

Abstract
An amine specific peptide derivatization strategy involving the use of novel isobaric stable isotope encoded ‘fixed charge’ sulfonium ion reagents, coupled with an analysis strategy employing capillary HPLC, ESI-MS, and automated data dependent ion trap CID-MS/MS, -MS3, and/or ETD-MS/MS, has been developed for the improved quantitative analysis of protein phosphorylation, and for identification and characterization of their site(s) of modification. Derivatization of 50 synthetic phosphopeptides with S,S′-dimethylthiobutanoylhydroxysuccinimide ester iodide (DMBNHS), followed by analysis using capillary HPLC-ESI-MS, yielded an average 2.5-fold increase in ionization efficiencies and a significant increase in the presence and/or abundance of higher charge state precursor ions compared to the non-derivatized phosphopeptides. Notably, 44% of the phosphopeptides (22 of 50) in their underivatized states yielded precursor ions whose maximum charge states corresponded to +2, while only 8% (4 of 50) remained at this maximum charge state following DMBNHS derivatization. Quantitative analysis was achieved by measuring the abundances of the diagnostic product ions corresponding to the neutral losses of ‘light’ (S(CH3)2) and ‘heavy’ (S(CD3)2) dimethylsulfide exclusively formed upon CID-MS/MS of isobaric stable isotope labeled forms of the DMBNHS derivatized phosphopeptides. Under these conditions, the phosphate group stayed intact. Access for a greater number of peptides to provide enhanced phosphopeptide sequence identification and phosphorylation site characterization was achieved via automated data-dependent CID-MS3 or ETD-MS/MS analysis due to the formation of the higher charge state precursor ions. Importantly, improved sequence coverage was observed using ETD-MS/MS following introduction of the sulfonium ion fixed charge, but with no detrimental effects on ETD fragmentation efficiency.
Keywords: Phosphopeptide fragmentation, Collision induced dissociation (CID), Electron transfer dissociation (ETD), Fixed charge
Introduction
Comprehensive phosphoproteome analysis requires the identification of phosphorylated proteins, localization of the specific sites of phosphorylation within these proteins, and quantification of the phosphorylation site occupancies under the specific biological environment [1-6]. To date, the success of such strategies have undoubtedly been facilitated by the application of tandem mass spectrometry (MS/MS) and associated sample fractionation/enrichment methods for phosphopeptide identification, characterization, and quantification [7-9]. However, when multiple potential sites of phosphorylation exist within a peptide sequence, unambiguous phosphorylation site characterization remains a considerable challenge.
Typically, phosphorylation sites are identified by MS/MS and/or -MS3 using either collision induced dissociation (CID) or electron transfer dissociation (ETD) as the ion activation techniques [8, 9]. Unfortunately, unambiguous phosphorylation site localization by using CID-MS/MS strategies in ion trap mass spectrometers (where ion activation occurs in the ms timescale) may be hindered by dominant facile neutral losses of 80 Da (HPO3) or 98 Da(H3PO4) from the phosphoaminoacid side chain [9-11], or intramolecular phosphate group ‘scrambling’ [12], due to the formation of strong intramolecular hydrogen bonding interactions between the phosphate group and the side chain of a protonated arginine or lysine residue that lower the barriers to side chain loss or rearrangement, particularly under ‘non-mobile’ or ‘partially mobile’ precursor ion proton mobility conditions. Numerous groups have employed ion trap CID-MS3 to subject the initial 98 Da neutral loss product ions to further dissociation to obtain additional structural information for phosphorylation site characterization [13-18]. However, competing pathways for the loss of 98 Da (i.e.; the combined losses of HPO3 and H2O from the phosphoamino acid side chain and an unmodified serine or threonine residue within the sequence, respectively), have been reported [12]. Therefore, as the extent to which this competing pathway is operative cannot be known a priori, MS3 methods may unfortunately not be used to localize phosphorylation site (s) without some ambiguity. To minimize the limitations associated with ion trap CID-MS/MS of protonated phosphopeptides therefore, precursor ions selected for dissociation should preferentially be in a ‘mobile’ protonation state (i.e.; charge states≥+2 for peptides formed by proteolysis with trypsin). ETD has an improved ability to localize phosphate groups [19-25], as no losses involving the phosphate side chain or phosphate group ‘scrambling’ are observed [9, 12, 19-25]. However, the effectiveness of ETD-MS/MS can be limited in that it also optimally requires the precursor ions to be highly multiply-charged (i.e.; ≥ 3+, where the ion/ion reaction cross-section is sufficiently large for efficient dissociation to occur) [19-21]. Finally, due to their acidic nature, there is the potential that ionization efficiencies and maximum precursor ion charge states of phosphopeptides can be lower compared to their nonphosphorylated counterparts, thereby limiting the sensitivity for their detection, and restricting the applicability of CID- and ETD-MS/MS to only a partial subset of the phosphopeptides expected to be found within complex phosphoproteomic mixtures [26-29].
To overcome these limitations, significant effort has recently been extended toward developing approaches to increase the ionization and charge states of phosphopeptide ions, and/or to alter their gas-phase fragmentation behaviors, for improved CID- and ETD-MS/MS performance. One approach has been to generate larger peptides capable of forming precursor ions by ESI that carry three or more positive charges, by using trypsin under conditions of limited proteolysis [25] or by using proteolytic enzymes with limited cleavage specificity, such as Lys-C [21, 22, 30], Glu-C [22], or Lys-N [31]. Another involves the addition of ‘supercharging’ reagents such as low concentrations of m-nitrobenzyl alcohol or sulfolane to the chromatographic elution or ESI solvents [32, 33], with a subsequent increase in peptide precursor ion charge states, ETD fragmentation efficiency, and database search scores for protein identification [32]. Finally, a variety of chemical derivatization strategies designed to introduce additional basic sites or fixed positive charges (e.g.; ammonium or phosphonium ion functional groups) have been developed and widely applied to proteome and phosphoproteome analysis studies using ESI-MS and CID- and/or ETD-MS/MS [34-48]. For example, Vasicek and Brodbelt have reported the formation of higher charge states and improved ETD-MS/MS fragmentation efficiency and sequence coverage upon alkylation of cysteine containing peptides with 3-(acrylamidopropyl)-trimethylammonium chloride [37]. For phosphopeptides, Xu et al. [39] and Zhang et al. [40] reported the use of 1-(2-pyrimidyl)piperazine for derivatization of the carboxyl groups, yielding enhanced ionization efficiencies and a shift toward higher peptide precursor ion charge states for subsequent ETD-MS/MS. The use of chemical derivatization is also attractive from the standpoint of being able to incorporate stable isotope labels for subsequent differential quantitative phosphoproteome analysis, by measuring the relative abundances of “light” and “heavy” labeled peptide precursor or product ions following acquisition of the ESI-MS or -MS/MS spectra, respectively [42-47]. However, while Thingholm et al. also reported significantly greater charging in ESI when using the commercially available isobaric Tag for Relative and Absolute Quantitation (iTRAQ) [42] and tandem mass tag (TMT) [43] reagents, a reduction in phosphopeptide identification efficiency was noted when using these tags with ion trap CID-MS/MS multistage activation and beam type (i.e., HCD) CID-MS/MS for quantitative analysis [48]. Pichler et al. also reported reduced CID-MS/MS identification rates using 8-plex iTRAQ and 6-plex TMT reagents compared with the 4-plex iTRAQ reagent [49]. Furthermore, while ion trap pulsed Q dissociation (PQD) or HCD fragmentation is typically employed for detection of the low m/z iTRAQ reporter ions for quantitation [50, 51], thereby overcoming the low mass cutoff limitation inherent to performing conventional CID-MS/MS in ion trap mass spectrometers, the fragmentation pathways for formation of the reporter ions using CID (as well as for ETD) are in competition with those for peptide backbone cleavage. Thus, when the reporter ions are formed at low relative abundance, the ability to perform accurate quantitation may be compromised. Finally, while ETD-MS/MS has been reported to identify a larger number of unique phosphopeptides than CID [23], use of the 4-plex iTRAQ reagent produces product ions that allow for only three channels of quantification, while peptides labeled with 8-plex iTRAQ tags produce reporter ions for only five channels of quantification [52, 53]. In the latter case, CID-MS3 of a characteristic ETD product ion at m/z 322 can be used to provide all eight iTRAQ reporter ions [53]. To address each of these issues, a dual ion activation strategy can be employed, whereby pulsed Q-dissociation or beam type CID (HCD)-MS/MS of a mass selected precursor ion is first used for phosphopeptide quantification, followed by ETD-MS/MS of the same precursor ion for sequence identification and phosphorylation site characterization [54, 55].
From the above discussion, it is clear that there remains room for the development of alternate chemical derivatization and associated analysis strategies that yield increased ionization efficiencies and precursor ion charge states, but without deleterious effects on the subsequent gas-phase fragmentation reactions and performance of CID- and ETD-MS/MS-based dual ion activation workflows employed for phosphopeptide identification, characterization, and quantitation. We have previously reported that peptides containing ‘fixed charge’ dialkylsulfonium ion derivatives located on selected functional groups of certain amino acids, including the thioether side chain of methionine (formed by reaction with phenacylbromide under acidic conditions) [56-58], the thiol side chain of cysteine (formed by reaction with 3-([N-bromoacetamido]propyl)-methylphenacylsulfonium bromide (BAPMPS)) [59], or the amino side chain of lysine or peptide N-termini (formed by reaction with S,S′-dimethylth-iobutanoylhydroxysuccinimide ester (DMBNHS)) [60] all undergo the exclusive neutral loss(es) of dialkylsulfide groups upon CID-MS/MS, independently of the amino acid composition and precursor ion charge state (i.e.; proton mobility), thereby enabling the selective identification of derivatized peptides from within complex mixtures. The use of stable isotope-containing methionine-specific fixed-charge derivatization reagents (i.e.; 2H5- or 13C6-labeled phenacylbromide) has previously been shown to facilitate the application of this approach toward the sensitive quantitative analysis of differential protein abundances, via ratiometric measurement of the relative abundances of the neutral loss product ions generated by CID-MS/MS dissociation of the light and heavy labeled peptide ions [56, 57]. Further structural interrogation of identified/quantified peptide ions has been achieved by subjecting the initial CID-MS/MS neutral loss product ion to CID-MS3 in a quadrupole ion trap mass spectrometer, or by energy resolved ‘pseudo’ MS3 in a triple quadrupole mass spectrometer [56-60]. Notably, the dissociation of a ‘monolinked’ N-terminal sulfonium ion derivatized phosphoserine containing peptide formed by reaction with the cross-linking reagent S-methyl 5,5’-thiodipentanoylhydroxysuccinimide was recently shown to result in preferential fragmentation at the site of the fixed charge sulfonium ion, while the phosphate group remained intact, indicating that the specificity associated with sulfonium ion fragmentation is not affected by competing losses involving the potentially labile phosphate side chain [61].
