

Abstract
Fluid shear stress generated by blood flow modulates endothelial cell function via specific intracellular signaling events. We showed previously that flow activated the phosphatidylinositol 3-kinase (PI3K), Akt, and endothelial nitric-oxide synthase (eNOS) via Src kinase-dependent transactivation of vascular endothelial growth factor receptor 2 (VEGFR2). The scaffold protein Gab1 plays an important role in receptor tyrosine kinase-mediated signal transduction. We found here that laminar flow (shear stress = 12 dynes/cm2) rapidly stimulated Gab1 tyrosine phosphorylation in both bovine aortic endothelial cells and human umbilical vein endothelial cells, which correlated with activation of Akt and eNOS. Gab1 phosphorylation as well as activation of Akt and eNOS by flow was inhibited by the Src kinase inhibitor PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) and VEGFR2 kinase inhibitors SU1498 and VTI, suggesting that flow-mediated Gab1 phosphorylation is Src kinase-dependent and VEGFR2-dependent. Tyrosine phosphorylation of Gab1 by flow was functionally important, because flow stimulated the association of Gab1 with the PI3K subunit p85 in a time-dependent manner. Furthermore, transfection of a Gab1 mutant lacking p85 binding sites inhibited flow-induced activation of Akt and eNOS. Finally, knockdown of endogenous Gab1 by small interference RNA abrogated flow activation of Akt and eNOS. These data demonstrate a critical role of Gab1 in flow-stimulated PI3K/Akt/eNOS signal pathway in endothelial cells.
Vascular endothelial cells, which form the inner lining of the blood vessel wall, are exposed to fluid shear stress, the dragging force generated by blood flow. Substantial evidence shows that flow modulates endothelial structure and function and is a major determinant of vascular remodeling, arterial tone, and atherogenesis (1, 2). Steady laminar flow has beneficial effects on endothelial cell function, including inhibition of platelet aggregation, low density lipoprotein uptake, adhesion molecule expression, and vascular smooth muscle cell proliferation, as well as enhancing endothelial cell survival (3). Many of these effects are related to increased nitric oxide (NO)1 production by endothelial cells exposed to flow (4). A decrease in the bioavailability of NO is a characteristic feature in patients with coronary artery disease (5) and aggravates the development of atherosclerotic lesions (6). Flow stimulates production of NO via endothelial nitric-oxide synthase (eNOS) both in cultured endothelial cells and in intact vessels (7–12). We and others have previously reported flow-stimulated phosphorylation of eNOS regulates its enzyme activity (10, 13–15), and phosphatidylinositol 3-kinase (PI3K) and its downstream serine/threonine protein kinase Akt (protein kinase B) mediate phosphorylation of eNOS at Ser-1179 (based on the bovine eNOS sequence and equivalent to human eNOS-Ser-1177) (11, 16, 17). Recently we have found that flow rapidly activates vascular endothelial growth factor receptor 2 (VEGFR2) in a ligand-independent but Src-kinase-dependent manner, which leads to activation of Akt and eNOS in endothelial cells (18). However, the proximal mechanisms for flow-mediated PI3K/Akt/eNOS signaling pathway via VEGFR2 remain unclear.
Scaffold proteins play an important role in receptor tyrosine kinase-mediated signal transduction pathways (19, 20). Gab1 (Grb2-associated binder 1) is a member of a family of docking proteins that includes Gab2 and Gab3 (21–23). Gab1-deficient mice die as embryos with multiple defects in placental, heart, skin, and muscle development (24, 25). In contrast, Gab2-deficient mice are viable, but have a defect in the mast cell lineages and in allergic reactions (26). Gab1 contains an amino-terminal PH domain, several proline-rich sequences, and multiple binding sites (phosphotyrosines) for SH2 domain-containing proteins. Gab1 rapidly becomes tyrosine-phosphorylated upon stimulation of appropriate cells with growth factors (27). Tyrosine-phosphorylated Gab1 binds multiple signaling molecules, including the PI3K subunit p85 (21–23). Gab1-p85 interaction is critical for PI3K activation in response to stimulation of several receptor tyrosine kinases, such as epithelial growth factor receptor, nerve growth factor receptor, and fibroblast growth factor receptor (28–31). Gab1 is highly expressed in endothelial cells (32), but its function and regulation has been little studied.
