

Abstract
Background
Tuber melanosporum, also known in the gastronomic community as “truffle”, features one of the largest fungal genomes (125 Mb) with an exceptionally high transposable element (TE) and repetitive DNA content (>58%). The main purpose of DNA methylation in fungi is TE silencing. As obligate outcrossing organisms, truffles are bound to a sexual mode of propagation, which together with TEs is thought to represent a major force driving the evolution of DNA methylation. Thus, it was of interest to examine if and how T. melanosporum exploits DNA methylation to maintain genome integrity.
Findings
We performed whole-genome DNA bisulfite sequencing and mRNA sequencing on different developmental stages of T. melanosporum; namely, fruitbody (“truffle”), free-living mycelium and ectomycorrhiza. The data revealed a high rate of cytosine methylation (>44%), selectively targeting TEs rather than genes with a strong preference for CpG sites. Whole genome DNA sequencing uncovered multiple TE-enriched, copy number variant regions bearing a significant fraction of hypomethylated and expressed TEs, almost exclusively in free-living mycelium propagated in vitro. Treatment of mycelia with 5-azacytidine partially reduced DNA methylation and increased TE transcription. Our transcriptome assembly also resulted in the identification of a set of novel transcripts from 614 genes.
Conclusions
The datasets presented here provide valuable and comprehensive (epi)genomic information that can be of interest for evolutionary genomics studies of multicellular (filamentous) fungi, in particular Ascomycetes belonging to the subphylum, Pezizomycotina. Evidence derived from comparative methylome and transcriptome analyses indicates that a non-exhaustive and partly reversible methylation process operates in truffles.
Keywords: DNA methylation, Tuber melanosporum, Ascomycete truffle, Pezizomycetes, Transposable elements, Whole-genome bisulfite sequencing, Methylome, Copy number variation, Transposon expression, 5-azacytidine, Genome plasticity
Data Description
Purpose of data acquisition
Genome-wide profiles of DNA methylation have recently emerged from methylome studies carried out on more than 20 eukaryotic organisms belonging to four different lineages [1-5]. Promoter and gene-body methylation marks are common in higher eukaryotes where they provide an additional level of gene regulation, while inactivation of transposon and other repeated elements appears to be the main purpose of DNA methylation in fungi (reviewed by [6]).
The black truffle (T. melanosporum) is a macrofungus and a highly appreciated gastronomic delicacy produced by an ectomycorrhizal ascomycetous symbiont found throughout southern Europe. It features one the largest genomes (125 Mb), with the highest transposable element (TE) and repetitive DNA content (>58%), amongst fungi that have been sequenced so far [7]. As obligate outcrossing organisms, truffles are bound to a sexual mode of propagation, which together with TEs, has been proposed to be a major force driving the evolution of DNA methylation [6,8]. T. melanosporum belongs to the Pezizales, a largely unexplored group of ascomycetes that includes Ascobolus immersus, a fungus relying on premeiotic DNA methylation as a means to control repetitive element proliferation [9,10].
In order to investigate how an extremely TE-rich organism, such as T. melanosporum exploits DNA methylation to cope with the more than 45,000 repeated elements that populate its genome [11], we have determined the genome-wide bisulfite sequencing (BS-seq) and RNA sequence (RNA-seq) profiles of the saprobic free-living mycelium (FLM), truffle fruitbody (FB) and ectomycorrhizal (ECM) developmental stages of this fungus (see Table 1 for a list of sequencing data and their descriptive statistics). To gain insight on the reversibility of DNA methylation and its effects on TE transcriptional activity, we determined and compared the BS-seq and RNA-seq profiles of untreated FLM and FLM treated with 5-azacytidine (5-aza), a DNA methylation inhibitor. Building on the above data, here we report the results of an in-depth investigation of the relationship between DNA methylation and transcription in genes and TEs, which led to the identification of novel transcripts from individual sample assemblies, as well as from the combined assembly. We also confirmed the relationship between copy number variation (CNV) and DNA methylation by whole-genome sequencing (WGS) of untreated genomic DNA. In this data note we describe how the samples were collected, processed for BS-seq and RNA-seq library construction and analysis, as well as subsequent data mining. We also provide an implementation of the Composite Repeat Induced Point mutation index (CRI), a dinucleotide frequency distribution analysis tool for evaluating the likelihood that DNA methylation in a given genome is induced by “Repeat-Induced Point mutation” (RIP) [12] (see “Availability and requirements of Software Used” for details).
Table 1.
