

Abstract
Changes in the expression and function of caveolin-1 (Cav-1) have been proposed as a pathogenic mechanism underlying many cardiovascular diseases. Cav-1 binds to and regulates the activity of numerous signaling proteins via interactions with its scaffolding domain. In endothelial cells, Cav-1 has been shown to reduce reactive oxygen species (ROS) production, but whether Cav-1 regulates the activity of NADPH oxidases (Nox), a major source of cellular ROS, has not yet been shown. Herein, we show that Cav-1 is primarily expressed in the endothelium and adventitia of pulmonary arteries (PA) and that Cav-1 expression is reduced in isolated PA from multiple models of pulmonary artery hypertension (PH). Reduced Cav-1 expression correlates with increased ROS production in the adventitia of hypertensive PA. In vitro experiments revealed a significant ability of Cav-1 and its scaffolding domain to inhibit Nox1-5 activity and it was also found that Cav-1 binds to Nox5 and Nox2 but not Nox4. In additional to post-translational actions, in primary cells, Cav-1 represses the mRNA and protein expression of Nox2 and Nox4 though inhibition of the NF-kB pathway. Lastly, in a mouse hypoxia model, the genetic ablation of Cav-1 increased the expression of Nox2 and Nox4 and exacerbated PH. Together, these results suggest that Cav-1 is a negative regulator of Nox function via two distinct mechanisms, acutely through direct binding and chronically through alteration of expression levels. Accordingly, the loss of Cav-1 expression in cardiovascular diseases such as PH may account for the increased Nox activity and greater production of ROS.
Keywords: Caveolin-1, NADPH oxidase, Reactive Oxygen Species, Pulmonary artery hypertension
Introduction
Caveolae or “little caves” are small flask-shaped invaginations of the plasma membrane that range in size from 50–100nm. In blood vessels, caveolae are present in the majority of cell types, including endothelial, fibroblast, macrophages and smooth muscle cells[1]. With distinct lipid and protein environments, caveolae are thought to be functionally important to cellular signaling, endocytosis, transcytosis, and the regulation of cell differentiation, proliferation and apoptosis [2]. There are 3 caveolin genes, Cav1-3, which encode transmembrane proteins of ~21KD-24kDa. Cav-1 is the primary coat protein of caveolae and loss of Cav-1 in genetic knockouts results in loss of the organelle [3]. Cav-1 is primarily expressed in endothelial cells and fibroblasts and has been shown to restrain cellular proliferation and migration, provide resistance to apoptosis and growth factor signaling [4]. Cav-2 is also present in caveolae and depends on Cav-1 for stability/expression [5]. Cav-3 is a muscle-specific gene primarily expressed in cardiac and skeletal myocytes [6].
Cav-1 can be functionally divided into three domains: N-terminal (101aa), transmembrane (33aa), and C-terminal (44aa) [4]. A well-defined region (82–101aa) has been termed the caveolin-scaffolding doman (CSD) and has been shown to bind a wide range of signaling molecules including G-protein subunits, receptor and non-receptor tyrosine kinases, endothelial nitric oxide synthase (eNOS), integrins, PKCs, growth factor receptors and small GTPases[4]. The binding of Cav-1 to a target proteins is usually inhibitory [4]. This has been well described for a number of proteins and the ability of the full length Cav-1 protein and the caveolin-1-scaffolding domain peptide to inhibit eNOS activity and NO production in endothelial cells, and impair endothelium-dependent relaxation in isolated blood vessels has been the subject of intense investigation[7].
Pulmonary Arterial Hypertension (PH) is a progressive, debilitating disease resulting from increased pulmonary vascular resistance. PH is characterized by excessive vascular cell proliferation, inward remodeling, vasoobliteration and a loss of compliance of the pulmonary blood vessels [8, 9]. It is well established that PH is associated with the overproduction of reactive oxygen species (ROS) and that ROS can contribute to the vascular dysfunction and stiffening observed in PH [10–14]. However, the mechanistic links underlying the increased production of ROS in PH remains poorly defined. The NADPH oxidases (Nox) are a major source of reactive oxygen species (ROS) and oxidative stress. There are 5 Nox genes (Nox1-Nox5), and vascular cells express Nox1, Nox2, Nox4 and Nox5 (Nox5 gene is not present in rodents genomes). Nox1 and 2 are activated by the protein-protein interaction of cytosolic subunits, including p47phox (NCF1), p67phox (NCF2), NOXA1 and NOXO1[15]. In contrast, Nox4 is thought to be constitutively active [11]. Increased expression of Nox2 and Nox4 has been reported in both mouse models of PH and human PH [16–18]. In mice, genetic deletion or pharmacological inhibition of Nox2 has been shown to reverse hypoxia-initiated PH [19, 20]. Another report by Green et al found that Nox4 inhibitor, GKT137831, attenuates PH in mice [21]. Cav-1 expression is decreased in both animal models of PH and human PH [22] and is thought to contribute to the pathogenesis. Indeed, the loss of Cav-1 has been shown to increase ROS levels in the vasculature and can promote cardiovascular diseases and cancer [23]. However, the ability of Cav-1 to regulate Nox enzymes is not yet known.
In the present study, we found that Cav-1 is primarily expressed in the endothelium and adventitia of pulmonary arteries (PA) and that the expression of Cav-1 is reduced in pulmonary arteries in multiple rat models of PH, including the monocrotaline (MCT), SUGEN/hypoxia (SU/HYP) and the Fawn Hooded Rat (FHR). We investigated the mechanisms by which Cav-1 regulates Nox activity using an integrated approach of heterologous expression systems, native cells and in vivo models. Collectively, our data support the concept that Cav-1 is a negative regulator of the activity and expression of Nox proteins and that loss of Cav-1 is an important regulatory pathway in PH.
Materials and Methods
Animal models of PH
Three rat models of pulmonary hypertension (PH) were employed. The monocrotaline (MCT)-model was induced by a single i.p. injection of MCT (60/mg/kg), which elicits a progressive and severe PH [24]. The SUGEN/Hypoxia (SU/HYP) model of PH results from injection of the VEGF receptor antagonist SU-5416 (20mg/kg, SQ) followed by 3 weeks of hypoxia (10% O2) and 10–11 weeks of normoxia (21% O2) as previously described[25]. The Fawn-hooded rat (FHR), a genetic model of PH spontaneously develops PH after 20 weeks of age[26]. Adult age-matched male Sprague-Dawley (SDR, 250–300g) rats were used as controls for all rat models of PH. One mouse model of PH was employed with WT and Cav-1−/− mice exposed to 10% oxygen (hypoxia) or room air (normoxia) in a clear plastic polypropylene chamber (20 × 20 × 30 inch) for 3 weeks. The chamber has ventilation holes and a small, quiet fan to provide forced circulation and instant homogenization of gases. The Animal Care and Use Committee at Georgia Regents University approved all procedures and protocols, and this study conformed to the Guide for the Care and Use of Laboratory Animals as published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). All animals were housed under temperature-controlled conditions (21–23°C), maintained on standard chow, allowed free access to food and water and exposed to a 12:12-h light-dark cycle. Post hemodynamic measurements, animals were euthanized by thoracotomy. Blood in the pulmonary vasculature was removed by PBS infusion through the pulmonary artery and the heart and lungs removed en bloc. The free wall of the right ventricle (RV), left ventricle (LV), and septum (S) were carefully dissected free and weighed individually to calculate the RV/LV+S ratio (Fulton index) as an index of RV hypertrophy.
Cell culture and Reagents
COS-7, human lung fibroblasts and bone marrow macrophages were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing L-glutamine, penicillin, streptomycin, and 10% (v/v) fetal bovine serum. Cells were transfected using Lipofectamine 2000 reagent (Invitrogen) as described previously [27–31]. Human lung microvascular endothelial cells (HLMVEC) were purchased from Lonza, and were grown in Endothelial Growth Medium-2-Microvessel (EGM-2MV) consisting of defined growth factors and supplemented with additional FBS up to 5% final concentration (Lonza). Cells were grown at 37 °C in 5% CO2 incubator and used from passage 2–6. Peritoneal macrophages from WT and Cav-1−/− mice were isolated as described [29]. In brief, 1 ml of thioglycolate was injected into each mouse 3–5 days prior to collection, peritoneal cells were collected by lavage, RBC lysed with hypotonic buffer and isolated macrophages cultured in complete DMEM. The caveolin-1 scaffolding domain peptide was purchased from Enzo life sciences (Farmingdale, NY). All chemicals were purchased from Sigma unless indicated otherwise.
