056 | 0.57687 | 0.94254 | 0.080339 |
| C6522-contig1.NC1-269 | 146 | 0.96999 | 1.5096 | 1.317 | 0.10647 |
| WS-2.0-GQ0024.B3.r-D12.1-239 | 200 | 0.989 | 1.9537 | 1.2692 | 0.10083 |
| WS-2.0-GQ0064.B3.r-I13.1-1236 | 240 | 0.9806 | 1.7036 | 1.2661 | 0.10062 |
| (B) | |||||
| 124_495 | 7 | 0.70594 | 0.38033 | −1.076 | 0.053519 |
| 13_496 | 17 | 1 | 1 | 1.6777 | 0.29291 |
| 234_171 | 45 | 0.70314 | 0.37449 | −0.98515 | 0.055809 |
| 295_78 | 62 | 0.78496 | 0.56232 | 0.87226 | 0.19316 |
| 45_1067 | 68 | 0.976 | 1.6091 | 1.0779 | 0.21397 |
| 50_135 | 70 | 0.86517 | 0.80733 | −1.2689 | 0.043362 |
| 68_286 | 76 | 1 | 1 | 2.7706 | 0.4658 |
Our second approach for identifying targets of local adaptation was to use Bayenv 2.0 (Coop et al. 2010) to test for associations between SNP allele frequencies and environmental variables. This approach complements the Fst outlier analysis, and the two analyses may produce different results for methodological or biological reasons (Keller et al. 2012). The Fst outlier analysis assumes an island model for the null distribution of neutral values, and the actual distribution of neutral Fst values will deviate from this expectation under other demographic scenarios such as the secondary contact in a hybrid zone (Schoville et al. 2012; Lotterhos & Whitlock 2014). Allele–environment associations are not sensitive to this problem. However, Fst outliers may reflect divergent selection and local adaptation due to unknown environmental drivers that are not tested in an allele–environment association approach. To overcome the lack of a set of control (noncandidate) SNPs, we built the control matrix using all 311 SNPs (G.Coop, personal communication). We also performed nonparametric Spearman rank correlation coefficient tests, as a measure of support for Bayes factors. SNPs highly ranked in Bayes factor lists (BF > 3; Eckert et al. 2010) with high correlation coefficients (ρ > 0.2) were strong candidates for divergent selection. Bayenv runs were carried out using 100 000 iterations (k) and a number of different seeds to ensure model convergence.
Both the BayeScan and Bayenv analyses were conducted on two sets of data. First, all candidate gene SNPs (311) were used to analyse populations from within the hybrid zone classified into elevational bands (350–600, 600–1600, 1600–1800 , 1800–2250 m, >2250 m). Secondly, a subset of SNPs (86) was used to study populations across the hybrid zone spanning a wide range from the pure Engelmann spruce populations in the south to the pure white spruce populations in the north. In the Bayenv analyses, SNP allele frequencies were tested for associations with 21 climatic variables from ClimateWNA (Wang et al. 2012) (Table 3).
Table 3.
Candidate gene single nucleotide polymorphisms that exhibit Fst outlier behaviour or that are associated with one or several environmental variables
| SNP ID | Fst outlier* | Environmental associations† | Annotation | Reference | Linkage group‡ | Position (cM)‡ |
|---|---|---|---|---|---|---|
| 208pg12875c | Yes | MWMT, SHM, EXT, Eref | Glycoside hydrolase family 28 protein/polygalacturonase (pectinase) | P. glauca | 8 | 74.547 |
| 295_78 | Yes | MWMT | No apical meristem | P. sitchensis | — | — |
| WS-2.0-GQ03105.B7-O12.3-654 | No | MWMT,MSP, SHM, Eref, CMD | Fructose-1,6-bisphosphatase | P. glauca | 10 | 10.469 |
| WS-2.0-GQ0064.B3.r-I13.1-1236 | Yes | MWMT | Acid phosphatase | P. glauca | 4 | 156.456 |
| 14_248 | No | MCMT,TD | ABC transporter | P. glauca | 5 | 27.6 |
| WS-2.0-GQ0041.BR-J07.2-36 | No | MAP, AHM, PAS | Unknown | P. glauca | 7 | 130.421 |
| 0_13680-contig2.C1-149 | No | MAP, AHM, PAS | Hypothetical protein | P. glauca | 2 | 102.3 |
| WS-2.0-GQ0021.BR.1-G04.1-641 | No | MAP, AHM, PAS | Unknown | P. glauca | 6 | 13 |
| WS-2.0-GQ0168.B3-N16.1-556 | No | SHM, CMD | Flavin reductase | P. glauca | 5 | 72.283 |
| 144_441 | No | CMD | Phytochrome 4 | P. patens | 6 | 26.2 |
| C2270-contig1.NC1-384 | Yes | — | CCAAT-binding transcription factor | P. glauca | 2 | 49.628 |
| C6522-contig1.NC1-269 | Yes | — | Unknown | — | — | — |
| WS-2.0-GQ0024.B3.r-D12.1-239 | Yes | — | Peroxisomal membrane protein | P. glauca | 8 | 20.264 |
| 69_753 | No | MAT,DD_0, DD_18 | CBL-interacting protein kinase | P. sitchensis | — | — |
| 68_286 | Yes | MWMT, MSP, AHM,SHM, DD5, DD18, EXT,Eref | Glycosyl hydrolase | P. sitchensis | — | — |
| 206_435 | No | MCMT,TD,DD_0 | Isoflavone reductase | P. sitchensis | — | — |
| 13_496 | Yes | MCMT,TD,DD5,bFFP,PAS | FK506-binding protein | P. sitchensis | — | — |
| 45_1067 | Yes | MCMT,Eref | Alpha-amylase | P. sitchensis | — | — |
| 288_628 | No | MAP, PAS | Late elongated hypocotyl | P. sitchensis | — | — |
| 288_302 | No | MAP, PAS | Late elongated hypocotyl | P. sitchensis | — | — |
Only outlier loci suggesting diversifying selection in the BayeScan analyses were considered.