Summary of sequencing data
| Sample | Library | Number of raw reads | Number of uniquely mapped | Mapability | Read length | Coverage per strand (X) |
|---|---|---|---|---|---|---|
|
FLM |
BS-seq |
122,903,319 |
78,983,505 |
64.26% |
100 |
31.61 |
|
FB |
BS-seq |
184,692,678 |
86,778,157 |
46.99% |
100 |
34.73 |
|
ECM |
BS-seq |
182,286,685 |
3,268,721 |
1.79% |
90 |
1.18 |
|
5-aza treated |
BS-seq |
99,836,913 |
41,349,427 |
70.48% |
51 |
8.44 |
|
5-aza untreated |
BS-seq |
84,315,198 |
35,348,536 |
70.19% |
51 |
7.21 |
|
FLM |
WG-seq |
57,386,669 |
45,239,191 |
78.83% |
51 |
9.23 |
|
FB |
WG-seq |
70,844,918 |
50,646,551 |
71.49% |
51 |
10.34 |
|
FLM |
RNA-seq |
68,287,049 |
63,751,666 |
93.36% |
50 |
N/A |
|
FB |
RNA-seq |
87,310,639 |
74,090,987 |
84.86% |
50 |
N/A |
|
5-aza treated |
RNA-seq |
72,050,137 |
60,644,746 |
84.17% |
51 |
N/A |
| 5-aza untreated | RNA-seq | 75,043,437 | 62,107,389 | 82.76% | 51 | N/A |
ECM: ECtoMycorrhiza; FB: FruitBody; FLM: Free-Living Mycelium.
Biological material
T. melanosporum (Vittad.) mycelium from the Mel28 strain, the same strain utilized for reference genome sequencing [7], was grown on 1% malt agar (Cristomalt-D, Difal, Villefranche-sur-Saône, France) for 5 weeks before harvesting. A field-collected mature fruitbody was used for the FB library. ECM tips were from common hazel (Corylus avellana L.) plantlets inoculated with a T. melanosporum mycelium slurry (Raggi Vivai, Cesena, Italy). For 5-aza treatment, T. melanosporum mycelia were grown in the dark at 23 °C in synthetic liquid medium as described [13]. Every 5 days 5-aza was added to mycelia at 10, 40, and 100 μM final concentrations (from a 10 mM stock solution in water) for 45 days (‘5-aza treated’), with the last addition made 24 hours before harvesting of mycelia and DNA/RNA extraction. The same volume of water, instead of 5-aza, was added to parallel control samples (‘5-aza untreated’).
Whole-genome bisulfite sequencing
Genomic DNA (gDNA) was extracted by grinding fruitbodies in liquid nitrogen followed by purification with the DNeasy Plant Mini kit (QIAGEN, Hilden, Germany). This was followed by extraction in 50% Phenol-50% extraction buffer (100 mM Tris–HCl pH 8.0, 100 mM NaCl, 20 mM EDTA, 1% SDS) at 65 °C for 10 min and centrifugation at 14,000 rpm for 10 min. The aqueous phase was transferred to new tubes and extracted twice with phenol:chloroform (1:1) and once with chloroform. Following ethanol precipitation, samples were resuspended in H2O. LiCl (2 M) precipitation was used to separate RNA (pellet) from gDNA (supernatant). gDNA was precipitated with ethanol and resuspended in H2O. Extracted gDNA was further purified with an additional phenol:chloroform extraction and sheared by sonication to generate DNA fragments in the 150–300 bp size range. Bisulfite treatment and library preparation were carried out as described [14], except that the EpiTect kit (QIAGEN) was utilized for bisulfite treatment. Two consecutive rounds of conversion were performed for a total of 10 hours. The resulting libraries were sequenced by Illumina sequencing technology (Hiseq 2000 sequencer; Illumina, San Diego, CA, USA).
Processing bisulfite converted reads
Bisulfite-converted reads were aligned to the reference genome (Tuber_melanosporum_v1.0) using BS Seeker 2 [15]. We achieved 32X, 35X and 1.2X coverage per strand for FB, FLM and ECM, respectively (see Table 1). The ECM sample contained substantial amounts of root cells from the hazelnut host tree (C. avellana), whose genome has not yet been determined. Even at this low coverage, we were able to delineate global, low-resolution methylation profiles for the symbiotic (ECM) stage. Genome-wide DNA methylation profiles were generated by determining methylation levels for each cytosine in the genome. Since bisulfite treatment coupled to PCR amplification converts unmethylated cytosines (Cs) to thymines (Ts), the methylation level at each cytosine was estimated as #C/(#C + #T), where #C is the number of methylated reads and #T is the number of unmethylated reads. The methylation level per cytosine serves as an estimate of the percentage of cells bearing a methylated cytosine at a specific locus.
For subsequent analysis we only considered cytosines that are covered by at least four reads (except for the ECM methylome, which has a considerably lower overall coverage). Table 2 shows that approximately 90% of the cytosines in FB and FLM were retained for analysis and that this percentage drops to approximately 75% for 5-aza treated and untreated. These values suggest our methylation profiles provide a good representation of the truffle methylomes (see Table 3 for the DNA methylation levels of genome, genes and transposable elements in individual BS-seq libraries).
