

Abstract
Asthma is a common heterogeneous disease with both genetic and environmental factors that affects millions of individuals worldwide. Activated type 2 helper T cells secrete a panel of cytokines, including IL-13, a central immune regulator of many of the hallmark type 2 disease characteristics found in asthma. IL-13 has been directly implicated as a potent stimulator of asthma induced airway remodeling. Although IL-13 is known to play a major role in the development and persistence of asthma, the complex combination of environmental and genetic origin of the disease obfuscate the solitary role of IL-13 in the disease. We therefore, used a genetically modified mouse model which conditionally overexpresses IL-13 in the lungs to study the independent role of IL-13 in the progression of asthma. Our results demonstrate IL-13 is associated with a systemic induction of genotoxic parameters such as oxidative DNA damage, single and double DNA strand breaks, micronucleus formation, and protein nitration. Furthermore we show that inflammation induced genotoxicity found in asthma extends beyond the primary site of the lung to circulating leukocytes and erythroblasts in the bone marrow eliciting systemic effects driven by IL-13 over-expression.
Keywords: IL-13, asthma, genotoxicity
1. Introduction
Asthma affects over 150 million individuals and is clinically diagnosed by a barrage of symptoms which include wheezing, coughing, and shortness of breath [1] [2]. Asthma can be subcategorized into two classes: allergic, and non-allergic asthma which constitute roughly 70% and 30% of cases respectively. Although there are almost no observable differences in the types of physiological changes that occur between the two subcategories, non-allergic asthmatics incur more severe and more frequent symptoms [3]. Airways of asthmatic individuals are distinguished through structural modifications, collectively called airway remodeling that includes bronchiolar inflammation, epithelial sloughing, goblet cell metaplasia, multiplied mucus glands, thickening of the lamina reticularis, increased airway smooth muscle mass, angiogenesis, and alterations in the extracellular matrix components. [4] [5]. Additionally, B lymphocytes, T lymphocytes, eosinophils, neutrophils, and macrophages also migrate to the airways, triggering the release of immunoglobulin E, leukotrienes, prostaglandins, histamines, and other chemical mediators leading to airway inflammation [6] [7].
In asthmatic individuals, T cells differentiate preferentially towards type 2 helper T cells (Th2) [8]. Th2 cells are thought to induce asthma through the secretion of many cytokines that activate inflammatory pathways both directly and indirectly [9]. Notably, Th2 cells secrete IL-13 which triggers STAT6 activation through activation of surface receptors present on eosinophils, mast cells, B lymphocytes, fibroblasts, and airway smooth muscle cells [10] [11] [12] [13] [14]. This activation leads to IgE synthesis, mucus hypersecretion, airway hyperreactivity, and tissue fibrosis [15]. Overexpression of IL-13 is necessary and sufficient to induce non-allergic asthma. [10] [15] [16].
Our lab previously published that intestinal mucosal inflammation leads to systemic genotoxicity in mice [17]. In this study we sought to determine if IL-13 induced asthma might also lead to systemic genotoxicity in mice. We used a genetically modified mouse model originally developed by the Elias lab [18], which allow for tetracycline activated conditional over-expression of IL-13 in the Clara Cells of the lungs. We then collected peripheral blood from these asthmatic mice and performed genotoxic assays throughout the progression of the disease.
2. Methods
2.1 Transgenic Mice
The CC10-rtTA-IL13 transgenic (TG) mouse is a well-characterized model of asthma [19]. Clara cell 10-kDa (CC10) gene promoter is used for conditionally expression of IL13 in the mouse lung inducible by doxycycline. CC10-rtTA-IL13 transgenic (TG) mice were generated in Dr. Talal Chatila lab David Geffen School of Medicine University of California Los Angeles, USA. Mice were housed and bred in an institutional specific pathogen free animal facility under standard conditions with a 12 hr light/dark cycle and fed a standard diet according to Animal Research Committee regulations at the University of California, Los Angeles.
2.2 Doxycycline treatment
10 TG mice and 9 WT littermate control mice were maintained on normal water until one month of age. After one month of age doxycycline was administered to the drinking water 2 g/L in 4% sucrose and kept in aluminum foil covered bottles to prevent light-induced degradation of doxycycline for a 3 week time period. The IL13 transgene exhibits baseline leakiness in the absence of doxycycline allowing transgenic mice to exhibit minor elevation of IL13 expression and minor allergic airway inflammation [50]. For this reasoning non-treated transgenic mice were not used as controls in our experiments. After the 3 week exposure to doxycycline all mice were euthanized and pathological assessments were conducted on the lungs.
2.3 Blood Collection
Peripheral blood was collected by facial vein puncture using 5 mm sterile lancets (Medipoint Inc. Mineola, NY) from experimental mice on specified days throughout the duration of treatment and on sacrifice day via terminal right ventricle cardiac puncture using a heparin-coated syringe (American Pharmaceutical Partners, Inc. Schaumburg, IL). Blood from each mouse was collected into EDTA-coated tubes (Sarstedt Aktiengesellschaft & Co., Numbrecht.
2.4 Immunofluorescence
50ul of whole peripheral blood was put into erythrocyte lysis buffer, cells were laid over poly -D-lysine-coated coverslips and fixed with 4% paraformaldehyde (Electron Microscopy Sciences) at room temperature as described previously [20]. Subsequently, cells were permeabilized with 0.5% Triton X-100 (Sigma), followed by 5 rinses in PBS. Blocking was done in aluminum-covered plates overnight at 4°C in 10% FBS. Coverslips were then incubated for 1 hour at room temperature with mouse anti-phospho-Histone H2A.X (Upstate, Temecula, CA) at a dilution of 1:400, or Mouse anti-8-oxoguanine clone 483.15 (Upstate, Temecula, CA) at 1:250 respectively. Coverslips were then rinsed with 0.1% Triton X-100. Following a second 10% FBS blocking, cells were stained with FITC-conjugated anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) at a dilution of 1:150 for samples with γH2AX primary and (1:200) for samples with 8-oxoguanine primary, respectively for 1 hour at room temperature. Coverslips were mounted onto slides using VECTASHIELD with DAPI (Vector Laboratories, Burlingame, CA). For both 8-oxoguanine and γH2AX assay analysis were done on a Zeiss automated microscope. At least 100 cells were counted per sample and cells with more than four distinct foci in the nucleus were considered positive for γH2AX [17] and cells that exhibited elevated fluorescent intensity compared to background were considered positive for 8-oxoguanine respectively. Apoptotic cells, which have an approximate 10-fold increase in nuclear foci in damaged cells, were not included in analyses [17, 21]. Statistical analyses were done using Poisson distribution 8-oxoguanine (STATA statistical analysis software) for γH2AX and using ANOVA and Tukey's post hoc test for 8-oxoguanine analysis (GraphPad Prism).
