

Abstract
OBJECTIVE
Pulmonary Hypertension (PH) is a progressive disease arising from remodeling and narrowing of pulmonary arteries (PA) resulting in high pulmonary blood pressure and ultimately right ventricular failure. Elevated production of reactive oxygen species (ROS) by NADPH oxidase 4 (Nox4) is associated with increased pressure in PH. However, the cellular location of Nox4 and its contribution to aberrant vascular remodeling in PH remains poorly understood. Therefore, we sought to identify the vascular cells expressing Nox4 in PA and determine the functional relevance of Nox4 in PH.
APPROACH AND RESULTS
Elevated expression of Nox4 was detected in hypertensive PA from 3 rat PH models and human PH using qRT-PCR, Western blot, and immunofluorescence. In the vascular wall, Nox4 was detected in both endothelium and adventitia and perivascular staining was prominently increased in hypertensive lung sections, colocalizing with cells expressing fibroblast and monocyte markers and matching the adventitial location of ROS production. Small molecule inhibitors of Nox4 reduced adventitial ROS generation and vascular remodeling as well as ameliorating right ventricular hypertrophy and non-invasive indices of PA stiffness in monocrotaline (MCT)-treated rats as determined by morphometric analysis and high resolution digital ultrasound. Nox4 inhibitors improved PH in both prevention and reversal protocols and reduced the expression of fibroblast markers in isolated PA. In fibroblasts, Nox4 over-expression stimulated migration and proliferation and was necessary for matrix gene expression.
CONCLUSIONS
These findings indicate that Nox4 is prominently expressed in the adventitia and contributes to altered fibroblast behavior, hypertensive vascular remodeling and the development of PH.
Keywords: pulmonary, Nox4, adventitia, vascular remodeling
Introduction
Pulmonary Hypertension (PH) is a progressive disease resulting from increased pulmonary vascular resistance. PH is resistant to current therapies and is characterized by excessive vascular cell proliferation, inward remodeling, rarefaction and a loss of compliance of the pulmonary blood vessels 1–3. Increased resistance to blood flow and more rigid blood vessels leads to failure of the right ventricle and eventual death. Further, PH is more frequent in women than men and untreated has a survival time of less than 5 years post diagnosis 4, 5.
Reactive oxygen species (ROS) have been proposed as a pathogenic mechanism underlying the vascular remodeling observed in PH. However, the source, cellular origin and functional significance of ROS in PH remain poorly defined. Elevated levels of ROS in PH are the net result of increased production and decreased degradation and there is evidence for both mechanisms in the etiology of elevated pulmonary pressure 6–10. The major intracellular sources of ROS include the mitochondria, aberrant oxygenase activity, and the NADPH oxidase family of oxidases (Nox) 11, 12. The human genome encodes five Nox isoforms and four of these, Nox1, Nox2, Nox4 and Nox5 are expressed in vascular cells (although Nox5 is not present in the genomes of rats and mice). In comparison to other sources of ROS, Nox enzymes are regarded as professional ROS generators and are capable of synthesizing high levels of ROS in a spatial and temporal manner. Nox1–4 are bound to p22phox and Nox1 and 2 are activated by binding numerous cytosolic subunits, including p47phox, p67phox or NOXO1 and NOXA1. In contrast, Nox4 is regarded as a constitutively active enzyme with ROS levels primarily controlled by changes in gene expression 13, 14. In mice, genetic deletion of Nox2 has been shown to reverse hypoxia-initiated PH 7, and Nox1 has been shown to be important for systemic hypertension 15, 16. Increased expression of Nox4 has been reported in both mouse models of PH and human PH 17–19. However, despite this knowledge, the functional significance of Nox4 in the development of PH is poorly understood. A recent publication by Green et al demonstrated the ability of combined Nox4/Nox1 inhibitors to ameliorate PH in mice 20. While that study supports the importance of Nox4 in the development of PH, it was performed in mice, which do not experience the advanced vascular remodeling observed in humans or rat models, and the investigators did not address the cell types expressing Nox4 in the vascular wall of pulmonary arteries.
Remodeled blood vessels in PH are characterized by increased stiffness 21, 22 secondary to collagen and elastin deposition, a process regulated by the adventitial fibroblast. The fibroblast, a primary cell type of the adventitia, contributes to the continual reorganization of the extracellular matrix via matrix deposition and secretion of growth factors, chemokines and inflammatory cytokines. Fibroblasts also influence and promote the inflammatory response by manipulating leukocyte recruitment, survival and behavior. In addition, a subset of circulating bone marrow derived cells termed fibrocytes that possess genetic markers and behaviors consistent with both fibroblasts and macrophages can also be found in the adventitia 23, 24. Nox enzymes and elevated ROS stimulate fibroblast proliferation 17, 25, 26, however, the contribution of specific Nox isoforms to adventitial proliferation and the development of PH is poorly defined.
The goal of the current study was to address the above deficiencies in our understanding of PH and provide new data on the regulation and functional significance of Nox4 in the pulmonary circulation. Collectively, our data support a significant and novel role for Nox4 in the pathogenesis of PH.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement
Results
Real-time RT-PCR was used to determine relative expression levels of Nox enzymes and associated subunits in isolated PA. In all three rat models of PH (FHR, MCT, and SU/HYP), Nox4 mRNA was significantly increased compared to the normotensive SDR (Fig. 1A–C). In addition, Western blot analysis revealed that Nox4 protein expression was significantly upregulated in PA from MCT-treated rats (Fig. 1D–E). Nox1 and Nox2 gene expression were also increased in PA from MCT-treated rats with PH (Supplemental Figs. IA; IIA). However, Nox1 mRNA expression was not increased in PA from the FHR or the SU/HYP rat or in hypoxic mouse lung (Supplemental Fig. IB–D). Screening of p22phox, p47phox, and p67phox subunits revealed no significant differences in mRNA expression between the normotensive (SDR) and hypertensive (MCT) PA (Supplemental Fig. IIC–E). However, there was a significant downregulation of p47phox and p67phox and NOXA1 and NOXO1 in SU/HYP PA (Supplemental Figs. IID–G). There was no change in the expression of p22phox, p47phox, and p67phox, NOXO1, NOXA1 between control rats and FHR (Supplemental Figs. IIH–L).