Here, we show that flow stimulates tyrosine phosphorylation of Gab1 in an Src kinase-dependent and VEGFR2-dependent manner in endothelial cells. Tyrosine phosphorylation of Gab1 is functionally important, because flow stimulates association of Gab1 with p85 and inhibiting Gab1 function and expression decreases flow-induced Akt and eNOS activation. These data demonstrate a critical role of Gab1 in flow-mediated PI3K/Akt/eNOS signaling in endothelial cells.
MATERIALS AND METHODS
Reagents
VTI (VEGF receptor tyrosine kinase inhibitor, 4-[(4′-chloro-2′-fluoro)phenylamino]-6,7-dimethoxyquinazoline), SU1498, PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine), and herbimycin A were purchased from Calbiochem. Anti-phospho eNOS antibody (Ser-1179 in bovine eNOS sequence, p-eNOS), anti-phospho-Akt antibody (Ser-473, p-Akt), and anti-Akt antibody were from Cell Signaling Technologies (Beverly, MA). Anti-eNOS monoclonal antibody was from BD Transduction Laboratories. Anti-phosphotyrosine 4G10 (pY-4G10), anti-PI3K p85 antibodies were from Upstate Biotechnology (Lake Placid, NY). Anti-Gab1 polyclonal antibody was from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture and Exposure to Flow
Bovine aortic endothelial cells (BAECs) were purchased from Clonetics (San Diego, CA) and were cultured in medium 199 supplemented with 10% fetal bovine serum (Invitrogen) (18). Human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical veins and grown in RPMI 1640 (Invitrogen) supplemented with 20% fetal bovine serum (HyClone), heparin (Sigma), and endothelial cell growth factors (isolated from bovine brain) (33). Confluent cells cultured in 60-mm dishes were serum-starved for 24 h and exposed to laminar flow (shear stress = 12 dyn/cm2) in a cone and plate viscometer (18). For the inhibitor studies, cells were pretreated with various inhibitors for 30 min in serum-depleted medium.
Gab1 Mutant and Transfection
Mammalian expression constructs for cDNA encoding FLAG-tagged human Gab1 construct, Gab1ΔPI3K (Y447F/Y472F/Y589F, lacking three PI3K binding sites) were created by site-specific mutagenesis as described previously (34). For transient transfection, BAECs were seeded in 60-mm dishes 24 h before transfection to yield a 60% confluent culture on the day of transfection. Transfection was performed using Lipofectamine, according to the supplier’s instructions described previously (35). 48 h after transfection, the cells were used for flow experiments.
Gab1 siRNA and Its Transfection
To determine the contribution of Gab1 in flow-stimulated signaling, we treated HUVECs with human Gab1 siRNA duplex obtained from Dharmacon, Inc. The sequences of specific siRNA against human Gab1 were sense GAGAGUGGAUUAU-GUUGUUUU and antisense 5′-PAACAACAUAAUCCACUCUCUU. The scrambled siRNA control was a nontargeting siRNA pool from Dharmacon, Inc. For transfection of siRNA, HUVECs were seeded into 60-mm dishes for 24 h about 80% confluence, and then transfection of siRNA was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Flow stimulation was performed 48 h after siRNA transfection.
Immunoprecipitation and Western Blot Analysis
Cells were harvested in lysis buffer (0.5% Triton X-100, 0.5% Nonidet P-40, 10 mM Tris, pH 7.5, 2.5 mM KCl, 150 mM NaCl, 30 mM β-glycerophosphate, 50 mM NaF, 1 mM Na3VO4, and 0.1% protease inhibitor mixture (Sigma) and clarified by centrifugation (18, 36). The protein concentration of the lysate was determined using the Bradford method (Bio-Rad, Hercules, California). Equal amounts of protein were incubated with specific antibody overnight at 4 °C with gentle rotation. Then protein A/G PLUS-agarose (Santa Cruz, California) was added and incubated for additional 2 h. Then the beads were washed extensively with lysis buffer, and immune complexes were eluted in SDS-PAGE sample buffer. Total immune complex samples or protein samples from total cell lysate were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with appropriate primary antibodies. After washing and incubating with secondary antibodies, immunoreactive proteins were visualized by using the ECL detection system (Amersham Biosciences). Where indicated, the membranes were stripped and reprobed with another antibody. Densitometric analyses of immunoblots were performed by National Institutes of Health Image. Results were normalized by arbitrarily setting the densitometry of control cells to 1.0.