Here, the use of novel isobaric stable isotope containing DMBNHS reagents for ‘global’ derivatization of peptide N-terminal amino and lysine functional groups, combined with ion trap CID-MS/MS analysis, are expected to serve as a useful strategy for enhanced phosphopeptide quantitative analysis, as the precursor ions of ‘light’ and ‘heavy’ isotope labeled sulfonium ion containing peptides will be exclusively converted to characteristic neutral loss products upon CID-MS/MS, i.e.; no ‘dilution’ of product ion abundance will be observed due to other competing fragmentation pathways. Furthermore, analogous to other reports involving the effects of fixed charge derivatization on ionization efficiencies and charge state distributions, the introduction of sulfonium ion fixed charge derivatives to phosphopeptides are expected to lead to improved ionization efficiencies and increase the abundance of high charge state precursor ions that are amenable to enhanced structural characterization and phosphorylation site localization by subsequently using automated data-dependent CID-MS3 or ETD-MS/MS analysis strategies.
Materials and Methods
Materials
All chemicals were analytical reagent (AR), or of a comparable or higher grade, and used without further purification. N-hydroxysuccinimide (NHS), N,N′-dicyclohexylcarbodimide (DCC) and anhydrous dimethyl sulfoxide (DMSO) (stored over 3-Å molecular sieves) were purchased from Fluka (Buchs, Switzerland). Sodium methanethiolate, γ-butyrolactone, γ-butyrolactone-d6, 4-bromobutyric acid, thiourea, iodomethane-d3 and tris(hydroxymethyl)aminomethane (Tris) were from Sigma-Aldrich (St. Louis, MO, USA). Sodium hydroxide, sodium phosphate dibasic (crystal), potassium phosphate monobasic (crystal), magnesium sulfate (anhydrous), and formic acid (FA) were purchased from Spectrum Chemicals (Gardena, CA, USA). Sodium chloride, hydrochloric acid, and potassium hydroxide were purchased from Columbus Chemical Industries (Columbus, WI, USA). Potassium chloride, sulfuric acid, N,N′-dimethyl formamide (DMF), and ethyl ether were purchased from Jade Scientific (Canton, MI, USA). Glacial acetic acid, dichloromethane, chloroform, methanol (anhydrous), ethanol, isopropyl alcohol, and ethyl acetate were purchased from Mallinckrodt Chemicals (Phillipsburg, NJ, USA). Iodomethane and acetonitrile were purchased from EMD Chemicals (San Diego, CA, USA). Trifluoroacetic acid (TFA) was purchased from Pierce (Rockford, IL, USA). All aqueous solutions were prepared by using deionized water purified by a Barnstead nanopure diamond purification system (Dubuque, IA, USA). The phosphoserine-containing peptide LFTGHPEpSLER was prepared ‘in house’ by manual stepwise Fmoc-based solid-phase peptide synthesis [11, 12]. All other phosphopeptides were synthesized either by Sigma-Genosys (The Woodlands, TX, USA) or by JPT Peptide Technologies GmbH (Berlin, Germany), and were used without further purification.
S,S′-Dimethylthiobutanoylhydroxysuccinimide Ester Iodide (DMBNHS)
DMBNHS (Scheme 1) was synthesized as previously described [60], starting with preparation of methylthiobutyric acid from γ-butyrolactone, followed by activation to the NHS-ester and finally alkylation with iodomethane. The product was stored in the dark until further use.
Scheme 1.
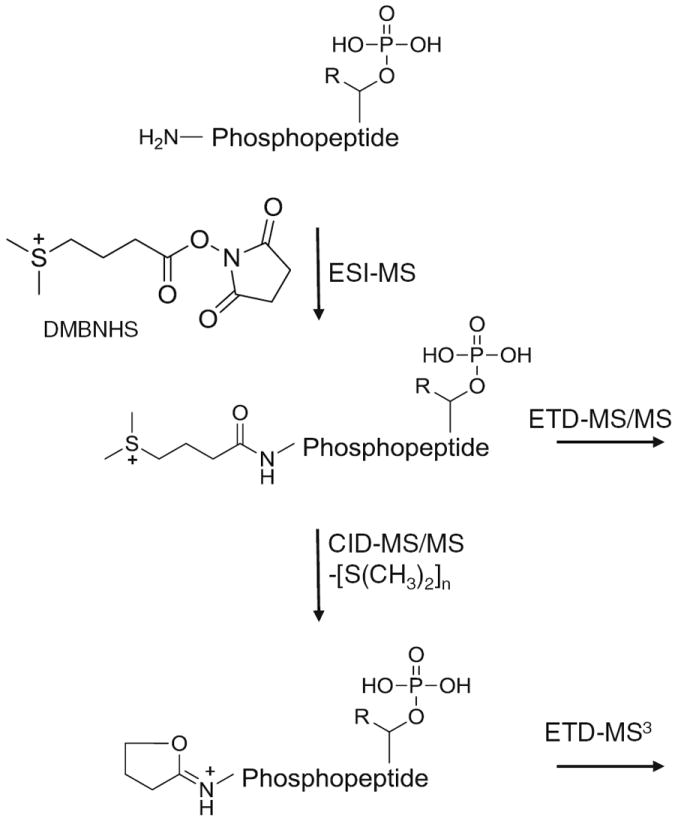
Structure and general strategy for the solution-phase derivatization, ESI-MS, and data dependent CID- and ETD-MS/MS or -MS3 fragmentation of DMBNHS labeled phosphopeptides
S,S′-Dimethylthio-d6-Butanoylhydroxysuccinimide Ester Iodide (D6-Light DMBNHS)
Using the same strategy as previously described [60] for the synthesis of DMBNHS, to a solution of 2.72 g sodium methanethiolate (34.9 mmol) in anhydrous DMSO (26 mL) under a N2 atmosphere, γ-butyrolactone-d6 (2.0 mL, 26.0 mmol) was added dropwise with stirring. The reaction was allowed to proceed at room temperature for 5 d, then 65 mL of 1 M HCl was added to the resulting slurry. Following extraction with diethyl ether (6×40 mL) and solvent evaporation under reduced pressure, the crude product was redissolved in 100 mL of dichloromethane and washed with H2O (3×25 mL). After drying with anhydrous MgSO4 and solvent evaporation in vacuo, a colorless oil of methylthio-d6-butyric acid 1 was obtained in 90% yield (3.04 g). 1H NMR (500 MHz, CDCl3): δ 2.06 (s, 3H), 11.27 (br, 1H). 2H NMR (500 MHz, CDCl3): δ 1.80 (s, 2H), 2.38 (s, 2H), 2.43 (s, broad, 2H). 1H-NMR spectra were obtained on a Varian Inova 500 MHz instrument and are reported in parts per million (ppm) relative to the solvents resonances (δ), with coupling constants (J) in Hertz (Hz). Then, 1.40 g of acid 1 (10.0 mmol) and 1.26 g of NHS (11.0 mmol) were dissolved in 2:1 vol/vol mixture of chloroform and dichloromethane (30 mL). After 5-min stirring at room temperature, DCC (2.27 g, 11.0 mmol) was added, and the resultant suspension was stirred over-night under a N2 atmosphere. The dicyclohexylurea (DCU) precipitate was then filtered and the solution was concentrated in vacuo. After resuspending the crude product in 8 mL of CH3CN, the remaining DCU was precipitated and removed by filtration. The solvent was then evaporated under reduced pressure to give methylthio-d6-butyric hydroxysuccinimide ester 2 as a yellow solid in quantitative yield. 1H NMR (500 MHz, CDCl3): δ 2.07 (s, 3H), 2.82 (s, 4H). Finally, a mixture of 2 (2.23 g, 9.4 mmol) and iodomethane (1.76 mL, 28.2 mmol) in CH3CN (16 mL) was allowed to react at room temperature in the dark for 2 d. After solvent evaporation under reduced pressure, the resultant yellow solid was washed with dichloromethane and dried in vacuo to give pale-yellow crystals of D6-light DMBNHS (Supplementary Scheme S1) in 86% yield (3.06 g). The compound was stored in the dark prior to future use. 1H NMR (500 MHz, CD3CN): δ 2.77 (s, 4H), 2.83 (s, 6H).
S,S′-d6-Dimethylthiobutanoylhydroxysuccinimide Ester Iodide (D6-Heavy DMBNHS)
Based on the method of Jessing [62], 4-bromobutyric acid (8.35 g, 50.0 mmol) and thiourea (5.70 g, 75.0 mmol) were dissolved in 100 mL of ethanol and refluxed overnight. The solvent was then evaporated under reduced pressure and 65 mL of 7.5 M NaOH (aq) was added. The mixture was refluxed at 90 °C under a N2 atmosphere. After a 16 h reaction, 2 M H2SO4 was added slowly while stirring in an ice bath until the pH=1. The resulting mixture was extracted 4 times with 60 mL of CH2Cl2, dried over anhydrous MgSO4, and concentrated in vacuo to give 5.27 g (88%) of thiobutyric acid 3 as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.33 (t, 1H, J=8.0 Hz), 1.93 (qui, 2H, J=7.0 Hz), 2.50 (t, 2H, J=7.0 Hz), 2.59 (q, 2H, J=7.0 Hz), 11.28 (s, broad, 1H). Then, using a procedure adapted from Crouch et al. [63], potassium hydroxide (1.31 g, 23.4 mmol) in 2 mL of methanol was added dropwise to 3 (1.08 g, 9 mmol) dissolved in 10 mL of methanol in an ice bath and stirred for 10 min. Iodomethane-d3 (672.4 μL, 10.8 mmol) was added slowly over 10 min and the solution was stirred at room temperature overnight. After evaporating the solvent under reduced pressure, 12 mL of H2O was added followed by 2 M HCl until the pH=1. The solution was extracted with 4×18 mL of diethyl ether. The organic extracts were combined and dried over anhydrous MgSO4. The solvent was removed in vacuo and 858.2 mg (70%) of d3-methylmercaptobutyric acid 4 was obtained as brown oil. 1H NMR (500 MHz, CDCl3): δ 1.92 (qui, 2H, J=7.0 Hz), 2.48 (t, 2H, J=7.5 Hz), 2.53 (t, 2H, J=7.5 Hz), 11.19 (s, broad, 1H). Then, under a N2 atmosphere, NHS (792.4 mg, 6.89 mmol) and 4 (858.2 mg, 6.26 mmol) were dissolved in a mixture of CHCl3 (15 mL) and CH2Cl2 (10 mL). The mixture was stirred for 5 minutes at room temperature, then DCC (1419.5 mg, 6.89 mmol) was added and a precipitate formed immediately. The resulting slurry was stirred in a N2 atmosphere. After 18 h, the mixture was filtered; the filtrate was collected and concentrated under reduced pressure. Then CH3CN (3 mL) was added and the precipitate was filtered out. The solvent was removed in vacuo to yield d3-methylmercaptobutyric hydroxysuccinimide ester 5 as a yellow solid with a quantitative yield. 1H NMR (500 MHz, CDCl3): δ 2.02 (qui, 2H, J=7.5 Hz), 2.58 (t, 2H, J=7.5 Hz), 2.74 (t, 2H, J=7.0 Hz), 2.82 (s, 4H). Finally, 5 (933.6 mg, 3.99 mmol) and d3-iodomethane (1.16 g, 7.98 mmol) were dissolved in CH3CN (8 mL) and stirred in the dark at room temperature for 2 d. The resulting solution was concentrated under reduced pressure. The resultant yellow solid was washed with dichloromethane and then dried in vacuo over night to yield 1.19 g (79%) of D6-heavy DMBNHS (Supplementary Scheme S2) as a white solid. The product was stored in the dark until further use. 1H NMR (500 MHz, CD3CN): δ 2.16 (qui, 2H, J=7.5 Hz), 2.77 (s, 4H), 2.85 (t, 2H, J=7.5 Hz), 3.30 (m, 2H).