Environmental associations based on Bayenv are as follows: Mean Annual Temperature (MAT), Precipitation as snow (PAS), Mean Warmest Month Temperature (MWMT), Summer Heat—Moisture Index (SHM), Continentality (TD), Annual Heat—Moisture Index (AHM), Mean Annual Precipitation (MAP), Mean Summer Precipitation (MSP), Degree-days below 0 °C (DD_0), Mean Coldest Month Temperature (MCMT), Eref (Hargreaves reference evaporation), CMD (Hargreaves climatic moisture deficit), Degree-days above 5 °C (DD5), bFFP (Julian date on which frost-free period starts), Degree-days below 18 °C (DD_18), Degree-days above 18 °C (DD18), Extreme maximum temperature over a 30-year period (EXT).
Taken from Pavy et al. (2012).
Results
Ecological niche modelling
Both general circulation models (CCM1 and GFDL) suggest that white and Engelmann spruce may have had the opportunity for contact as early as the Last Glacial Maximum at 21 000 YBP in the southern part of the Rocky Mountains (Figs 2 and 3 and S1, Supporting information). Models were evaluated against modern sample plot species occurrence, fossil pollen and macrofossil data records since the Last Glacial Maximum using the area under the receiver-operating characteristic (AUC). Evaluations of Engelmann and white spruce with 55 744 modern sample plots showed good model fit with AUC values of 0.80 and 0.88, respectively. Validations with 2651 fossil pollen and macrofossil data since the Last Glacial Maximum for both species resulted in average AUCs of 0.62 and 0.61 for the CCM1 and GFDL model, respectively (Table S2, Supporting information). Model sensitivity for all periods and both species was generally lower and specificity generally very high, indicating that models were better predictors of species absences than species presences. This is to be expected, given the limited number of validation points available for each species, particularly in the periods immediately following the Last Glacial Maximum.
Fig 2.

Glacial re-colonization patterns of Picea glauca, P. engelmannii and their hybrids from 21 000 to 14 000 years before present based on climate niche modelling (CCM1 model) and palaeoclimate data. Years before present are in bold at the top of the graph. P. engelmannii is in green, P. glauca is in blue and the hybrid zone is in brown. Species frequency levels were divided equally by thirds: 0.33 (Low), 0.66 (Med), 1.00 (High). Laurentian and Cordilleran ice sheets are shown as inserts in each of the maps (Dyke et al. 2002).
Fig 3.

Postglacial re-colonization patterns of Picea glauca, P. engelmannii and their hybrids based on climate niche modelling (CCM1 model) and palaeoclimate data. Years before present are in bold at the top of the graph. P. engelmannii is in green, P. glauca is in blue and the hybrid zone is in brown. Species frequency levels were divided equally by thirds: 0.33 (Low), 0.66 (Med), 1.00 (High). Laurentian and Cordilleran ice sheets are shown as inserts in each of the maps (Dyke et al. 2002).
Genomic admixture and hybrid index
High levels of admixture and introgression were found within the contact zone between white and Engelmann spruce, where most individuals had hybrid ancestry. Both the admixture and the hybrid index analysis showed asymmetry in introgression towards Engelmann spruce, meaning that in general in the hybrid zone, there were more hybrid individuals with a higher genetic contribution from Engelmann spruce than from white spruce (Fig. 1). The histogram of hybrid classes showed that the majority of the hybrids (60%) had a hybrid index higher than 0.6, weighted towards Engelmann spruce ancestry, while the classes between 0.8 and 0.9 (putative Engelmann spruce advanced generation backcrosses) accounted for 14% of the trees. Hybrid classes between 0.1 and 0.2 (putative white spruce advanced generation backcrosses) were relatively rare (0.8%) in the zone (Fig. S2, Supporting information). The frequency distribution of hybrid classes suggests this is an old hybrid zone.
The low levels of linkage disequilibrium in hybridizing populations relative to parental populations also suggest that this is an ancient hybrid zone, in which advanced generation hybrid genotypes predominate and recombination is widespread. Based on 351 pairwise comparisons between informative sites across 23 genes, we found an overall mean LD (r2) of 0.41. There was no evidence for excess LD in individuals with intermediate genetic ancestry (mean r2 = 0.32) in comparison with LD estimates for parentals (mean r2 = 0.43), suggesting little or no recent admixture in the hybrid zone (Table S3, Supporting information). In fact, very few F1 hybrids were previously identified in this hybrid zone (De La Torre et al. 2014).