DNA constructs
Plasmid DNA encoding Nox5β (AF325189), Nox1, Nox3 and Nox4 have been described previously [27, 31]. The full length human Cav-1α construct was obtained from OriGene (Rockville, MD). Cav-1Δ was generated by deleting bp 244–303 using the following primers F: 5′-cacagttttttgctgtctgccctctttggc-3′ and R: 5′-gacagcaaaaaactgtgtgtcccttctgg-3′.
Oxidized DNA ELISA
Quantitation of the levels of ROS-modified deoxyguansine residues (8-OHdG) was achieved by ELISA (Cell Biolabs) according to manufacturer’s instructions. In brief, genomic DNA was isolated (Qiagen) from control or MCT-treated rat (4wk) lungs. DNA was converted to nucleosides and centrifuged at 6000g for 5 minutes and the supernatant used to quantify 8-OHdG levels.
RT-PCR and real time PCR
cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad) and used to assess relative gene expression using real time RT-PCR (Bio-Rad iQ SYBR Green) using the following primes. Rat Nox2: CCCTTTGGTACAGCCAGTGAAG-AT (forward), CAATCCCAGCTCCCACTAACATCA (reverse). Rat Nox4: CTGCATCTGTCCTGAACCTCAA (forward), TCTCCTGCTAGGGACCTTCTGT (reverse). Rat GAPDH: GACATGCCGCCTGGAGAAAC (forward), AGCCCAGGATGCCCTTTAGT (reverse). Mouse Cav-1: TCTACAAGCCCAACAACAAGG (forward), AGGAAG GAGAGAATGGCAAAG (reverse). Mouse Nox2: GCTGGGATCACAGGAATTGT (forward), GGTGATGACCACCTTTTGCT (reverse). Mouse Nox4: TGTTGCATGTTTCAGGTGGT (forward), TGGAACTTGGGTTCTTCCAG (reverse). Mouse GAPDH: AGGTCATCCCAGAGCTGAACG (forward), CACCCTGTTGCTGTAGCCGTAT (reverse). Human Cav-1: TTCGCCATTCTCTCTTTCCT (forward), CAGCTTCAAAGAGTGGGTCA (reverse). Human Nox1: AAGCCGACAGGCCACAGAT (forward), GTCACATACTCCACTGTCGTGTTTC (reverse). Human Nox2: GCAGCCTGCCTGAATTTCA (forward), TGAGCAGCACGCACTGGA (reverse). Human Nox4: CTTCCGTTGGTTTGCAGATT (forward), TGGGTCCACAACAGA AAACA (reverse). Human GAPDH: AGAAGGCTGGGGCTCATTTG (forward), AGGGGCCATCCACAGTCTTC (reverse).
Co-immunoprecipitation and Western blotting analysis
Cells were lysed on ice in 20mM Tris-HCl (pH 7.4), 1% Triton X-100, 100mM NaCl, 1mM Na3VO4, 10mM NaF, and 1% protease inhibitor cocktail (Sigma). Soluble extracts were incubated for 2 h at 4°C with relevant antibodies: anti-HA (Roche Applied Science) and a negative isotype control mouse immunoglobulin (IgG) (Santa Cruz Biotechnology), anti-Cav-1 (Cell signaling), anti-Nox2 (BD), anti-Nox4 (Epitomics) or complexes precipitated with protein A/G agarose (Santa Cruz Biotechnology). Western blotting was performed as described previously [32–36] using anti-HA (Roche), anti-Cav-1 (Cell signaling), anti-Cav-2 (Thermo Scientific), anti-Cavin (Santa Cruz Biotechnology), anti-Nox2 (BD, sigma, abcam), anti-Nox4 (Epitomics), and anti-GAPDH antibodies (Santa Cruz Biotechnology).
Proximity ligation assay for co-localization
An in situ Proximity Ligation Assay (PLA, Duolink, Sigma) was performed to assess whether endogenous Cav-1 (Cell signaling) and Nox2 (BD) co-localize in mouse macrophages. PLA reactions were performed according to the manufacturer’s instructions using primary antibodies (anti-Cav-1, 1:100 and anti-Nox2, 1:100), followed by a pair of oligonucleotide-labeled secondary antibodies. In this assay, the PLA probes create a positive signal only when the epitopes of the target proteins are in close proximity (<40 nm). The signal from each of the detected pair of PLA probes was then counted using fluorescence microscopy (excitation: BP545/25, emission: BP605/70). For negative controls, one primary antibody or both primary antibodies were omitted.
Confocal microscopy
To determine the location and expression of specific cellular markers in blood vessels, both normotensive and/or PH lung sections were stained with α-actin (Abcam; 1:700 dilution) for 30 minutes before being double-stained with antibodies against either Cav-1 (cell signaling; 1:500 dilution), FAP (Santa Cruz; 1:500 dilution), Nox5[34] and the marker of ROS production, 8-hydroxydeoxyguanosine, (Thermo Scientific; 1:200 dilution) for 30 minutes. Immunofluorescence-labeled lung sections were examined using a Zeiss LSM 510 laser scanning confocal microscope.
Genetic silencing of Cav-1
The siRNA targeting Cav-1 (siRNA ID: s2446) was obtained from Applied Biosystems. Validated control and targeting siRNA were transfected into cells using siPORT™ Amine (Applied Biosystems) as described [29].
Measurement of reactive oxygen species
NADPH oxidase-derived superoxide and hydrogen peroxide production were measured by L-012 and Amplex red assay as described previously and net increases in ROS were determined by subtracting levels from cells not expressing Nox genes [27–29, 31, 34, 37–39].
Statistical Analysis
Data were reported as mean ± SE and statistical analyses were performed using Instat software (GraphPad Software Inc., San Diego, CA). Comparisons between two groups were made with an unpaired student’s t-test or for multiple comparisons an ANOVA with a Bonferroni post hoc test. Differences were considered as significant at p < 0.05.
Results
PH is associated with reduced vascular expression of Cav-1 and dependent proteins and increased ROS production
To determine the cellular location of Cav-1 expression in pulmonary arteries, we performed immunofluorescence staining of lung sections from vehicle and MCT-treated rats. In control rats, Cav-1 was expressed in both endothelial cells and in the adventitia. In MCT-treated rats with PH, the staining for Cav-1 was significantly reduced (Fig. 1A). To confirm these findings using more quantitative methods, we next assessed Cav-1 mRNA and protein levels using real time PCR and Western blotting. In all three rat models of PH (FHR, MCT, and SU/HYP), we found reduced protein levels of Cav-1 in isolated PA (Fig. 1B). In the MCT model, we found reduced levels of Cav-1 protein and mRNA, but also other genes dependent on Cav-1 expression, Cav-2, Cavin-1 and PV-1 (Fig. 1C and Supplemental Fig. 1). We next assessed the levels of ROS production in hypertensive PA using immunofluorescence imaging for 8-hydroxydeoxyguanosine, a DNA nucleoside that is generated by ROS and used as an in vivo footprint of ROS production. We found 8-hydroxydeoxyguanosine staining was robustly increased in PA from 4-week MCT-treated rats (Fig. 2A–B). The highest signal was observed in the adventitia, which overlapped with the fibroblast marker, fibroblast activation protein (FAP). We further found that Nox2 and Nox4 mRNA and protein levels were significantly increased in isolated PA (Fig. 2C–D), suggesting that the elevated levels of ROS in hypertensive PA may derive from increased expression and or activity of Nox proteins.
Figure 1. Pulmonary hypertension reduces Cav-1 and caveolar protein expression.