2.5 Micronucleus assay
3μl of whole blood were spread on a microscope slide and stained with Wright-Giemsa solution (Sigma-Aldrich, St. Louis, MO). At least 4000 erythrocytes were counted according to published recommendations [22]. MN were counted and scored with an Olympus Ax70 (Tokyo, Japan) at 100X magnification. Statistical analysis was done using repeated measures ANOVA followed by Tukey's post-tests (GraphPad Prism).
2.6 DNA single strand breaks
Oxidative DNA damage and DNA strand breaks were measured in peripheral blood cells using the alkaline comet assay. Peripheral blood was collected before doxycycline administration (day 0), and on days 3, 6, 9, 12, 15, 18 and 21 days of doxycycline treatment. Blood was diluted 1:1 with RPMI + 20% DMSO, slowly frozen and stored at -80°C until the assay was performed. The comet assay was done as described previously [23]. Briefly, cells were mixed with low melting-point agarose, and placed in triplicate onto normal agarose layed over gelbond (Lonza Inc. Rockland, ME). The gel was immersed in lysis buffer (2.5 M NaCl, 0.1 M EDTA, 10 mM Tris, 1% Triton, and 10% DMSO), then alkaline electrophoresis buffer (0.3 M NaOH, 1 mM EDTA). After 20 minutes in the electrophoresis buffer at 4°C, the gel was run for 45 minutes at 300 mA, allowed to dry and then stained with SYBR Gold (Molecular Probes). Comet tail-moments were analyzed using CASP (Comet Assay Software Project, http://casp.sourceforge.net/). To measure oxidative DNA damage, the comet assay was modified to include an incubation step with hOGG1 (New England Biolabs, Ipswich, MA). As described previously, embedded cells were incubated with hOGG1 (1:300 in NEBuffer1 and BSA) at 37°C for 30 minutes following the lysis step [24]. Tail-moments were normalized to a control to account for inter-experimental variability. Statistical analyses were done using ANOVA (GraphPad Prism).
2.7 ELISA analysis
Serum was separated from blood by centrifugation on all mice immediately before doxycycline administration (Day0) and on days 3, 5,7,10,13,16,18, and 21. After collection serum samples were aliquoted into micro-centrifuge tubes and kept at -20°C until analysis. Sandwich ELISAs were conducted on samples that were diluted 1:10. Analysis of total Mouse IgE was done according to manufacturer's instructions (BD biosciences). Each sample was done in triplicate and analyzed using the relative standard curve method optical density vs. concentration. Statistical analysis was done using linear mixed model with repeated measurements nested within each mouse. (STATA statistical analysis software)
2.8 Gene expression analysis
Lungs from WT and IL-13 mice at the end of the 3-week doxycycline exposure mice were perfused and lavaged before immersion into RNAlater (Qiagen). Lungs were kept at 4°C for 24 hours then transferred to -80°C until RNA was isolated using the RNeasy Mini kit according to manufacturer's instructions (Qiagen). cDNA was synthesized using SuperscriptIII (Invitrogen) according to manufacturer's recommendations. Quantitative realtime PCR was performed on an ABI Prism 7500 gene expression system (Applied Biosystems) using Taqman gene expression assays for H2AX, 8-oxoguanine, IL-13, IL-4, IL-5, CCL-11/Eotaxin, TGF-β, Tnfα, and Gapdh was used as an internal control. Each reaction was done in triplicate and analyzed using the relative standard curve method.
2.9 Bronchoalveolar lavage (BAL)
The amount of lung inflammation caused by increased infiltration of immune-circulating cells was assessed by BAL. Briefly, mice were euthanized the trachea was isolated via blunt dissection and small caliber tubing was inserted and secured in the airway. A volume of 1 ml of 1X PBS was flushed and removed 3 times successively from the lungs of WT and IL-13 mice until 3 mL of BAL fluid was collected. BAL fluid was centrifuged at 1600 rpm at 4°C for 10 minutes. After centrifugation cells were re-suspended in 200ul of 1X PBS. A 1:1 ratio of cells to trypan blue was put into a hemocytometer and the number of viable cells was counted. After cell viability was assessed 200 ul of remaining cells were put into cytospin and spun at 400 RPM for 5 minutes. Slides were removed from cytospin and allowed to air dry. After drying the slides were stained using (Thermo Kwik Diff staining kit) using manufactures staining recommendation. Slides were allowed to air dry overnight and were mounted with paramount and a coverslip and allowed to dry. Finally at least 200 cells were differentiated by light microscopy based on conventional morphological criteria for each animal.
2.10 Immunohistochemistry
γH2A.X, 8-Hydroxyguanosine, and Nitrotyrosine stains were done on lung tissue of (WT) and IL-13 mice. The slides were placed in xylene to remove paraffin, then a series of ethanol washes. After a wash in tap water, the slides were incubated in 3% Hydrogen peroxide / methanol solution for 10 minutes. The slides were then washed in distilled water, and incubated for 25 minutes in Citrate Buffer pH6 (Invitrogen Corporation) at 95 degrees Celsius using a vegetable steamer. Next, the slides were brought to room temperature, rinsed with PBST (Phosphate Buffered Saline containing 0.05% Tween-20), then incubated at room temperature for 1 hour with Anti-gamma H2A.X (phosphor S139) antibody (Abcam, ab22551), 2 hours with anti-8-Hydroxyguanosine antibody (Abcam, ab48508), or for 45 minutes with Nitrotyrosine (Millipore, 06-284) at the dilutions of 1: 50 for γH2A.X, and 8-Hydroxyguanosine antibodies and at1:200 for the Nitrotyrosine antibody, respectively. The slides were then rinsed with PBST, and were incubated with Dako EnVision+ System –HRP Labelled Polymer Anti-Mouse (Dako, K4001) at room temperature for 30 minutes. Subsequently after a rinse with PBST, the slides were incubated with DAB (3,3′-Diaminobenzidine) for visualization. Finally, the slides were washed in tap water, counterstained with Harris' Hematoxylin, dehydrated in ethanol, and mounted with media.
3. Results
Serum IgE levels are induced in IL-13 Mice. Increased level of IgE is a major factor in the etiology of asthma and is found abundantly in the serum of both human and murine models of asthma [25] [26] [27]. Therefore we assessed the levels of IgE in our transgenic IL-13 over-expression model. We observed that the levels of IgE were higher in the transgenic mice 12 days after IL-13 over-expression and were consistently higher throughout the experiment until terminal day 21 (Fig. 1). Although IL-13 mice exhibited higher IgE levels this induction was only significant at day 21 at **,p<0.007 IL-13 Mice exhibit increased inflammation of the lung. To understand the role IL-13 induced lung inflammation plays in the genotoxicity and progression of asthma we compared the lungs of WT and IL-13 animals. In (Fig.2A and 2C) WT animals exhibit no increase of inflammation. In contrast IL-13 mice (Fig. 2B and 2D) exhibit significant inflammation of the lung characterized in the airways bronchiolar lumen. IL-13 mice showed increased inflammatory cells migration in the bronchiolar epithelium, marked hyperplasia, goblet cell metaplasia, and eosinophilic intracytoplasmic inclusion bodies present in clara cells. Thus IL-13 increases inflammation via recruitment of inflammatory cells migrating into airway spaces.