Despite high levels of introgression, both Engelmann spruce and white spruce remain genetically well differentiated, as evidenced by the results from STRUCTURE, TESS, PCA and Fst analyses. Delta K showed a peak for K = 2, each cluster representing one of the pure species (Fig. S3, Supporting information). The PCA analysis showed similar results, but the hybrids formed a separate cluster from pure species (Fig. S4, Supporting information). The model formed by the first three principal components was significantly correlated with elevation (R2 = 0.52, F = 282.38, P < 0.0001). The Fst analysis showed a greater genetic distance between pure species (0.208) than among white spruce and hybrids (0.09± 0.04) and among Engelmann spruce and hybrids (0.08 ± 0.02).
Detection of adaptive loci
Twenty loci showed some evidence of divergent selection. Of these, six loci were identified by both the Fst outlier analysis and the Bayenv gene–environment association analysis, three loci were identified by the Fst outlier analysis only, and 11 were identified by Bayenv only (Tables 2 and 3). Bayes factors (BF) for nine Fst outlier loci showing diversifying selection ranged from 3 to 47.51, with most of the values ranging from 3 to 10 (Table S4, Supporting information). The majority of Fst outlier loci indicated diversifying selection; only three outlier loci suggested balancing selection with low Fst estimates and negative α values (Table 2, Fig. 4). The three Fst outlier loci without significant associations with climate (C2270-contig1.NC1-384, C6522-contig1.NC1-269 and WS-2.0-GQ0024.B3.r-D12.1-239) showed relatively strong correlations with one or several environmental variables (ρ > 0.6); however, their BF values did not exceed our cut-off of 3.
Fig 4.
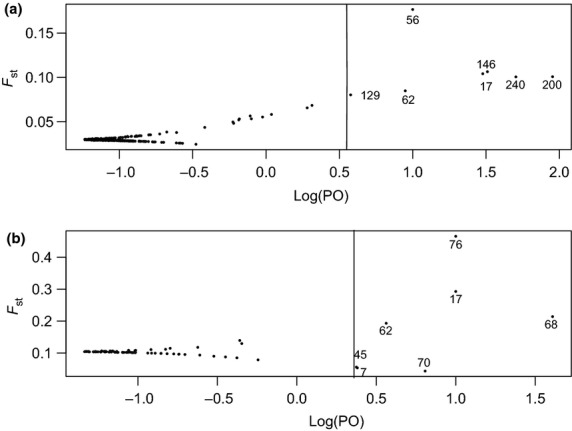
Results of Bayesian outlier detection analysis (a) within contact zone using 311 single nucleotide polymorphism (SNP) loci and (b) across hybrid zone, using 86 SNP loci. Estimate of Fst plotted against transformed P-values, where PO = p/(1-p). Loci (full circles) at the right of the vertical line showed significant deviations from neutrality.
The association analysis detected 11 potentially locally adaptive loci that were not detected by the Fst outlier test. These loci showed strong correlations with both mean annual temperature and precipitation as snow, variables strongly associated with elevational gradients that were previously identified as important in shaping adaptive variation, fitness and genetic structure within the hybrid zone (De La Torre et al. 2014).
Most of the SNPs identified as outliers or associated with environmental variables differed between analyses within the hybrid zone only and analyses that spanned both allopatric parental species and hybrid populations, probably reflecting different selection pressures acting along elevational and latitudinal gradients. Fst outliers may reflect divergent selection due to unknown environmental drivers that have not been identified and therefore were not detected by our allele–environment association approach. Adaptive loci were widely distributed in the genome in seven of the 12 linkage groups and pointed towards important functions in cold adaptation such as signal transduction, transcription regulation and carbohydrate metabolism (Table 3).
Discussion
Glacial and postglacial re-colonization patterns
Demographic processes during past glacial cycles have greatly influenced current species distributions and have played a role in the local adaptation and speciation of several North American species (Shafer et al. 2010). White spruce and Engelmann spruce are closely related species likely diverged in allopatry during the Pliocene (∼5 MYA), according to a recent Picea phylogeny based on nuclear, plastid and mitochondrial sequences (Lockwood et al. 2013). During the Pleistocene, the Laurentide and the Cordilleran ice sheets, which covered much of Canada and northwestern United States, led to the displacement of both white and Engelmann spruce south of their present distribution (Shafer et al. 2010). According to our palaeoclimatic modelling, these species most recently had the potential to come into secondary contact by 21 000 YBP, in the southern Rocky Mountains in Colorado and Wyoming, considerably south and east of the current hybrid zone. At 21 000 YBP, the ice sheets reached their largest extension and the white spruce and Engelmann spruce ranges were at their most restricted, suggesting the likelihood of one or several other episodes of secondary contact before 21 000 YBP (Fig. 2). The difference in timing of formation between the Laurentide Ice Sheet (27 000–30 000 YBP) and the Cordilleran Ice Sheet (19 000–22 000 YBP) also suggest the chance for secondary contact before 21 000 YBP in British Columbia (Dyke et al. 2002).