Table 2.
Number of cytosines included in the methylation analysis
| Number of cytosines* (% genome coverage) | FLM | FB | ECM | 5-aza treated | 5-aza untreated |
|---|---|---|---|---|---|
|
CG |
7,682,955 (92.58%) |
7,444,625 (89.71%) |
55,380 (0.67%) |
6,594,758 (79.47%) |
6,147,635 (74.08%) |
|
CHG |
9,111,984 (93.50%) |
8,829,674 (90.61%) |
52,152 (0.54%) |
7,739,358 (79.42%) |
7,054,610 (72.39%) |
| CHH | 34,258,661 (91.68%) | 32,954,550 (88.19%) | 152,076 (0.41%) | 28,932,890 (77.43%) | 25,963,097 (69.48%) |
ECM: ECtoMycorrhiza; FB: FruitBody; FLM: Free-Living Mycelium.
*cytosines with reads coverage > = 4.
Table 3.
Average methylation levels of genome, genes, and transposable elements
| Methylation level* | FLM | FB | ECM | 5-aza treated | 5-aza untreated | |
|---|---|---|---|---|---|---|
|
Genome |
CG |
30.3% |
28.9% |
6.4% |
26.4% |
25.3% |
|
CHG |
8.1% |
8.9% |
3.4% |
7.3% |
8.3% |
|
|
CHH |
10.1% |
10.3% |
3.3% |
8.8% |
9.9% |
|
|
Gene |
CG |
0.64% |
0.87% |
0.84% |
0.68% |
0.68% |
|
CHG |
0.36% |
0.61% |
0.82% |
0.39% |
0.41% |
|
|
CHH |
0.39% |
0.63% |
0.95% |
0.43% |
0.44% |
|
| TE |
CG |
70.51% |
69.71% |
59.16% |
72.87% |
72.89% |
|
CHG |
14.85% |
16.50% |
15.31% |
13.99% |
16.97% |
|
| CHH | 17.27% | 17.85% | 17.30% | 16.11% | 19.24% | |
ECM: ECtoMycorrhiza; FB: FruitBody; FLM: Free-Living Mycelium.
*The methylation level is estimated based on all cytosines with reads coverage > = 4 in each sample.
BS-seq data coverage (i.e., read depth) across the genome revealed CNVs between FLM and FB. Specifically, we identified 107 genomic regions with significant CNV (defined as a 100 kb window with a |Log coverage ratio (FLM/FB)| ≥0.3), corresponding to 7.3% of the genome. One hundred and two (95%) of these regions were independently confirmed by standard Illumina sequencing performed on non-bisulfite-treated FLM genomic DNA. This high coincidence rate supports the notion that BS-seq data are unbiased for CNV calling.
RNA-seq library preparation and data analysis
Total RNA was extracted as described above, dissolved in H2O after LiCl precipitation and purified with the RNeasy Plant Mini kit (QIAGEN) followed by on-column DNase I digestion as per manufacturer’s instructions. RNA integrity was verified with a Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), which yielded RNA integrity number (RIN) scores of 7.0 and 6.5 for FB and FLM, respectively. RNA was quantified with the Qubit RNA BR Assay kit (Life technologies, Carlsbad, CA, USA) and 1 μg of purified RNA was utilized as starting material for library construction, which was carried out with the Illumina TruSeq RNA Sample Preparation kit as per manufacturer's instructions. Libraries were sequenced with an Illumina HiSeq 2000 system using 50-bp single-end reads.
The reads were mapped against transcripts of genes and TEs using Tophat [16] allowing up to two mismatches, and only unique alignments were kept. The quality of alignments was checked using FastQC [17]. The resulting alignment files were processed with the HTSeq program (version 0.5.4p3, [18]) to create a gene matrix for downstream analysis. Only reads mapped entirely to a single gene or TE were kept for further analysis. In our FB and FLM datasets, approximately 75% of genes are covered with a depth of at least 10 reads (see Figure 1A). In another set of data comparing 5-aza treated versus untreated FLM samples, about 90% of genes are covered by 10 or more reads (see Figure 1B), suggesting a satisfactory completeness of our data coverage. Reads corresponding to similar, but not identical repeats were also mapped, albeit with a lower ability to be mapped compared to a regular (i.e., distinct sequence element) alignment. The Venn diagrams in Figures 1C and 1D show that 55-60% of TEs are covered in both samples in the two comparisons. In total, 63,742,213 and 74,109,065 reads from FB and FLM, respectively, were uniquely mapped, corresponding to an overall 93.34% and 84.88% mapping. Expression levels were computed as Reads Per Kilobase of exon per Million reads mapped (RPKM) [19]. Gene length was calculated as the union length of all possible exons.
Figure 1.