Fig.1.
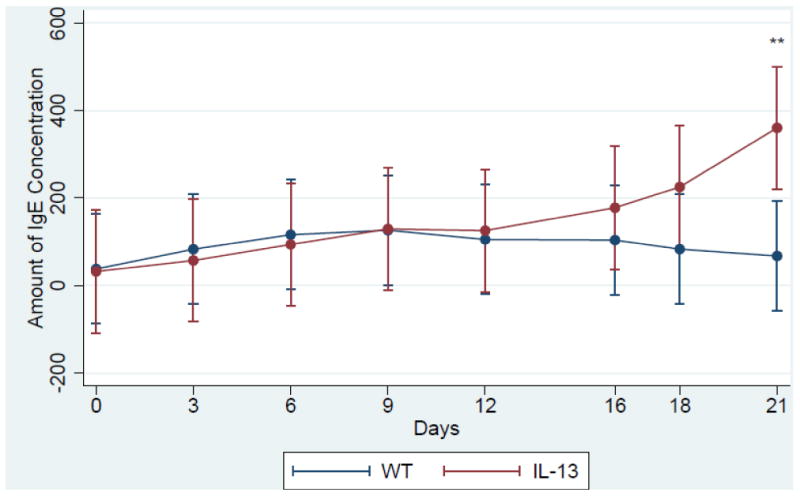
IgE concentration assessed via sandwhich ELISA. **indicates p<, 0.007 n= 5 in both IL-13 and WT animals. Assay was performed with triplicate blood samples from each WT (n=5) and Il-13 (n=5) mouse. (color)
Fig. 2. IL-13 over-expression induces lung inflammation in asthmatic mice.
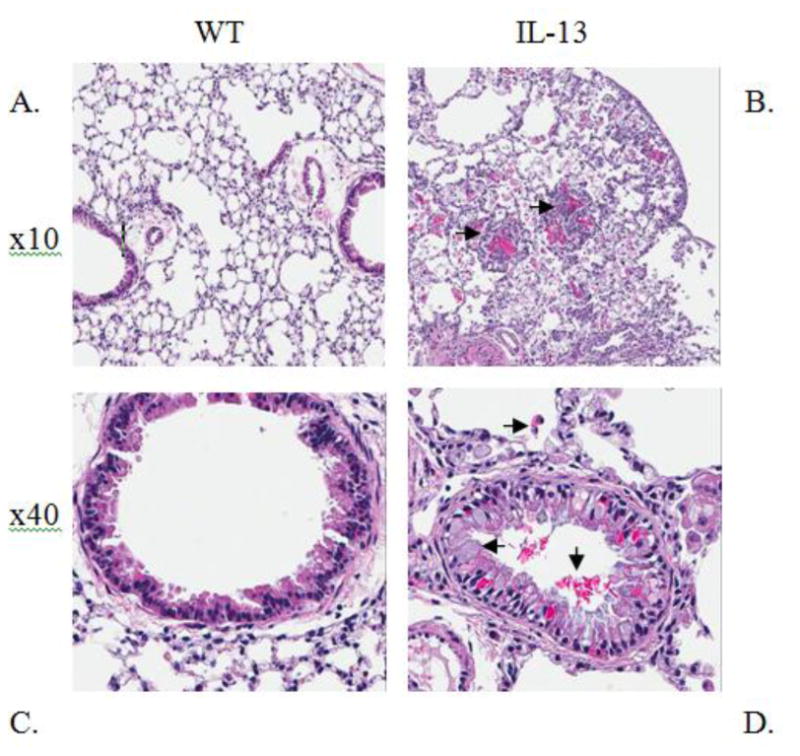
Representative lung histology Hematoxylin & Eosin (H&E) staining at indicated magnifications. (A) 10x image of Wild type (WT) lung and Interleukin 13 over expressed mice (B), both at one month old. Arrows in (B) 10x image indicate formation of granuloma metafoci surrounding eosinophilic crystals. 40x image of WT (C) and (D) 40x image of IL-13 mice. Arrows in (D) indicate eosinophil migration, goblet cell metaplasia, and eosinophilic crystal formation in bronchial lumen. n=9 for WT and n=10 for IL-13 mice. (color)
IL-13 mice exhibit increased immune cell infiltration in BAL fluid. To delineate what immune cells may be implicated in the persistent inflammatory and genotoxic response present in the asthma mouse model we determined the cellular composition of the BAL fluid in both the WT and IL-13 mice. IL-13 animals had a near 5-fold increase in eosinophil presence in BAL fluid compared to WT animals at p<0.04 (Fig. 3). IL-13 mice also exhibited significantly more circulation of neutrophils in the BAL fluid at ***, p<0.0004 compared to WT littermates. IL-13 mice also exhibited an increase induction of lymphocytes although this observation was not significantly different from WT animals.
Fig.3. Inflammatory cell composition of bronchial alveolar lavage fluid (BALF).
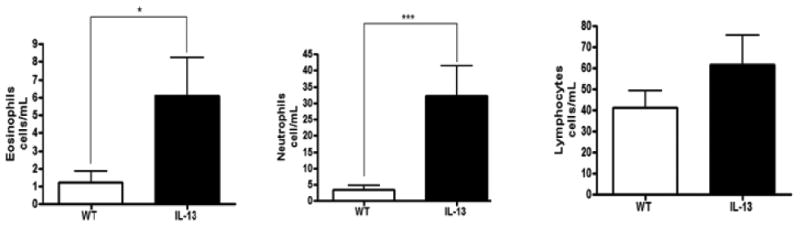
Differential cell analysis were determined by light microscopic evaluation n=9 for WT, and n=10 for IL-13 animals,* indicates p<0.05,*** indicates p<0.0004 respectively analysis were conducted using two tailed Student's unpaired T-test with Mann-Whitney determination.
IL-13 mice exhibit persistent inflammation induced immune response. To elucidate the role IL-13 plays in up-regulation of inflammation in the lung we assessed key mediators of asthma Fig. 4A-4C, inflammatory disease Fig.4D-F, and genotoxicity Fig. 4G via quantitative real-time PCR. As a measurement of further efficacy of our tissue specific asthma model we measured IL-13 levels in the lungs of both groups of experimental mice. The asthma mouse model exhibited an expected significant increased gene expression of IL-13 at **, p<0.001 compared to WT animals. IL-4, TNF-α, IL-5 were significantly up-regulated in IL-13 mice at *,p<0.01 compared to WT animals. IL-13 mice also displayed a marked increase of TGF-β Fig.1-6E at **,p<0.001 compared to WT mice. Cc11 transcript levels in IL-13 mice were slightly higher but did not show a significant increase compared to WT mice Fig.1-6G These data are indicative of a chronically activated innate immune response present in IL-13 mice compared to WT mice.