Derivatization and Sample Preparation
Phosphopeptides (see Supplementary Table S1) were dissolved in phosphate-buffered saline (800 mg of NaCl, 217 mg of Na2HPO4 ·7H2O, 20 mg of KCl, and 20 mg of KH2PO4 per 100 mL, pH 7.5) to a concentration of 10 μM of each peptide. DMBNHS was dissolved in DMF and added to the phosphopeptide solution at a 200 to 400-molar excess of reagent to primary amine [the final DMF volume was 10% (vol/vol)]. The reaction was allowed to proceed for 30 min at room temperature in the dark then was quenched by the addition of a 160-molar excess of Tris (0.5 M, pH 8.26 at 25 °C) with incubation at room temperature for 10 min to ensure complete deactivation of the derivatization reagent. In order to compare the ionization efficiencies of underivatized and DMBNHS derivatized phosphopeptides, equal amounts of underivatized phosphopeptides were then spiked into the solutions, which was diluted to a final individual peptide concentration of 0.2 μM with 3% acetic acid/5% CH3CN prior to HPLC-MS analysis.
For a proof-of-principle demonstration of quantitative analysis, the same reaction conditions were employed for isobaric differential isotope labeling of a 6-phosphopeptide mixture (no. 3, 7, 22, 24, 27, and 44 in Table S1), using either D6-light DMBNHS or D6-heavy DMBNHS reagent. 40 μL of peptide solution (25 μM each peptide) was combined with 4 μL of reagent solution (0.4 M in DMF) to maintain a 200-molar excess of reagent to primary amine group. Following incubation and quenching, the D6-light and D6-heavy DMBNHS labeled phosphopeptide mixtures were pooled in molar ratios of 5:1, 2:1, 1:1, 1:2, and 1:5. Each solution was then diluted into 3% acetic acid/5% CH3CN prior to HPLC-MS analysis.
HPLC-ESI-MS, CID-MS/MS, -MS3, and ETD-MS/MS
HPLC-MS, -MS/MS, and -MS3 analysis was performed using either an LTQ or LTQ XL linear quadrupole ion trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled with a Paradigm MS4 capillary RP-HPLC system (Michrom Bioresources, Auburn, CA, USA) equipped with an Advance nESI source. Phosphopeptide mixtures were separated on a 200 μm i.d. × 50 mm fused silica column packed with Magic C18 (3 μm, 200Å; Michrom Bioresources). Gradient elution was performed linearly over a 45-min period from 95% solvent A (0.1% formic acid in H2O) to 50% solvent B (0.1% formic acid in CH3CN) at a flow rate of 2 μL min−1. The ion transfer tube of the mass spectrometer was set at 180 °C and the spray voltage was maintained at 2.0 kV. All MS and MS/MS spectra were collected automatically using methods created with the Xcalibur software (Thermo, San Jose, CA, USA) and all spectra were recorded in centroid mode. For CID-MS/MS and -MS3 analysis using the LTQ, the top three most abundant precursor ions from each MS scan above a threshold of 10,000 counts were selected for CID-MS/MS using standard conditions. The isolation window was maintained at 2.0m/z and the normalized collision energy was set at 25%. Then, automated CID-MS3 was performed using a data-dependent constant neutral loss scan mode (DDCNL) of operation, as previously described [60]. When target neutral losses (see Supplementary Tables S2 and S3) within an m/z variance of ±0.5 were detected above a threshold of 10,000, CID-MS3 was automatically initiated to further fragment the most abundant neutral loss product ion. Dynamic exclusion was enabled to allow a maximum of five analyses of the selected m/z within 30 s before it was placed on a dynamic exclusion list for a period of 10 s. Alternatively, using the LTQ XL operated in an ‘Nth order double play with ETD’ mode of operation, the top five precursor ions from each MS scan above a threshold abundance of 10,000 counts were selected for sequential CID-MS/MS and ETD-MS/MS analysis, with the isolation window maintained at 2.0m/z. The normalized collision energy was set at 30% for CID experiments. ETD experiments were carried out by using fluoranthene anions as the electron-transfer reagent with optimized reaction times of 30 ms and ETcaD enabled.
Data Analysis
The relative abundance of phosphopeptide ions was determined from the full mass spectra averaged over the corresponding chromatographic peaks. The total ionization efficiency was calculated as the sum of the relative abundances of each observed peptide ion charge state. Quantitative analysis was achieved by calculating the ratio of “light” and “heavy” labeled neutral loss [i.e., S(CH3)2 and S(CD3)2] product ion abundances determined following HPLC-CID-MS/MS analysis of D6-light and D6-heavy DMBNHS labeled phosphopeptides for all available charge states. Multiple neutral losses were observed from multiply modified phosphopeptides. In some cases, other neutral losses such as H2O and NH3 were also observed along with the target neutral losses. In these cases, the summed abundances from all neutral loss product ions were used for determination of the ratio. During ETD-MS/MS, neutral losses of S(CH3)2 or S(CD3)2 were observed from the intact charge reduced precursor ions. In these cases, the same calculations as those described above were applied for relative quantitative analysis.
Results and Discussion
Evaluation of ESI-MS Ionization Efficiencies and Charge State Distributions of Phosphopeptide Ions upon DMBNHS Derivatization
To determine the effect of DMBNHS derivatization on ESI-MS ionization efficiencies and charge state distributions compared to their non-derivatized forms, 50 ‘tryptic’ phosphopeptides whose sequences, phosphorylation site locations, number of phosphorylation sites, peptide mass, peptide charge states, potential for missed cleavages etc.; are representative of in vivo phosphopeptides previously reported in the literature were synthesized, subjected to reaction with DMBNHS, mixed with an equal amount of unmodified peptide, then analyzed by HPLC-ESI-MS. To avoid signal suppression among co-eluting phosphopeptides, the 50 peptides were divided into 10 groups prior to analysis. Some peptides were present in multiple mixtures in order to evaluate inter-sample variances. The normalized and absolute abundances of each phosphopeptide are shown in Figure 1 and in the inset to Figure 1, respectively, in an order corresponding to the observed chromatographic elution times of the underivatized peptides. Triplicate analysis was found to result in less than 20% variation in abundance for each phosphopeptide.
Figure 1.
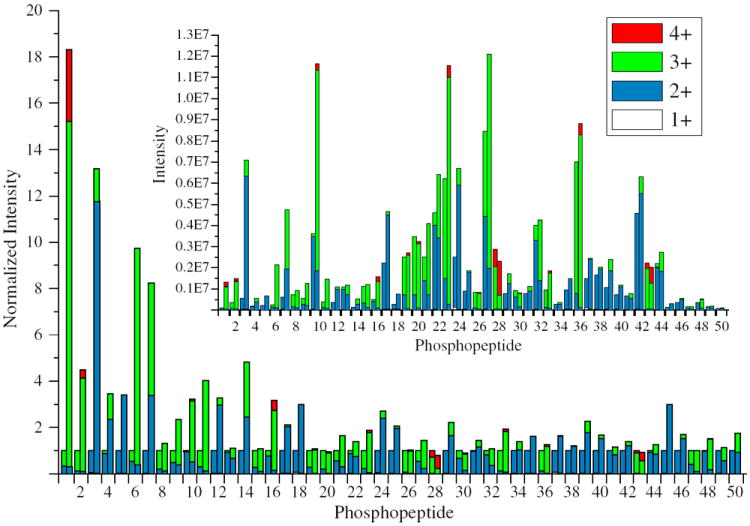
Normalized intensities and charge state distributions of 50 underivatized and DMBNHS derivatized phosphopeptides following HPLC-MS analysis. The sequence of each phosphopeptide is listed in Table S1. For each peptide, the underivatized and derivatized peptide data are shown on the left and right hand side of each column, respectively. The inset shows the measured intensities for each peptide
An average 2.5-fold increase in total ESI-MS ionization efficiency was observed for the derivatized peptides compared with their non-derivatized forms. Interestingly, an average 1.8-fold enhancement was observed for singly modified phosphopeptides (i.e., those containing only a single amino group at the N-terminus), while an average 3.5-fold enhancement was observed for doubly modified species (i.e., those containing N-terminal amino and lysine side chain ε-amino groups), indicating an accumulation effect from the presence of multiple fixed charge derivatives. It was observed that DMBNHS labeled phosphopeptides generally had longer retention times than their unlabeled counterparts, likely due to the increased hydrophobicity of the alkyl chain that was present within the DMBNHS label (s). It was also observed that earlier-eluting phosphopeptides generally demonstrated a greater enhancement in ionization efficiency compared to later-eluting ones, suggesting that the magnitude of enhancement in ionization efficiency upon DMBNHS labeling was related to the phosphopeptide’s hydrophobicity [64]. For example, for phosphopeptides 1 (ETEEQDpSDSAEQGDPAGEGK) and 50 (AFLpSPPTLLEGPLR), which eluted first and last from the reversed-phase C18 column among the 50 peptides examined here, the doubly modified peptide 1 experienced an 18.3-fold increase in total ionization efficiency (with a significant difference in elution time between the modified and unmodified peptide of +6.06 min); while there was only a 1.76-fold increase for the singly modified peptide 50 (with an essentially unchanged difference in elution time between the modified and unmodified peptide of −0.36 min). Typically, there is a linear relationship between peptide retention time in RP-HPLC and its hydrophobicity index [65]; hence, it may be concluded that the more hydrophilic phosphopeptides tend to be more effective for improved ionization efficiency by fixed charge DMBNHS labeling, and that the contribution of DMBNHS derivatization to improved phosphopeptide ionization efficiency and charge state distribution is not simply due to the presence of the positively charged sulfonium ion, but also from the presence of the hydrophobic propyl chain within the tag attached to the modified amino group. In addition, due to their typical longer retention times, labeled phosphopeptides are eluted into the mass spectrometer with a higher fraction of organic solvent compared to their unlabeled forms, which may also facilitate improved ESI efficiencies [66].