Figure 1. Nox4 expression is increased in rat hypertensive pulmonary arteries (PA).

Nox4 mRNA (measured by qRT-PCR (ΔΔCt) normalized to 18S) is significantly increased in PA from FHR (24week), MCT (4week), and SU/HYP (14week) (1A–C). Western blot analysis shows that Nox4 protein expression is upregulated in 4 week MCT-treated rat PA (1D–E). * Significantly different from SDR/ vehicle controls, p < 0.05 (n = 5–6).
To address the functional contribution of elevated Nox4 expression in the development of PH, we treated control and MCT-rats with three different Nox4 inhibitors. To confirm efficacy, we measured ROS in a cell type expressing Nox4. Inhibitor VCC588646 (A), VCC202273 (C) and GKT136901 (G) decreased Nox4 activity in a concentration-dependent manner (Fig. 2A–C). ROS production was completely inhibited by the flavoprotein inhibitor, diphenyleneiodonium (DPI) and by catalase (CAT), a scavenger of hydrogen peroxide (Fig. 2C). Morphometric analysis of PA revealed significant medial remodeling by 4 weeks in MCT-treated rats that was abrogated by both Nox4 inhibitor VCC588646 (A) and VCC202273 (C) (Fig. 2D–E). The relative activity of Nox4 inhibitors VCC588646 (A) and VCC202273 (C) against Nox1 activity was tested in a HEK cell line expressing Nox1, NOXO1 and NOXA1 as previously described27. Both inhibitors reduced Nox1 activity but at concentrations higher than for Nox4 (Supplemental Fig. III).
Figure 2. Efficacy of three distinct Nox4 inhibitors and effect on PA remodeling in vivo.

(A) Nox4 activity in the presence of inhibitor VCC588646 (A), (B) Nox4 activity with VCC202273 (C), and (C) Nox4 activity with inhibitor GKT136901 (n=5) (D) H & E staining of lung segments from rats treated with vehicle and MCT (60mg/kg, IP) for 4wks in presence/absence of Nox4 inhibitor A or C. Magnification 40X, (E) quantitative PA morphometric analysis of the effect of inhibitor A or C, * Significantly different from Vehicle, # Significantly different from MCT, p < 0.05 (n = 5–6).
The effect of Nox4 inhibition on cardiac remodeling and indices of cardiopulmonary function is shown in Fig. 3 and extended to Supplemental Fig. IV. MCT significantly increased RV hypertrophy (RV/LV+S), which was inhibited with VCC202273 (C) (Fig. 3A) and also VCC588646 (A) and GKT136901 (G) (Supplemental Fig. IVA–B). Changes in right ventricular function (RVSP; RVmax dp/dt), which were elevated in the MCT-treated lungs, were also abrogated by Nox4 inhibition (Fig. 3B–C; Supplemental Fig. IVC). Non-invasive assessment of RV remodeling using digital ultrasound also revealed significant time-dependent increases in RV thickness in MCT-treated rats which were reduced by Nox4 inhibitors (Fig. 3D–E; Supplemental Fig. IVD–E). Cardiac output was decreased at 4-week post MCT-treatment and improved with Nox4 inhibition (Fig. 3F). There was evidence of PA remodeling as determined by analysis of PA hemodynamics including a reduction in the velocity time integral (VTI), pulmonary ejection time (PET) and pulmonary artery acceleration time (PAAT) (Fig. 3G–I; Supplemental Fig. IVF–I). Nox4 inhibition reduced RV thickness, increased cardiac output (CO), and increased VTI, PET and PAAT (Fig. 3E–I, Supplemental Fig. IVD–I), which collectively suggest a reduction in vessel stiffness (remodeling) and improved cardiac performance. Importantly, in all of the end points measured, all 3 inhibitors of Nox4 demonstrated significant efficacy in ameliorating the structural and functional changes with PH.
Figure 3. Effect of Nox4 inhibition on cardiac remodeling and cardiopulmonary functional indices in MCT-treated rats.

MCT significantly increases right ventricular (RV) hypertrophy as measured by the Fulton Index (RV/LV+S), which is inhibited by VCC202273 (C) (3A). Right ventricular function indices (RVSP; RVmax dp/dt), are abrogated by Nox4 inhibition (3B–C). Non-invasive assessment of RV remodeling using the Vevo 2100 reveals significant time dependent increases in RV thickness in MCT-treated rats (3D–E). 4-week post MCT-treatment shows evidence of reduced PA elasticity as determined by analysis of PA hemodynamics using the velocity time integral (VTI) and pulmonary artery acceleration time (PAAT) parameters as well as a decrease in cardiac output (CO) (3F–H). Inhibition of Nox4 by VCC202273 (C) reduced RV thickness, increased CO, and increased VTI, PAAT and PET (3D–I). * Significantly different from Vehicle, # significantly different from MCT, p < 0.05 (n = 5–6).
To assess whether inhibition of Nox4 can reverse, attenuate or halt the progression of experimental PH in MCT-treated rats with established PH, we measured time-dependent changes in RV and PA remodeling via ultrasound. Reversal protocols were employed as they are more relevant to the clinical predicament and also can determine whether the target of interest remains functionally important in the later stages of disease and whether pre-existing endpoints of PH can be improved. The Nox4 inhibitor VCC202273 (C) was administered to MCT-treated rats upon the first detectable increase in RV thickness, which occurred at 3-weeks post MCT treatment (Fig.3E). As shown in Fig. 4, VCC202273 (C) (1mg/kg/day) significantly attenuated the progression of compensatory RV hypertrophy (Fig.4A), VTI (Fig. 4B) and PAAT (Fig. 4C) in MCT-treated rats by weeks 5 and 6 post-MCT treatment. At the conclusion of the experiment (6 weeks), invasive and post mortem endpoints were recorded (RVSP and Fulton Index). Nox4 inhibition attenuated the MCT-induced increases in RVSP and Fulton Index at 6 weeks post initiation of MCT-treatment (Fig.4D–E). While there was no indication that Nox4 inhibition promoted reversal of PH beyond pre-treatment levels, there was significant evidence for the attenuation of progression of the disease.