Statistical Analysis
Group differences were analyzed using the standard Student’s t test. All values are expressed as means ± S.E. p < 0.05 was considered statistically significant.
RESULTS
Flow Stimulates Rapid Tyrosine Phosphorylation of Gab1 in Endothelial Cells
To gain insight into flow-mediated signaling events downstream of VEGFR2 (18), we studied tyrosine phosphorylation of Gab1. BAECs were exposed to flow for varying times and harvested for analysis of Gab1 phosphorylation. Tyrosine phosphorylation of Gab1 occurred within 2 min, peaked at 15 min (5.7 ± 0.6-fold increase), was sustained for 30 min (Fig. 1, A (upper panel) and B), and returned to near baseline by 120 min after stimulation (not shown). There was no significant change in Gab1 protein levels during these experiments (Fig. 1A, lower panel). Consistent with the results reported previously (18), both Akt and eNOS were time-dependently phosphorylated in response to flow in BAECs (Fig. 1C).
Fig. 1. Flow stimulates phosphorylation of Gab1, Akt, and eNOS in BAECs.

BAECs were exposed to flow (shear stress = 12 dyn/cm2) for the time periods indicated. A, cell lysates were immunoprecipitated (IP) with anti-Gab1 antibody, and the tyrosine phosphorylation of Gab1 (pY-Gab1) was detected by immunoblotting (IB) with anti-phosphotyrosine antibody 4G10, pY(4G10). The total amount of Gab1 in immunoprecipitates was determined by reprobing the same blots with anti-Gab1 antibody. B, quantitative analysis of protein tyrosine phosphorylation for Gab1. Results were normalized by arbitrarily setting the densitometry of control cells (time = 0) to 1.0 (n = 4). C, phosphorylation of Akt and eNOS in cell lysates were analyzed by immunoblotting with phosphospecific antibodies, phospho-Akt (Ser-473) and phospho-eNOS (Ser-1179). The same blots were stripped and reprobed with antibodies detecting total Akt and eNOS to monitor equal loading of samples.
Flow also stimulated tyrosine phosphorylation of Gab1 in HUVECs in a time-dependent manner (Fig. 2, A and B), although slower compared with BAECs. Akt and eNOS were time-dependently phosphorylated in response to flow in HUVECs (Fig. 2C). The time course for Gab1 phosphorylation is correlated with that for Akt and eNOS activation by flow in both BAECs and HUVECs (Figs. 1 and 2), suggesting that Gab1 phosphorylation might be involved in Akt and eNOS activation by flow in endothelial cells.
Fig. 2. Flow stimulates phosphorylation of Gab1, Akt, and eNOS in HUVECs.

HUVECs were exposed to flow (shear stress = 12 dyn/cm2) for the time periods indicated. Phosphorylation of Gab1, Akt, and eNOS in cell lysates were analyzed by immunoprecipitation (IP) and immunoblotting (IB) as mentioned in Fig. 1. A, tyrosine phosphorylation of Gab1 and total amount of Gab1. B, quantitative analysis of protein tyrosine phosphorylation for Gab1 (n = 3). C, phosphorylation of Akt and eNOS and total Akt and eNOS.
Src Kinases and VGEFR2 Are Involved in Flow-induced Gab1 Tyrosine Phosphorylation
To characterize the tyrosine kinase responsible for flow-induced Gab1 phosphorylation, we first utilized the Src tyrosine kinase inhibitors, herbimycin A and PP2. Treatment with 1 μM herbimycin A abrogated flow-mediated Gab1 tyrosine phosphorylation (Fig. 3A). Herbimycin A also abolished activation of Akt and eNOS by flow (Fig. 3A). Similarly, 10 μM PP2 inhibited flow-mediated Gab1 tyrosine phosphorylation (Fig. 3B). Treatment with PP2 inhibited activation of Akt and eNOS as well (Fig. 3B). These results strongly suggested that Src family tyrosine kinases play an important role in flow-induced Gab1 tyrosine phosphorylation as well as activation of Akt and eNOS in endothelial cells.