(A) Relative distribution of Cav-1 and α-SMA expression in control and hypertensive pulmonary arteries (PA). Immunofluorescence images of sections of lungs from vehicle, 4-wk MCT (60mg/kg, IP) using antibodies recognizing rat Cav-1 and α-actin. (B) Loss of Cav-1 and other caveolar protein expression in isolated PA from FHR, SUGEN/hypoxia (SU/Hyp) and MCT- induced hypertensive PA (n=3). (C) mRNA expression levels of Cav-1, Cav-2, Cavin-1 and PV-1 in PA from MCT rats were measured by qRT-PCR (ΔΔCt) normalized to GAPDH. * different from Vehicle, p < 0.05 (n = 5–6).
Figure 2. PH is associated with increased vascular ROS production and Nox2 and Nox4 expression.

(A) Localization of in vivo reactive oxygen species (ROS) in pulmonary arterioles. Sections of control and 4wk MCT-treated rat lungs were co-stained for fibroblast activation protein (FAP), 8 Hydroxy-2′dexoyguanosine (ROS marker) and DAPI. (B) ROS production was quantified by an Oxidative DNA Damage ELISA kit using genomic DNA isolated from control and 4wk MCT-treated rat lungs. Nox2 and Nox4 protein (C) and mRNA (D) expression were measured in isolated PA from MCT rats. Results are representative of at least 3–5 separate experiments. * different from Vehicle, p < 0.05 (n = 5–6).
Cav-1 negatively regulates Nox-derived ROS in vitro
The inverse correlation observed between the elevated ROS and elevated Nox levels versus the reduced expression of Cav-1 in PA from rats with PH promoted us to investigate whether Cav-1 can directly influence the ability of NADPH oxidases to produce ROS. We found that increased expression of Cav-1 decreased ROS from Nox1, 2, 3, 4, and 5 (Fig. 3 A–E). Fusion of cell penetrating peptide sequences (antennapedia) to the Cav-1 scaffolding domain (aa82–101 has been shown to mimic the intracellular actions of Cav-1 in suppressing the activity of protein binding partners of Cav-1 that contain a Cav-1 binding motif[40, 41]. We observed that the Cav-1 scaffolding peptide reduced ROS production from Nox1, 2, 3 and 5. However, the Cav-1 peptide failed to inhibit Nox4-dependent ROS production (Fig. 3F–J). In contrast, reducing the expression of endogenous Cav-1 using siRNA in human lung fibroblasts or in Cav-1 knockout macrophages led to the increased ROS production (Fig. 3K–L). Cholesterol depletion (Supplemental Fig. 2A) using methyl-β-cyclodextrin (CD) increased, whereas cholesterol loading decreased Nox2-dependent superoxide in mouse macrophages. The specificity of L-012 and Amplex Red for the Nox-dependent production of superoxide and hydrogen peroxide is shown in Supplementary Fig. 2B–E.
Figure 3. Cav-1 negatively regulates ROS production from NADPH oxidases in both heterologous expression systems and native cells.

(A–E) COS-7 cells were co-transfected with cDNAs encoding Nox1, 3, 4, 5 and either Cav-1 or a non-specific control gene (RFP), or peritoneal macrophages (source of Nox2) were transfected with either Cav-1 or RFP. Superoxide production was monitored by L-012-mediated chemiluminescence and hydrogen peroxide (H2O2) was measured using the Amplex® Red assay with excitation of 530–560 nm and emission detection at ~590 nm. (F–J) COS-7 cells or peritoneal macrophages expressing Nox1-Nox5 were treated with vehicle or the Cav-1 scaffolding domain peptide for 24 hrs and ROS production measured as described. (K) Knockdown of Cav-1 using siRNA (10, 30nM) in human lung fibroblasts increases ROS production. (L) Macrophages isolated from Cav-1−/− mice produce significantly higher amount of ROS compared to that of WT mice. (M) Cav-1 directly regulates ROS production from Nox5. COS-7 cells were co-transfected with cDNAs encoding Nox5 and either Cav-1 or a non-specific control gene (RFP). The activity of Nox5 in cell-free extracts was determined following injection of NADPH (arrow). * different from Control, WT or Vehicle, p < 0.05 (n = 5–6).
To determine whether Cav-1 can directly regulate Nox activity versus effects on other signaling moieties, we measured Nox5 activity in an isolated enzyme activity with supplemental cofactors with and without co-expression of Cav-1[27, 42]. Nox5 was used because it is a prototypical Nox enzyme that does not require the assembly of other protein subunits for activation[43]. Partially purified Nox5 was incubated with maximal cofactors (FAD, calcium) and NADPH was given (arrow) to initiate superoxide production. We found that in addition to its inhibitory actions in intact cells, Cav-1 significantly decreased Nox5-dependent superoxide in an isolated enzyme activity assay (Fig. 3M). Co-expression of Cav-1Δ, which lacks the scaffolding domain, was unable to modify Nox5 activity at the multiple concentrations used (Supplemental Fig. 3A). Against eNOS, Cav-1 but not the deletion mutant was able to reduce NO production (Supplemental Fig. 3B). Furthermore, the increased expression of Cav-1 did not noticeably alter the location of Nox5 (Supplemental Fig. 4A). Collectively these results support the ability of the scaffolding domain of Cav-1 to regulate the post-translational activity of Nox enzymes.
Nox5 and Nox2, but not Nox4, are Cav-1 binding proteins
In a heterologous expression system in COS-7 cells, Cav-1 dose-dependently inhibited superoxide production from cells expressing Nox5 without modifying the level of Nox5 expression (Fig. 4A). Similarly, the Cav-1 scaffolding domain peptide suppressed superoxide (Fig. 3J) without altering Nox5 expression (Fig. 4B). This data suggests a post-translational mechanism of Nox inhibition and we next determined whether Cav-1 can function as an allosteric regulator and bind to Nox5. Using a co-IP approach, we found evidence for a strong physical association between Nox5 and Cav-1 (Fig. 4C–D). Using a similar approach we found that Nox2 also binds to Cav-1 (Fig. 4E). Previous studies have shown that calcium-influx promotes the dissociation of Cav-1 from certain binding partners including eNOS. Therefore we investigated whether increasing intracellular calcium alters the degree of binding between Nox2 and Cav-1. We found that the amount of Nox2 bound to Cav-1 was decreased in cells exposed to ionomycin (Figure 4F), or in Nox2-dependent immune complexes treated with calcium (Figure 4G). The co-localization of endogenous Nox2 and Cav-1 was confirmed using an in situ proximity ligation assay in murine macrophages (Fig. 4H). Due to the inability of the Cav-1 peptide to inhibit Nox4 (Fig. 3I), we pursued additional experiments in human lung fibroblasts which express Nox4. In contrast to Nox2 and Nox5, we found that in fibroblasts, Nox4 does not directly interact with Cav-1 using a co-IP approach. In support of this, immunofluorescence staining suggests that in fibroblasts, Nox4 and Cav-1 reside in different intracellular locations (Fig. 4I–J). Together, these data suggest that Cav-1 can inhibit ROS from Nox2 and Nox5 by direct enzyme binding. As Nox5 is absent from rodent genomes, we next determined whether Nox5 is expressed in human PA and therefore may have relevance to PH. Lung sections from patients with PH were stained with antibodies to Nox5 and smooth muscle actin. Robust staining for Nox5 was observed in the medial and adventitial layers of PA(Supplemental Fig. 4B).
Figure 4. Binding and post-translational regulation of Nox5 and Nox2 by Cav-1.