Figure 4. Inhibition of Nox4 slows the progression of established pulmonary hypertension.

Rats with established MCT-induced pulmonary hypertension were treated with VCC202273 (C) (1mg/kg/day) starting from week 3 through week 6. Inhibition of Nox4 reduced the thickness of the right ventricle (4A) and improved velocity time integral (VTI, 4B) and pulmonary artery acceleration time (PAAT, 4C) in weeks 4–6 post-MCT treatment. Endpoint measurements of RVSP and Fulton index show a reduction in right ventricular hypertrophy in rats treated with VCC202273 (C) (4D–E). * Significantly different from Vehicle/Control, # significantly different from MCT, p < 0.05 (n = 6 for each group).
To identify the regions of the blood vessel wall and cell types expressing Nox4 in hypertensive PA, we performed immunofluorescence staining of lung sections from MCT-treated rats and in lungs from normal (control) and human PH. In control rat PA, Nox4 was detected in the intima (endothelial cells) and cells of the adventitia (Fig. 5; top panel). In 4-week MCT-treated rats, using two different antibodies that are selective for Nox4 (Epitomics and Abcam), there was a dramatic increase in Nox4-positive cells in the adventitia (Fig. 5; MCT, left panels). In PA from MCT-treated rats, Nox4-positive cells were detected in the remodeled medial layer but this was largely in areas devoid of α-actin expression (Fig. 5, MCT; right panels). Nox4 exhibited a staining pattern in sections of human lung from individuals with normal and elevated pulmonary blood pressure (Fig. 5, lower panels) that was consistent with the MCT-rat model. In lung sections from animals treated with the Nox4 inhibitor VCC202273 (C), MCT-stimulated vascular remodeling and Nox4 expression were significantly attenuated (Fig. 5; MCT+C). Further, a time course of Nox4 expression in MCT-treated rats revealed increased Nox4 expression and progressive medial remodeling at 2 and 3-weeks post MCT treatment (Supplemental Fig. VI). Negative controls for the fluorescent secondary antibodies were performed using non-immune IgG in human lung sections and were without significant staining as shown in Supplemental Fig. VII.
Figure 5. Nox4 is primarily expressed in the adventitia and endothelium of rat and human PA.

Confocal images of lung sections from control, experimental PH (4-week MCT), and human PAH (IPAH undergoing lung transplant). Sections were stained with Nox4 and α-actin antibodies. Nox4 is highly expressed in the intima (endothelial cells) and cells of the adventitia in 4-week MCT-treated rats and human PH PA. There is an abundance of Nox4-expressed cells present in the remodeled medial layer but devoid of α-actin expression in the MCT and human PH PA. In the presence of Nox4 VCC202273 (C), (MCT + C), Nox4 expression is similar to vehicle-treated PA in the MCT-treated group.
The location of ROS production in hypertensive PA was performed by immunofluorescence imaging for 8-hydroxydeoxyguanosine (8-OHdG), a DNA nucleoside that is generated by ROS. 8-OHdG is used as an in vivo footprint of ROS production and increased staining was detected in PA from 4-week MCT-treated rats. The highest signal was observed in the adventitia, which overlapped significantly with the fibroblast marker, fibroblast activating protein (FAP) (Fig. 6A; MCT). A less pronounced increase in ROS levels was also observed in the media of hypertensive PA, and was in proportion with the lower levels of Nox4 seen in the media compared to the adventitia. ROS levels in both the medial and adventitial layers were decreased in sections from rats treated with the Nox4 inhibitor VCC202273 (C) (Fig. 6A; MCT+C) suggesting that the elevated ROS production in hypertensive PA derives from increased Nox4 expression. In MCT-treated PA, there was significant overlap between Nox4-positive cells in the adventitia and cells expressing fibroblast markers (cellular fibronectin, CD90) as well as the monocytic cell marker CD11B (Fig. 6B). These data are supported by Western blot analysis of PA from control and 4-week MCT-treated rats revealing increased protein expression of fibroblast markers such as CD90, cellular fibronectin (cellFN1), periostin, tenascin N, and vimentin (Fig. 7A–B, densitometry shown in Supplemental Fig. VA). The effect of Nox4 inhibition on fibroblast and inflammatory markers was also determined in PA isolated from control, MCT and MCT-treated rats treated with VCC202273 (C) using the reversal protocol (see Methods). Three weeks exposure to a Nox4 inhibitor that was initiated after 3 weeks of MCT, resulted in prominent decreases in fibroblast markers (CD90, cellFN1, vimentin, periostin, and thrombospondin4) as well as a marker of pericytes (NG2) compared to MCT treatment alone (Fig. 7B; with densitometry shown in Supplemental Fig. VB). A time course of Nox4 expression in isolated lung segments is shown in Supplemental Fig. VI. These data, in conjunction with the functional indices of PAH (Fig. 3 and Supplemental Fig. IV), provide strong evidence for the pharmacological efficacy of Nox4 inhibitors and the importance of this pathway in the progression of PH.
Figure 6. Nox4 expression and reactive oxygen species (ROS) production is localized in the adventitia.

(A) Sections of control and 4-week MCT-treated rat lungs costained for fibroblast activation protein (FAP), 8 Hydroxy-2'dexoyguanosine (ROS marker) and DAPI. (B) Co-staining for Nox4 and cellular fibronectin, CD90 and CD11b ROS production is elevated in PA adventitia from 4-week MCT-treated rats, which overlapped significantly with the fibroblast marker fibroblast activating protein (FAP) (A; MCT). ROS are decreased to control (vehicle) levels by the Nox4 inhibitor VCC202273 (C) (A; MCT+C). In MCT-treated PA, there is significant overlap between Nox4-positive cells in the adventitia and cells expressing fibroblast markers (cellular fibronectin, CD90) as well as the monocytic cell marker CD11B (B).
Figure 7. Increased expression of fibroblast markers in hypertensive PA and effect of Nox4 inhibition in established PH.