Fig. 3. Src kinases are involved in flow-induced phosphorylation of Gab1, Akt, and eNOS in endothelial cells.
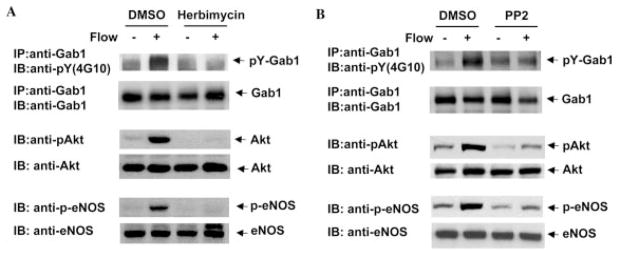
BAECs were pretreated with vehicle (Me2SO (DMSO), or 1 μmol/liter herbimycin (A), or 10 μmol/liter PP2 (B) for 30 min before exposure to flow for 15 min. Phosphorylation of Gab1, Akt, and eNOS in cell lysates were detected by immunoprecipitation (IP) and immunoblotting (IB) with phosphospecific antibodies. The same blots were stripped and reprobed with antibody detecting total Gab1, Akt, and eNOS to monitor equal loading of samples. Results are representative for three independent experiments.
We showed previously that VEGFR2 tyrosine phosphorylation by flow was Src kinase-dependent, and VEGFR2 was required for flow-mediated PI3K/Akt/eNOS signaling (18). To examine the role of VEGFR2 in flow-mediated Gab1 tyrosine phosphorylation, we studied the effects of SU1498 and VTI, two structurally different VEGFR2 kinase inhibitors. Treatment with 10 μM VTI significantly inhibited flow-mediated Gab1 tyrosine phosphorylation (Fig. 4A). VTI also significantly inhibited activation of Akt and eNOS by flow (Fig. 4A). Similar effects were observed when cells were treated with SU1498 (Fig. 4B). These results indicate that Gab1 tyrosine phosphorylation by flow is Src kinase- and VEGFR2 kinase-dependent, similar to flow-induced activation of Akt and eNOS (18).
Fig. 4. VEGFR2 mediates flow-induced phosphorylation of Gab1, Akt, and eNOS in endothelial cells.

BAECs were pretreated with vehicle (Me2SO (DMSO), or 10 μmol/liter VTI (A), or 10 μmol/liter SU1498 (B) for 30 min before exposure to flow for 15 min. Phosphorylation of Gab1, Akt, and eNOS in cell lysates were detected by immunoprecipitation (IP) and immunoblotting (IB) with phosphospecific antibodies. The same blots were stripped and reprobed with antibody detecting total Gab1, Akt, and eNOS to monitor equal loading of samples. Results are representative for three independent experiments.
Gab1 Tyrosine Phosphorylation by Flow Mediates Activation of Akt and eNOS in Endothelial Cells
To gain insight into the functional significance of Gab1 tyrosine phosphorylation in flow-mediated signaling events, we determined whether flow increases the association of Gab1 with PI3K in endothelial cells as reported for growth factor-stimulated cells (28–31). BAECs were exposed to flow for various times indicated in Fig. 5A, and cell lysates were immunoprecipitated with anti-Gab1 antibody and immunoblotted with anti-p85 antibody. Immunoprecipitation of Gab1 after exposure of BAECs to flow showed a significant increase in co-immunoprecipitated p85 (Fig. 5A),
Fig. 5. Tyrosine phosphorylation of Gab1 is required for flow-induced activation of Akt and eNOS in endothelial cells.

A, BAECs were exposed to flow for the time periods indicated. Cell lysates were immunoprecipitated (IP) with anti-Gab1 antibody. The PI3K subunit p85 was detected in the immunoprecipitates by immunoblotting (IB) with anti-p85 antibody. The total amount of Gab1 in immunoprecipitates was determined by reprobing the same blots with anti-Gab1 antibody. B and C, BAECs were transfected with pcDNA as control or Gab1ΔPI3K for 48 h and then exposed to flow for 15 min. Phosphorylation of Akt and eNOS in cell lysates were analyzed by immunoblotting with phosphospecific antibodies. The amount of Akt, eNOS, and transfected FLAG-Gab1ΔPI3K were detected by reprobing the same blots with anti-Akt, anti-eNOS, and anti-FLAG antibodies. Representative immunoblots (B) and quantitative analysis of protein phosphorylation for Akt and eNOS (C) were shown (n = 3).