In addition to an increase in overall ionization efficiencies, another important outcome of DMBNHS derivatization was an increase in precursor ion charge states and/or abundances (see Table S1 for the observed abundances and % charge state abundances of each unlabeled and labeled phosphopeptide). A summary of the average % charge state intensities following HPLC-ESI/MS of the 50 underivatized and DMBNHS derivatized phosphopeptides is shown in Table 1, along with separate summaries for the singly and doubly modified phosphopeptides. Twenty-two of the 50 phosphopeptides (44%) yielded precursor ions with maximum charge states corresponding to +2 in their underivatized states, 26 had precursor ions with maximum charge states corresponding to +3, and two had precursor ions with maximum charge states corresponding to +4. In contrast, following DMBNHS derivatization, only 4 of the 50 phosphopeptides yielded precursor ions whose maximum charge states were still +2, while 33 had precursor ions with maximum charge states corresponding to +3, and 13 had precursor ions with maximum charge states corresponding to +4. Overall, higher charge states were observed for 29 of the 50 labeled phosphopeptides following derivatization. For example, for phosphopeptide no. 39 (pSPGAPGPLTLK) that gave an overall 2.25-fold increase in ionization efficiency upon labeling, only singly and doubly charged precursor ions were observed before derivatization (Figure 2a) while the doubly DMBNHS modified form of the peptide (Figure 2b) yielded abundant doubly and triply charged species. Similarly, phosphopeptide no. 23 (VGEEEHVpYSFPNK) gave an overall 1.87-fold increase in ionization, with an increase from doubly and triply charged precursor ions in the underivatized state, to doubly, triply, and quadruply charged precursor ion species upon derivatization (see Supplementary Figure S1A and B, respectively). The data in Table 1 indicates that doubly-modified peptides gave rise to a greater increase in maximum charge states upon derivatization, which is consistent with the results described above regarding the overall increases in ionization efficiency for doubly versus singly DMBNHS modified phosphopeptides.
Table 1.
Average % Charge State Intensities Following HPLC-ESI/MS of Underivatized and DMBNHS Derivatized Phosphopeptides
| All phosphopeptides (50) | Average % charge state intensities | |||
|---|---|---|---|---|
| 1+ | 2+ | 3+ | 4+ | |
| Unlabeled | 0.85±1.39a | 70.13±35.27 | 28.24±34.58 | 0.79±4.29 |
| Labeled | 0.06±0.12 | 46.33±38.67 | 49.87±35.94 | 3.75±11.45 |
| Ratio (labeled/unlabeled) | 0.071 | 0.66 | 1.77 | 4.75 |
| Phosphopeptides containing 1 modifiable site (30) | Average % | |||
| 1+ | 2+ | 3+ | 4+ | |
| Unlabeled | 1.03±1.57 | 74.40±35.10 | 23.26±33.28 | 1.31±5.51 |
| Labeled | 0.09±0.15 | 59.01±37.69 | 36.91±33.90 | 3.99±14.34 |
| Ratio (labeled/unlabeled) | 0.087 | 0.79 | 1.59 | 3.05 |
| Phosphopeptides containing 2 modifiable sites (20) | Average % | |||
| 1+ | 2+ | 3+ | 4+ | |
| Unlabeled | 0.57±1.04 | 63.72±35.43 | 35.71±35.99 | 0.00±0.00 |
| Labeled | 0.00±0.01 | 27.30±32.45 | 69.31±30.22 | 3.39±4.89 |
| Ratio (labeled/unlabeled) | 0.00 | 0.43 | 1.94 | |
Errors indicate±standard deviation
Figure 2.
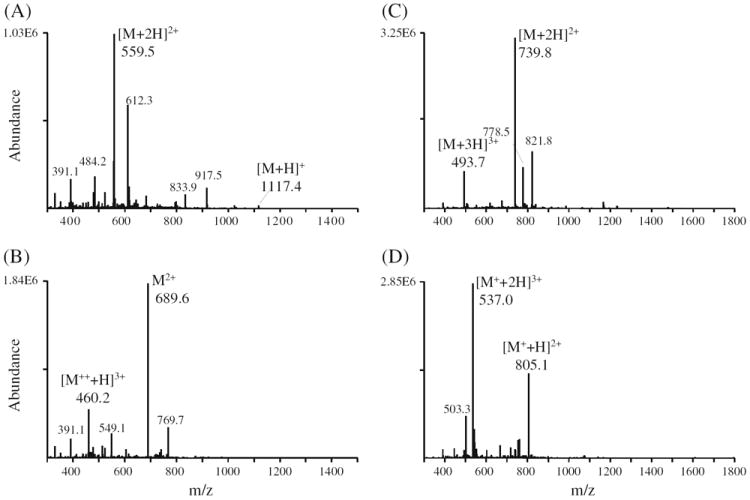
HPLC-ESI-MS spectra of underivatized and DMBNHS derivatized phosphopeptide no. 39 (pSPGAPGPLTLK) [panels (A) and (B), respectively] and phosphopeptide no. 32 (pYATPQVIQAPGPR) [panels (C) and (D), respectively]
Importantly, for the 21 phosphopeptides whose maximum charge states were not observed to increase upon labeling, an increase in the abundance of the highest charge state present was observed in each case. For example, Figure 2c and d show the spectra obtained following HPLC-ESI-MS analysis of the unmodified and singly DMBNHS modified phosphopeptide no. 32 (pYATPQVIQAPGPR). For this peptide, although only a very modest 1.06-fold increase in overall ionization efficiency was observed, a 3.98-fold increase in the abundance of the triply charged precursor (i.e., the highest observed charge state) from 7.15E5 to 2.85E6 was still achieved. Similarly, for phosphopeptide no. 43 (LGHPEALSAGpTGSPQPPSFTYAQQR), (see Supplementary Figure S1C and D, respectively) the abundance of the highest observed quadruply charged precursor ion charge state was increased from 2.50E5 to 7.38E5 (2.95-fold), despite a minor decrease in overall ionization efficiency (0.91-fold) upon derivatization.
Fixed Charge Sulfonium Ion Derivatization for Enhanced Collision Induced Dissociation (CID) and Electron Transfer Dissociation (ETD) Characterization of Phosphopeptide Ions
CID-MS/MS and -MS3 Analysis of DMBNHS Labeled Phosphopeptides
CID-MS/MS and -MS3 spectra of the singly DMBNHS modified phosphopeptide no. 43 (LGHPEALSAGpTGSPQPPSFTYAQQR) are shown in Figure 3a and b, respectively. Consistent with previous studies on the gas-phase fragmentation reactions of fixed charge sulfonium ion containing peptides, the exclusive neutral loss of dimethylsulfide [S (CH3)2] was observed upon dissociation of the triply charged [M+ + 2H]3+ precursor ion [56-61]. Notably, also consistent with previous results, the lability of the sulfonium ion derivative to undergo cleavage resulting in the specific neutral loss of S(CH3)2 was found to essentially completely suppress any competing loss of H3PO4 from the phosphothreonine side chain [61]. CID-MS3 of the neutral loss product ion from Figure 3a (see Figure 3b) gave rise to a range of sequence-type product ions resulting from cleavages along the peptide backbone, from which the peptide sequence and site of phosphorylation could potentially be derived, together with some relatively low abundance non-sequence-type ions corresponding to losses of the phosphate group and/or water from the precursor or sequence-type product ions. It is expected that as the proton affinity of the product ion structure formed via the loss of dimethylsulfide during CID-MS/MS (previously proposed to be a cyclic iminohydrofuran (IHF) moiety [58, 60]) will be higher than that of an amino group [60], the neutral loss product ion that is initially formed may have decreased proton mobility compared to that of the unmodified peptide of the same charge state. Decreased proton mobility within IHF-modified phosphopeptides could lead to more abundant loss of the phosphate group or increase the potential for phosphate group ‘scrambling’ under CID-MS3 conditions [12], which therefore could limit the extent of unambiguous sequence information for subsequent phosphopeptide identification and characterization. However, for the DMBNHS derivatized phosphopeptide no. 43, the appearance of the CID-MS3 spectrum was very similar to that obtained by CID-MS/MS of the unmodified triply protonated ([M+3H]3+) precursor ion shown in Figure 3c, demonstrating that in this case, the initial product ion is stable to further dissociation involving the IHF-moiety, and had only a limited influence on CID-MS3 product ion formation compared to the unmodified peptide.
Figure 3.
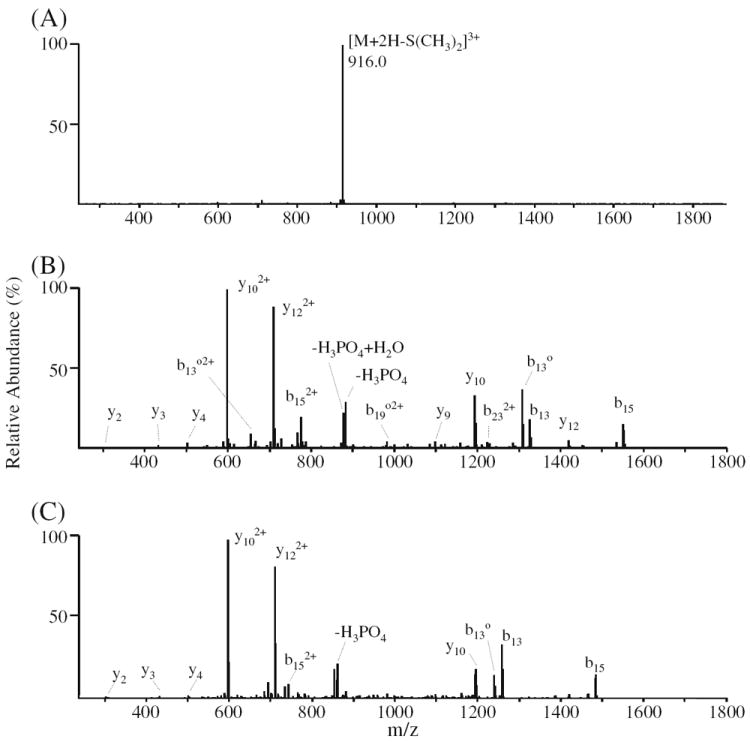
ESI-MSn analysis of phosphopeptide no. 43 (LGHPEALSAGpTGSPQPPSFTYAQQR). (A) CID-MS/MS of the [M+ + 2H]3+ precursor ion from the singly derivatized phosphopeptide. (B) CID-MS3 of the m/z 916.0 neutral loss product ion from panel (B). (C) CID-MS/MS of the [M+3H]3+ precursor ion from the underivatized phosphopeptide. Δ=−98 Da (−H3PO4 or − (HPO3+H2O)); o=−18 Da (H2O)
CID-MS/MS of the doubly modified phosphotyrosine-containing peptide no. 23 (VGEEEHVpYSFPNK) yielded two product ions corresponding to the characteristic sequential exclusive neutral losses of one and two S(CH3)2 molecules, thereby providing diagnostic information regarding the presence of a lysine residue within the peptide, in addition to the α-amino group (Figure 4a). Similar characteristic double neutral losses were observed for all peptides that were doubly DMBNHS modified (another example is shown in Supplementary Figure S2A for the doubly modified phosphoserine-containing peptide no. 46 (ASpSLEDLVLK)). The presence of these diagnostic double neutral losses could easily be utilized for pre- or post-search filtering to improve the specificity of database search procedures for large scale protein sequence analysis, analogous to that previously described using the characteristic immonium ions formed by CID-MS/MS [67], or the diagnostic neutral loss product ions formed by ETD-MS/MS [68]. CID of the m/z 584.5 product ion from Figure 4a resulted in an MS3 product ion spectrum (Figure 4b) containing an abundant series of both b- and y-type ions and substantially greater sequence coverage compared to that obtained by CID-MS/MS of the unmodified peptide, which was dominated by a single y122+ product ion (Figure 4c). This is likely due to a change in either the relative location of the ionizing protons between the two precursor ions, that altered the propensity for cleavage of particular amide bonds, or more likely due to an increase in the relative basicity of the N-terminal peptide fragments resulting from the presence of the IHF-moiety at the former α-amino group within the DMBNHS derivatized peptide. In contrast, for the doubly DMBNHS modified phosphopeptide no. 46, CID-MS3 of the product ion formed via the initial double neutral loss of two S(CH3)2 groups (Supplementary Figure S2B) resulted in a spectrum with essentially the same sequence ion coverage compared to the unmodified peptide (Supplementary Figure S2C), but with substantially more highly abundant H3PO4 neutral loss and H3PO4+H2O neutral loss product ions. In this case, the dominant loss of H3PO4, likely formed due to an overall decrease in proton mobility or proton location within the initial neutral loss product ion formed by MS/MS, increases the possibility of phosphate group ‘scrambling’ [12]. Therefore, caution should be used in employing CID-MS3 as part of an analysis strategy coupled with DMBNHS labeling for the identification and characterization of unknown phosphopeptides.