Coverage of RNA-seq libraries in genes and transposable elements. A: histogram of read coverage in genes between FB and FLM libraries, and B: between 5-aza untreated and treated libraries. C: Venn diagram of read coverage in TE between FB and FLM libraries, and D: between 5-aza untreated and treated libraries. FB: FruitBody; FLM: Free-Living Mycelium.
Methods
Prediction of novel genes
We used the RNA-seq datasets listed in Table 1 (FB, FLM, FLM treated/untreated with 5-aza) to predict novel genes not included in the original annotation [20]. The prediction pipeline was based on a previously published approach [21]. First, reads were aligned to the reference genome (truffle v1.0 assembly) using TopHat [16], which resulted in the mapping of non-annotated regions. A guided transcript assembly using Cufflinks [22] with the truffle v1.0 assembly was then carried out for each sample. The resulting individual transcript assemblies were compared with the reference assembly using Cuffcompare [22] to identify novel transcripts.
A transcript is deemed novel if it does not overlap any known transcript in truffle v1.0 and its FPKM (Fragments Per Kilobase of exon per Million fragments mapped) value is at least four in the 95% FPKM confidence interval with a class code “u” in the output of Cuffcompare (i.e., unknown, intergenic transcript). Finally, to claim these as novel genes, the overlapping novel transcripts are clustered into a single interval, i.e., the longest transcript, that spans across all the overlapping transcripts. The clusters of overlapping novel transcripts are considered novel genes if their sequences are homologous to any known protein (BlastX analysis via Blast2GO) [23]. After removal of 11 extremely long genes (≥36,573 bp, the longest known gene in truffles, see Additional file 1: Figures S1 and S2 for the length distributions), we identified a total of 614 novel genes, 200 to 500 of which present in each sample (Table 4). A complete list of novel genes along with expression levels and predicted protein functions are available in Additional file 2: Table S1.
Table 4.
Prediction of genes in RNA-seq datasets
| Sample | Reference assembly | Number of novel genes |
|---|---|---|
| FB |
Truffle v1.0 (7,496 genes) |
282 |
| FLM |
328 |
|
| 5-aza treated |
466 |
|
| 5-aza untreated |
440 |
|
| Merged | 614 |
FB: FruitBody; FLM: Free-Living Mycelium.
DNA methylation and transcription analysis
To investigate the relationship between DNA methylation and transcription we performed a joint analysis of the BS-seq and RNA-seq data on genes and TEs. The results highlight a very different pattern between genes and TEs. As shown in Figure 2A, genes are mostly methylation depleted in all contexts, with no clear difference in methylation levels between genes expressed at high, medium or low levels. This argues against a possible involvement of gene body methylation in gene expression regulation. On the other hand, silenced TEs are generally highly (70-80%) methylated, and a clear anti-correlation is apparent between DNA transcription and methylation for the expressed TEs, thus suggesting TE expression regulation by DNA methylation (Figure 2B).
Figure 2.
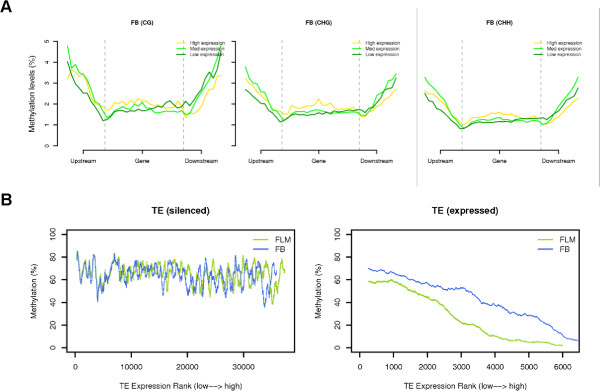
Integrative analysis of DNA methylation and transcription in genes and transposable elements. A: Meta-gene plots of CG/CHG/CHH methylation level in high/med/low expression genes. B: Methylation level of TEs ranked by the expression level from low to high in silenced (bottom left) and expressed (bottom right) TEs. TE: Transposable elements.
Transcriptome analysis of truffle mycelia treated with 5-aza
We recently showed that mycelia treated with the demethylating agent 5-aza exhibit a 5-aza concentration-dependent transition to a fluffy-like phenotype [11]. 5-aza treated mycelia also displayed decreased global methylation levels compared to untreated samples. A reactivation of TE transcription was also reported. Here we performed a functional analysis of the genes differentially expressed between untreated and 5-aza treated mycelia using Blast2GO [23]. As revealed by this analysis, untreated vs. 5-aza treated differential transcripts are enriched in genes involved in the oxidation-reduction process and transmembrane transport. The other genes code for transcription factors or for proteins involved in metabolism, response to stimuli, and mycelium development (Table 5). In addition to transcriptional changes, we also identified 68 novel genes/transcripts that are only present in the treated, but not in the untreated sample. Figure 3 shows a novel gene identified in treated, but not in untreated mycelium. Together with our previous findings, this result further supports the existence of an epigenetic regulatory system operating in response to environmental stress exposure in truffles.