Fig.4. Assessment of cytokine panel in lung mRNA measured by quantitative real-time PCR.
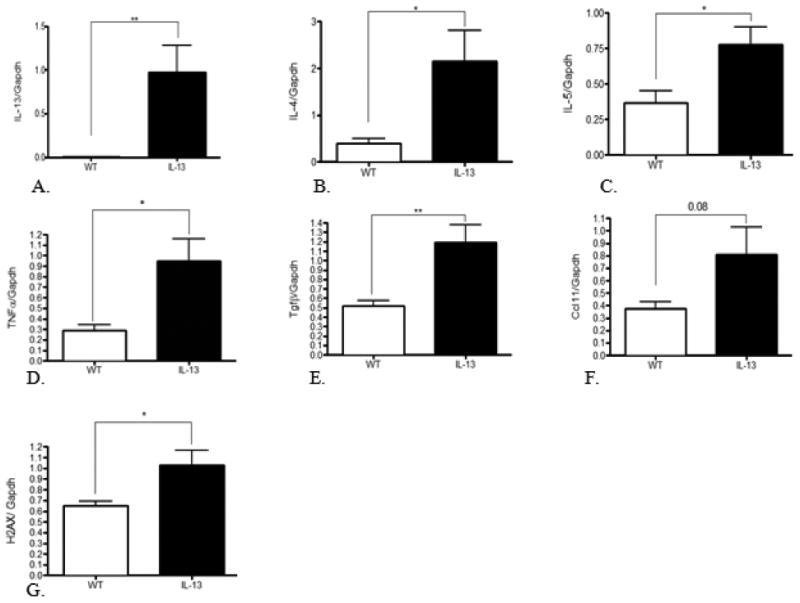
Mean expression divided by Gapdh, the internal control gene. * indicates p<0.01, ** indicates p<0.001, analysis were conducted using two tailed Student's unpaired T-test. n=9 for WT animals and n=10 for IL-13 animals.
Fig.6. Persistent genotoxicity measured via inflammation induced 8-oxoguanine and double stranded breaks measured via γH2AX in peripheral blood.
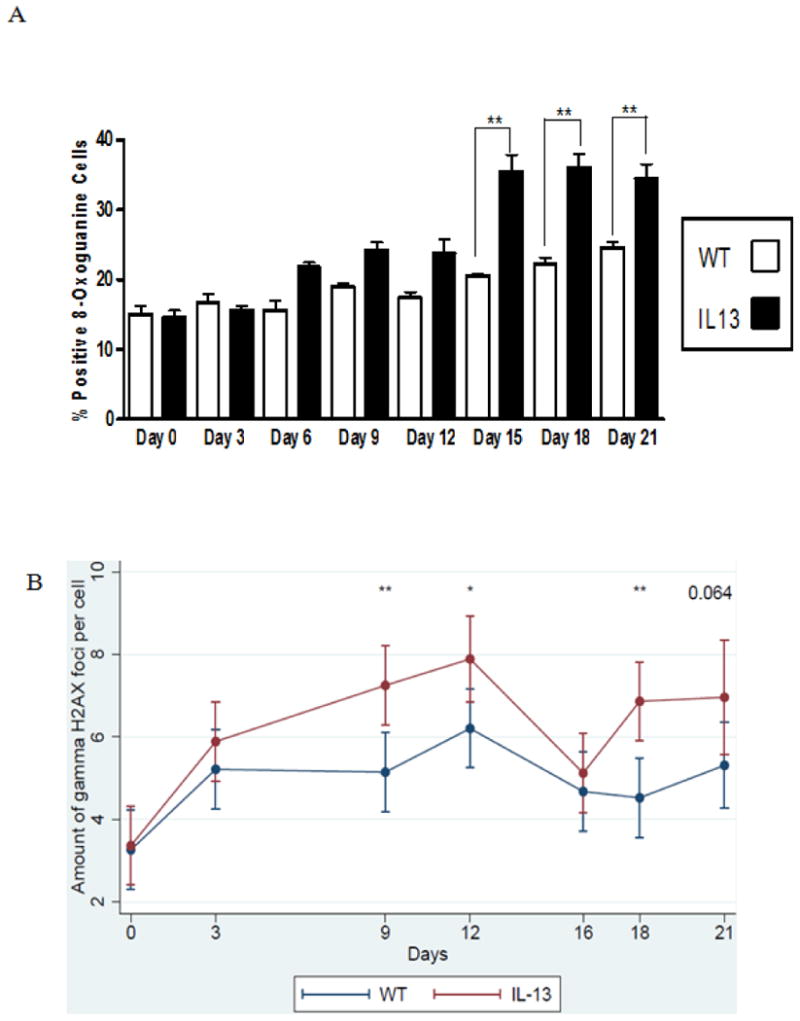
A.) Percent positive cells for 8-oxoguanine induction in white blood cells. Presence of 8-oxoguanine was confirmed by immunofluorescence. Positive cells stain brightly green compared to no immunofluorescent staining for negative cells. White bars indicate Wild type (WT) animals and black bars indicate IL-13 animals. Data represent mean ± SEM. Statistical analyses were done using ANOVA testing and Tukey's post hoc analysis. n=5 in all groups. ** indicates p<0.001.B Assessment of double strand breaks measured via γH2AX assay, were counted per cell using fluorescent microscopy before doxycycline administration at Day 0 and after doxycycline administration at days 3,9,12,16,18 and day 21 using a linear mixed model to determine genotoxic accumulation over time. * indicates p<0.02, ** indicates p<0.002 n=5 for WT and IL-13 animals. (color)
IL-13 Mice have increased staining of markers of genotoxicity in lung tissue. To further assess asthma induced genotoxicity, the lungs of WT and IL-13 mice were stained with γH2AX, anti-8- Hydroxyguanosine, and Nitrotyrosine antibodies. IL-13 mice exhibited increased staining with markers of genotoxicity in the lungs in comparison to WT animals (Fig. 5).
Fig.5. Staining of markers of genotoxicity and oxidative protein damage in lung tissue as measured by immunohistochemistry.