Figure 4.
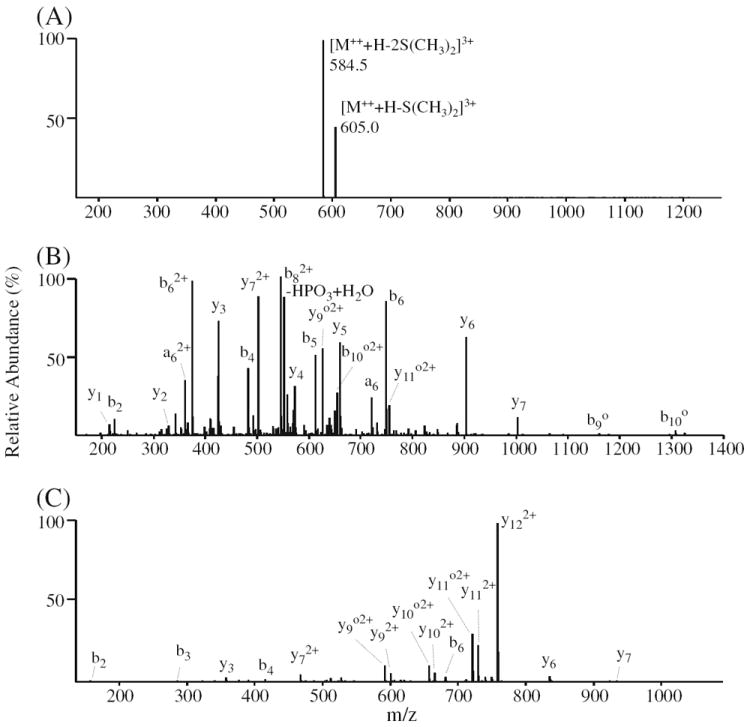
ESI-MSn analysis of phosphopeptide no. 23 (VGEEEHVpYSFPNK). (A) CID-MS/MS of the [M++ + H]3+ precursor ion from the doubly derivatized phosphopeptide. (B) CID-MS3 of the m/z 584.5 double neutral loss product ion from panel (A). (C) CID-MS/MS of the [M+3H]3+ precursor ion from the underivatized phosphopeptide. Δ=−98 Da [−H3PO4 or –(HPO3+H2O)]; o= −18 Da (H2O)
ETD-MS/MS Analysis of DMBNHS Labeled Phosphopeptides
At present, the ETD fragmentation behavior of fixed charge sulfonium ion containing peptides has not been explored. Here, since ETD fragmentation efficiency is highly dependent on the charge density of the precursor ions, and typically a precursor ion charge state of at least 3+ is required to obtain high quality ETD-MS/MS spectra (recall that a favorable shift from doubly to triply charged precursor ions was observed upon DMBNHS labeling; see Table 1), the ETD fragmentation behavior of triply charged phosphopeptide ions were of most interest to this study. A representative example of the ETD-MS/MS fragmentation for the triply charged precursor ions of a singly DMBNHS labeled and unlabeled phosphopeptide (peptide no. 43, LGHPEAL-SAGpTGSPQPPSFTYAQQR) are shown in Figure 5a and b, respectively, while Figure 6a and b show representative ETD-MS/MS spectra for a doubly DMBNHS labeled and unlabeled phosphopeptide (peptide no. 23, VGEEEHVpYSFPNK).
Figure 5.
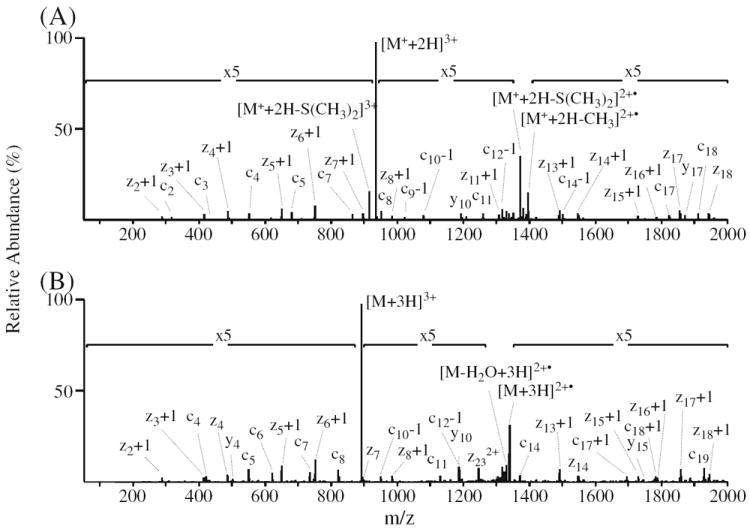
ETD-MS/MS analysis of phosphopeptide no. 43 (LGHPEALSAGpTGSPQPPSFTYAQQR). (A) ETD-MS/MS of the [M+ + 2H]3+ precursor ion from the singly derivatized phosphopeptide. (B) ETD-MS/MS of the [M+3H]3+ precursor ion from the underivatized phosphopeptide
Figure 6.
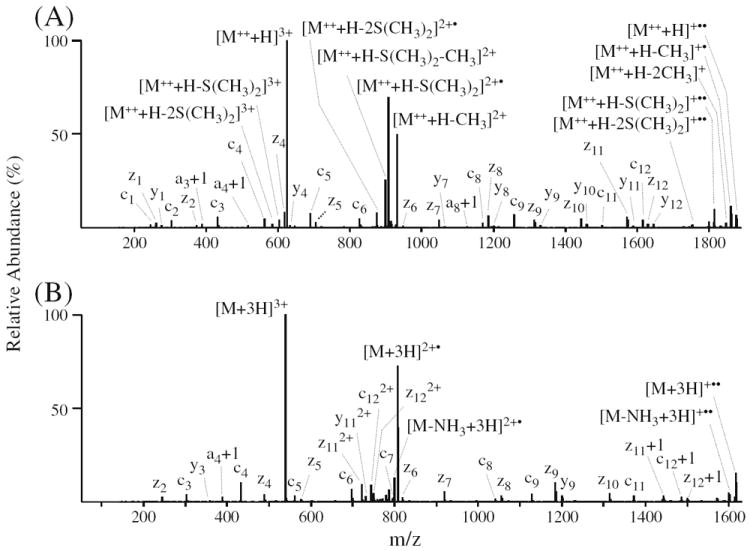
ETD-MS/MS analysis of phosphopeptide no. 23 (VGEEEHVpYSFPNK). (A) ETD-MS/MS of the [M++ + H]3+ precursor ion from the doubly derivatized phosphopeptide. (B) ETD-MS/MS of the [M+3H]3+ precursor ion from the underivatized phosphopeptide
Notably, the fragmentation efficiency and z- and c-type product ion distributions and abundances observed following ETD of the triply charged singly DMBNHS labeled phosphopeptide 43 in Figure 5a were essentially identical to that of the unlabeled peptide in Figure 5b. A mass shift of 130 Da observed on all c-type product ions confirmed that the modification was located at the amino group of the N-terminus. The appearance of a low mass c-ion (e.g., c2) in the product ion spectrum of the labeled peptide, which was not present in the spectrum of the unlabeled peptide, provided the potential to improve the overall sequence coverage for phosphopeptide characterization. Similarly increased numbers of low mass c-type ions in the ETD spectra of DMBNHS labeled peptides were observed for other phosphopeptides examined here (see below). The absence of the doubly charged z232+ ion in the spectrum of the derivatized peptide is probably due to the reduced number of ionizing protons present within the labeled peptide (i.e., one of the three charge carriers is the fixed charge sulfonium ion instead of a proton). Li et al. have previously reported a greatly reduced abundance of backbone fragments obtained via ECD-MS/MS of peptides labeled with 2,4,6-trimethylpyridine (TMP); the authors reasoned that the pyridinium ion can form a stable radical upon capture of the electron, such that further N–Cα dissociations are suppressed [69]. However, enhanced sequence coverage has been reported following ECD of phosphonium labeled glycopeptides and phosphopeptides [70], as well as following ETD of trimethylammonium labeled cysteine peptides [37]. This might be attributable to the different recombination energies of the introduced fixed charge containing functional groups compared to that of protonated basic groups within the peptides [69, 70].
Compared with the unlabeled phosphopeptide 43 (Figure 5b), the most significant difference observed in the ETD-MS/MS spectrum of the DMBNHS labeled peptide is the presence of abundant methyl radical and dimethylsulfide neutral losses from the charge reduced intact doubly charged precursor ion (Figure 5a). Side-chain losses or entire losses of the tag have been a common observation from previous reports regarding dissociation of fixed charge derivatized peptides by electron driven techniques [37, 69, 70]. These fragmentations likely result from initial capture of the electron by the fixed charge functional group residing at either aromatic rings or aliphatic chains. Similarly, the transfer of an electron from the ETD anionic reagent to the fixed charge sulfonium ion derivative in the current study may result in formation of a sulfur radical (see Supplementary Scheme S3), which triggers subsequent homolytic cleavage of the adjacent C–S bonds, and resulting in methyl radical or dimethylsulfide group losses. The presence of sequence ions resulting from peptide backbone N – Cα cleavage and from non-sequence ion side-chain losses resulting from cleavage at the sulfonium ion moiety indicates competition for the electron between the protonated peptide and the fixed charge tag. Interestingly, a low abundance dimethylsulfide neutral loss (3.2% relative base peak abundance) was also observed from the intact precursor ion, similar to that observed from CID-MS/MS of DMBNHS labeled phosphopeptide. This loss is most likely generated by resonant collisional activation during precursor ion isolation, or by the supplemental collisional activation applied in the ETD experiments [71].