The climatic changes associated with the Last Glacial Maximum led to the displacement of plant and animal species into several ice age refugia (Shafer et al. 2010). White spruce appears to have survived the Last Glacial Maximum in two refugia, the unglaciated Yukon Valley, north from the ice front and the Appalachian Mountains Wisconsin refugium (Anderson et al. 2006). After the last glaciation, by 11 000 YBP, white spruce was distributed as far north as Alaska and had re-established its transcontinental range. While the white spruce range expanded, the Engelmann spruce range likely contracted. Engelmann spruce populations, which were more abundant and occupied lower elevations in the Rocky Mountains and the Great Basin during the Pleistocene, became fragmented when the temperature rose during the Xerothermic Period by 11 000 YBP (Ledig et al. 2006).
As a result of white spruce and Engelmann spruce range expansions and contractions, it appears likely that the hybrid zone moved west and then north, reaching British Columbia by 14 000 YBP following the recession of the Cordilleran and Laurentide Ice Sheets (Figs 2 and 3). This concurs with previous biogeographical reports (Pellatt et al. 2001; Ledig et al. 2006). After 14 000 YBP, the hybrid zone had available climatic habitat to expand its range in latitude and longitude until reaching its current northern and eastern expanse (Fig. 3).
The current phylogeographical structure of several other species in British Columbia and the Northwestern United States also appear to result from secondary contact between populations expanding from glacial refugia south or north of the Cordilleran ice sheet. For this reason, eastern British Columbia is considered a geographical ‘hotspot’ of hybrid zones, due to the number of plant and animal contact zones (Maroja et al. 2007; Irwin et al. 2009).
The climate niche modelling also had some limitations. Both the CCM1 and the GFDL model indicated climatic habitat for Engelmann spruce rather than white spruce in Alaska between 21 000 and 14 000 YBP. This inconsistency with previous genetic reports of a white spruce refugium (Anderson et al. 2006) may be due to fewer data points and lower resolution in the Alaska ecoregion in comparison with other ecoregions or due to white spruce ecotypes in Alaska that had a different climatic niche than those elsewhere in the species range (Aitken et al. 2008).
Genomic admixture
Extensive admixture and introgression was observed in the contact zone, with most alleles being shared by white spruce, Engelmann spruce and their hybrids. Despite this extensive introgression, pure species remained well differentiated from each other, supporting the view that white spruce and Engelmann spruce are recognizably distinct species. Our results point towards a genetic architecture shaped primarily by past introgression events, because little evidence for recent admixture has been found in the hybrid zone. The frequency distribution of hybrid classes supports the results of the palaeoclimatic modelling, indicating this is an ancient hybrid zone, in which advanced generation hybrid genotypes dominate and recombination is widespread. The lower levels of LD in hybrid populations than in parental species also suggest that hybridization has been ongoing for many generations, but we recognize this analysis, limited to 23 genes, lacks power. Nonetheless, recombination takes many generations to break up chromosomal blocks derived from each parental species; therefore, LD in hybrids is expected to decay faster with increasing number of backcrosses generations (Lexer et al. 2007). Reduced rates of current admixture between species may be a consequence of strong environmental selection causing locally adapted hybrids and parentals (De La Torre et al. 2014), or other early-evolving endogenous or exogenous isolating mechanisms (Andrew & Rieseberg 2013).
Results of the genomic analysis indicated a strong asymmetry in introgression towards Engelmann spruce, which may be explained by demographic processes during range expansion and contraction (Excoffier et al. 2009). If a species expands its range into that of a congener, and the two species hybridize, this would also produce a moving hybrid zone during the expansion (Buggs 2007). This suggests that introgression may have first occurred between Engelmann spruce as the local species and white spruce as the invading species, contributing to the asymmetrical patterns of introgression observed.
Functional roles of genes exhibiting outlier behaviour
Results of the Fst outlier and environmental association analyses identified a small percentage of candidate SNP loci that deviate from selective neutrality (6.4% of loci were significant in each analysis alone, with 1.9% common to both analyses). The small number of loci identified as putatively adaptive is typical of recent genome scans in plant and animal species (Strasburg et al. 2012). As we used all of our candidate loci to determine the population covariance matrix for Bayenv and the underlying neutral distribution of Fst, our results are likely somewhat more conservative than using independent, non-candidate or non-coding loci. However, the Fst outlier analysis may still result in false positives due to secondary contact and introgression between these species resulting in neutral population structure (Lotterhos & Whitlock 2014).
Loci suggesting diversifying selection that were both Fst outliers and associated with environmental variables were linked to signal transduction, transcription regulation and carbohydrate metabolism, all of which can be important gene functions in local adaptation to climate. Immunophilins (SNP 13_496) are intracellular receptors for the immunosupressants FK506 and rapamycin, which inhibit different signalling pathways required for T-cell activation (Luan et al. 1996). Studies in Arabidopsis and rice have suggested that FK506 homologs are encoded by a small gene family in higher plants. Leaf-specific immunophilins are regulated by light in fava beans, suggesting they may play a role in plants light signal transduction (Luan et al. 1996).