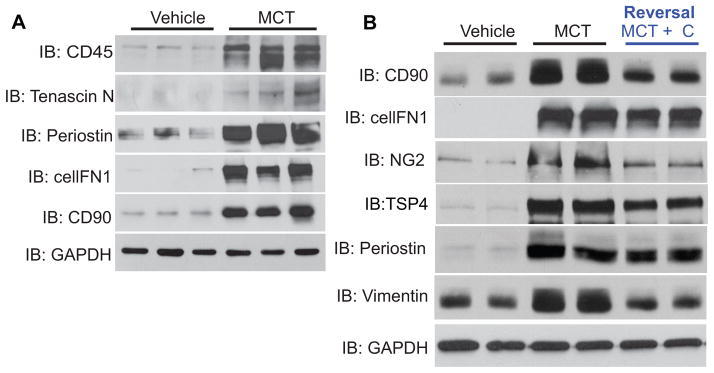
Western blots of PA isolated from control and 4 week MCT-rats (A). MCT-rats were treated with Nox4 inhibitor VCC202273 (C) starting at week 3 post MCT and continuing through week 6 in a reversal protocol (see Methods, B). (TSP4 = thrombospondin-4; NG2 = chondroitin sulfate proteoglycan). (n = 3–4 per group).
To assess whether Nox4 modifies fibroblast function we transduced human lung fibroblasts with an adenovirus expressing Nox4. Nox4-transduced fibroblasts exhibited a robust increase in cellular proliferation as demonstrated by real-time changes in electrical impedance using Electric Cell-substrate Impedance Sensing arrays (ECIS), as shown in Fig. 8A. In addition, fibroblasts transduced with the Nox4-adenovirus displayed increased cell proliferation (total cell number, Fig. 8B), and the number of viable cells as measured via the 3-(4,5-dimethylthiazol2-yl)-2,5-diphenyletrazolium bromide (MTT) assay (Fig. 8C). Further, using anti CD90-immunomagnetic isolation, fibroblasts were isolated from control or 4-week MCT-treated rat lungs. Western blot analysis revealed the expected enrichment of CD90-positive cells along with increased expression of Nox4 relative to the loading control, Hsp90 (Fig. 8D). Fibroblasts isolated from PA of MCT-treated rats displayed a high level of proliferation that was decreased by the Nox4 inhibitor VCC202273 (C) (Fig. 8E). Adenoviral delivery of Nox4 also stimulated an increase in fibroblast motility (Fig. 8F) compared to cells treated with a control virus. These data show that increased Nox4 expression alters fibroblast behavior, favoring a pro-proliferatory and pro-migratory phenotype. Lung fibroblasts exposed to the pro-fibrotic growth factor TGF-β1 robustly increased Nox4 mRNA and protein levels as well as cellular fibronectin in a time-dependent manner (Fig. 8G). To determine the relative ability of vascular cell types to express Nox4 in response to TGF-β1, we exposed human cultured pulmonary endothelial (HPAEC), human pulmonary arterial vascular smooth muscle cells (HPAVSMC), and human lung fibroblasts to TGF-β1 for 24h. The phenotype of each cell type was confirmed using relevant cell specific markers. We found the greatest induction of Nox4 in fibroblasts > HPAEC ≫ HPAVSMC (Fig. 8H). We next determined if Nox4 was important for the ability of TGF-β1 to induce profibrotic changes. In human fibroblasts, Nox4 siRNA decreased Nox4 expression and subsequently prevented the expression of the TGF-β1-dependent genes, fibronectin and ACTA (smooth muscle actin) (Fig. 8I).
Figure 8. Nox4 stimulates fibroblast proliferation and migration and TGF-β1 expression.

(A) Electrical impedance (ECIS) of human lung fibroblasts in the presence/absence of Nox4 adenovirus. Nox4-transduced fibroblasts exhibit a robust increase in cellular proliferation in real time using ECIS. (B) Nox4 increases fibroblast cell number, and (C) the number of viable cells using MTT assay. (D) Magnetic bead isolation of CD90 positive fibroblasts from lungs of control and MCT-treated rats. Western blot analysis revealed enrichment of CD90-positive cells with increased expression of Nox4 relative to the loading control, Hsp90. (E) Fibroblasts (p1) isolated from PA of MCT-treated rats displayed robust proliferation that is decreased by Nox4 inhibitor VCC202273 (C). (F) Adenoviral delivery of Nox4 stimulated an increase in fibroblast motility. (G) 24h. exposure to TGF-β1 robustly increased both Nox4 mRNA and protein levels in human lung fibroblasts while contemporaneously increasing the synthesis of cellular fibronectin. (H) Expression of cell specific markers (eNOS; H-caldesmon; CD90) in cultured HPAEC, HPAVSMC and fibroblasts; TGF-β1 increased CD90 and Nox4 expression in fibroblasts. (I) Transfection of fibroblasts with Nox4 siRNA decreased Nox4 expression and subsequently prevented the expression of TGF-β1-dependent genes such as fibronectin and ACTA. * Significantly different from Lac Z, 0, Control, 0 hr, and Cont siRNA; + Significantly different from Cont siRNA + TGFβ p<0.05 (n=3–6 per group).
Discussion
A positive correlation between increased pulmonary artery ROS production and the elevated blood pressure of PH was observed over 2 decades ago and in the time since has been confirmed by numerous independent groups 6, 7, 19, 28, 29. While the consensus is for elevated ROS levels in PH, the source of the ROS and its functional contribution to pathologic remodeling of pulmonary blood vessels remain less well defined. The major goal of the current study was to determine the relative importance of Nox isoforms, a major source of cellular ROS, to the etiology of PH.