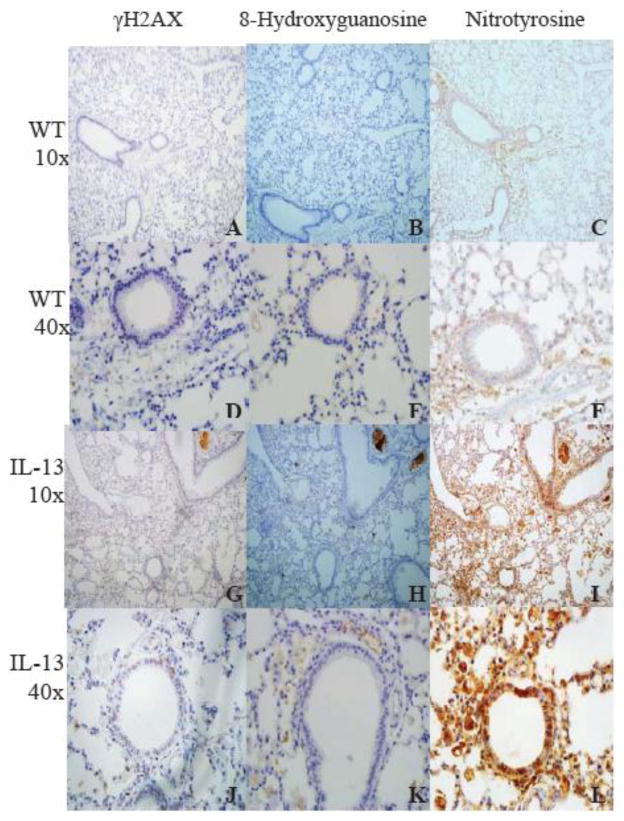
Markers of double stranded breaks (A-J), reactive oxygen species (B-K), and inflammation (C-L) induced genotoxicity were stained in WT and IL-13 mice. Lung tissue in IL-13 mice (G-L) exhibited increased staining in all genotoxic parameters in comparison to WT mice (A-F). n=9 for WT animals and n=10 for IL-13 animals. (color)
IL-13 Mice show systemically elevated reactive oxygen species induced genotoxicity and double stranded breaks in peripheral blood. Because IL-13 is a major mediator of allergic asthma and induces higher levels of inflammation in asthmatic mice [28], we hypothesized that inflammation induced systemic DNA damage would be more prevalent in IL-13 compared to wild type (WT) mice. Increase of asthma induced genotoxicity was assessed in peripheral white blood cells as a systemic measurement of DNA damage. 8-oxoguanine is a mutagenic lesion caused by the interaction of a reactive oxygen species to DNA that causes G:C to T:A transversion mutations during replication [29]. Percent positive 8-oxoguanine staining in peripheral white blood cells was assessed using fluorescent microscopy. Blood was taken on day 0 as an assessment of baseline levels of 8-oxoguanine induction between both WT and IL-13 groups. After 6 days of doxycycline presence in drinking water IL-13 mice exhibited a slight increase of 8-oxoguanine immuno-staining compared to wild type mice this induction persisted and became statistically significant at **,p<0.001 at day 15 and remained elevated throughout the 21 day exposure to doxycycline (Fig.6A). As a measure of the amount of genotoxicity caused by an accumulation of DNA double strand breaks the γH2AX assay was assessed in the WT and IL-13 animals. H2AX is a member of the histone H2A protein family and becomes rapidly phosphorylated in presence of a DNA damaging event [30]. This rapid phosphorylation causes recruitment of DNA repair proteins to the site of the break and is detectable by specific antibodies to γH2AX. In Fig. 6B we assessed the amount of double strand breaks present in the WT and IL-13 animals. Transgenic animals exhibited an increase in the amount of double strand breaks occurring at every time point after over-expression. This induction of γH2AX was significant on day 9 at **, p<0.002, at day 12 at *, p<0.02, and at day 18 day at **,p<0.001 respectively. A nearly significant induction of γH2AX at P=0.064 on day 21 was also observed in the IL-13 mice
IL-13 mice have systemic single strand breaks and persistent systemic genotoxicity that induces comet and micronucleus formation in peripheral blood leukocytes respectively. The in vivo micronucleus assay was conducted in mature normochromatic erythrocytes circulating in the peripheral blood to determine chromosomal damage. Micronuclei in erythrocytes/erythroblasts from the peripheral blood or bone marrow have been induced in the presence of chromosome breaks, spindle abnormalities, or structurally abnormal chromosomes [22] [29]. Mature micronucleated normochromatic erythrocytes represent the final developmental stage of erythroblasts containing micronuclei stemming in the bone marrow, and permit the simultaneous study of the generation and elimination of micronucleated erythrocytes [29] [32]. Blood was taken on day 0 as an assessment of baseline levels of 8-oxoguanine induction between both WT and IL-13 groups. After 6 days of doxycycline presence in drinking water IL-13 mice exhibited a significant increase of micronuclei formation in peripheral blood compared to WT animals at *, p<0.05. This statistically significant induction at *, p<0.05 of micronuclei persisted until terminal date Day 21 (Fig.7A).
Fig.7. IL-13 over-expression induced single stranded breaks and micronucleated cells in peripheral blood.
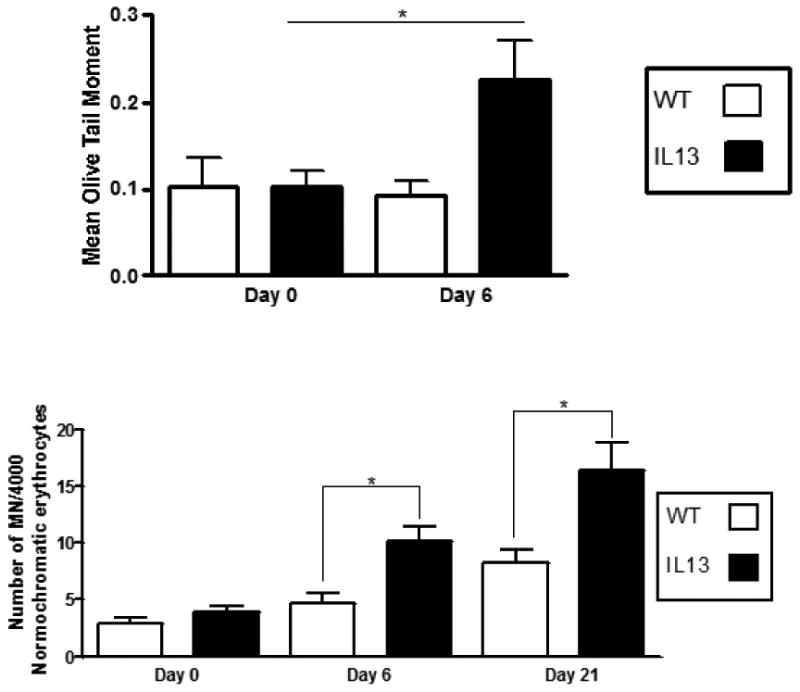
A.) Assessment of single strand breaks were measured via comet assay before doxycycline administration at Day 0 and after doxycycline administration at days 6. At least 100 olive tail moments were counted via fluorescent microscopy and assessed using CASP software. White bars indicate Wild type (WT) animals and black bars indicate IL-13 animals. Data represent mean ± SEM. Statistical analyses were done using ANOVA testing and Tukey's post hoc analysis. * indicates p<0.05 n=5 for WT and IL-13 animals. B.) Number of micronucleated cells per 4000 normorchromatic erythrocytes. Presence of micronuclei were confirmed by light microscope at 100X. White bars indicate Wild type (WT) animals and black bars indicate IL-13 animals. Data represent mean ± SEM. Statistical analyses were done using ANOVA testing and Tukey's post hoc analysis. n=9 for WT and n=10 for IL-13.* indicates p<0.05.