No apical meristem (Nam) genes (SNP 295_78) are part of the NAC domain proteins, which are plant-specific transcriptional factors known to play important role in plant developmental processes (Hu et al. 2010). It has been suggested that NAC genes may also play an important role in wood formation and secondary cell wall biosynthesis in Populus trichocarpa (Hu et al. 2010).
Carbohydrates play an important role in protecting cellular membranes from freezing injury by reducing osmotic potential across the membranes and by stabilizing them (Holliday et al. 2008). Triggered by the cessation of growth and the onset of dormancy, carbohydrate metabolism is modified towards the accumulation of storage compounds, cryoprotective or dehydration-protective solutes and reductive co-factors, increasing cold hardiness in Sitka spruce (Picea sitchensis) (Dauwe et al. 2012). In this study, we have found two loci with functions related to carbohydrate metabolism, SNP 45_1067 (alpha-amylase) and SNP 68_286 (glycosyl hydrolase). Sugar content and cold hardiness are positively correlated in several tree species as Scots pine (Pinus silvestris), lodgepole pine (Pinus contorta), Norway spruce (Picea abies) and red spruce (Picea rubens) (Ogren et al. 1997).
Environmental gradients and local adaptation
The Engelmann-white spruce hybrid zone is structured along elevational gradients, and many climatic and other environmental factors vary with elevation and with latitude in this topographically heterogeneous region. For example, trees at lower elevations experience hotter, drier summers and colder winters, while those at high elevations experience deeper winter snowpacks, shorter growing seasons and colder summers, with frost events not infrequent during the growing season (De La Torre et al. 2014). SNPs showing significant associations with environmental factors reflect the complexity of local adaptation to various environmental factors. Some loci are associated primarily with climatic variables reflecting summer temperatures and moisture deficits. For example, SNPs 208pg12875c, 295_78 and WS-2.0-GQ03105.B7-012.3_654 are associated with mean warmest month temperature, summer heat–moisture index or climate-moisture deficit or mean summer precipitation. Others SNPs (e.g. WS-2.0-GQ0041.BR_J07.2-36) are predominantly associated with precipitation-related variables such as mean annual precipitation and precipitation as snow. Still others (e.g. SNPs 13_496, 45_1067 and 206_435) are significantly associated with estimates of winter temperatures, with the mean coldest month temperature, continentality and degree-days below freezing as associated environmental variables. These associated SNPs are likely within or linked to genes that affect different traits involved in local adaptation to different environmental factors across these steep environmental gradients in southern and central British Columbia.
Maintenance of species boundaries in spruce
This study contributes to the long-standing debate on the maintenance of white spruce and Engelmann spruce species identities despite hybridization, by viewing the evolutionary relationships between these two species based on the genic view of speciation (Wu 2001). White and Engelmann spruce appear to have highly porous genomes, yet a small number of widely distributed genes under selection likely counteract the homogenizing effects of gene flow and prevent introgression in surrounding regions, maintaining species differences despite interspecific gene flow.
The maintenance of species integrity between hybridizing species has been reported for several plant and animal species, challenging the view that species differentiation is a genome-wide phenomenon. Sambatti et al. (2012) found that a large number of small divergent genomic regions between hybridizing species Helianthus annuus and H. petiolaris maintained species differences despite extensive interspecific gene flow. DeFaveri et al. (2013) found that selection acting on multiple loci widely and heterogeneously distributed across the genome has contributed to the divergence between stickleback populations despite high levels of gene flow. Even in more advanced stages of speciation, sympatric species may share parts of their genomes as a consequence of ongoing gene flow, as has been seen in Heliconius butterfly species (Martin et al. 2013).
When analysing allele frequency differentials between white spruce, Engelmann spruce and hybrids, we found that most alleles freely cross species barriers, whereas a small number of them are restricted to one parental species and their hybrids, with greater divergence between species. A subset of these loci apparently impermeable to introgression between species was identified in the outlier and the environmental association analyses, suggesting they are under selection or linked to loci under selection, and in relation to adaptive differences between white, Engelmann spruce and their hybrids. There are likely many more such loci across the genome as we studied a relatively small number of candidate loci. These putatively adaptive loci point to some important gene functions in adaptation to winter regimes.
Within this spruce hybrid complex, both parental species and their hybrids are locally adapted to different environments found along elevational gradients, in which key factors for survival are adaptation to the length of the growing seasons and the depth of the snowpack (De La Torre et al. 2014). The presence of locally adapted populations and apparently reduced rates of current interspecific gene flow suggest that hybrid populations in this complex may be in the early stages of ecological speciation, with hybrid populations undergoing the transition from local adaptation to incipient homoploid species (Andrew & Rieseberg 2013). Further studies of fine-scale linkage and the size and distribution of haplotype blocks from parental species using newly available genomic tools will help to elucidate this process.