To examine the role of Gab1 in flow-induced activation of Akt and eNOS, the downstream signaling molecules of PI3K, we transfected BAECs with the mutant Gab1ΔPI3K (Y434F, Y343F, and Y243F) lacking PI3K binding sites (21, 28), which has dominant negative effect on growth factor-induced the recruitment and activation of PI3K (21, 28). Although transfection efficiency is ~40% in BAECs, overexpressed Gab1ΔPI3K significantly decreased Akt activation in response to flow (Fig. 5, B and C), indicating that Gab1/PI3K interaction is responsible for Akt activation. Moreover, eNOS activation was also attenuated by overexpression of Gab1ΔPI3K in endothelial cells (Fig. 5, B and C).
Knockdown of Gab1 by siRNA Inhibited Flow-induced Activation of Akt and eNOS in Endothelial Cells
To further support the critical role of Gab1 in flow-induced PI3K/Akt/eNOS signaling, we knocked down endogenous Gab1 in endothelial cells using siRNA. Transfection of HUVECs with human Gab1 siRNA significantly reduced endogenous Gab1 expression, whereas control siRNA had no effect (Fig. 6A). Gab1 siRNA is specific for targeting Gab1, because expression of β-actin, Akt, and eNOS were not changed (Fig. 6A). Decreasing Gab1 expression by siRNA significantly inhibited flow-induced activation of Akt and eNOS in HUVECs (Fig. 6, A and B).
Fig. 6. Knockdown Gab1 by siRNA inhibits flow-induced activation of Akt and eNOS in endothelial cells.

HUVECs were transfected with scrambled siRNA (control) or human Gab1 siRNA for 48 h, and then exposed to flow for 30 min. A, cell lysates were analyzed by immunoblotting (IB) with anti-Gab1 antibody to confirm the knockdown effect of siRNA. β-Actin was detected by reprobing the same blots with anti-β-actin antibodies. Phosphorylation of Akt and eNOS in cell lysates were analyzed by immunoblotting with anti-phospho-Akt and anti-phospho-eNOS antibodies. The amounts of Akt and eNOS were detected by reprobing the same blots with anti-Akt and anti-eNOS antibodies. Representative immunoblots (A) and quantitative analysis of protein phosphorylation for eNOS (B) were shown (n = 3).
DISCUSSION
The major findings of the present study are that flow stimulates tyrosine phosphorylation of Gab1 in a Src kinase-dependent and VEGFR2-dependent manner, and that tyrosine-phosphorylated Gab1 is required for flow-induced activation of Akt and eNOS in endothelial cells. We found that Gab1 is rapidly tyrosine-phosphorylated in both BAECs and HUVECs in response to flow, which are correlated with activation of Akt and eNOS. Inhibition of Src kinases or VEGFR2 kinase with specific inhibitors significantly reduced flow-stimulated tyrosine phosphorylation of Gab1 and activation of Akt and eNOS. Furthermore, flow stimulated association of Gab1 with the PI3K subunit p85 in a time-dependent manner, and transfection of Gab1 mutant lacking p85 binding sites into endothelial cells inhibited flow-mediated activation of Akt and eNOS. Finally, knockdown of Gab1 by siRNA attenuated flow-induced activation of Akt and eNOS in endothelial cells. This is the first report to show a critical role of Gab1, a scaffold adaptor protein, in the fluid shear stress-mediated PI3K/Akt/eNOS pathway in endothelial cells.