Given the inability of any single animal model of PH to faithfully emulate the complexity of human PH, we first determined the expression of individual Nox isoforms in multiple rat models including 2 inducible and one genetic model. Our studies were conducted primarily in rat models as they exhibit robust, irreversible pulmonary vascular remodeling which is a defining characteristic of the human disease30, 31. Changes in vascular gene expression were selectively measured in isolated pulmonary arteries (PA, down to the 4th branch). In PA, we found significantly increased expression of Nox4 mRNA in all three rat models. This correlated with increased protein expression in the MCT-model and suggests that Nox4 is a potential source of the elevated ROS in hypertensive PA. This data is consistent with prior studies in mice, rats and in humans with PH 18–20, 32, 33. In the MCT model, we observed increased expression of Nox1 and Nox2 mRNA, which is also consistent with previous reports 34, 35. However, Nox1 levels were not elevated in PA from the fawn hooded rat, a genetic model of PH or in the rat and mouse hypoxia models. Further, in all three rat PH models, no increase in the expression of the multiple accessory proteins that support the post-translational activation of Nox 1–3 was observed. The significance of this is not known as the activity of Nox1–3 dependent on stimulus-dependent assembly of a functional oxidase. In contrast, the elevated expression of Nox4 has additional significance as it is the only Nox isoform that is constitutively active. This means that increases in Nox4 gene expression translate to increased ROS13. Collectively, these findings support elevated Nox4 expression in hypertensive PA as a common variable in multiple rodent models of PH and in humans.
A major obstacle in the identification of functional roles for Nox isoforms in PH has been the lack of appropriate tools, particularly in the rat. To investigate the significance of Nox4 in PH, we employed 3 structurally distinct Nox4 inhibitors 36, 37. This was done to ensure greater selectivity towards a common target (Nox4) and to minimize any possible non-specific effects of individual inhibitors. The efficacy of these inhibitors against Nox4 activity was first confirmed in vitro and then in vivo in the MCT rat model of PH. This model was selected based on the expeditious time course of PH (3–4weeks versus the 14 and 24weeks for SU/Hyp and the FHR). We initially determined if inhibiting Nox4 would prevent the development of PH in MCT-treated rats (day 1 treatment). Our objective in these studies was to determine if the increased Nox4 expression observed contributes to the induction of PH. We found that Nox4 inhibitors were effective at preventing PH as determined by indices of right ventricular remodeling and non-invasive in vivo measurements of right ventricle thickness, cardiac output and remodeling of PA (VTI, PET, PAAT). Our data are consistent with those reported recently by Green et al in a mouse hypoxia model of PH. In that study, a related Nox4 inhibitor (GKT137831) improved indices of ventricular remodeling and PA wall thickening but did not decrease RVSP20. However, a major limitation of the mouse model is the absence of significant vascular remodeling. Our study revealed that Nox4 inhibitors were effective at preventing the robust hypertrophic remodeling of PA in MCT-treated rats with PH. A further distinction of our study is that we also employed Nox4 inhibitors in protocols to assess the ability to reverse established PH. This approach is more clinically relevant as the early stages of PH are generally silent, difficult to detect and rarely treated. We found that Nox4 inhibitors were effective at halting the progression of PH. However, at the dose of inhibitor used and the time points studied, we did not observe an ability to fully halt the progression of the measured parameters of PH to levels below that of the pre-treatment 3 week MCT-treated rats. The reason for this is not yet known. In isolated PA from rats with PH, we observed near complete reversal of some indices such as the expression CD90 and vimentin with Nox4 inhibition. However, incomplete reversal was seen with others including cellular fibronectin, thrombospondin 4 and periostin. The reasons for diversity in the ability of Nox4 inhibitors to reverse changes in the expression of specific genes in PA, and the inability to completely stop or reverse the progression of PH await further investigation. It remains possible that higher doses of the Nox4 inhibitors or combination therapy with established therapeutics for PH (ET antagonists, PDE5 inhibitors, etc.) may enable more complete reversal of established PH. Germane to the overall thesis of this study, we found that inhibition of Nox4 was able to decrease the expression of genes in hypertensive PA that are considered to be cellular markers of fibroblasts.
To determine a cellular mechanism by which Nox4 influences the progression of PH, we first performed immunofluorescence imaging in isolated lung sections. Nox4 expression was detected in both endothelium and adventitia of PA, and perivascular staining was prominently increased in animals and humans with PH. In the adventitia, Nox4 was detected in cell types expressing markers consistent with fibroblasts and monocytes. The perivascular expression of Nox4 also matched the location of ROS production and the markers of fibroblasts and monocytic cells. A surprising observation was the relative absence of Nox4 in pulmonary artery vascular smooth muscle. Adventitial staining was confirmed using 2 distinct Nox4 antibodies and in hypertensive PA, the cell types expressing Nox4 in the medial layer did not overlap with those staining for smooth muscle actin. These results are supported by the higher levels of Nox4 detected in fibroblasts immuno-isolated from MCT-treated rat lungs and are consistent with previous reports showing elevated Nox4 in perivascular fibroblasts from individuals with IPAH 17. Nox4 has been detected in most cell types with higher levels of expression seen in fibroblasts, particularly in the setting of pulmonary fibrosis 17, 26, 38, 39. Also consistent with previous studies, we observed that human PAVSMC in culture have detectable levels of Nox4, which are increased in the presence of TGF-β1. However, these levels are less robust than those observed in fibroblasts.
The functional relevance of Nox4 in adventitial cells is not well described. The tunica externa or adventitia is a loosely defined collection of cells including fibroblasts and immune cells, collagen and elastic fibers that encircle the tunica media and intima layers of the blood vessel40. The adventitia orchestrates inflammation and vascular proliferation in response to injury, atherosclerosis and pulmonary and systemic hypertension41. The fibroblast is a primary cell type of the adventitia, responsible for the continual reorganization of the extracellular matrix via matrix deposition and secretion of growth factors, chemokines and inflammatory cytokines42. Aberrant vascular remodeling in PH occurs through increased inflammation, proliferation and fibrosis, processes that collectively yield more muscular and less compliant PA 1. Fibroblasts have key roles in these actions and actively secrete matrix, growth factors and promote the inflammatory response by manipulating leukocyte recruitment and behavior43, 44. We observed that increased expression of Nox4, in the absence of other stimuli, was sufficient to increase fibroblast migration and proliferation and treatment of fibroblasts isolated from PA from MCT-treated rats with Nox4 inhibitors reduced cellular proliferation. Similarly, silencing Nox4 in fibroblasts decreased the ability of TGF-β to increase matrix and induce contractile gene expression, which is consistent with other reports 26, 39, 45. A subset of bone marrow derived cells termed fibrocytes can also be found in the adventitia which have characteristics of both fibroblasts and macrophages23, 46. While we observed significant overlap of Nox4 staining with fibroblast and macrophage markers, it is not yet known if Nox4 is expressed in fibrocytes. In isolated PA from MCT-treated rats, we found increased expression of numerous markers that have been used to identify fibroblasts including CD90, cellular fibronectin, periostin, vimentin, and fibroblast activation protein (FAP). A role for Nox4 in regulating perivascular fibroblast behavior in PH is supported by results showing that Nox4 inhibition decreases the expression of the fibroblast markers in isolated PA from rats with established PH. These data suggest an ability of Nox4 to regulate the number and behavior of adventitial fibroblasts in animals with PH.