The alkaline comet assay is a gel electrophoresis assay that allows the detection of single and double strand breaks, and alkali labile sites at the single cell level [17]. Asthma is an inflammatory disease that produces large amounts of reactive oxygen species (ROS) [32, 33]. Interaction of ROS with DNA may result in mutagenic oxidative base modifications such as 8-hydroxydeoxyguanosine (8-oxo-dGuo) and induce DNA strand breaks [32]. Transgenic animals exhibited an increase in the amount of single strand breaks occurring after 6 days of IL-13 over-expression compared to all other groups where no increase was observed. This induction of strand breaks was significant for indicated groups at *, p<0.05. (Fig.7B)
4 Discussion
Asthma is a chronic obstructive lung disease characterized by chronic inflammation of the airways and recurrent bronchospasms ranging from mild to debilitating [34]. It is well established that Interleukin-13 serves as a major mediator of the asthmatic process [16]. This study is the first to assess IL-13 role in genotoxicity concomitantly with the inflammatory asthmatic process. Our study also demonstrates for the first time that this genotoxicity extends beyond the primary site of the lung to circulating leukocytes and erythroblasts in the bone marrow eliciting systemic effect in peripheral blood driven by IL-13 overexpression.
IL-13 mice exhibited an asthmatic phenotype consistent with previous data. We found a significant increase in IgE. IL-13 mice had sub-epithelial eosinophilic infiltration, peribronchiolar, and perivascular lymphoid infiltration, free floating eosinophilic crystals, many surrounded by aggregates of macrophages, giant cells and neutrophil PMNs [4,16, 35]. IL-13 mice showed a significant influx of eosinophils, a cellular hallmark of asthma [36], a significant increase in neutrophils, and non-significant increase of lymphocytes in the bronchoalveolar lavage fluid. Transgenic mice had increased gene expression of IL-13, IL-4, IL-5,TNFα, Tgfβ, γH2AX, and Ccl11 produced in the lungs [37] [28] [38] [39]. There was a significant increase in γH2AX levels in the lungs of the IL-13 mice and an induced yet non-significant increase in 8-oxoguanine. In IL-13 mice, we detected a significant increase of single and double stranded breaks in the peripheral blood and lung, a significant induction of micronucleus formation in the normochromatic erythrocytes present in the peripheral blood leukocytes, as well as increased staining of markers of genotoxicity in the lung [29].
We utilized the well characterized inducible over-expression CC10-rtTA-IL13 transgenic (TG) mouse to elucidate the effect interleukin-13 may have in the genotoxicity of asthma. Recent work has shown that IL-13 signaling is mediated by the type-2 IL-4 receptor, which consists of the IL-4R alpha and IL-13R alpha 1 chains [15] [41], yet IL-13 alone is necessary and sufficient to render the major pathophysiological effects of asthma [16]. IL-4 along with IL-13 is a key mediator of inflammation, has an overlapping biological function as IL-13, yet has a distinct role in asthma progression [15]. IL-4 is best known for its role for defining the Th2 phenotype of lymphocytes in asthma, but also exacerbating the asthmatic phenotype by increasing airway hyperresponsiveness, eosinophil recruitment, and mucus over-production [16] [35]. We next investigated mRNA levels of CCL-11/eotaxin. There was not a significant increase in CCL-11/eotaxin transcript present in the lung mRNA, however, we did observe a significant increase in IL-5 lung mRNA transcript. A possible explanation to the induced yet nonsignificant increase in CCL11/eotaxin transcript may be found in the work of Humbles et. al. [41]. These data suggest, in corroboration with Conroy, et. al. [36] that migrating eosinophils in the bronchoalveolar lavage fluid may stimulate release of IL-5, but not CCL11/eotaxin.
With regards to the genotoxicity of IL-13 induced asthma we discovered an increase in the amount of γH2AX and 8-oxoguanine in the blood and lungs of these mice. Phosphorylation of histone H2A to form γH2AX in the presence of a DNA damaging event is used as a biomarker of cellular response to DSBs and has a potential for monitoring DNA damage and repair in human and mice [42] [43]. 8-oxo-7,8- dihydroguanine (8oxoG) is an abundant ROS induced lesion that when accumulated has been associated with numerous diseases, including cancer [17] [44] [45]. We did observe a slight increase of 8oxoG in the WT animals which most likely can be attributed to the repeated blood draws that caused a moderate increase in the production of stress related ROS- induced DNA damage similarly found in Westbrook, et. al [46]. As a measure of ongoing DNA damage Westbrook, et. al [17] show that an accumulation of double-strand breaks can lead to chromosome breaks and micronucleus formation.
Perturbations to erythroblasts in the bone marrow may be a humoral effect of inflammation-associated DNA damage, as with the peripheral leukocytes. We suggest that increased inflammation in our experimental mice causes a significant induction of migratory cells that preferentially release pro-inflammatory cytokines at sites of inflammation. This re-circulating pool of activated cells may recruit more effector cells, which come into contact with erythroblasts in the bone marrow causing the observed clastogenicity.
To evaluate the inflammatory cell composition we determined the differential cell percentages. The increased prevalence of neutrophils over that of eosinophils in the bronchoalveolar lavage fluid may depict the presence of a more chronic asthmatic phenotype, an idea supported by Kamath et. al [47]. Moreover this significant influx of both neutrophils and eosinophils may help generate the enhanced systemic genotoxic response found in the blood. This observation of an induction of γH2AX mRNA levels further confirms increased genotoxicity in IL-13 mice and correlates with our hypothesis of systemic genotoxicity.