Acknowledgments
We thank Barry Jaquish for granting access to the Ministry of Forests breeding materials; Jean Bousquet, Nathalie Pavy and Nathalie Isabel for providing linkage data; Loren Rieseberg for useful comments on previous manuscripts drafts; and Lisa Erdle and Christine Chourmouzis for help with sample collection. This work was supported by Genome British Columbia, the Forest Genetics Council of British Columbia, and the Province of British Columbia Ministry of Forests and Natural Resources Operations (funding to S.N.A.), by the Natural Science and Engineering Research Council of Canada (NSERC; Discovery grant to S.N.A.); and by UBC tuition fee awards, UBC University Graduate Fellowship (UGF), UBC Four Year Fellowship and B.C government Pacific Century Graduate Scholarship to A.R. De La Torre.
This study is part of the PhD thesis of A.R.D.L.T. on the genetic structure, admixture and local adaptation in the Picea glauca x P. engelmannii hybrid zone. A.R.D.L.T. is broadly interested in the genetics and genomics of tree species. D.R. is a postdoctoral scholar interested in evaluating species distributions under different environments through modelling. S.N.A. is a forest geneticist broadly interested in quantitative genetics, population genomics, local adaptation to climate and climate change outcomes for tree populations.
Data accessibility
SNP and climate data are available at Dryad doi:10.5061/dryad.7h65f.
Supporting Information
Additional supporting information may be found in the online version of this article.
Annotation and location in the P.glauca genome of 311 candidate gene SNPs used in this study.
Validation statistics for P. engelmannii and P. glauca model projections for each time period within each GCM.
Estimates of pairwise linkage disequilibrium between informative nucleotide sites across 23 genes in 132 individuals in the Picea glauca x P. engelmannii hybrid zone.
Results of the environmental association analysis using Bayenv (a) within the hybrid zone using 311 SNPs; and (b) across the hybrid zone using 86 SNPs.
Glacial and postglacial re-colonization patterns of Picea glauca, P.engelmannii and their hybrids from 21 000 years ago to the present day, based on climate niche modeling (GFDL model) and palaeoclimate data.
Histogram of genotypic classes based on Introgress hybrid index, showing a higher percentage of advanced generation hybrids in the hybrid zone.
Analysis of population structure in the Picea glauca x P. engelmannii hybrid zone using SNP markers.
Plot of the first two principal components (PC1 and PC2) of a principal components analysis for 86 SNP loci in the Picea glauca x P. engelmanniii hybrid zone.
References
- Abbott R, Albach D, Ansell S, et al. Hybridization and speciation. Journal of Evolutionary Biology. 2013;26:229–246. doi: 10.1111/j.1420-9101.2012.02599.x. [DOI] [PubMed] [Google Scholar]
- Aitken SN, Yeaman S, Holliday JA, Wang T, Curtis-McLane S. Adaptation, migration or extirpation: climate change outcomes for tree populations. Evolutionary Applications. 2008;1:95–111. doi: 10.1111/j.1752-4571.2007.00013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LL, Feng Sheng H, Nelson DM, Petit RJ, Paige KN. Ice-age endurance: DNA evidence of a white spruce refugium in Alaska. PNAS. 2006;103:12447–12450. doi: 10.1073/pnas.0605310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew RL, Rieseberg LH. Divergence is focused on few genomic regions early in speciation: incipient speciation of sunflower ecotypes. Evolution. 2013;67–9:2468–2482. doi: 10.1111/evo.12106. [DOI] [PubMed] [Google Scholar]
- Araujo MB, Guisan A. Five (or so) challenges for species distribution modelling. Journal of Biogeography. 2006;33:1677–1688. [Google Scholar]
- Arnold ML. Natural Hybridization and Evolution. Oxford: Oxford University Press; 1997. [Google Scholar]
- Barton NH, Hewitt GM. Analysis of hybrid zones. Annals of Review of Ecology and Systematic. 1985;16:113–148. [Google Scholar]
- Buggs RJA. Empirical study of hybrid zone movement. Heredity. 2007;99:301–312. doi: 10.1038/sj.hdy.6800997. [DOI] [PubMed] [Google Scholar]
- Bush ABG, Philander SGH. The climate of the last glacial maximum: results from a coupled atmosphere-ocean general circulation model. Journal of Geophysical Research-Atmospheres. 1999;104:24509–24525. [Google Scholar]
- Chapman MA, Hiscock SJ, Filatov DA. Genomic divergence during speciation driven by adaptation to altitude. Molecular Biology and Evolution. 2013;30:2553–2567. doi: 10.1093/molbev/mst168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Durand E, Forbes F, Francois O. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Molecular Ecology Notes. 2007;7:747–756. [Google Scholar]
- Coop G, Witonsky D, Di Rienzo A, Pritchard JK. Using environmental correlations to identify loci underlying local adaptation. Genetics. 2010;185:1411–1423. doi: 10.1534/genetics.110.114819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauwe R, Holliday JA, Aitken SN, Mansfield SD. Metabolic dynamics during autumn cold acclimation within and among populations of Sitka spruce (Picea sitchensis) New Phytologist. 2012;194:192–205. doi: 10.1111/j.1469-8137.2011.04027.x. [DOI] [PubMed] [Google Scholar]