Table 5.
Differentially expressed genes between untreated and 5-aza treated mycelia
| Gene* | Description | RPKM No aza | RPKM aza | Fold Change | adj. p value |
|---|---|---|---|---|---|
|
Oxidation-reduction process
| |||||
| GSTUMT00004482001 |
Flavin-binding monooxygenase-like protein |
8 |
258 |
33.80 |
3.51E-09 |
| GSTUMT00009102001 |
Extracellular dioxygenase |
18 |
196 |
10.71 |
1.33E-09 |
| GSTUMT00002834001 |
Alpha-ketoglutarate-dependent taurine dioxygenase |
47 |
414 |
8.84 |
4.30E-09 |
| GSTUMT00006862001 |
Alcohol dehydrogenase 1 |
4355 |
34021 |
7.81 |
4.81E-08 |
| GSTUMT00005793001 |
Bilirubin oxidase |
1590 |
8041 |
5.06 |
2.28E-05 |
| GSTUMT00000137001 |
Cytochrome P450 |
3 |
17 |
5.06 |
5.85E-04 |
| GSTUMT00006911001 |
Multicopper oxidase |
19 |
90 |
4.64 |
4.87E-03 |
| GSTUMT00008228001 |
Zinc-binding oxidoreductase |
34 |
147 |
4.35 |
3.20E-04 |
| GSTUMT00002706001 |
Alpha-ketoglutarate-dependent sulfonate dioxygenase protein |
4 |
14 |
3.61 |
3.51E-02 |
| GSTUMT00000322001 |
Fatty acid oxygenase |
137 |
48 |
0.35 |
4.81E-02 |
| GSTUMT00000528001 |
Short-chain alcohol dehydrogenases |
334 |
110 |
0.33 |
1.22E-02 |
| GSTUMT00007846001 |
FAD binding domain-containing protein |
172 |
56 |
0.33 |
1.54E-02 |
| GSTUMT00002653001 |
Pyridoxal reductase |
104 |
31 |
0.30 |
5.83E-03 |
| GSTUMT00000530001 |
FAD binding domain-containing protein |
61 |
18 |
0.30 |
8.39E-03 |
| GSTUMT00012134001 |
Dopa -dioxygenase |
4704 |
1368 |
0.29 |
3.10E-03 |
| GSTUMT00000158001 |
Ferritin ribonucleotide reductase-like protein |
54 |
15 |
0.28 |
4.52E-03 |
| GSTUMT00002858001 |
3-oxoacyl-(acyl-carrier-protein) reductase |
22 |
5 |
0.25 |
8.53E-03 |
| GSTUMT00012200001 |
Malate synthase |
71 |
15 |
0.21 |
2.41E-03 |
| GSTUMT00001645001 |
Zinc-binding alcohol |
219 |
34 |
0.16 |
1.06E-06 |
| GSTUMT00006980001 |
Alcohol dehydrogenase |
1022 |
146 |
0.14 |
1.36E-07 |
| GSTUMT00007117001 |
D-isomer specific 2-hydroxyacid dehydrogenase |
4 |
0.4 |
0.10 |
4.66E-02 |
|
Transmembrane transport
| |||||
| GSTUMT00001488001 |
Amino acid permease |
163 |
737 |
4.53 |
9.07E-03 |
| GSTUMT00003668001 |
Carboxylic acid transport protein |
20 |
82 |
4.03 |
6.04E-03 |
| GSTUMT00010777001 |
Mate efflux family protein |
16 |
66 |
4.03 |
1.22E-02 |
| GSTUMT00008972001 |
Lactose permease |
16 |
64 |
4.02 |
7.12E-03 |
| GSTUMT00001400001 |
MFS efflux transporter |
86 |
295 |
3.44 |
5.58E-03 |
| GSTUMT00000284001 |
Mg2+ transporter family-like protein |
1082 |
366 |
0.34 |
2.56E-02 |
| GSTUMT00008966001 |
MFS general substrate transporter |
27 |
9 |
0.34 |
4.85E-02 |
| GSTUMT00000070001 |
MFS multidrug transporter |
119 |
30 |
0.25 |
1.20E-03 |
| GSTUMT00004586001 |
ABC transporter |
32 |
5 |
0.17 |
3.72E-02 |
| GSTUMT00005001001 |
MFS drug efflux |
663 |
84 |
0.13 |
4.47E-08 |
|
Transcription factors
| |||||
| GSTUMT00005836001 |
Fungal transcriptional regulatory protein |
1 |
18 |
17.23 |
1.91E-09 |
| GSTUMT00010369001 |
STE12-like transcription factor |
1 |
7 |
12.50 |
1.93E-04 |
| GSTUMT00004615001 |
Binuclear zinc transcription factor |
2 |
10 |
6.40 |
1.95E-04 |
| GSTUMT00004613001 |