In conclusion, we propose that the key asthmatic mediator interleukin-13, increases important elements of the inflammatory response including ROS derived oxidative stress causing an induction in genotoxicity that has wide reaching systemic genotoxic effects, such as oxidative DNA damage, single and double DNA strand breaks, micronucleus formation, and protein nitration in the peripheral blood. Two potential explanations for the local inflammation and systemic genotoxicity are described by Westbrook, et. al[17]. In the first model, innate immune cells activated by inflammation release reactive species that damage circulating leukocytes in the periphery. In the second model, inflammatory cytokines are responsible for systemic genotoxicity through cytokine receptor mediated production of free radicals that damage distant leukocytes. These models are not mutually exclusive, making the second model more likely as it has been shown that injection of cytokines causes systemic genotoxicity in mice [46]. We further suggest increased immune cell infiltration and inflammation byproducts as a possible culprit to this genotoxic induction. The induction of systemic strand breaks which are prevalent in many types of cancer were found to be significantly increased in our IL-13 induced asthma model. Recent studies also point to the fact that asthmatic patients have higher cancer risk [48] [49] [50]. Previous studies in human asthmatics also identified increased strand breaks produced during the direct interaction of ROS with DNA or during the repair process of damaged DNA [32] [52]. In summary, asthma is associated with systemic genotoxicity through single and double DNA strand breaks, oxidative DNA damage, protein nitration, and micronucleus formation. This study further implicates IL-13 as a potential therapeutic target for other pulmonary diseases involving carcinogenesis. In addition, systemic genotoxicity might be a convenient blood marker for the extent and severity of asthma.
Highlights.
We use doxycycline treated CC10-rtTA-IL13 transgenic mice as a model for asthma.
Il-13 overexpression in mouse lungs elicits genotoxicity in peripheral blood cells.
Genotoxicity in peripheral blood cells include oxidative DNA damage.
Genotoxicity in peripheral blood cells include single and double strand DNA breaks.
Genotoxicity in peripheral blood cells include micronucleus formation.
Acknowledgments
Aaron Chapman- designed research, performed research, analyzed data, and wrote the paper. Daniel Malkin- designed research, performed research, analyzed data, and wrote the paper Jessica Camacho- performed research, analyzed data, aided in scientific discussions. Robert Schiestl- Principal Investigator. We thank Dr. Talal Chatila (Harvard University) and Dr. Brigitte Gomperts for donating IL-13 over-expressing mice. We also thank Dr. Nora Rozengurt, Dr. Petra Wise, Ko Kiehle and Ngan Doan for mouse pathological assessment, aid in initial experimental setup, and mouse IHC staining respectively. We also thank Ana Scuric, Aleksandra Durisic, Nami Moradi, Kristin Yamada Ph.D., Sarkis Aroyan, Jared Liu, Ryan Bakhit, and ATS statistical consultants for experimental and statistical input. This work was supported by a grant from the National Institute for Allergy and Infectious Disease, NIH R56AI094756-0.
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akinbami LJ, Moorman JE, Liu X. Asthma prevalence, health care use, and mortality: United States,2005-2009. Natl Health Stat Report. 2011:1–14. [PubMed] [Google Scholar]
- 2.Miller AL. The etiologies, pathophysiology, and alternative/complementary treatment of asthma. Altern Med Rev. 2001;6:20–47. [PubMed] [Google Scholar]
- 3.Romanet-Manent S, Charpin D, Magnan A, Lanteaume A, Vervloet D. Allergic vs nonallergic asthma: what makes the difference? Allergy. 2002;57:607–613. doi: 10.1034/j.1398-9995.2002.23504.x. [DOI] [PubMed] [Google Scholar]
- 4.Fireman P. Understanding asthma pathophysiology. Allergy Asthma Proc. 2003;24:79–83. [PubMed] [Google Scholar]
- 5.Hyde DM, Miller LA, Schelegle ES, et al. Asthma: a comparison of animal models using stereological methods. European Respiratory Review. 2006;15:122–135. [Google Scholar]
- 6.Bradley BL, Azzawi M, Jacobson M, et al. Eosinophils, T-lymphocytes, mast cells, neutrophils, and macrophages in bronchial biopsy specimens from atopic subjects with asthma: Comparison with biopsy specimens from atopic subjects without asthma and normal control subjects and relationship to bronchial hyperresponsiveness. Journal of Allergy and Clinical Immunology. 1991;88:661–674. doi: 10.1016/0091-6749(91)90160-p. [DOI] [PubMed] [Google Scholar]
- 7.Henderson WR, Lewis DB, Albert RK, et al. The importance of leukotrienes in airway inflammation in a mouse model of asthma. The Journal of Experimental Medicine. 1996;184:1483–1494. doi: 10.1084/jem.184.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zimmermann N, King NE, Laporte J, et al. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–1874. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 10.Akdis M, Burgler S, Crameri R, et al. Interleukins, from 1 to 37, and interferon-γ: Receptors, functions, and roles in diseases. Journal of Allergy and Clinical Immunology. 2011;127:701–721. e770. doi: 10.1016/j.jaci.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 11.Chatila TA. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol Med. 2004;10:493–499. doi: 10.1016/j.molmed.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol. 2000;105:1063–1070. doi: 10.1067/mai.2000.107604. [DOI] [PubMed] [Google Scholar]
- 13.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 14.Medoff BD, Thomas SY, Luster AD. T cell trafficking in allergic asthma: the ins and outs. Annu Rev Immunol. 2008;26:205–232. doi: 10.1146/annurev.immunol.26.021607.090312. [DOI] [PubMed] [Google Scholar]
- 15.Munitz A, Brandt EB, Mingler M, Finkelman FD, Rothenberg ME. Distinct roles for IL-13 and IL-4 via IL-13 receptor alpha1 and the type II IL-4 receptor in asthma pathogenesis. Proc Natl Acad Sci U S A. 2008;105:7240–7245. doi: 10.1073/pnas.0802465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wills-Karp M, Luyimbazi J, Xu X, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 17.Westbrook AM, Wei B, Braun J, Schiestl RH. Intestinal mucosal inflammation leads to systemic genotoxicity in mice. Cancer Res. 2009;69:4827–4834. doi: 10.1158/0008-5472.CAN-08-4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Z, Ma B, Homer RJ, Zheng T, Elias JA. Use of the tetracycline-controlled transcriptional silencer (tTS) to eliminate transgene leak in inducible overexpression transgenic mice. J Biol Chem. 2001;276:25222–25229. doi: 10.1074/jbc.M101512200. [DOI] [PubMed] [Google Scholar]