- De La Torre AR, Wang T, Jaquish B, Aitken SN. Adaptation and exogenous selection in a Picea glauca x Picea engelmannii hybrid zone: implications for forest management under climate change. New Phytologist. 2014;201:687–699. doi: 10.1111/nph.12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFaveri J, Jonsson PR, Merila J. Heterogeneous genomic differentiation in marine threespine sticklebacks: adaptation along an environmental gradient. Evolution. 2013;67–9:2530–2546. doi: 10.1111/evo.12097. [DOI] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1987;12:13–15. [Google Scholar]
- Durand E, Jay F, Gaggiotti OE, Francois O. Spatial inference of admixture proportions and secondary contact zones. Molecular Biology and Evolution. 2009;26:1963–1973. doi: 10.1093/molbev/msp106. [DOI] [PubMed] [Google Scholar]
- Dyke AS, Andrews JT, Clark PU, et al. The laurentide and Innuitian ice sheets during the last glacial maximum. Quaternary Science Reviews. 2002;21:9–31. [Google Scholar]
- Earl DA, VonHoldt BM. Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetic Resources. 2011;4:359–361. [Google Scholar]
- Eckert AJ, Bower AD, Gonzalez-Martinez SC, Wegrzyn JL, Neale DB. Back to nature: ecological genomics of loblolly pine (Pinus taeda Pinaceae) Molecular Ecology. 2010;19:3789–3805. doi: 10.1111/j.1365-294X.2010.04698.x. [DOI] [PubMed] [Google Scholar]
- Elith J, Leathwick JR. Species distribution models: ecological explanation and prediction across space and time. Annual Review of Ecology, Evolution and Systematics. 2009;40:677–697. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the program STRUCTURE: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Foll M, Petit R. Genetic consequences of range expansions. Annual Review of Ecology and Evolution. 2009;40:481–501. [Google Scholar]
- Feder JL, Flaxman SM, Egan SP, Comeault AA, Nosil P. Geographic mode of speciation and genomic divergence. Annual Review of Ecology, Evolution, and Systematics. 2013;44:4.1–4.25. [Google Scholar]
- Foll M, Gaggiotti OE. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180:977–993. doi: 10.1534/genetics.108.092221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompert Z, Buerkle AC. Introgress: a software package for mapping components of isolation in hybrids. Molecular Ecology Resources. 2010;10:378–384. doi: 10.1111/j.1755-0998.2009.02733.x. [DOI] [PubMed] [Google Scholar]
- Haselhorst M, Buerkle CA. Population genetic structure of Picea engelmannii P. glauca and their previously unrecognized hybrids in the central Rocky Mountains. Tree Genetics and Genomes. 2013;9:669–681. [Google Scholar]
- Holliday JA, Ralph SG, White R, Bohlmann J, Aitken SN. Global monitoring of autumn gene expression within and among phenotypically divergent populations of Sitka spruce (Picea sitchensis) New Phytologist. 2008;178:103–122. doi: 10.1111/j.1469-8137.2007.02346.x. [DOI] [PubMed] [Google Scholar]
- Hu R, Qi G, Kong Y, et al. Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biology. 2010;10:145. doi: 10.1186/1471-2229-10-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DE, Brelsford A, Toews DPL, MacDonald C, Phinney M. Extensive hybridization in a contact zone between MacGillivray's warblers Oporornis tolmiei and mourning warblers O. philadelphia detected using molecular and morphological analyses. Journal of Avian Biology. 2009;40:539–552. [Google Scholar]
- Keller S, Levsen N, Olson MS, Tiffin P. Local adaptation in the flowering-time gene network of balsam poplar, Populus balsamifera L. Molecular Biology and Evolution. 2012;29:3143–3152. doi: 10.1093/molbev/mss121. [DOI] [PubMed] [Google Scholar]
- Kutzbach J, Gallimore R, Harrison S, et al. Climate and biome simulations for the past 21 000 years. Quaternary Science Reviews. 1998;17:473–506. [Google Scholar]
- Ledig FT, Hodgkiss PD, Johnson DR. The structure of genetic diversity in Engelmann spruce and a comparison with blue spruce. Canadian Journal of Botany. 2006;84:1806–1828. [Google Scholar]
- Lexer C, Buerkle CA, Joseph JA, Heinze B, Fay MF. Admixture in European populus hybrid zones makes feasible the mapping of loci that contribute to reproductive isolation and trait differences. Heredity. 2007;98:74–84. doi: 10.1038/sj.hdy.6800898. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Lockwood JD, Aleksic JM, Zou J, Wang J, Liu J, Renner SS. A new phylogeny for the genus Picea from plastid, mitochondrial, and nuclear sequences. Molecular Phylogenetics and Evolution. 2013;69:717–727. doi: 10.1016/j.ympev.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Lotterhos K, Whitlock M. Evaluation of demographic history and neutral parameterization on the performance of Fst outlier tests. Molecular Ecology. 2014 doi: 10.1111/mec.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan S, Kudla J, Gruissem W, Schreiber SL. Molecular characterization of a FKBP-type immunophilin from higher plants. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6964–6969. doi: 10.1073/pnas.93.14.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroja LS, Bogdanowicz SM, Wallin KF, Raffa KF, Harrison RG. Phylogeography of spruce beetles (Dendroctonus rufipennis Kirby) (Curculionidae Scolytinae) in North America. Molecular Ecology. 2007;16:2560–2573. doi: 10.1111/j.1365-294X.2007.03320.x. [DOI] [PubMed] [Google Scholar]