Gab1 has multiple tyrosine phosphorylation sites that serve as binding sites for the SH2 domains of PI3K, phospholipase C-γ, SHP2, and CrkL (27, 28, 37). Gab1 is tyrosine-phosphorylated in response to many growth factors and cytokines, resulting in activation of both the Ras/MAPK and PI3K/Akt signaling cascades (21–23). Here we show for the first time that mechanotransduction via fluid shear stress rapidly induces Gab1 tyrosine phosphorylation in endothelial cells. In recognizing the significance of tyrosine phosphorylation induced on Gab1 by flow, the critical issue was to determine which one or more tyrosine kinases are responsible for this phosphorylation event. We have previously shown that Src kinases and VEGFR2 are implicated in the cellular response to flow (18), therefore we evaluated the putative role of Src kinases and VEGFR2 in flow-induced Gab1 phosphorylation using selective inhibitors, herbimycin, PP2, VTI, and SU1498, respectively. These experiments show that these inhibitors significantly attenuated tyrosine phosphorylation of Gab1 and phosphorylation of Akt and eNOS by flow, indicating that Src kinases and VEGFR2 participate in flow-induced Gab1 phosphorylation as well as activation of Akt and eNOS in endothelial cells.
We previously showed that flow induced PI3K/Akt/eNOS pathway through Src kinases and VEGFR2 in endothelial cells (18), but it is still not clear whether flow-stimulated VEGFR2 recruits and activates PI3K directly. VEGFR2 has several potential PI3K binding sites, many of them have been shown to be involved in activation of PI3K and Akt (38), but none of them has been clearly shown to directly recruit p85 of PI3K (39, 40). In this report, we show that activation of VEGFR2 by flow induces PI3K-Akt-eNOS activation in endothelial cells through the tyrosine phosphorylation of the docking protein Gab1. Based on our data published previously (18) and the results from this study, we propose that flow stimulates activation of Src kinases and transactivates VEGFR2. VEGFR2 activation results in recruitment and tyrosine phosphorylation of the scaffold adaptor Gab1. Phosphorylation of Gab1 leads to recruitment of PI3K, and the association of Gab1 with PI3K is required for activation of Akt, which induces eNOS activation and subsequently NO production in endothelial cells. It is well documented that VEGF stimulates VEGFR2 and PI3K/Akt/eNOS signaling in endothelial cells (16, 41). Therefore, it will be interesting to know whether the function of Gab1 is specific for mechanosignaling or involved in signal transduction of VEGFR2 activated by both flow and VEGF. The role of Gab1 in VEGF-induced signaling is under investigation.
In summary, our data show that Src kinase- and VEGFR2-dependent Gab1 tyrosine phosphorylation by flow mediates activation of Akt and eNOS in endothelial cells. Because endothelial-derived NO from eNOS is an important mediator for maintaining endothelial normal function and a negative regulator of vascular inflammation (4, 5, 42), we suggest that flow-mediated Gab1 phosphorylation may be involved in atheroprotective effect of laminar flow.
Footnotes
This work was supported by American Heart Association Grant 0235480T (to Z.-G. J.) and National Institutes of Health Grant HL64839 (to B. C. B.).
The abbreviations used are: NO, nitric oxide; eNOS, endothelial nitric-oxide synthase; PI3K, phosphatidylinositol 3-kinase; VEGFR2, vascular endothelial growth factor receptor 2; Gab1, Grb2-associated binder 1; siRNA, small interference RNA; BAEC, bovine aortic endothelial cell; HUVEC, human umbilical vein endothelial cell; VTI, VEGF receptor tyrosine kinase inhibitor; SU1498, 4-[(4′-chloro-2′-fluoro)phenylamino]-6,7-dimethoxyquinazoline); PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine; Akt, protein kinase B.