(A) COS-7 cells were co-transfected with cDNAs encoding Nox5 and either progressively higher amounts of Cav-1 or a non-specific control gene (RFP), and superoxide production was monitored by L-012-mediated chemiluminescence. The relative expression of Nox5, Cav-1 and GAPDH was determined by immunoblotting with anti-HA, anti-Cav-1 and anti-GAPDH antibodies. (B) COS-7 cells expressing Nox5 were treated with Vehicle or Cav-1 scaffolding domain peptide for 24 hrs, cell lysates were immunoblotted for Nox5 and GAPDH. (C) COS-7 cells were co-transfected with Nox5 and Cav-1 and 48 h later, cell lysates were immunoprecipitated using either a negative isotype control mouse immunoglobulin (IgG) or a monoclonal antibody selective for Cav-1. Immune complexes were immunoblotted for Cav-1, or Nox5. (D) The reverse experiment was performed and lysates from Nox5 and Cav-1 transfected COS-7 cells were immunoprecipitated using control IgG or anti-Nox5 antibodies and Immunoblotted for Cav-1, or Nox5. (E) Cell lysates from murine macrophages were immunoprecipitated using either a negative isotype control mouse immunoglobulin (IgG) or a monoclonal antibody selective for Cav-1. Immune complexes were immunoblotted for Cav-1, or Nox2. (F) Macrophages were treated with or without ionomycin (1μM) for 30mins, and cells lysates were immunoprecipitated using a monoclonal antibody selective for Nox2. Immune complexes were immunoblotted for Nox2, or Cav-1. (G) Nox2 was immunoprecipitated from macrophage lysates and incubated in PBS, Calcium (100μM), or Calcium (100μM)+CaM (1μM) for 30mins. Immune complexes were washed and immunoblotted for Nox2, or Cav-1. (H) Representative images of proximity ligation assays (PLA) for the co-localization of Cav-1 and Nox2 in murine macrophages. (I) Cell lysates from human lung fibroblasts were immunoprecipitated using either a negative isotype control mouse immunoglobulin (IgG) or a monoclonal antibody selective for Nox4. Immune complexes were immunoblotted for Nox4, p22, or Cav-1. (J) Immunofluorescence images of lung fibroblasts using antibodies recognizing Cav-1 and Nox4. Results are representative of at least 3–5 separate experiments. * different from Control or Vehicle, p < 0.05 (n = 5–6).
Cav-1 regulates Nox expression in native cells
To assess whether Cav-1 modifies endogenous Nox gene expression in native cells, we transduced murine macrophages and human lung fibroblasts with either a negative control (RFP) or an adenovirus expressing Cav-1. Macrophages transduced with the Cav-1 adenovirus exhibited a robust decrease in Nox2 protein expression. In equivalent experiments in fibroblasts, increased expression of Cav-1 decreased Nox4 expression (Fig. 5A, B). In contrast, macrophages isolated from Cav-1−/− mice had significantly higher levels of Nox2 expression compared to WT mice (Fig. 5C) and reducing the expression of Cav-1 via a siRNA approach in human lung fibroblasts resulted in increased Nox4 protein expression in a dose-dependent manner (Fig. 5D).
Figure 5. Cav-1 alters the expression of Nox proteins in native cells.
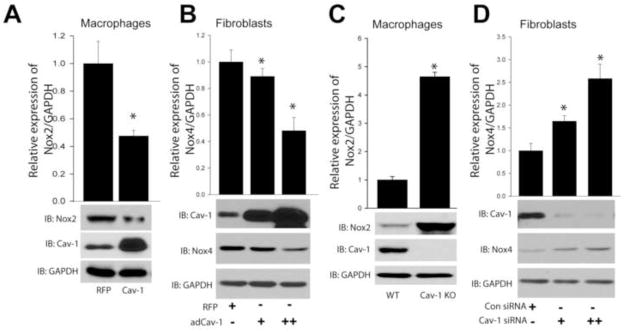
(A) Macrophages were transduced with either Cav-1 adenovirus (30MOI) or a non-specific control gene (RFP) (30MOI) for 48hrs, and cell lysates were immunoblotted for Nox2, Cav-1, and GAPDH. (B) Human lung fibroblasts were exposed to increasing amounts of Cav-1 adenovirus (10 and 30MOI) and cell lysates immunoblotted for Nox4, Cav-1, and GAPDH. (C) Macrophages from WT or Cav-1 knockout mice were isolated, and cell lysates immunoblotted for Nox2, Cav-1, and GAPDH. (D) Cav-1 expression was silenced using siRNA (10(+), 30(++) nM) in human lung fibroblasts, and cell lysates immunoblotted for Nox4, Cav-1, and GAPDH. Results are representative of at least 3–5 separate experiments.
To further explore the mechanisms underlying the ability of Cav-1 to influence the gene expression of Nox2 and Nox4, we next examined mRNA levels. Consistent with its ability to negatively regulate Nox protein expression, Cav-1−/− macrophages had significantly higher levels of Nox2 mRNA expression under both basal and LPS-stimulated conditions compared to WT macrophages (Fig. 6A). Increased expression of Cav-1 via adenovirus decreased the ability of LPS to stimulate increased Nox2 mRNA levels (Fig. 6B). However, there was no significant difference between RFP and Cav-1 transduced cells under unstimulated conditions, suggesting that Cav-1 has a greater influence on LPS-stimulated signaling pathway that influences Nox2 gene transcription. This ability of Cav-1 was observed in other cell types and in HLMVEC, the decreased expression of Cav-1 via siRNA, increased Nox1, Nox2, and Nox4 expression in response to LPS (Fig. 6C). To further delineate the underlying mechanisms by which Cav-1 regulates Nox mRNA expression, we transduced mouse macrophages with either RFP or Cav-1 adenoviruses and stimulated with LPS. The increased expression of Cav-1 significantly inhibited NF-kB activation as reflected by the reduction in p65 phosphorylation (Fig. 6D). To determine whether the regulation of Nox expression is dependent on NF-κB in macrophages and HLMVECs, we employed LPS to stimulate and BAY-11-7082 to selectively inhibit NF-κB. We found that the protein and mRNA expression of Nox1, 2 and 4 in macrophages and HLMVECs was increased by LPS and decreased by the inhibitor of NF-κB, BAY-11-7082 (Fig. 6E and Supplemental Fig. 5A–B).
Figure 6. Cav-1 regulates Nox mRNA expression in macrophages and endothelial cells.

(A) Peritoneal macrophages were isolated from wild type mice or Cav-1−/− mice and treated with or without LPS (100ng/ml, overnight), and relative levels of mRNA expression of Cav-1 and Nox2 were determined by qRT-PCR (ΔΔCt) normalized to GAPDH. (B) Peritoneal macrophages were isolated from wild type mice were transduced with either a non-specific control gene (RFP) (30MOI) or a Cav-1 adenovirus (30MOI) for 24hrs, and then treated with or without LPS (100ng/ml, overnight) and mRNA expression level of Nox2 were measured by qRT-PCR (ΔΔCt) normalized to GAPDH. (C) HLMVECs were transfected with a validated negative control siRNA (60nM) or Cav-1 siRNA (60nM) for 24hrs. Cells were then exposed to LPS (100ng/ml, overnight), and mRNA expression levels of Cav-1, Nox1, Nox2 and Nox4 were measured by qRT-PCR (ΔΔCt) normalized to GAPDH. (D) Mouse macrophages were transduced with either control adenovirus (RFP, 30 MOI) or Cav-1 adenovirus (30 MOI) for 48hrs, stimulated with LPS (1μg/ml) and cell lysates immunoblotted for p65, phosphorylation p65, Cav-1, and GAPDH. (E) Mouse macrophages were treated with or without LPS (100ng/ml) and BAY-11-7082 (20μM) for 12 hours, and cell lysates immunoblotted for Nox2 and GAPDH. * p < 0.05 (n = 6).
In lungs from Cav-1−/− mice exposed to hypoxia to induce PH, both Nox2 and Nox4 mRNA and protein expression are significantly upregulated suggesting that the reciprocal relationship observed in vitro is also relevant in vivo (Fig. 7A–B). To assess the functional effects of Cav-1 loss, we compared hypoxia-driven pulmonary hypertension in WT and Cav-1−/− mice. The loss of Cav-1 in Cav-1−/− mice, resulted in more significant PH as evaluated by compensatory hypertrophy of the RV (Fig. 7C). These data suggest that the increased severity of PH due to the loss of Cav-1 might result, at least in part, from the upregulation of Nox-derived ROS production.
Figure 7. Cav-1 knockout mice exhibit increased Nox2 and Nox4 mRNA and protein expression and increased pulmonary hypertension.