Similar results were also obtained when comparing the ETD-MS/MS spectra of the triply charged doubly labeled and unlabeled phosphotyrosine containing peptide VGEEEHVpYSFPNK (no. 23), as shown in Figure 6a and b, respectively. The addition of a second sulfonium ion containing tag did not cause any noticeable differences in the peptide backbone cleavage efficiency or product ion abundances. However, an overall increase in sequence coverage was achieved due to the appearance of low mass c1, c2, and z1 ions. Similar to that discussed above, doubly charged product ions (e.g., z112+, y112+, c122+, and z122+) observed as abundant products in the ETD-MS/MS spectrum of the unlabeled peptide were not observed in the product ion spectrum for the doubly DMBNHS labeled peptide. Multiple neutral losses of abundant methyl radical and dimethylsulfide groups from the charge reduced intact precursor ions of the doubly DMBNHS labeled peptide were observed (Figure 6a). For the neutral loss products arising from the initial charge reduced doubly charged product, the loss of a second tag [i.e., −S(CH3)2 or −CH3• in addition to the loss of a first S(CH3)2 group] was probably a result of the applied supplemental collisional activation employed during ETD experiments, since only one electron was transferred to the peptide precursor ion. However, for the singly charged neutral loss product ions arising from the charge reduced singly charged intact precursor, secondary electron transfer likely induces cleavage on the second sulfonium ion tag. This is consistent with the fact that double CH3• losses were mainly found in the charge reduced singly charged intact precursor ions.
Another example is shown in Supplementary Figure S3A for ETD-MS/MS of the triply charged precursor ion of the doubly DMBNHS labeled phosphoserine containing peptide pSPGAPGPLTLK (no. 39). For the unmodified form of this peptide, ETD-MS/MS could only be obtained for the doubly protonated precursor ion, since a triply protonated precursor ion charge state was not observed in the MS spectrum (see Supplementary Figure S3B). Notably, only very limited and low abundance sequence ions were observed in the latter case, consistent with previous reports on the typically limited dissociation efficiency of low charge state precursor ions by ETD. Clearly, the formation of higher charge state precursor ions by using sulfonium ion derivatization enables enhanced dissociation efficiencies and increased sequence-coverage by using ETD, and allows access to a broader range of phosphopeptide sequences (up to 44% of the total) that may otherwise be inaccessible to analysis using this technique.
Fixed Charge Sulfonium Ion Derivation Coupled with Isobaric Stable Isotope Labeling for Enhanced Collision Induced Dissociation (CID) and Electron Transfer Dissociation (ETD) Quantitative Phosphopeptide Analysis
As described above, the introduction of sulfonium ion fixed charge derivatives to phosphopeptides provides improved ionization efficiencies and increased abundance high charge state precursor ions that are amenable to enhanced structural characterization and phosphorylation site localization by using ETD-MS/MS analysis strategies. Furthermore, the dissociation of sulfonium ion labeled phosphopeptides gave rise to exclusive S(CH3)2 neutral loss product ions upon CID-MS/MS. In order to apply this desirable ionization and gas-phase fragmentation behavior toward the sensitive differential quantitative analysis of phosphopeptide abundances, via ratiometric measurement of the relative abundances of the characteristic neutral loss product ions generated by CID-MS/MS dissociation of ‘light’ and ‘heavy’ labeled peptide ions, the novel isobaric stable isotope labeled sulfonium ion containing reagents S,S′-dimethylthio-d6-butanoylhydroxysuccinimide ester iodide (D6-light DMBNHS) and S,S′-d6-dimethylthiobutanoylhydroxysuccinimide ester iodide (D6-heavy DMBNHS) (Scheme 2) were prepared by the introduction of six deuteriums into (1) the butyl chain of the DMBNHS structure, via the reaction of d6-γ-butyrolactone with sodium methanethiolate, followed by esterification with NHS and alkylation with iodomethane, or (2) the dimethylsulfide moiety of the DMBNHS structure, via the reaction of 4-bromobutyric acid with thiourea, followed by alkylation with d3-iodomethane, esterification with NHS and final alkylation with d3-iodomethane, respectively. Upon CID-MS/MS of a precursor ion containing the two isobaric peptides labeled with these two reagents, product ions corresponding to the exclusive neutral losses of S(CH3)2 (62 Da) and S(CD3)2 (68 Da) are expected (Scheme 2), which may be used for differential quantification of phosphopeptide abundances between two samples. Then, the same precursor ion can be automatically subjected to ETD-MS/MS to obtain the necessary structural information for phosphopeptide sequence identification and phosphorylation site localization.
Scheme 2.
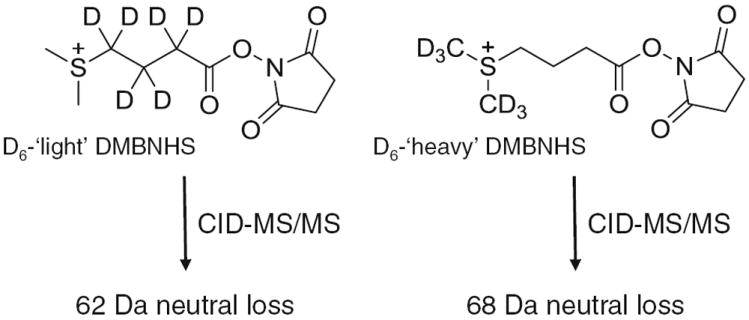
Structures and CID-MS/MS fragmentation reactions of the stable isotope labeled DMBNHS reagents S,S′-dimethylthio-d6-butanoylhydroxysuccinimide ester iodide (D6-light DMBNHS) and S,S′-d6-dimethylthiobutanoylhydroxysuccinimide ester iodide (D6-heavy DMBNHS)
In a proof of principle demonstration of this approach, a mixture of six synthetic phosphopeptides (no’s. 3, 7, 22, 24, 27, and 44 in Table S1) were differentially labeled using either the D6-light or D6-heavy DMBNHS reagents, then pooled in a range of ratios from 5:1 to 1:5 and subjected to analysis by LC-MS and data dependent CID- and ETD-MS/MS. Importantly, the D6-light and D6-heavy DMBNHS labeled phosphopeptides co-eluted from the reversed- phase C18 column, appearing as single chromatographic peaks for each peptide (Figure 7a). Due to their isobaric nature, differentially labeled peptides also appeared as single peaks in the MS spectra (an example is shown in Figure 7b for peptide no. 27), thereby providing an enhanced peak capacity for their detection. Following acquisition of each MS scan, precursor ions were then subjected to data dependent MS/MS analysis using an ‘Nth order double play with ETD’ mode of operation, whereby the top five precursor ions from each MS scan above a threshold abundance of 10,000 counts were selected for sequential CID-MS/MS and ETD-MS/MS.
Figure 7.
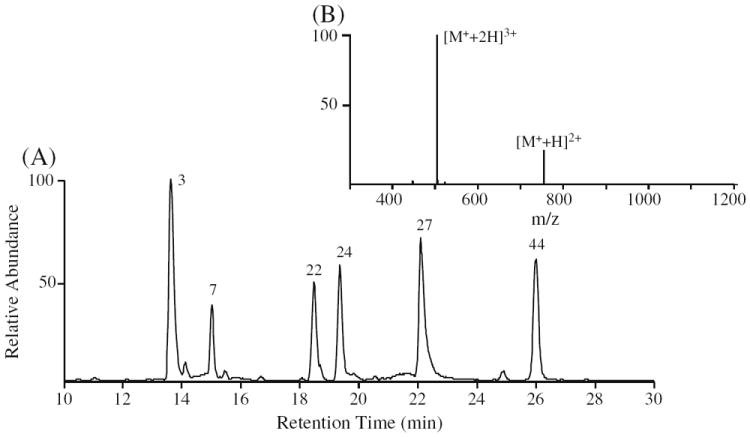
RP-HPLC ESI-MS analysis of D6-light DMBNHS and D6-heavy DMBNHS differentially labeled phosphopeptides. (A) Base peak chromatogram of a representative 6-phosphopeptide mixture differentially labeled with D6-light DMBNHS and D6-heavy DMBNHS at a ratio of 1:1. The sequence of each phosphopeptide is listed in Table S1. (B) Mass spectrum obtained by ESI analysis of the singly derivatized phosphopeptide no. 27 (LFTGHPEpSLER)
Representative spectra are shown in Figure 8a and b for dissociation of the triply charged singly labeled phosphoserine containing peptide LFTGHPEpSLER (no. 27), labeled with a 1:1 ratio of D6-light and D6-heavy DMBNHS reagent, using CID-MS/MS (Figure 8a) and ETD-MS/MS (Figure 8b). As expected, two product ions were exclusively formed upon CID-MS/MS, corresponding to the neutral losses of S(CH3)2 (62 Da) and S(CD3)2 (68 Da), respectively (inset to Figure 8a). A similar result was also observed by CID-MS/MS of the doubly charged precursor ion (data not shown). It can be seen from Figure 8a that the relative abundance of the neutral loss product ion resulting from dissociation of the D6-light labeled phosphopeptide is lower than the expected value. This is due to the presence of a D5-light DMBNHS impurity resulting from the use of a 98 atom %D labeled γ-butyrolactone-d6 starting material for our synthesis. Although a kinetic isotope effect on the fragment ion yields associated with the “light” and “heavy” dimethylsulfide loss could also potentially contribute to this discrepancy, fragmentation efficiencies regarding D6-light and D6-heavy DMBNHS and their differentially labeled peptides were not systematically examined in this study, However, after taking into account the presence of the impurity, the correct relative abundance of the S(CH3)2 neutral loss product ion could be readily determined. It is known that using deuterated isotope labels can cause some chromatographic separation of “light” and “heavy” labeled peptides [72], potentially resulting in deviations to quantitative analysis depending on the time point across the chromatographic peak at which a given precursor ion is selected for analysis. However, the use of isobaric deuterated reagents should diminish or eliminate any shift in retention time of labeled peptides during LC separation. In the current study, some of the D6-light labeled peptides were observed to elute slightly earlier than their D6-heavy labeled counterparts, but, the effect of this on accurate quantification was minimized by simply averaging all the CID-MS/MS scans (repeat count=5) that were acquired for a given precursor ion across each chromatographic peak. Following analysis of the various mixtures of light- and heavy-labeled phosphopeptide no. 27, from both its 3+ and 2+ precursor ion charge states, a linear relationship (R2=0.9999) between the measured and expected ratios (5:1, 5:1, 2:1, 1:1, 1:2, and 1:5) was obtained (see Supplementary Figure S4). Indeed, for each of the six differentially labeled phosphopeptides in the mixture, an excellent agreement between measured and expected values was obtained. A plot showing the average ratio for all peptides, determined from both their 3+ and 2+ charge states, is shown in Supplementary Figure S5 (R2=0.9986). Note that although 3+ charge states were observed for all the peptides upon DMBNHS labeling, the most abundant charge state for peptides no. 3, 22, 24, and 44 remained 2+; hence these ions provided the greatest sensitivity for quantification in these cases.
Figure 8.