GAL4 domain-containing protein |
90 |
402 |
4.49 |
1.03E-02 |
| GSTUMT00003504001 |
C2H2 finger domain |
1522 |
467 |
0.31 |
4.98E-03 |
|
Metabolic process
| |||||
| GSTUMT00008986001 |
Glycoside hydrolase family 61 protein |
0.2 |
5 |
25.60 |
1.10E-04 |
| GSTUMT00001850001 |
Endo- -beta-glucanase eng1 |
94 |
1499 |
16.02 |
4.71E-13 |
| GSTUMT00012011001 |
Glycoside hydrolase family 28 protein |
7 |
57 |
7.73 |
3.97E-04 |
| GSTUMT00005539001 |
Dynamin family protein |
27 |
195 |
7.18 |
2.59E-05 |
| GSTUMT00010778001 |
Agmatinase 1 |
230 |
1316 |
5.72 |
1.70E-05 |
| GSTUMT00011981001 |
Acetyltransferase |
298 |
1376 |
4.62 |
1.62E-04 |
| GSTUMT00007852001 |
Alpha amylase |
166 |
704 |
4.25 |
6.60E-04 |
| GSTUMT00003032001 |
Polysaccharide deacetylase |
1222 |
4444 |
3.64 |
3.18E-03 |
| GSTUMT00001583001 |
Pirin |
273 |
968 |
3.54 |
3.99E-03 |
| GSTUMT00005439001 |
Glycerophosphoryl diester phosphodiesterase |
214 |
758 |
3.54 |
1.08E-02 |
| GSTUMT00001660001 |
Glycoside hydrolase family 16 protein |
109 |
374 |
3.43 |
6.66E-03 |
| GSTUMT00006417001 |
Fatty acid activator |
417 |
1410 |
3.38 |
5.16E-03 |
| GSTUMT00007213001 |
Tat pathway signal sequence |
74 |
239 |
3.25 |
1.98E-02 |
| GSTUMT00002512001 |
Gnat family N-acetyltransferase |
99 |
311 |
3.14 |
2.83E-02 |
| GSTUMT00000969001 |
Branched-chain amino acid aminotransferase |
840 |
270 |
0.32 |
9.18E-03 |
|
Response to stimuli/mycelium development
| |||||
| GSTUMT00004040001 |
Regulator of G protein signaling domain protein |
54 |
209 |
3.84 |
1.40E-03 |
| GSTUMT00007138001 |
Osmolarity two-component sensor histidine kinase SLN1 |
401 |
1134 |
2.83 |
4.81E-02 |
| GSTUMT00004012001 |
Annexin |
72 |
23 |
0.33 |
1.38E-02 |
|
Others
| |||||
| GSTUMT00004681001 |
RNase P RPR2 RPP21 subunit domain-containing protein |
52 |
339 |
6.53 |
9.70E-07 |
| GSTUMT00002939001 |
Allergenic cerato-platanin ASP F13 |
361 |
1730 |
4.80 |
5.46E-05 |
| GSTUMT00010365001 |
Putative endo-glucanase |
8 |
39 |
4.66 |
1.93E-02 |
| GSTUMT00006611001 |
Protoglobin protein |
999 |
3827 |
3.83 |
3.11E-03 |
| GSTUMT00006718001 |
Glutathione S-transferase |
228 |
80 |
0.35 |
3.10E-02 |
| GSTUMT00008071001 |
XAA-pro dipeptidase |
1272 |
413 |
0.32 |
1.27E-02 |
| GSTUMT00001676001 |
Arrestin (or S-antigen) N-terminal domain protein |
96 |
31 |
0.32 |
1.22E-02 |
| GSTUMT00006632001 | P-loop containing nucleoside triphosphate hydrolase | 3053 | 724 | 0.24 | 2.20E-04 |
*One hundred fifteen genes undergo a significant change in expression levels (adjusted p value <0.05) upon 5-aza treatment. Functional annotation of the differentially expressed genes/sequences (either up- or down-regulated) was performed with the Blast2GO suite. Fifty-three genes code for hypothetical proteins. The remaining 62 genes were classified into distinct functional categories. “Oxidation-reduction process” and “transmembrane transport” appear as enriched GO terms (Fisher exact test p = 2.6E-4 and p = 7.9E-3, respectively) among differentially expressed genes.
Figure 3.
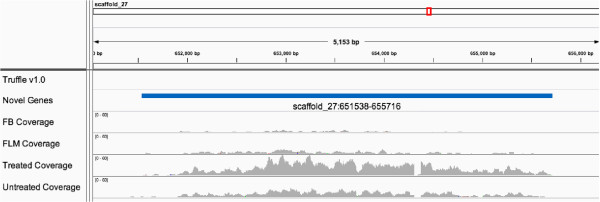
Screenshot of a novel gene identified in 5-aza treated mycelia.