(A) Protein expression of Nox2 and Nox4 in whole lung lysates from WT or Cav-1−/− mice exposed to hypoxia was determined by Western blot. (B) Nox2 and Nox4 mRNA expression were determined by qRT-PCR (ΔΔCt) normalized to GAPDH in lung tissue from WT or Cav-1−/− mice exposed to hypoxia. *different from wild type, p < 0.05 (n = 3). (C) Hypoxia-induced PH is enhanced in Cav1−/− mice, * versus normoxia, + versus WT p<0.05 (n=3–5).
Discussion
Cav-1 is an important regulatory molecule that has been implicated in the pathogenesis of atherosclerosis[44], inflammation[40], fibrosis [45] and pulmonary arterial hypertension [22]. In addition to its structural role in caveolae, Cav-1 binds to and regulates the activity and intracellular location of numerous signaling molecules. Given the wide range of cellular functions influenced by Cav-1, the mechanisms by which it influences disease pathways are complex and incompletely understood. The ability of Cav-1 to regulate eNOS activity has been well established, but another important mechanism contributing to each of the above listed disease processes is an overabundance of ROS production[9, 46]. In the current study, we show that Cav-1 is a negative regulator of ROS production from Nox1-5 and this is achieved by a multitude of mechanisms including enzyme binding and the inhibition of endogenous Nox gene expression. We further show that isolated PA from rodent models of PH has reduced Cav-1, increased Nox expression and increased levels of ROS. Moreover, mice harboring a genetic deletion of Cav-1 have increased Nox expression and more pronounced PH compared to WT mice. Collectively these data identify Cav-1 as a negative regulator of Nox enzymes which is consistent with data from human and animal models of PH showing reduced Cav-1 expression and both increased Nox expression and ROS production.
Increased production of ROS in the pulmonary vasculature has been widely reported in both clinical and experimental PH[47, 48]. In our study, we observed elevated ROS in the lungs and pulmonary arteries from animals with PH. Nox isoforms are major sources of cellular ROS production[15] and we observed higher levels of expression of both Nox2 and Nox4 in hypertensive PA which is consistent with previously published data [21, 49]. The mechanisms underlying the increased Nox expression and activity in PH are not fully elucidated. While an association between the loss of Cav-1 and increased ROS production has been reported[50, 51], the ability of Cav-1 to directly influence Nox activity has not yet been shown. To determine whether Cav-1 can directly impact Nox activity and ROS production, we employed a heterologous expression system in vitro. We found that upregulation of Cav-1 decreased the ability of Nox1-5 to produce ROS. To better reveal the mechanisms underlying the inhibitory actions of Cav-1, we employed the caveolin-1 scaffolding domain peptide (aa82–101) linked to a cell penetrating sequence[40]. Treatment of Nox expressing cells with the Cav-1 peptide reduced superoxide production from Nox1, 2, 3 and 5. In native vascular cells, we pursued loss of function experiments using Cav-1 siRNA and Cav-1 knockout cells and gain of function experiments using a Cav-1 expressing adenovirus. We found that reducing the expression of Cav-1 increased the production of ROS in human lung fibroblasts and macrophages. In contrast, increased expression of Cav-1 decreased ROS production in macrophages. Without directly influencing Cav-1 expression, we also show that manipulation of cholesterol levels with cyclodextrin can influence Nox activity. Collectively, these data support a functional ability of Cav-1 to regulate Nox activity.
Cav-1 has been shown to bind to numerous signaling proteins, many of which are concentrated in plasma membrane caveolae or localized near endomembranes where Cav-1 is also expressed. The binding of Cav-1 to other proteins is mediated via interactions between its scaffolding domain and a Cav-1 binding domain that is present in many of the established binding partners of Cav-1. The ability of Cav-1 to reduce Nox5 activity without significantly altering protein expression suggested a post-translational mechanism of regulation. There was no significant change in the intracellular location of Nox5 when co-expressed with Cav-1 and this is further supported by the ability of Cav-1 to reduce Nox5 activity in isolated enzyme assays. We found using a co-IP approach that Cav-1 is present in complexes with Nox5 which is similar to that described for other proteins that are regulated by Cav-1 including eNOS[7]. Co-IP and proximity ligation assays also revealed that Nox2 is a Cav-1 binding protein. Together, these data suggest that Cav-1 can directly inhibit Nox5 and Nox2 activity via allosteric regulation. Given the ability of the Cav-1 scaffolding domain peptide to reduce the activity of Nox1-3 and 5, we anticipate that Cav-1 regulates the activity of Nox1 and Nox3 via a similar mechanism. However, this paradigm is not true for all Nox enzymes. We found that the Cav-1 scaffolding domain peptide failed to inhibit Nox4 activity. To determine a mechanism, in lung fibroblasts that express Nox4 endogenously, we found that in contrast to Nox2 and Nox5, Cav-1 does not appear to bind to Nox4 in co-IP experiments or reside within the same intracellular location. These data suggest that binding is necessary to inhibit enzyme activity. In previous studies we have found that hsp90 binds to Nox1-3 and 5 and is necessary for superoxide production. However, hsp90 does not bind to Nox4 which is regarded as a constitutively active Nox isoform [28, 29]. Binding of hsp90 to eNOS has been shown to regulate Cav-1 binding[41], however, it is not yet known whether hsp90 can similarly influence the binding of Cav-1 to Nox enzymes. There are numerous studies showing that other proteins such as calmodulin can negatively influence Cav-1 binding[5]. We found that the amount of Cav-1 bound to Nox2 was dependent on the level of calcium as both ionomycin exposure in intact cells and increasing the calcium-concentration in Nox2-dependent immune complexes induced the dissociation of bound Cav-1. How this occurs is not yet known, but the calcium-dependent regulation of Cav-1 binding may account, at least in part, for the established ability of calcium ionophores to increase the activity of Nox2[52]. The ability of the scaffolding domain alone to regulate the activity of Nox1-3,5 and the inability of a mutant of caveolin-1 lacking this domain to influence Nox5 activity suggests that aa82–101 are crucial. However, the region(s) on Nox2 and Nox5 that are responsible for binding to Cav-1 are not yet known. Within the extreme C-terminus of all Nox enzymes is a motif that conforms to the caveolin binding motif (x xxxx, where is an aromatic and x any amino acid). However, mutation of this domain completely eliminates Nox activity suggesting it has other vital functions (data not shown). It has also been shown that Cav-1 is required for the stimulus dependent activation of Nox enzymes. Cav-1 contributes to AngII-induced activation of Rac1 and translocation of the Nox subunit p47 and Nox2 activation in endothelial cells[53, 54]. To isolate the direct effects of Cav-1 on Nox activity our study focused on basal (unstimulated) ROS production and also utilized Nox5, which does not require the assembly of cytosolic subunits for ROS production. While data from these experiments supports a direct role for Cav-1 in regulating Nox activity, these effects might be negated by additional effects of Cav-1 that decrease receptor activation and signaling and therefore the stimulus-dependent activation of Nox enzymes. The mechanisms underlying the ability of full length Cav-1 to negatively regulate Nox4 expression in COS-7 cells remain unclear. It is increasingly apparent that the regulation of Nox4 is more complex and incompletely understood than previously appreciated. There are reports showing that Nox4 is constitutively active and that it primarily emits hydrogen peroxide due to the rapid conversion of superoxide. Others have shown stimulus dependent regulation of Nox4 and the production of superoxide[11]. These discrepancies may be context dependent and one variable that may be very important to the regulation of Nox4 is its subcellular localization. In our experiments using human lung fibroblasts, we did not find evidence to support a protein: protein interaction between Nox4 and Cav-1. This data is consistent with their subcellular distribution in different subcellular compartments with Nox4 predominantly nuclear and Cav-1 in cytoplasmic membranes. In COS-7 cells, Nox4 is primarily expressed in the endoplasmic reticulum which is similar to that described in endothelial cells[55, 56]. This would place Nox4 and Cav-1 in closer proximity and perhaps allow them to physically interact.