- 19.Zheng T, Zhu Z, Wang Z, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstine JV, Nahas S, Gamo K, et al. Constitutive phosphorylation of ATM in lymphoblastoid cell lines from patients with ICF syndrome without downstream kinase activity. DNA Repair (Amst) 2006;5:432–443. doi: 10.1016/j.dnarep.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Muslimovic A, Ismail IH, Gao Y, Hammarsten O. An optimized method for measurement of gamma-H2AX in blood mononuclear and cultured cells. Nat Protoc. 2008;3:1187–1193. doi: 10.1038/nprot.2008.93. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi M, Tice RR, MacGregor JT, et al. In vivo rodent erythrocyte micronucleus assay. Mutat Res. 1994;312:293–304. doi: 10.1016/0165-1161(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 23.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 24.Smith CC, O'Donovan MR, Martin EA. hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis. 2006;21:185–190. doi: 10.1093/mutage/gel019. [DOI] [PubMed] [Google Scholar]
- 25.Hamelmann E, Tadeda K, Oshiba A, Gelfand EW. Role of IgE in the development of allergic airway inflammation and airway hyperresponsiveness--a murine model. Allergy. 1999;54:297–305. doi: 10.1034/j.1398-9995.1999.00085.x. [DOI] [PubMed] [Google Scholar]
- 26.Mora J, Riggs EK, Fu J, et al. Expression of the high affinity IgE receptor by neutrophils of individuals with allergic asthma is both minimal and insensitive to regulation by serum IgE. Clin Immunol. 2009;132:132–140. doi: 10.1016/j.clim.2009.03.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang XH, Zhao W, Liu SG, Feng XP. Correlation of IL-4 and IL-13 gene polymorphisms with asthma and total serum IgE levels. Zhonghua Jie He He Hu Xi Za Zhi. 2009;32:161–164. [PubMed] [Google Scholar]
- 28.Elias JA, Zheng T, Lee CG, et al. Transgenic modeling of interleukin-13 in the lung. Chest. 2003;123:339s–345s. [PubMed] [Google Scholar]
- 29.Westbrook AM, Schiestl RH. Atm-deficient mice exhibit increased sensitivity to dextran sulfate sodium-induced colitis characterized by elevated DNA damage and persistent immune activation. Cancer Res. 2010;70:1875–1884. doi: 10.1158/0008-5472.CAN-09-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonner WM, Redon CE, Dickey JS, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinheider G, Neth R, Marquardt H. Evaluation of nongenotoxic and genotoxic factors modulating the frequency of micronucleated erythrocytes in the peripheral blood of mice. Cell Biol Toxicol. 1986;2:197–211. doi: 10.1007/BF00117712. [DOI] [PubMed] [Google Scholar]
- 32.Hasbal C, Aksu BY, Himmetoglu S, et al. DNA damage and glutathione level in children with asthma bronchiale: effect of antiasthmatic therapy. Pediatr Allergy Immunol. 2010;21:e674–678. doi: 10.1111/j.1399-3038.2009.00959.x. [DOI] [PubMed] [Google Scholar]
- 33.Luzina IG, Keegan AD, Heller NM, Rook GA, Shea-Donohue T, Atamas SP. Regulation of inflammation by interleukin-4: a review of “alternatives”. J Leukoc Biol. 2012;92:753–764. doi: 10.1189/jlb.0412214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bosse Y, Lemire M, Poon AH, et al. Asthma and genes encoding components of the vitamin D pathway. Respir Res. 2009;10:98. doi: 10.1186/1465-9921-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grunig G, Warnock M, Wakil AE, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conroy DM, Humbles AA, Rankin SM, et al. The role of the eosinophil-selective chemokine, eotaxin, in allergic and non-allergic airways inflammation. Mem Inst Oswaldo Cruz. 1997;92(Suppl 2):183–191. doi: 10.1590/s0074-02761997000800024. [DOI] [PubMed] [Google Scholar]
- 37.Ma B, Liu W, Homer RJ, et al. Role of CCR5 in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol. 2006;176:4968–4978. doi: 10.4049/jimmunol.176.8.4968. [DOI] [PubMed] [Google Scholar]
- 38.Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 39.Vanoirbeek JA, Tarkowski M, De Vooght V, Nemery B, Hoet PH. Immunological determinants in a mouse model of chemical-induced asthma after multiple exposures. Scand J Immunol. 2009;70:25–33. doi: 10.1111/j.1365-3083.2009.02263.x. [DOI] [PubMed] [Google Scholar]
- 40.Mentink-Kane MM, Wynn TA. Opposing roles for IL-13 and IL-13 receptor alpha 2 in health and disease. Immunol Rev. 2004;202:191–202. doi: 10.1111/j.0105-2896.2004.00210.x. [DOI] [PubMed] [Google Scholar]
- 41.Humbles AA, Conroy DM, Marleau S, et al. Kinetics of eotaxin generation and its relationship to eosinophil accumulation in allergic airways disease: analysis in a guinea pig model in vivo. J Exp Med. 1997;186:601–612. doi: 10.1084/jem.186.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valdiglesias V, Giunta S, Fenech M, Neri M, Bonassi S. gammaH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res. 2013 doi: 10.1016/j.mrrev.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto ML, Reliene R, Oshima J, Schiestl RH. Effects of human Werner helicase on intrachromosomal homologous recombination mediated DNA deletions in mice. Mutat Res. 2008;644:11–16. doi: 10.1016/j.mrfmmm.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 44.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Fortini P, Pascucci B, Parlanti E, D'Errico M, Simonelli V, Dogliotti E. 8-Oxoguanine DNA damage: at the crossroad of alternative repair pathways. Mutat Res. 2003;531:127–139. doi: 10.1016/j.mrfmmm.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 46.Westbrook AM, Wei B, Braun J, Schiestl RH. Intestinal inflammation induces genotoxicity to extraintestinal tissues and cell types in mice. Int J Cancer. 2011;129:1815–1825. doi: 10.1002/ijc.26146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamath AV, Pavord ID, Ruparelia PR, Chilvers ER. Is the neutrophil the key effector cell in severe asthma? Thorax. 2005;60:529–530. doi: 10.1136/thx.2005.043182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boffetta P, Ye W, Boman G, Nyren Lung cancer risk in a population-based cohort of patients hospitalized for asthma in Sweden. Eur Respir J. 2002;19:127–133. doi: 10.1183/09031936.02.00245802. [DOI] [PubMed] [Google Scholar]
- 49.Brown DW, Young KE, Anda RF, Felitti VJ, Giles WH. Re: asthma and the risk of lung cancer. findings from the Adverse Childhood Experiences (ACE) Cancer Causes Control. 2006;17:349–350. doi: 10.1007/s10552-005-0420-5. [DOI] [PubMed] [Google Scholar]
- 50.Garcia Sanz MT, Gonzalez Barcala FJ, Alvarez Dobano JM, Valdes Cuadrado L. Asthma and risk of lung cancer. Clin Transl Oncol. 2011;13:728–730. doi: 10.1007/s12094-011-0723-9. [DOI] [PubMed] [Google Scholar]
- 51.Zeyrek D, Cakmak A, Atas A, Kocyigit A, Erel O. DNA damage in children with asthma bronchiale and its association with oxidative and antioxidative measurements. Pediatr Allergy Immunol. 2009;20:370–376. doi: 10.1111/j.1399-3038.2008.00780.x. [DOI] [PubMed] [Google Scholar]