The composite RIP index (CRI) computation
The composite RIP index (CRI, [12]; see below ‘Availability and requirements of Software Used’) was implemented to measure CA = > TA enrichment within repeated sequences from Tuber and other organisms’ genomes. The Composite RIP Index (CRI) was calculated as (RIP product)-(RIP substrate) [12]. The RIP product score measures the frequency of RIP products taking into account potential false positives due to local density, while the RIP substrate score measures the depletion of RIP targets and their reverse complement (e.g., CpA, and TpG).
To evaluate and compare CRI values in different organisms, we predicted repeats in T. melanosporum, Neurospora crassa, Uncinocarpus reesii, Aspergillus nidulans and Saccharomyces cerevisiae. Repeat annotations were predicted from the corresponding genome sequences [24-27] by RepeatMasker [28]. Further details on CRI implementation, including raw data, processed data, scripts (python and R) and output files can be accessed at [29].
Conclusions
We provide a comprehensive genomic, epigenomic and transcriptomic sequencing data resource for the black truffle T. melanosporum. The use of a reversible, rather than an irreversible mechanism (such as RIP) to cope with the multitude of repeated transposable elements that populate the T. melanosporum genome resembles the situation in more complex organisms, including humans, and is in line with the view of a “generative” (i.e., genome-shaping) rather than a purely “destructive” role of transposable elements [30]. More targeted follow-up studies built upon the results of the present work may uncover variations of the DNA methylation and transposon activity profiles associated to functionally interpretable (presumably adaptive) modifications of gene expression. If extended to T. melanosporum specimens from different geographic areas, epigenomic analyses, such as the one described in this work, may shed light on the relationships between DNA methylation, transposon-mediated genome shaping and commercially relevant organoleptic properties, such as aroma. In addition to their general evolutionary biological implications, our findings may thus provide a new mechanistic layer to explain intraspecific variability.
Availability and requirements of Software Used
Implementation of the Composite RIP Index
•Project name: The composite RIP index (CRI) computation
•Project home page:https://github.com/wwliao/critool/
•Operating system(s): Platform-independent
•Programming language: Python and R
•Other requirements: Python 2.7, R
•License: MIT
•Any restrictions to use by non-academics: None
Availability of supporting data
For all sequencing data, in addition to those already described in [11] (GSE49700) we provide several formats to facilitate downstream data analysis: BS-seq data is presented in several formats -- a BAM file of alignments, ATCGmap for SNP calling and coverage analysis, CGmap for methylation analysis and a wiggle file for data visualization on genome browsers, such as IGV [31] (see Additional file 1: Table S1). For WG-seq data, BAM and ATCGmap are provided. For RNA-seq data, we provide BAM files and novel genes detected from the merged transcriptome assembly (see Additional file 2: Table S1). All the datasets presented here are publicly available in the GigaScience repository, GigaDB [19].
Abbreviations
5-aza: 5-azacytidine; 5mC: 5-methylcytosine; CNV: Copy Number Variation; CRI: Composite RIP Index; DMT: DNA Methyl Transferase; ECM: ECtoMycorrhiza; FB: FruitBody; FLM: Free-Living Mycelium; gDNA: genomic DNA; RIP: Repeat-Induced Point mutation; RPKM: Reads Per Kilobase per Million mapped reads; TE: Transposable Element.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BM and MM have been in charge of truffle material manipulation, DNA and RNA extraction, and prepared the libraries. BM, PYC and WWL performed BS-sequencing and the bioinformatic analysis. AJ and DL performed RNA-seq analysis. SO and MP planned and supervised the study and wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Histogram of gene length in Truffle v1.0. Figure S2. Histogram of gene length of the 614 novel genes. Table S1. Data file formats.
Novel genes with expression levels and predicted protein functions.
Contributor Information
Pao-Yang Chen, Email: paoyang@gate.sinica.edu.tw.
Barbara Montanini, Email: barbara.montanini@unipr.it.
Wen-Wei Liao, Email: wwliao@gate.sinica.edu.tw.
Marco Morselli, Email: mmorselli@ucla.edu.
Artur Jaroszewicz, Email: arturtjaro@gmail.com.
David Lopez, Email: davidlopez@ucla.edu.
Simone Ottonello, Email: simone.ottonello@unipr.it.
Matteo Pellegrini, Email: matteop@mcdb.ucla.edu.
Acknowledgements
This work was supported by a grant from Academia Sinica to PYC and by grants from the Fondazione Cariparma and the Ministry of Education, University and Research (MIUR) to SO and by a grant from the Institute for Genomics and Proteomics at UCLA to MP.