In PH, not only is ROS production increased, but this occurs alongside an increase in Nox expression. Whether Cav-1 can regulate the expression of endogenous Nox proteins in native cells has not been addressed directly. Studies by Price et al showed that NF-kB is activated in individuals with PH, and that activated NF-kB is a driver of inflammation in hypertensive PA[57]. It has also been shown that Cav-1 can negatively regulate the activation of NF-kB in endothelial cells and the increase in lung inflammation in response to LPS [58, 59] and viral infection [60]. Loss of Cav-1 has been shown to exacerbate inflammation and oxidative stress in a number of animal models[51, 61], but the role of Nox enzymes has not yet been appreciated. In our study, we found that decreased levels of Cav-1 coincided with increased Nox2 and Nox4 expression in macrophages and fibroblasts, respectively. Gain of function experiments showed that increased expression of Cav-1 results in the downregulation of Nox2 and Nox4 expression. In HLMVEC, qRT-PCR revealed that mRNA expression of Nox1, Nox2 and Nox4 are robustly increased by LPS in Cav-1 deficient cells. NF-kB activation has been shown to regulate the expression of Nox enzymes [62, 63] and the ability of Cav-1 to suppress this pathway is one potential mechanism by which it can regulate the expression of endogenous Nox proteins. Collectively, these results suggest that not only does Cav-1 suppress the post-translational activity of Nox enzymes, but it also negatively regulates gene expression.
Cav-1 is essential for the formation of flask shaped plasma membrane invaginations termed caveolae. Oligomers of Cav-1 coat the surface of caveolae and together with the cooperation of other caveolar proteins such as Cav-2 and Cavin-1, they give the organelle is characteristic geometry. The physical association of Cav-1 with Cavin-1 and Cav-2 is important in the formation of caveolae and the stability/expression of both proteins is conferred by binding to Cav-1 [4, 7]. In control pulmonary arteries, we detected Cav-1 expression in endothelial cells and also in perivascular cells. Endothelial cells are well documented to express high levels of Cav-1[64], and other cell types in the adventitia including fibroblasts and macrophages are also known to express Cav-1[65, 66]. Interestingly, the media or smooth muscle cell layer was absent significant Cav-1 expression. Previous studies have shown that smooth muscle cells express Cav-1 [67, 68] but the majority of studies have used cultured cells where VSMC are proliferating and de-differentiated compared to those populating intact blood vessels. In animals with pulmonary hypertension, we observed a decrease in immunofluorescence staining for Cav-1 which was confirmed by Western blotting in multiple rat models including the FHR, MCT and SU/HYP. These results are in agreement with previous studies and also in human PH[22]. In addition to Cav-1 we also determined the expression levels of other caveolar proteins including Cav-2, Cavin-1 and PV-1 by Western blotting and mRNA. We observed a significant decrease in Cav-2, Cavin-1 and PV-1 in hypertensive PA. These results suggest a generalized loss of caveolar proteins in PH. Reduced expression of Cav-1 is associated with hyperproliferative disorders including cancer and fibrosis and the complete loss of Cav-1 in knockout mice is sufficient to cause PH suggesting it has an important regulatory role in pathogenic mechanisms[1, 22].
Interestingly, the primarily cellular location of Cav-1 in pulmonary blood vessels was the endothelium and adventitia/perivascular cells. The functional importance of Cav-1 in cells types of the adventitia is also likely to contribute to the development of PH. The adventitia encircles the inner layers of the blood vessel and is a loose assortment of matrix proteins and cells including fibroblasts, which express Nox4, and immune cells such as macrophages, that express Nox2 [69]. Fibroblasts express high levels of Cav-1 [70]. Loss of Cav-1 in knockout mice leads to severe fibroproliferative diseases [71, 72] and in humans, Cav-1 polymorphisms correlate with a higher incidence of lung fibrosis [73]. Increased Cav-1 expression confers the opposite phenotype of reduced fibroblast proliferation, signaling and matrix protein and metalloprotease expression [45, 74]. In macrophages, Cav-1 is also expressed and is an established negative regulator of inflammation [75, 76]. The ability of Cav-1 to regulate Nox expression was also shown in vivo in Cav-1 knockout mice, where increased Nox2 and Nox4 expression was observed in lung tissue. Elevated ROS have been shown to promote both increased inflammation and fibrosis [44, 77] and ability of Cav-1 to negatively regulate Nox activity and expression, as outlined in the current study, suggest that this mechanism may contribute to the inflammation and fibrosis that drive the vascular remodeling in PH. This contention is supported, albeit indirectly, by data showing that PH is more severe in Cav-1 knockout out mice. Numerous other studies have direct evidenced supporting the ability of increased Nox enzyme activity to contribute to the pathogenesis of PH. In mice, genetic deletion or pharmacological inhibition of Nox2 has been shown to reverse hypoxia-initiated PH [19, 20]. Another report by Green et al found that the Nox1/4 inhibitor, GKT137831, attenuates PH in mice [21].
In summary, our study has revealed two mechanisms by which Cav-1 can regulate Nox-dependent ROS production; through acute post-translational modification and more chronically by restraining mRNA and protein expression. By decreasing the activity and expression of Nox enzymes, our study highlights a new role by which Cav-1 may influence inflammation and fibrosis. The ability of Cav-1 to antagonize ROS production may have important ramifications in diseases such as pulmonary hypertension which have an abundance of inflammation and fibrosis. Indeed, targeting the reduced Cav-1 expression levels in PH maybe an important therapeutic consideration.
Supplementary Material
Highlights.
Cav-1 is expressed in the endothelium and adventitia of PA, and is reduced in 3 models of PAH.
Reduced Cav-1 correlates with increased ROS and Nox2, 4 expression in hypertensive PA.
Cav-1 negatively regulates Nox1-3, 5 and Cav-1 was present in immune complexes with Nox5 and Nox2.
Cav-1 influences the mRNA and protein expression of Nox2 and Nox4 and inhibits NF-kB activation.
Genetic ablation of Cav-1 increased the levels of Nox2, 4 and exacerbated hypoxia-induced PAH.