- Martin SH, Dasmahapatra KK, Nadeau NJ, et al. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Research. 2013;23:1817–1828. doi: 10.1101/gr.159426.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore WS. An evaluation of narrow hybrid zones in vertebrates. Quarterly Review of Biology. 1977;52:263–277. [Google Scholar]
- Namroud M-C, Beaulieu J, Juge N, Laroche J, Bousquet J. Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Molecular Ecology. 2008;17:3599–3613. doi: 10.1111/j.1365-294X.2008.03840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogren E, Nilsson T, Sundblad LG. Relationship between respiratory depletion of sugars and loss of cold hardiness in coniferous seedlings over-wintering at raised temperatures: Indications of different sensitivities of spruce and pine. Plant Cell and Environment. 1997;20:247–253. [Google Scholar]
- Pavy N, Pelgas B, Laroche J, Rigault P, Isabel N, Bousquet J. A spruce gene map infers ancient plant genome reshuffling and subsequent slow evolution in the gymnosperm lineage leading to extant conifers. BMC Genomics. 2012;10:84. doi: 10.1186/1741-7007-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. GENALEX 6: Genetic Analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes. 2006;6:288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellatt M, Hebda R, Mathewes R. High-resolution holocene vegetation history and climate from hole 1034b, odp leg 169s, Saanich Inlet, Canada. Marine Geology. 2001;174:211–226. [Google Scholar]
- Porth I, Hamberger B, White R, Ritland K. Defense mechanisms against herbivory in Picea: sequence evolution and expression regulation of gene family members in the phenylpropanoid pathway. BMC Genomics. 2011;12:608. doi: 10.1186/1471-2164-12-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotyped data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2013. Available at: http://www.R-project.org. [Google Scholar]
- Rajora O, Dancik B. Population genetic variation, structure, and evolution in Engelmann spruce, white spruce, and their natural hybrid complex in Alberta. Canadian Journal of Botany. 2000;78:768–780. [Google Scholar]
- Roberts DR, Hamann A. Method selection for species distribution modeling: are temporally or spatially independent evaluations necessary? Ecography. 2012;35:792–802. [Google Scholar]
- Sambatti JBM, Strasburg JL, Ortiz-Barrientos D, Baack EJ, Rieseberg LH. Reconciling extremely strong barriers with high levels of gene exchange in annual sunflowers. Evolution. 2012;66:1459–1473. doi: 10.1111/j.1558-5646.2011.01537.x. [DOI] [PubMed] [Google Scholar]
- Schoville SD, Bonin A, Francois O, Lobreaux S, Melodelima C, Manel S. Adaptive genetic variation on the landscape: methods and cases. Annual Review of Ecology, Evolution and Systematics. 2012;43:23–43. [Google Scholar]
- Shafer ABA, Cullingham CI, Cote SD, Coltman DV. Of glaciers and refugia: a decade of study sheds new light on the phylogeography of northwestern North America. Molecular Ecology. 2010;19:4589–4621. doi: 10.1111/j.1365-294X.2010.04828.x. [DOI] [PubMed] [Google Scholar]
- Strasburg JL, Sherman NA, Wright KM, Moyle LC, Willis JH, Rieseberg LH. What can patterns of differentiation across plant genomes tell us about adaptation and speciation? Philosophical Transaction of the Royal Society B. 2012;367:364–373. doi: 10.1098/rstb.2011.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Hamann A, Spittlehouse DL, Murdock T. ClimateWNA - high-resolution spatial climate data for western North America. Journal of Applied Meteorology and Climatology. 2012;51:16–29. [Google Scholar]
- Wu CI. The genic view of the process of speciation. Journal of Evolutionary Biology. 2001;14:851–865. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Annotation and location in the P.glauca genome of 311 candidate gene SNPs used in this study.
Validation statistics for P. engelmannii and P. glauca model projections for each time period within each GCM.
Estimates of pairwise linkage disequilibrium between informative nucleotide sites across 23 genes in 132 individuals in the Picea glauca x P. engelmannii hybrid zone.
Results of the environmental association analysis using Bayenv (a) within the hybrid zone using 311 SNPs; and (b) across the hybrid zone using 86 SNPs.
Glacial and postglacial re-colonization patterns of Picea glauca, P.engelmannii and their hybrids from 21 000 years ago to the present day, based on climate niche modeling (GFDL model) and palaeoclimate data.
Histogram of genotypic classes based on Introgress hybrid index, showing a higher percentage of advanced generation hybrids in the hybrid zone.
Analysis of population structure in the Picea glauca x P. engelmannii hybrid zone using SNP markers.
Plot of the first two principal components (PC1 and PC2) of a principal components analysis for 86 SNP loci in the Picea glauca x P. engelmanniii hybrid zone.
Data Availability Statement
SNP and climate data are available at Dryad doi:10.5061/dryad.7h65f.