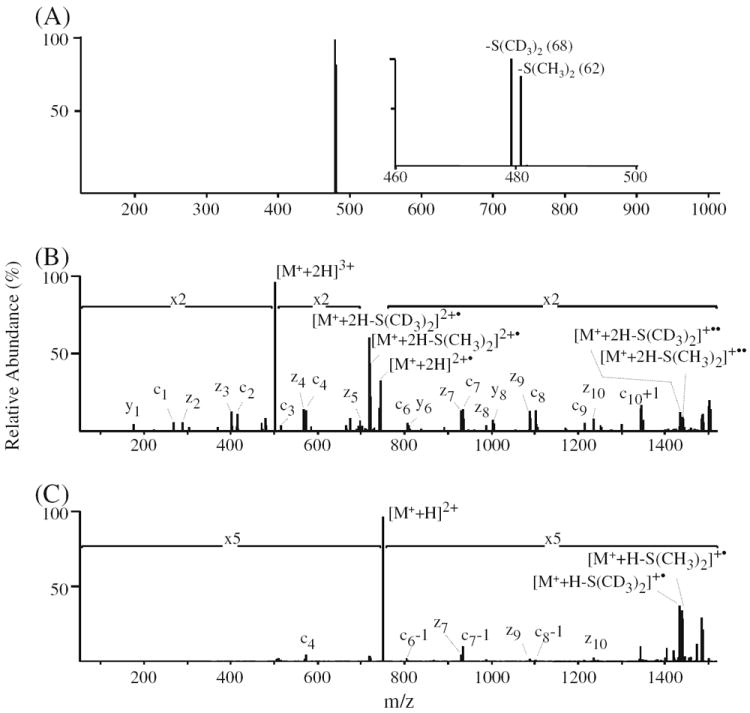
CID- and ETD-MS/MSanalysis of phosphopeptide no. 27 (LFTGHPEpSLER) differentially labeled with D6-light DMBNHS and D6-heavy DMBNHS at a ratio of 1:1. (A) CID-MS/MS of the [M+ + 2H]3+ precursor ion. The inset shows an expanded m/z region around the observed exclusive S(CH3)2 and S(CD3)2 neutral loss product ions. (B) ETD-MS/MS of the [M+ + 2H]3+ precursor ion. (C) ETD-MS/MS of the [M+ +H]2+ precursor ion
Consistent with the results obtained by ETD-MS/MS of the natural abundance DMBNHS labeled phosphopeptides, the ETD-MS/MS spectra of the triply charged singly labeled phosphopeptide no. 27 provided a clear series of z- and c-type product ions from which the peptide sequence and site of phosphorylation could be readily and unambiguously determined, as well as moderately abundant neutral losses of S (CH3)2 and S(CD3)2 from the charge reduced intact doubly and singly charged precursor ions (Figure 8b). An abundance ratio of 0.99:1 was measured from these ions (after correction for the presence of the D5-light DMBNHS impurity), which is very close to the expected ratio of 1:1, suggesting that ETD-generated sulfonium ion tag specific neutral loss product ions may also be used for quantitative analysis. Similarly, although ETD-MS/MS of the doubly charged singly labeled phosphopeptide no. 27 (Figure 8c) provided only limited sequence information due to its poor dissociation efficiency, quantitative information regarding the ratio of light- to heavy-labeled peptide could readily be obtained (1.02:1) by measuring the abundances of the diagnostic S(CH3)2 and S(CD3)2 neutral loss product ions. From the data obtained by ETD-MS/MS of the +3 and +2 charge states, a linear relationship between the measured and expected ratios (R2=0.9999) was obtained, comparable to that determined from the CID-MS/MS experiments (Supplementary Figure S6). Thus, the use of ETD-MS/MS alone can provide simultaneous identification and relative quantification of phosphopeptides without need for prior CID-MS/MS, However, it is expected that this would be a less sensitive approach for quantification compared that obtained using CID, due to only a fraction of the precursor ion abundance being directed toward the required sulfonium ion specific neutral loss fragmentation pathway, particularly for higher charge state precursor ions. Therefore, for future studies involving the application of these reagents to large-scale phosphoproteome analysis, a dual ion activation strategy is recommended whereby ion trap CID-MS/MS is first used for phosphopeptide quantification, followed by ETD-MS/MS for sequence identification and phosphorylation site characterization.
Conclusions
The results described here demonstrate that the introduction of fixed charge sulfonium ion moieties to the primary amine groups (N-termini and lysine side chains) of phosphopeptides leads to improved ionization efficiencies and increased and/or higher abundance charge state precursor ions. Upon CID-MS/MS, exclusive neutral loss(es) of dimethylsulfide are observed, independently of the proton mobility of the phosphopeptide, while keeping the phosphate group(s) intact. The relative abundances of ‘light’ versus ‘heavy’ neutral loss product ions from isobaric stable isotope encoded DMBNHS labeled phosphopeptides during CID-MS/MS or ETD-MS/MS enables differential quantitative analysis, whereas subsequent ETD-MS/MS of the intact precursor ion(s) allows phosphopeptide sequence identification and phosphorylation site characterization. In comparison to other derivatization methods, the DMBNHS labeling approach has the advantage of neutral loss specificity during collision induced gas-phase fragmentation along with an enhanced quantitation capability using both CID-MS/MS and ETD-MS/MS techniques. These results are therefore highly suggestive of the great potential of this reagent for further applied studies involving large scale quantitative analysis or protein post-translational modifications.
Supplementary Material
Acknowledgments
The authors acknowledge that support for this work was provided by funding from the National Science Foundation (CAREER: CHE-0547940).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-011-0190-0) contains supplementary material, which is available to authorized users.
References
- 1.Mann M, Ong S-E, Grønborg M, Steen H, Jensen ON, Pandey A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002;20:261–226. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 2.Reinders J, Sickmann A. State-of-the-art in phosphoproteomics. Proteomics. 2005;5:4052–4061. doi: 10.1002/pmic.200401289. [DOI] [PubMed] [Google Scholar]
- 3.Paradela A, Albar JP. Advances in the analysis of protein phosphorylation. J Proteome Res. 2008;7:1809–1818. doi: 10.1021/pr7006544. [DOI] [PubMed] [Google Scholar]
- 4.Nita-Lazar A, Saito-Benz H, White FM. Quantitative phosphoproteomics by mass spectrometry: Past, present, and future. Proteomics. 2008;8:4433–4443. doi: 10.1002/pmic.200800231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thingholm TE, Jensen ON, Larsen MR. Analytical strategies for phosphoproteomics. Proteomics. 2009;9:1451–1468. doi: 10.1002/pmic.200800454. [DOI] [PubMed] [Google Scholar]
- 6.Grimsrud PA, Swaney DL, Wenger CD, Beauchene NA, Coon JJ. Phosphoproteomics for the masses. ACS Chem Biol. 2010;5:105–119. doi: 10.1021/cb900277e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn JD, Reid GE, Bruening ML. Techniques for phosphopeptide enrichment prior to analysis by mass spectrometry. Mass Spectrom Rev. 2010;29:29–54. doi: 10.1002/mas.20219. [DOI] [PubMed] [Google Scholar]
- 8.Boersema PJ, Mohammed S, Heck AJ. Phosphopeptide fragmentation and analysis by mass spectrometry. J Mass Spectrom. 2009;44:861–878. doi: 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- 9.Palumbo AM, Smith SA, Kalcic CL, Dantus M, Stemmer PM, Reid GE. Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrom Rev. 2011 doi: 10.1002/mas.20310. in press. [DOI] [PubMed] [Google Scholar]
- 10.Annan RS, Carr SA. Phosphopeptide analysis by matrix-assisted laser desorption time-of-flight mass spectrometry. Anal Chem. 1996;68:3413–3421. doi: 10.1021/ac960221g. [DOI] [PubMed] [Google Scholar]
- 11.Palumbo AM, Tepe JJ, Reid GE. Mechanistic insights into the multistage gas-phase fragmentation behavior of phosphoserine- and phosphothreonine- containing peptides. J Proteome Res. 2008;7:771–779. doi: 10.1021/pr0705136. [DOI] [PubMed] [Google Scholar]
- 12.Palumbo AM, Reid GE. Evaluation of gas-phase rearrangement and competing fragmentation reactions on protein phosphorylation site assignment using CID-MS/MS and MS3. Anal Chem. 2008;80:9735–9747. doi: 10.1021/ac801768s. [DOI] [PubMed] [Google Scholar]
- 13.Schlosser A, Pipkorn R, Bossemeyer D, Lehmann WD. Analysis of protein phosphorylation by combination of elastase digest and neutral loss tandem mass spectrometry. Anal Chem. 2001;73:170–176. doi: 10.1021/ac000826j. [DOI] [PubMed] [Google Scholar]
- 14.Chang EJ, Archambault V, McLachlin DT, Krutchinsky AN, Chait BT. Analysis of protein phosphorylation by hypothesis-driven multiple-stage mass spectrometry. Anal Chem. 2004;76:4472–4483. doi: 10.1021/ac049637h. [DOI] [PubMed] [Google Scholar]
- 15.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal Chem. 2004;76:3590–3598. doi: 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- 16.Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 17.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 18.Ulintz PJ, Yocum AK, Bodenmiller B, Aebersold R, Andrews PC, Nesvizhskii A. Comparison of MS(2)-only, MSA, and MS(2)/MS (3) methodologies for phosphopeptide identification. J Proteome Res. 2009;8:887–899. doi: 10.1021/pr800535h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiesner J, Premsler T, Sickmann A. Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics. 2008;8:4466–4483. doi: 10.1002/pmic.200800329. [DOI] [PubMed] [Google Scholar]
- 21.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance evaluation of electron transfer dissociation. Mol Cell Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2199–2204. doi: 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swaney DL, McAlister GC, Coon JJ. Decision tree–driven tandem mass spectrometry for shotgun proteomics. Nat Method. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swaney DL, Wenger CD, Thomson JA, Coon JJ. Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2009;106:995–1000. doi: 10.1073/pnas.0811964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng N, Zhang J, Zong C, Wang Y, Lu H, Yang P, Wang W, Young GW, Wang Y, Korge P, Lotz C, Doran P, Liem DA, Apweiler R, Weiss JN, Duan H, Ping P. Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Mol Cell Proeomt. 2011 doi: 10.1074/mcp.M110.000117. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonewald LF, Bibbs L, Kates SA, Khatri A, McMurray JS, Medzihradszky KF, Weintraub ST. Study on the synthesis and characterization of peptides containing pY. J Peptide Res. 1999;53:161–169. doi: 10.1034/j.1399-3011.1999.00014.x. [DOI] [PubMed] [Google Scholar]
- 27.Janek K, Wenschuh H, Bienert M, Krause E. Phosphopeptide analysis by positive and negative ion MALDI MS. Rapid Commun Mass Spectrom. 2001;15:1593–1599. doi: 10.1002/rcm.417. [DOI] [PubMed] [Google Scholar]
- 28.Gropengiesser J, Varadarajan BT, Stephanowitz H, Krause E. The relative influence of phosphorylation and methylation on responsiveness of peptides to MALDI and ESI mass spectrometry. J Mass Spectrom. 2009;44:821–831. doi: 10.1002/jms.1581. [DOI] [PubMed] [Google Scholar]
- 29.Steen H, Jebanathirajah JA, Rush J, Morrice N, Kirschner MW. Phosphorylation analysis by mass spectrometry: Myths, facts, and the consequences for qualitative and quantitative measurements. Mol Cell Proteom. 2006;5:172–181. doi: 10.1074/mcp.M500135-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JEP, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taouatas N, Drugan MM, Heck AJR, Mohammed S. Straightforward ladder sequencing of peptides using a Lys-N metalloendopeptidase. Nat Methods. 2008;5:405–407. doi: 10.1038/nmeth.1204. [DOI] [PubMed] [Google Scholar]