Acknowledgments
The authors gratefully acknowledge the technical assistance of Louise Meadows and Yevgeniy Kovalenkov. This work was supported by GRU Pilot Study Research Program Award (PSRP) 0053A (SAB and DF), GRU Extramural Success Award (ESA) 00006A (SAB and DF), R01-HL-092446 (DF), P01-HL-0101902 (DF), and by the Postdoctoral Fellowship from the American Heart Association 13POST14800025 (FC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Williams TM, Lisanti MP. The Caveolin genes: from cell biology to medicine. Annals of medicine. 2004;36:584–595. doi: 10.1080/07853890410018899. [DOI] [PubMed] [Google Scholar]
- 2.Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 3.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 4.Liu P, Rudick M, Anderson RG. Multiple functions of caveolin-1. J Biol Chem. 2002;277:41295–41298. doi: 10.1074/jbc.R200020200. [DOI] [PubMed] [Google Scholar]
- 5.Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem. 1997;272:18522–18525. doi: 10.1074/jbc.272.30.18522. [DOI] [PubMed] [Google Scholar]
- 6.Carotenuto F, Minieri M, Monego G, Fiaccavento R, Bertoni A, Sinigaglia F, Vecchini A, Carosella L, Di Nardo P. A diet supplemented with ALA-rich flaxseed prevents cardiomyocyte apoptosis by regulating caveolin-3 expression. Cardiovascular research. 2013 doi: 10.1093/cvr/cvt211. [DOI] [PubMed] [Google Scholar]
- 7.Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovascular research. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahmoud AM, Dutta D, Lavery L, Stephens DN, Villanueva FS, Kim K. Noninvasive detection of lipids in atherosclerotic plaque using ultrasound thermal strain imaging: in vivo animal study. Journal of the American College of Cardiology. 2013;62:1804–1809. doi: 10.1016/j.jacc.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circulation research. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 10.Wong CM, Bansal G, Pavlickova L, Marcocci L, Suzuki YJ. Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid Redox Signal. 2013;18:1789–1796. doi: 10.1089/ars.2012.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen F, Haigh S, Barman S, Fulton DJ. From form to function: the role of Nox4 in the cardiovascular system. Front Physiol. 2012;3:412. doi: 10.3389/fphys.2012.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fike CD, Dikalova A, Slaughter JC, Kaplowitz MR, Zhang Y, Aschner JL. Reactive oxygen species-reducing strategies improve pulmonary arterial responses to nitric oxide in piglets with chronic hypoxia-induced pulmonary hypertension. Antioxid Redox Signal. 2013;18:1727–1738. doi: 10.1089/ars.2012.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freund-Michel V, Guibert C, Dubois M, Courtois A, Marthan R, Savineau JP, Muller B. Reactive oxygen species as therapeutic targets in pulmonary hypertension. Ther Adv Respir Dis. 2013;7:175–200. doi: 10.1177/1753465812472940. [DOI] [PubMed] [Google Scholar]
- 14.Veit F, Pak O, Egemnazarov B, Roth M, Kosanovic D, Seimetz M, Sommer N, Ghofrani HA, Seeger W, Grimminger F, Brandes RP, Schermuly RT, Weissmann N. Function of NADPH Oxidase 1 in Pulmonary Arterial Smooth Muscle Cells After Monocrotaline-Induced Pulmonary Vascular Remodeling. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2012.4904. [DOI] [PubMed] [Google Scholar]
- 15.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 16.Kim BY, Han MJ, Chung AS. Effects of reactive oxygen species on proliferation of Chinese hamster lung fibroblast (V79) cells. Free Radic Biol Med. 2001;30:686–698. doi: 10.1016/s0891-5849(00)00514-1. [DOI] [PubMed] [Google Scholar]
- 17.Bellini A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. 2007;87:858–870. doi: 10.1038/labinvest.3700654. [DOI] [PubMed] [Google Scholar]
- 18.Wedgwood S, Lakshminrusimha S, Czech L, Schumacker PT, Steinhorn RH. Increased p22(phox)/Nox4 expression is involved in remodeling through hydrogen peroxide signaling in experimental persistent pulmonary hypertension of the newborn. Antioxid Redox Signal. 2013;18:1765–1776. doi: 10.1089/ars.2012.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norton CE, Broughton BR, Jernigan NL, Walker BR, Resta TC. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid Redox Signal. 2013;18:1777–1788. doi: 10.1089/ars.2012.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox) American journal of physiology. Lung cellular and molecular physiology. 2006;290:L2–10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 21.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. American journal of respiratory cell and molecular biology. 2012;47:718–726. doi: 10.1165/rcmb.2011-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathew R. Pathogenesis of pulmonary hypertension: a case for caveolin-1 and cell membrane integrity. Am J Physiol Heart Circ Physiol. 2014;306:H15–25. doi: 10.1152/ajpheart.00266.2013. [DOI] [PubMed] [Google Scholar]
- 23.Trimmer C, Sotgia F, Whitaker-Menezes D, Balliet RM, Eaton G, Martinez-Outschoorn UE, Pavlides S, Howell A, Iozzo RV, Pestell RG, Scherer PE, Capozza F, Lisanti MP. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: a new genetically tractable model for human cancer associated fibroblasts. Cancer Biol Ther. 2011;11:383–394. doi: 10.4161/cbt.11.4.14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 25.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 26.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 27.Chen F, Fulton DJ. An inhibitor of protein arginine methyltransferases, 7,7′-carbonylbis(azanediyl)bis(4-hydroxynaphthalene-2-sulfonic acid (AMI-1), is a potent scavenger of NADPH-oxidase-derived superoxide. Mol Pharmacol. 2010;77:280–287. doi: 10.1124/mol.109.061077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen F, Pandey D, Chadli A, Catravas JD, Chen T, Fulton DJ. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid Redox Signal. 2011;14:2107–2119. doi: 10.1089/ars.2010.3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen F, Yu Y, Qian J, Wang Y, Cheng B, Dimitropoulou C, Patel V, Chadli A, Rudic RD, Stepp DW, Catravas JD, Fulton DJ. Opposing actions of heat shock protein 90 and 70 regulate nicotinamide adenine dinucleotide phosphate oxidase stability and reactive oxygen species production. Arterioscler Thromb Vasc Biol. 2012;32:2989–2999. doi: 10.1161/ATVBAHA.112.300361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elms S, Chen F, Wang Y, Qian J, Askari B, Yu Y, Pandey D, Iddings J, Caldwell RB, Fulton DJ. Insights into the arginine paradox: evidence against the importance of subcellular location of arginase and eNOS. Am J Physiol Heart Circ Physiol. 2013;305:H651–666. doi: 10.1152/ajpheart.00755.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandey D, Chen F, Patel A, Wang CY, Dimitropoulou C, Patel VS, Rudic RD, Stepp DW, Fulton DJ. SUMO1 negatively regulates reactive oxygen species production from NADPH oxidases. Arterioscler Thromb Vasc Biol. 2011;31:1634–1642. doi: 10.1161/ATVBAHA.111.226621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen F, Dang YH, Yan CX, Liu YL, Deng YJ, Fulton DJ, Chen T. Sequence-length variation of mtDNA HVS-I C-stretch in Chinese ethnic groups. J Zhejiang Univ Sci B. 2009;10:711–720. doi: 10.1631/jzus.B0920140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen YJ, Liu YL, Zhong Q, Yu YF, Su HL, Toque HA, Dang YH, Chen F, Xu M, Chen T. Tetrahydropalmatine protects against methamphetamine-induced spatial learning and memory impairment in mice. Neurosci Bull. 2012;28:222–232. doi: 10.1007/s12264-012-1236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pandey D, Patel A, Patel V, Chen F, Qian J, Wang Y, Barman SA, Venema RC, Stepp DW, Rudic RD, Fulton DJ. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol. 2012;302:H1919–1928. doi: 10.1152/ajpheart.00910.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y, Xing B, Dang YH, Qu CL, Zhu F, Yan CX. Microinjection of valproic acid into the ventrolateral orbital cortex enhances stress-related memory formation. PLoS One. 2013;8:e52698. doi: 10.1371/journal.pone.0052698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma XC, Jiang D, Jiang WH, Wang F, Jia M, Wu J, Hashimoto K, Dang YH, Gao CG. Social isolation-induced aggression potentiates anxiety and depressive-like behavior in male mice subjected to unpredictable chronic mild stress. PLoS One. 2011;6:e20955. doi: 10.1371/journal.pone.0020955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen F, Lucas R, Fulton D. The subcellular compartmentalization of arginine metabolizing enzymes and their role in endothelial dysfunction. Front Immunol. 2013;4:184. doi: 10.3389/fimmu.2013.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pandey D, Fulton DJ. Molecular regulation of NADPH oxidase 5 via the MAPK pathway. Am J Physiol Heart Circ Physiol. 2011;300:H1336–1344. doi: 10.1152/ajpheart.01163.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian J, Chen F, Kovalenkov Y, Pandey D, Moseley MA, Foster MW, Black SM, Venema RC, Stepp DW, Fulton DJ. Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S-nitrosylation. Free Radic Biol Med. 2012;52:1806–1819. doi: 10.1016/j.freeradbiomed.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, Sessa WC. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nature medicine. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- 41.Gratton JP, Fontana J, O’Connor DS, Garcia-Cardena G, McCabe TJ, Sessa WC. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J Biol Chem. 2000;275:22268–22272. doi: 10.1074/jbc.M001644200. [DOI] [PubMed] [Google Scholar]