TGF-β1, an autocrine growth factor implicated in the pathophysiological vascular remodeling in PH 47, 48, robustly increased both Nox4 mRNA and protein levels in human lung fibroblasts. When comparing the ability of TGF-β1 to drive Nox4 expression in PAEC, PAVSMC and lung fibroblasts, we observed the greatest upregulation of Nox4 in fibroblasts with less in VSMC and none in PAEC. These data suggest that other mechanisms regulate Nox4 expression in endothelial cells. Indeed, numerous stimuli have been shown to modulate Nox4 expression including hypoxia, angiotensin II, cAMP, PKC etc14 and it is possible that these signaling pathways contribute in varying degrees to the elevated Nox4 expression observed in PH. Our data is in agreement with previous studies 32, 49, and others have shown that TGF-β1 can upgregulate Nox4 expression in other cell types including human cardiac fibroblasts, airway smooth muscle and vascular smooth muscle32, 38, 50. The functional effects of Nox4 in vascular smooth muscle are similar to those reported in fibroblasts and include altered signaling, increased ability to proliferate and migrate 32, 33 and may reflect the altered phenotype of smooth muscle cells in culture. A role for fibroblasts in pathologic remodeling in PH is supported by studies in transgenic mice with fibroblast specific activation of TGF-β1 signaling. These mice develop mild PH with medial hypertrophy, inflammation and fibrosis 51. While this study strongly supports a role for fibroblast TGF-β1 signaling in aberrant pulmonary vascular remodeling, PH can be further exacerbated with additional stress on the endothelium and reflects the important contributions of multiple cell types in the development of PH.
In addition to Nox4, other Nox isoforms have been implicated in the development of PH. A recent study by Veit et al., reported increased Nox1 expression in isolated pulmonary microvessels and cultured PAVSMC isolated from MCT-treated rats. They did not observe increased expression of Nox4 (or Nox2) in cultured PA-VSMC from MCT-treated rats35. The low expression of Nox4 in PAVSMC is in agreement with our study showing that in intact PA, Nox4 expression is low in the medial (smooth muscle) layer relative to the prominent expression of Nox4 observed in adventitial and endothelial cells. The importance of cell type to the expression of individual Nox isoforms is further emphasized in a study from Li et al., who have shown increased Nox4, but not Nox1, expression in adventitial fibroblasts isolated from humans with IPAH17. However, a potential role for Nox1 is important to address as the inhibitors we have used to determine a role for Nox4 in PAH also inhibit Nox137. In our study, we also found that Nox1 expression was increased in the MCT-model of PAH, but expression levels were not increased in rat or mouse hypoxia models or in the FHR. The functional importance of Nox1 was recently assessed in a study by Iwata et al. using Nox1 knockout mice52. Contrary to expectations, the loss of Nox1 did not protect against PH but instead promoted PH. At least in the mouse hypoxia model, Nox1 is proposed to repress PAVSMC proliferation through actions on potassium channels. Further studies are needed to define a functional role for Nox1 in rodent models and establish its importance in human PH. In contrast, Nox2 knockout mice are protected from PH 7, 18. How Nox2 contributes to PH is not fully understood. It is highly expressed in immune cells such as macrophages and emits superoxide rather than the preferential generation of hydrogen peroxide from Nox4. It may directly influence the contractile responses of PA53 but it has also been shown that Nox2 knockout mice fail to upregulate Nox4 in response to hypoxia 18. In contrast to Nox4, Nox2 requires cytosolic subunits for activation. We did not observe increased expression of these subunits in hypertensive PA, but it is not known whether this is important for assembly of a functional oxidase in PH. In our study, a functional role for Nox4 can be deduced from the use of the Nox4/1 inhibitor GKT136901 which is not selective for Nox2 54 and also from other studies using GKT137831 which has been shown to be effective in a mouse model of PH20.
Nox4 has gained considerable attention as a primary source of ROS, and cellular proliferation in the pathogenesis of both idiopathic pulmonary fibrosis and pulmonary hypertension. The results of the current study are in agreement with both concepts and connect Nox4 as a common variable in fibroblasts (and other perivascular cells) that contributes to the remodeling of hypertensive pulmonary arterioles. The remodeling of blood vessels requires the participation of all three layers and while numerous studies have proposed a central role for endothelial cells (inside out remodeling), it has also been shown that vascular remodeling can be driven by changes in the adventitia (i.e. ‘outside in’). In both humans and animal models of PH, prominent inflammation, activation and restructuring of the adventitia is observed 55. The adventitial location of Nox4 is therefore highly suited to orchestrate the changes in vascular inflammation and matrix deposition that are widely observed in PH. Current therapies for PH are ineffective in the long term and new therapeutic strategies are needed., Our study supports the effectiveness of Nox4 inhibitors in halting the progression of experimental PH, although (as previously addressed), this strategy was unable to completely reverse the cardiopulmonary functional and morphometric changes that have already occurred. It remains to be determined whether this is a limitation of modalities that target Nox4 or whether a combination of therapeutic approaches will have superior efficacy.
Supplementary Material
Significance.