- 32.Kjeldsen F, Giessing A, Ingrell CR, Jensen ON. Peptide sequencing and characterization of post-translational modifications by enhanced ion-charging and liquid chromatography electron-transfer dissociation tandem mass spectrometry. Anal Chem. 2007;79:9243–9252. doi: 10.1021/ac701700g. [DOI] [PubMed] [Google Scholar]
- 33.Lomeli SH, Peng IX, Yin S, Ogorzalek Loo RR, Loo JA. New reagents for increasing ESI multiple charging of proteins and protein complexes. J Am Soc Mass Spectrom. 2010;21:127–131. doi: 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leitner A, Lindner W. Chemistry meets proteomics: the use of chemical tagging reactions for MS-based proteomics. Proteomics. 2006;6:5418–5434. doi: 10.1002/pmic.200600255. [DOI] [PubMed] [Google Scholar]
- 35.Leitner A, Lindner W. Chemical tagging strategies for mass spectrometry-based phospho-proteomics. Methods Mol Biol. 2009;527:229–243. doi: 10.1007/978-1-60327-834-8_17. [DOI] [PubMed] [Google Scholar]
- 36.Mirzaei H, Regnier F. Enhancing electrospray ionization efficiency of peptides by derivatization. Anal Chem. 2006;78:4175–4183. doi: 10.1021/ac0602266. [DOI] [PubMed] [Google Scholar]
- 37.Vasicek L, Brodbelt JS. Enhanced electron transfer dissociation through fixed charge derivatization of cysteines. Anal Chem. 2009;81:7876–7884. doi: 10.1021/ac901482s. [DOI] [PubMed] [Google Scholar]
- 38.Madsen JA, Brodbelt JS. Simplifying fragmentation patterns of multiply charged peptides by N-terminal derivatization and electron transfer collision activated dissociation. Anal Chem. 2009;81:3645–3653. doi: 10.1021/ac9000942. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y, Zhang L, Lu H, Yang P. Mass spectrometry analysis of phosphopeptides after peptide carboxy group derivatization. Anal Chem. 2008;80:8324–8328. doi: 10.1021/ac801220c. [DOI] [PubMed] [Google Scholar]
- 40.Zhang L, Xu Y, Lu H, Yang P. Carboxy group derivatization for enhanced electron-transfer dissociation mass spectrometric analysis of phosphopeptides. Proteomics. 2009;9:4093–4097. doi: 10.1002/pmic.200800963. [DOI] [PubMed] [Google Scholar]
- 41.Hennrich ML, Boersema PJ, van den Toorn H, Mischerikow N, Heck AJ, Mohammed S. Effect of chemical modifications on peptide fragmentation behavior upon electron transfer induced dissociation. Anal Chem. 2009;81:7814–7822. doi: 10.1021/ac901108g. [DOI] [PubMed] [Google Scholar]
- 42.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteom. 2004;4:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 44.Phanstiel D, Unwin R, McAlister GC, Coon JJ. Peptide quantification using 8-plex isobaric tags and electron transfer dissociation tandem mass spectrometry. Anal Chem. 2009;81:1693–1698. doi: 10.1021/ac8019202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sachon E, Mohammed S, Bache N, Jensen ON. Phosphopeptide quantitation using amine-reactive isobaric tagging reagents and tandem mass spectrometry: application to proteins isolated by gel electrophoresis. Rapid Commun Mass Spectrom. 2006;20:1127–1134. doi: 10.1002/rcm.2427. [DOI] [PubMed] [Google Scholar]
- 46.Viner RI, Zhang T, Second T, Zabrouskov V. Quantification of post-translationally modified peptides of bovine α-crystalline using tandem mass tags and electron transfer dissociation. J Proteoms. 2009;72:874–885. doi: 10.1016/j.jprot.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 47.Nilsson CL, Dillon R, Devakumar A, Shi SD, Greig M, Rogers JC, Krastins B, Rosenblatt M, Kilmer G, Major M, Kaboord BJ, Sarracino D, Rezai T, Prakash A, Lopez M, Ji Y, Priebe W, Lang FF, Colman H, Conrad CA. Quantitative phosphoproteomic analysis of the STAT3/IL-6/HIF1α signaling network: An initial study in GSC11 glioblastoma stem cells. J Proteome Res. 2010;9:430–443. doi: 10.1021/pr9007927. [DOI] [PubMed] [Google Scholar]
- 48.Thingholm TE, Palmisano G, Kjeldsen F, Larsen MR. Undesirable charge-enhancement of isobaric tagged phosphopeptides leads to reduced identification efficiency. J Prot Res. 2010;9:4045–4052. doi: 10.1021/pr100230q. [DOI] [PubMed] [Google Scholar]
- 49.Pichler P, Köcher T, Holzmann J, Mazanek M, Taus T, Ammerer G, Mechtler K. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Anal Chem. 2010;82:6549–6558. doi: 10.1021/ac100890k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bantscheff M, Boesche M, Eberhard D, Matthieson T, Sweetman G, Kuster B. Robust and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer. Mol Cell Proteom. 2008;7:1702–1713. doi: 10.1074/mcp.M800029-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang F, Wu S, Stenoien DL, Zhao R, Monroe ME, Gritsenko MA, Purvine SO, Polpitiya AD, Tolić N, Zhang Q, Norbeck AD, Orton DJ, Moore RJ, Tang K, Anderson GA, Pasa-Tolić L, Camp DG, Smith RD. Combined pulsed-Q dissociation and electron transfer dissociation for identification and quantification of iTRAQ-labeled phosphopeptides. Anal Chem. 2009;81:4137–4143. doi: 10.1021/ac802605m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han H, Pappin DJ, Ross PL, McLuckey SA. Electron transfer dissociation of iTRAQ labeled peptide ions. J Proteome Res. 2008;7:3643–3648. doi: 10.1021/pr8001113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phanstiel D, Zhang Y, Marto JA, Coon JJ. Peptide and protein quantification using iTRAQ with electron transfer dissociation. J Am Soc Mass Spectrom. 2008;19:1255–1262. doi: 10.1016/j.jasms.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mischerikow N, van Nierop P, Li KW, Bernstein HG, Smit AB, Heck AJ, Altelaar AF. Gaining efficiency by parallel quantification and identification of iTRAQ-labeled peptides using HCD and decision tree guided CID/ETD on an LTQ Orbitrap. Analyst. 2010;135:2643–2652. doi: 10.1039/c0an00267d. [DOI] [PubMed] [Google Scholar]
- 55.Boja ES, Phillips D, French SA, Harris RA, Balaban RS. Quantitative mitochondrial phosphoproteomics using iTRAQ on an LTQ-Orbitrap with high energy collision dissociation. J Proteome Res. 2009;8:4665–4675. doi: 10.1021/pr900387b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reid GE, Roberts KD, Simpson RJ, O’Hair RAJ. Selective Identification and quantitative analysis of methionine containing peptides by charge derivatization and tandem mass spectrometry. J Am Soc Mass Spectrom. 2005;16:1131–1150. doi: 10.1016/j.jasms.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 57.Froelich JM, Kaplinghat S, Reid GE. Automated neutral loss and data dependent energy resolved “Pseudo MS3” for the targeted identification, characterization, and quantitative analysis of methionine- containing peptides. Eur J Mass Spectrom. 2008;14:219–229. doi: 10.1255/ejms.931. [DOI] [PubMed] [Google Scholar]
- 58.Amunugama M, Roberts KD, Reid GE. Mechanisms for the selective gas-phase fragmentation reactions of methionine side chain fixed charge sulfonium ion containing peptides. J Am Soc Mass Spectrom. 2006;17:1631–1642. doi: 10.1016/j.jasms.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 59.Roberts KD, Reid GE. Leaving group effects on the selective gas phase fragmentation reactions of cysteine side chain fixed charge containing peptide ions. J Mass Spectrom. 2007;42:187–198. doi: 10.1002/jms.1150. [DOI] [PubMed] [Google Scholar]
- 60.Zhou X, Lu Y, Wang W, Borhan B, Reid GE. ‘Fixed charge’ Chemical derivatization and data dependant multistage tandem mass spectrometry for mapping protein surface residue accessibility. J Am Soc Mass Spectrom. 2010;21:1339–1351. doi: 10.1016/j.jasms.2010.03.047. [DOI] [PubMed] [Google Scholar]
- 61.Lu Y, Tanasova M, Borhan B, Reid GE. An ionic reagent for controlling the gas-phase fragmentation reactions of cross-linked peptides. Anal Chem. 2008;80:9279–9287. doi: 10.1021/ac801625e. [DOI] [PubMed] [Google Scholar]
- 62.Jessing M, Brandt M, Jensen KJ, Christensen JB, Boas UJ. Org Chem. 2006;71:6734–6741. doi: 10.1021/jo060687r. [DOI] [PubMed] [Google Scholar]
- 63.Crouch NP, Adlington RM, Baldwin JE, Lee M-H, MacKinnon CH. Tetrahedron. 1997;53:6993–7010. [Google Scholar]
- 64.Kulevich SE, Frey BL, Kreitinger G, Smith LM. Alkylating tryptic peptides to enhance electrospray ionization mass spectrometry analysis. Anal Chem. 2010;82:10135–10142. doi: 10.1021/ac1019792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krokhin OV, Craig R, Spicer V, Ens W, Standing KG, Beavis RC, Wilkins JA. Mol Cell Proteom. 2004;3:908–919. doi: 10.1074/mcp.M400031-MCP200. [DOI] [PubMed] [Google Scholar]
- 66.Frahm JL, Bori ID, Comins DL, Hawkridge AM, Muddiman DC. Achieving augmented limits of detection for peptides with hydrophobic alkyl tags. Anal Chem. 2007;79:3989–3995. doi: 10.1021/ac070558q. [DOI] [PubMed] [Google Scholar]
- 67.Hohmann LJ, Eng JK, Gemmill A, Klimek J, Vitek O, Reid GE, Martin DB. Quantification of the compositional information provided by immonium ions on a quadrupole-TOF mass spectrometer. Anal Chem. 2008;80:5596–5606. doi: 10.1021/ac8006076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia QW, Lee MV, Rose CM, Marsh AJ, Hubler SL, Wenger CD, Coon JJ. Characterization and diagnostic value of amino acid side chain neutral losses following electron-transfer dissociation. J Am Soc Mass Spectrom. 2011;22:255–264. doi: 10.1007/s13361-010-0029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li X, Cournoyer JJ, Lin C, O’Connor PB. The effect of fixed charge modifications on electron capture dissociation. J Am Soc Mass Spectrom. 2008;19:1514–1526. doi: 10.1016/j.jasms.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chamot-Rooke J, van der Rest G, Dalleu A, Bayc S, Lemoineb J. The combination of electron capture dissociation and fixed charge derivatization increases sequence coverage for O-glycosylated and O-phosphorylated peptides. J Am Soc Mass Spectrom. 2007;18:1405–1413. doi: 10.1016/j.jasms.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 71.Swaney DL, McAlister GC, Wirtala M, Schwartz JC, Syka JEP, Coon JJ. Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal Chem. 2007;79:477–485. doi: 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang R, Regnier FE. Minimizing resolution of isotopically coded peptides in comparative proteomics. J Proteome Res. 2002;1:139–147. doi: 10.1021/pr015516b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.