- 42.Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ. Novel mechanism of activation of NADPH oxidase 5. calcium sensitization via phosphorylation. J Biol Chem. 2007;282:6494–6507. doi: 10.1074/jbc.M608966200. [DOI] [PubMed] [Google Scholar]
- 43.Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal. 2009;11:2443–2452. doi: 10.1089/ars.2009.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001;53:135–159. [PubMed] [Google Scholar]
- 45.Tourkina E, Richard M, Gooz P, Bonner M, Pannu J, Harley R, Bernatchez PN, Sessa WC, Silver RM, Hoffman S. Antifibrotic properties of caveolin-1 scaffolding domain in vitro and in vivo. American journal of physiology. Lung cellular and molecular physiology. 2008;294:L843–861. doi: 10.1152/ajplung.00295.2007. [DOI] [PubMed] [Google Scholar]
- 46.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 47.Wedgwood S, Black SM. Role of reactive oxygen species in vascular remodeling associated with pulmonary hypertension. Antioxid Redox Signal. 2003;5:759–769. doi: 10.1089/152308603770380061. [DOI] [PubMed] [Google Scholar]
- 48.Nozik-Grayck E, Stenmark KR. Role of reactive oxygen species in chronic hypoxia-induced pulmonary hypertension and vascular remodeling. Adv Exp Med Biol. 2007;618:101–112. doi: 10.1007/978-0-387-75434-5_8. [DOI] [PubMed] [Google Scholar]
- 49.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circulation research. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 50.Karuppiah K, Druhan LJ, Chen CA, Smith T, Zweier JL, Sessa WC, Cardounel AJ. Suppression of eNOS-derived superoxide by caveolin-1: a biopterin-dependent mechanism. Am J Physiol Heart Circ Physiol. 2011;301:H903–911. doi: 10.1152/ajpheart.00936.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell cycle. 2010;9:2201–2219. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 52.Cockeran R, Theron AJ, Steel HC, Matlola NM, Mitchell TJ, Feldman C, Anderson R. Proinflammatory interactions of pneumolysin with human neutrophils. J Infect Dis. 2001;183:604–611. doi: 10.1086/318536. [DOI] [PubMed] [Google Scholar]
- 53.Lobysheva I, Rath G, Sekkali B, Bouzin C, Feron O, Gallez B, Dessy C, Balligand JL. Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:2098–2105. doi: 10.1161/ATVBAHA.111.230623. [DOI] [PubMed] [Google Scholar]
- 54.Zuo L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW. Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after angiotensin II type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arterioscler Thromb Vasc Biol. 2005;25:1824–1830. doi: 10.1161/01.ATV.0000175295.09607.18. [DOI] [PubMed] [Google Scholar]
- 55.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. Journal of the American College of Cardiology. 2008;52:1803–1809. doi: 10.1016/j.jacc.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Molecular and cellular biology. 2010;30:3553–3568. doi: 10.1128/MCB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Price LC, Caramori G, Perros F, Meng C, Gambaryan N, Dorfmuller P, Montani D, Casolari P, Zhu J, Dimopoulos K, Shao D, Girerd B, Mumby S, Proudfoot A, Griffiths M, Papi A, Humbert M, Adcock IM, Wort SJ. Nuclear Factor kappa-B Is Activated in the Pulmonary Vessels of Patients with End-Stage Idiopathic Pulmonary Arterial Hypertension. PLoS One. 2013;8:e75415. doi: 10.1371/journal.pone.0075415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tiruppathi C, Shimizu J, Miyawaki-Shimizu K, Vogel SM, Bair AM, Minshall RD, Predescu D, Malik AB. Role of NF-kappaB-dependent caveolin-1 expression in the mechanism of increased endothelial permeability induced by lipopolysaccharide. J Biol Chem. 2008;283:4210–4218. doi: 10.1074/jbc.M703153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garrean S, Gao XP, Brovkovych V, Shimizu J, Zhao YY, Vogel SM, Malik AB. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol. 2006;177:4853–4860. doi: 10.4049/jimmunol.177.7.4853. [DOI] [PubMed] [Google Scholar]
- 60.Wang XM, Nadeau PE, Lin S, Abbott JR, Mergia A. Caveolin 1 inhibits HIV replication by transcriptional repression mediated through NF-kappaB. J Virol. 2011;85:5483–5493. doi: 10.1128/JVI.00254-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuan K, Huang C, Fox J, Gaid M, Weaver A, Li G, Singh BB, Gao H, Wu M. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB) J Biol Chem. 2011;286:21814–21825. doi: 10.1074/jbc.M111.237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manea A, Manea SA, Gafencu AV, Raicu M. Regulation of NADPH oxidase subunit p22(phox) by NF-kB in human aortic smooth muscle cells. Archives of physiology and biochemistry. 2007;113:163–172. doi: 10.1080/13813450701531235. [DOI] [PubMed] [Google Scholar]
- 63.Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, Fan TH, Mitchell PO, Sutliff RL, Hart CM. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. American journal of respiratory cell and molecular biology. 2009;40:601–609. doi: 10.1165/rcmb.2008-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 65.Gargalovic P, Dory L. Caveolin-1 and caveolin-2 expression in mouse macrophages. High density lipoprotein 3-stimulated secretion and a lack of significant subcellular co-localization. J Biol Chem. 2001;276:26164–26170. doi: 10.1074/jbc.M011291200. [DOI] [PubMed] [Google Scholar]
- 66.Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, Ifedigbo E, Xu X, Oury TD, Kaminski N, Choi AM. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. The Journal of experimental medicine. 2006;203:2895–2906. doi: 10.1084/jem.20061536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, Thistlethwaite PA, Miyanohara A, Farquhar MG, Yuan JX, Insel PA. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J. 2007;21:2970–2979. doi: 10.1096/fj.07-8424com. [DOI] [PubMed] [Google Scholar]
- 68.Peterson TE, Guicciardi ME, Gulati R, Kleppe LS, Mueske CS, Mookadam M, Sowa G, Gores GJ, Sessa WC, Simari RD. Caveolin-1 can regulate vascular smooth muscle cell fate by switching platelet-derived growth factor signaling from a proliferative to an apoptotic pathway. Arterioscler Thromb Vasc Biol. 2003;23:1521–1527. doi: 10.1161/01.ATV.0000081743.35125.05. [DOI] [PubMed] [Google Scholar]
- 69.Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circulation research. 2001;89:1111–1121. doi: 10.1161/hh2401.100844. [DOI] [PubMed] [Google Scholar]
- 70.Capozza F, Cohen AW, Cheung MW, Sotgia F, Schubert W, Battista M, Lee H, Frank PG, Lisanti MP. Muscle-specific interaction of caveolin isoforms: differential complex formation between caveolins in fibroblastic vs. muscle cells. American journal of physiology. Cell physiology. 2005;288:C677–691. doi: 10.1152/ajpcell.00232.2004. [DOI] [PubMed] [Google Scholar]
- 71.Del Galdo F, Lisanti MP, Jimenez SA. Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol. 2008;20:713–719. doi: 10.1097/bor.0b013e3283103d27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Castello-Cros R, Whitaker-Menezes D, Molchansky A, Purkins G, Soslowsky LJ, Beason DP, Sotgia F, Iozzo RV, Lisanti MP. Scleroderma-like properties of skin from caveolin-1-deficient mice: implications for new treatment strategies in patients with fibrosis and systemic sclerosis. Cell cycle. 2011;10:2140–2150. doi: 10.4161/cc.10.13.16227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kastelijn EA, van Moorsel CH, Kazemier KM, Roothaan SM, Ruven HJ, Kwakkel-van Erp JM, van de Graaf EA, Zanen P, van Kessel DA, Grutters JC. A genetic polymorphism in the CAV1 gene associates with the development of bronchiolitis obliterans syndrome after lung transplantation. Fibrogenesis Tissue Repair. 2011;4:24. doi: 10.1186/1755-1536-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Volonte D, Zhang K, Lisanti MP, Galbiati F. Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol Biol Cell. 2002;13:2502–2517. doi: 10.1091/mbc.01-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang XM, Kim HP, Song R, Choi AM. Caveolin-1 confers antiinflammatory effects in murine macrophages via the MKK3/p38 MAPK pathway. American journal of respiratory cell and molecular biology. 2006;34:434–442. doi: 10.1165/rcmb.2005-0376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang PX, Murray TS, Villella VR, Ferrari E, Esposito S, D’Souza A, Raia V, Maiuri L, Krause DS, Egan ME, Bruscia EM. Reduced caveolin-1 promotes hyperinflammation due to abnormal heme oxygenase-1 localization in lipopolysaccharide-challenged macrophages with dysfunctional cystic fibrosis transmembrane conductance regulator. J Immunol. 2013;190:5196–5206. doi: 10.4049/jimmunol.1201607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nature medicine. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.