References
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M, Jacobsen SE. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- Zemach A, Zilberman D. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr Biol. 2010;20:R780–785. doi: 10.1016/j.cub.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Martin F, Kohler A, Murat C, Balestrini R, Coutinho PM, Jaillon O, Montanini B, Morin E, Noel B, Percudani R, Porcel B, Rubini A, Amicucci A, Amselem J, Anthouard V, Arcioni S, Artiguenave F, Aury JM, Ballario P, Bolchi A, Brenna A, Brun A, Buee M, Cantarel B, Chevalier G, Couloux A, Da Silva C, Denoeud F, Duplessis S, Ghignone S. et al. Perigord black truffle genome uncovers evolutionary origins and mechanisms of symbiosis. Nature. 2010;464:1033–1038. doi: 10.1038/nature08867. [DOI] [PubMed] [Google Scholar]
- Bestor TH. Sex brings transposons and genomes into conflict. Genetica. 1999;107:289–295. doi: 10.1023/A:1003990818251. [DOI] [PubMed] [Google Scholar]
- Faugeron G. Diversity of homology-dependent gene silencing strategies in fungi. Curr Opin Microbiol. 2000;3:144–148. doi: 10.1016/S1369-5274(00)00066-7. [DOI] [PubMed] [Google Scholar]
- Percudani R, Trevisi A, Zambonelli A, Ottonello S. Molecular phylogeny of truffles (Pezizales: Terfeziaceae, Tuberaceae) derived from nuclear rDNA sequence analysis. Mol Phylogenet Evol. 1999;13:169–180. doi: 10.1006/mpev.1999.0638. [DOI] [PubMed] [Google Scholar]
- Montanini B, Chen PY, Morselli M, Jaroszewicz A, Lopez D, Martin F, Ottonello S, Pellegrini M. Non-exhaustive DNA methylation-mediated transposon silencing in the black truffle genome, a complex fungal genome with massive repeat element content. Genome Biol. 2014;15:411. doi: 10.1186/s13059-014-0411-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis ZA, Honda S, Khlafallah TK, Jeffress JK, Freitag M, Mohn F, Schubeler D, Selker EU. Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa. Genome Res. 2009;19:427–437. doi: 10.1101/gr.086231.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanini B, Moretto N, Soragni E, Percudani R, Ottonello S. A high-affinity ammonium transporter from the mycorrhizal ascomycete Tuber borchii. Fungal Genet Biol. 2002;36:22–34. doi: 10.1016/S1087-1845(02)00001-4. [DOI] [PubMed] [Google Scholar]
- Feng S, Rubbi L, Jacobsen SE, Pellegrini M. Determining DNA methylation profiles using sequencing. Methods Mol Biol. 2011;733:223–238. doi: 10.1007/978-1-61779-089-8_16. [DOI] [PubMed] [Google Scholar]
- Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen PY, Pellegrini M. BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics. 2013;14:774. doi: 10.1186/1471-2164-14-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FastQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- HTSeq: Analysing high-throughput sequencing data with Python. http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html. [DOI] [PMC free article] [PubMed]
- Chen PY, Montanini B, Liao WW, Morselli M, Jaroszewicz A, Lopez D, Ottonello S, Pellegrini M. A comprehensive resource of genomic, epigenomic and transcriptomic sequencing data of black truffle, Tuber melanosporum. GigaScience Database. 2014. http://dx.doi.org/10.5524/100101. [DOI] [PMC free article] [PubMed]
- MycorWeb: Tuber melanosporum DB. http://mycor.nancy.inra.fr/IMGC/TuberGenome/download.php?select=anno.
- Weikard R, Hadlich F, Kuehn C. Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genomics. 2013;14:789. doi: 10.1186/1471-2164-14-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute: Download Sequence - Neurospora crassa. http://www.broadinstitute.org/annotation/genome/neurospora/MultiDownloads.html.
- Broad Institute: Download Sequence - Uncinocarpus reesii. http://www.broadinstitute.org/annotation/genome/uncinocarpus_reesii.3/MultiDownloads.html.
- Broad Institute: Saccharomyces cerevisiae RM11-1a Database. http://www.broadinstitute.org/annotation/genome/saccharomyces_cerevisiae.
- Broad Institute: Download Sequence - Aspergilli. http://www.broadinstitute.org/annotation/genome/aspergillus_group/MultiDownloads.html.
- RepeatMasker. http://www.repeatmasker.org.
- Computing Composite RIP index (CRI) in repetitive sequences. https://github.com/wwliao/critool.
- Fedoroff NV. Presidential address. Transposable elements, epigenetics, and genome evolution. Science. 2012;338:758–767. doi: 10.1126/science.338.6108.758. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histogram of gene length in Truffle v1.0. Figure S2. Histogram of gene length of the 614 novel genes. Table S1. Data file formats.
Novel genes with expression levels and predicted protein functions.