This study is novel in that we address the importance of Nox4 in Pulmonary Hypertension (PH) using a comprehensive pharmacological approach in vivo with novel Nox4 inhibitors, identifying previously under studied perivascular cell types expressing Nox4 in multiple animal models and in hypertensive human pulmonary arteries. We also address the functional significance of Nox4 in adventitial fibroblasts. The traditional approach to study vascular changes in PH has been to focus on endothelial and smooth muscle cells (i.e. ‘inside out’). Our studies diverge from this theme and suggest an ‘outside in’ (adventitial) process of vascular remodeling that is mediated by Nox4, which although has physiological roles in the endothelium may also have pathologic importance in fibroblasts. Nox4 has been shown to be important in the pathogenesis of other fibrotic conditions such as pulmonary fibrosis, but our study reveals a role for Nox4 in vascular fibrosis associated with PH.
Acknowledgments
The authors gratefully acknowledge the technical assistance of Louise Meadows, Yevgeniy Kovalenkov, and Stephen Haigh.
Sources of funding
This work was supported by Georgia Regents University (GRU) Pilot Study Research Program Award (PSRP) 0053A (SAB and DF), GRU Extramural Success Award (ESA) 00006A (SAB and DF), R01-HL-68026 (SAB), R01-HL-092446 (DF and DS), P01-HL-0101902 (SB, JC, and DF), R01-HL-60190 (SB), and R01-HL-67841 (SB).
Non-standard Abbreviations and acronyms
- PH
pulmonary hypertension
- ROS
reactive oxygen species
- Nox
NADPH oxidase
- PA
pulmonary arteries
- MCT
monocrotaline
- FHR
Fawn hooded rat
- SU/HYP
Sugen-hypoxia
- RV
right ventricle
- RVSP
right ventricular systolic pressure
- VTI
velocity time integral
- PAAT
pulmonary artery acceleration time
- PET
pulmonary ejection time
- SDR
Sprague dawley rat
- CO
cardiac output
- FAP
fibronectin activating protein
- ECIS
Electric Cell-substrate Impedance Sensing
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- HPAEC
human pulmonary arterial endothelial cells
- HPAVSMC
human pulmonary arterial vascular smooth muscle cells
- Cell FN1
cellular fibronectin,
- TSP4
thrombospondin-4
- NG2
chondroitin sulfate proteoglycan
Footnotes
Disclosures
None
References
- 1.Tuder RM, Stacher E, Robinson J, Kumar R, Graham BB. Pathology of pulmonary hypertension. Clin Chest Med. 2013;34:639–650. doi: 10.1016/j.ccm.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 2.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 3.Crosswhite P, Sun Z. Molecular mechanisms of pulmonary arterial remodeling. Molecular medicine. 2014;20:191–201. doi: 10.2119/molmed.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cogolludo A, Moreno L, Villamor E. Mechanisms controlling vascular tone in pulmonary arterial hypertension: Implications for vasodilator therapy. Pharmacology. 2007;79:65–75. doi: 10.1159/000097754. [DOI] [PubMed] [Google Scholar]
- 5.Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982–2006. Eur Respir J. 2007;30:1103–1110. doi: 10.1183/09031936.00042107. [DOI] [PubMed] [Google Scholar]
- 6.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 7.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: Role of superoxide and nadph oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 8.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. Nadph oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol. 2009;297:L596–607. doi: 10.1152/ajplung.90568.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masri FA, Comhair SA, Dostanic-Larson I, Kaneko FT, Dweik RA, Arroliga AC, Erzurum SC. Deficiency of lung antioxidants in idiopathic pulmonary arterial hypertension. Clin Transl Sci. 2008;1:99–106. doi: 10.1111/j.1752-8062.2008.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121:2661–2671. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lassegue B, Griendling KK. Nadph oxidases: Functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 13.Nisimoto Y, Jackson HM, Ogawa H, Kawahara T, Lambeth JD. Constitutive nadph-dependent electron transferase activity of the nox4 dehydrogenase domain. Biochemistry. 2010;49:2433–2442. doi: 10.1021/bi9022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen F, Haigh S, Barman S, Fulton DJ. From form to function: The role of nox4 in the cardiovascular system. Front Physiol. 2012;3:412. doi: 10.3389/fphys.2012.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin ii-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 16.Basset O, Deffert C, Foti M, Bedard K, Jaquet V, Ogier-Denis E, Krause KH. Nadph oxidase 1 deficiency alters caveolin phosphorylation and angiotensin ii-receptor localization in vascular smooth muscle. Antioxid Redox Signal. 2009;11:2371–2384. doi: 10.1089/ars.2009.2584. [DOI] [PubMed] [Google Scholar]
- 17.Li S, Tabar SS, Malec V, Eul BG, Klepetko W, Weissmann N, Grimminger F, Seeger W, Rose F, Hanze J. Nox4 regulates ros levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid Redox Signal. 2008;10:1687–1698. doi: 10.1089/ars.2008.2035. [DOI] [PubMed] [Google Scholar]
- 18.Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, Fan TH, Mitchell PO, Sutliff RL, Hart CM. The role of nadph oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol. 2009;40:601–609. doi: 10.1165/rcmb.2008-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mittal M, Roth M, Konig P, et al. Hypoxia-dependent regulation of nonphagocytic nadph oxidase subunit nox4 in the pulmonary vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 20.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The nox4 inhibitor, gkt137831, attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol. 2012;47:718–726. doi: 10.1165/rcmb.2011-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coflesky JT, Jones RC, Reid LM, Evans JN. Mechanical properties and structure of isolated pulmonary arteries remodeled by chronic hyperoxia. Am Rev Respir Dis. 1987;136:388–394. doi: 10.1164/ajrccm/136.2.388. [DOI] [PubMed] [Google Scholar]
- 22.Sanz J, Kariisa M, Dellegrottaglie S, Prat-Gonzalez S, Garcia MJ, Fuster V, Rajagopalan S. Evaluation of pulmonary artery stiffness in pulmonary hypertension with cardiac magnetic resonance. JACC Cardiovasc Imaging. 2009;2:286–295. doi: 10.1016/j.jcmg.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 23.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Molecular medicine. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 24.Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: Emerging effector cells in chronic inflammation. Nat Rev Immunol. 2011;11:427–435. doi: 10.1038/nri2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rey FE, Pagano PJ. The reactive adventitia: Fibroblast oxidase in vascular function. Arterioscler Thromb Vasc Biol. 2002;22:1962–1971. doi: 10.1161/01.atv.0000043452.30772.18. [DOI] [PubMed] [Google Scholar]
- 26.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. Nadph oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen F, Pandey D, Chadli A, Catravas JD, Chen T, Fulton DJ. Hsp90 regulates nadph oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid Redox Signal. 2011;14:2107–2119. doi: 10.1089/ars.2010.3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhoades RA, Packer CS, Roepke DA, Jin N, Meiss RA. Reactive oxygen species alter contractile properties of pulmonary arterial smooth muscle. Can J Physiol Pharmacol. 1990;68:1581–1589. doi: 10.1139/y90-241. [DOI] [PubMed] [Google Scholar]
- 29.Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, Muller B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol. 2006;148:714–723. doi: 10.1038/sj.bjp.0706779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 31.Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, Taraseviciene-Stewart L, Sung Y, Kraskauskas D, Farkas D, Conrad DH, Nicolls MR, Voelkel NF. A brief overview of mouse models of pulmonary arterial hypertension: Problems and prospects. Am J Physiol Lung Cell Mol Physiol. 2012;302:L977–991. doi: 10.1152/ajplung.00362.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces nox4 nad(p)h oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 33.Ismail S, Sturrock A, Wu P, Cahill B, Norman K, Huecksteadt T, Sanders K, Kennedy T, Hoidal J. Nox4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: The role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am J Physiol Lung Cell Mol Physiol. 2009;296:L489–499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Csiszar A, Labinskyy N, Olson S, Pinto JT, Gupte S, Wu JM, Hu F, Ballabh P, Podlutsky A, Losonczy G, de Cabo R, Mathew R, Wolin MS, Ungvari Z. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension. 2009;54:668–675. doi: 10.1161/HYPERTENSIONAHA.109.133397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veit F, Pak O, Egemnazarov B, Roth M, Kosanovic D, Seimetz M, Sommer N, Ghofrani HA, Seeger W, Grimminger F, Brandes RP, Schermuly RT, Weissmann N. Function of nadph oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid Redox Signal. 2013;19:2213–2231. doi: 10.1089/ars.2012.4904. [DOI] [PubMed] [Google Scholar]
- 36.Borbely G, Szabadkai I, Horvath Z, Marko P, Varga Z, Breza N, Baska F, Vantus T, Huszar M, Geiszt M, Hunyady L, Buday L, Orfi L, Keri G. Small-molecule inhibitors of nadph oxidase 4. J Med Chem. 2010;53:6758–6762. doi: 10.1021/jm1004368. [DOI] [PubMed] [Google Scholar]
- 37.Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P. First in class, potent, and orally bioavailable nadph oxidase isoform 4 (nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2010;53:7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 38.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. Nad(p)h oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 39.Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. Nox4/nadph oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates tgfbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010;65:733–738. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: From innocent bystander to active participant. Circ Res. 2001;89:1111–1121. doi: 10.1161/hh2401.100844. [DOI] [PubMed] [Google Scholar]
- 41.Majesky MW, Dong XR, Hoglund V, Mahoney WM, Jr, Daum G. The adventitia: A dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol. 2011;31:1530–1539. doi: 10.1161/ATVBAHA.110.221549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648. doi: 10.1016/j.cardiores.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Enzerink A, Vaheri A. Fibroblast activation in vascular inflammation. J Thromb Haemost. 2011;9:619–626. doi: 10.1111/j.1538-7836.2011.04209.x. [DOI] [PubMed] [Google Scholar]
- 45.Chan EC, Peshavariya HM, Liu GS, Jiang F, Lim SY, Dusting GJ. Nox4 modulates collagen production stimulated by transforming growth factor beta1 in vivo and in vitro. Biochemical and biophysical research communications. 2013;430:918–925. doi: 10.1016/j.bbrc.2012.11.138. [DOI] [PubMed] [Google Scholar]
- 46.Bellini A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. 2007;87:858–870. doi: 10.1038/labinvest.3700654. [DOI] [PubMed] [Google Scholar]
- 47.Ma W, Han W, Greer PA, Tuder RM, Toque HA, Wang KK, Caldwell RW, Su Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. The Journal of clinical investigation. 2011;121:4548–4566. doi: 10.1172/JCI57734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Long L, Crosby A, Yang X, Southwood M, Upton PD, Kim DK, Morrell NW. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: Potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation. 2009;119:566–576. doi: 10.1161/CIRCULATIONAHA.108.821504. [DOI] [PubMed] [Google Scholar]
- 49.Thannickal VJ, Fanburg BL. Activation of an h2o2-generating nadh oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 50.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27:42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derrett-Smith EC, Dooley A, Gilbane AJ, Trinder SL, Khan K, Baliga R, Holmes AM, Hobbs AJ, Abraham D, Denton CP. Endothelial injury in a transforming growth factor beta-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis and rheumatism. 2013;65:2928–2939. doi: 10.1002/art.38078. [DOI] [PubMed] [Google Scholar]
- 52.Iwata K, Ikami K, Matsuno K, et al. Deficiency of nox1/nicotinamide adenine dinucleotide phosphate, reduced form oxidase leads to pulmonary vascular remodeling. Arterioscler Thromb Vasc Biol. 2014;34:110–119. doi: 10.1161/ATVBAHA.113.302107. [DOI] [PubMed] [Google Scholar]
- 53.Norton CE, Broughton BR, Jernigan NL, Walker BR, Resta TC. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires egfr-dependent activation of nad(p)h oxidase 2. Antioxid Redox Signal. 2013;18:1777–1788. doi: 10.1089/ars.2012.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garrido-Urbani S, Jemelin S, Deffert C, Carnesecchi S, Basset O, Szyndralewiez C, Heitz F, Page P, Montet X, Michalik L, Arbiser J, Ruegg C, Krause KH, Imhof BA. Targeting vascular nadph oxidase 1 blocks tumor angiogenesis through a pparalpha mediated mechanism. PloS one. 2011;6:e14665. doi: 10.1371/journal.pone.0014665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 2006;21:134–145. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.