References
- 1.Davies PF. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gimbrone MA, Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G. Ann N Y Acad Sci. 2000;902:230–239. doi: 10.1111/j.1749-6632.2000.tb06318.x. discussion 239–240. [DOI] [PubMed] [Google Scholar]
- 3.Traub O, Berk BC. Arterioscler Thromb Vasc Biol. 1998;18:677–685. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- 4.Tedgui A, Mallat Z. Circ Res. 2001;88:877–887. doi: 10.1161/hh0901.090440. [DOI] [PubMed] [Google Scholar]
- 5.Zeiher A. Lancet. 1996;348:s10–s12. doi: 10.1016/s0140-6736(96)98004-6. [DOI] [PubMed] [Google Scholar]
- 6.Moroi M, Zhang L, Yasuda T, Virmani R, Gold HK, Fishman MC, Huang PL. J Clin Invest. 1998;101:1225–1232. doi: 10.1172/JCI1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubanyi GM, Romero JC, Vanhoutte PM. Am J Physiol. 1986;19:H1145–H1149. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- 8.Kuchan MJ, Frangos JA. Am J Physiol. 1994;266:C628–C636. doi: 10.1152/ajpcell.1994.266.3.C628. [DOI] [PubMed] [Google Scholar]
- 9.Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH. Circ Res. 1994;74:349–353. doi: 10.1161/01.res.74.2.349. [DOI] [PubMed] [Google Scholar]
- 10.Corson MA, James NL, Latta SE, Nerem RM, Berk BC, Harrison DG. Circ Res. 1996;79:984–991. doi: 10.1161/01.res.79.5.984. [DOI] [PubMed] [Google Scholar]
- 11.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 12.Go YM, Boo YC, Park H, Maland MC, Patel R, Pritchard KA, Jr, Fujio Y, Walsh K, Darley-Usmar V, Jo H. J Appl Physiol. 2001;91:1574–1581. doi: 10.1152/jappl.2001.91.4.1574. [DOI] [PubMed] [Google Scholar]
- 13.Michel T, Li GK, Busconi L. Proc Natl Acad Sci U S A. 1993;90:6252–6256. doi: 10.1073/pnas.90.13.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayajiki K, Kindermann M, Hecker M, Fleming I, Busse R. Circ Res. 1996;78:750–758. doi: 10.1161/01.res.78.5.750. [DOI] [PubMed] [Google Scholar]
- 15.Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. J Biol Chem. 2002;277:3388–3396. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 16.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG, Berk BC, Aebersold R, Corson MA. J Biol Chem. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- 18.Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Circ Res. 2003;93:354–363. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 19.Pawson T, Scott JD. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 20.Schlessinger J. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Rohrschneider LR. FEBS Lett. 2002;515:1–7. doi: 10.1016/s0014-5793(02)02425-0. [DOI] [PubMed] [Google Scholar]
- 22.Nishida K, Hirano T. Cancer Sci. 2003;94:1029–1033. doi: 10.1111/j.1349-7006.2003.tb01396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu H, Neel BG. Trends Cell Biol. 2003;13:122–130. doi: 10.1016/s0962-8924(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 24.Itoh M, Yoshida Y, Nishida K, Narimatsu M, Hibi M, Hirano T. Mol Cell Biol. 2000;20:3695–3704. doi: 10.1128/mcb.20.10.3695-3704.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sachs M, Brohmann H, Zechner D, Muller T, Hulsken J, Walther I, Schaeper U, Birchmeier C, Birchmeier W. J Cell Biol. 2000;150:1375–1384. doi: 10.1083/jcb.150.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu H, Saito K, Klaman LD, Shen J, Fleming T, Wang Y, Pratt JC, Lin G, Lim B, Kinet JP, Neel BG. Nature. 2001;412:186–190. doi: 10.1038/35084076. [DOI] [PubMed] [Google Scholar]
- 27.Holgado-Madruga M, Emlet DR, Moscatello DK, Godwin AK, Wong AJ. Nature. 1996;379:560–564. doi: 10.1038/379560a0. [DOI] [PubMed] [Google Scholar]
- 28.Holgado-Madruga M, Moscatello DK, Emlet DR, Dieterich R, Wong AJ. Proc Natl Acad Sci U S A. 1997;94:12419–12424. doi: 10.1073/pnas.94.23.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodrigues GA, Falasca M, Zhang Z, Ong SH, Schlessinger J. Mol Cell Biol. 2000;20:1448–1459. doi: 10.1128/mcb.20.4.1448-1459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R, Neel BG. Mol Cell Biol. 2002;22:4062–4072. doi: 10.1128/MCB.22.12.4062-4072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamothe B, Yamada M, Schaeper U, Birchmeier W, Lax I, Schlessinger J. Mol Cell Biol. 2004;24:5657–5666. doi: 10.1128/MCB.24.13.5657-5666.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osawa M, Masuda M, Kusano K, Fujiwara K. J Cell Biol. 2002;158:773–785. doi: 10.1083/jcb.200205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin ZG, Lungu AO, Xie L, Wang M, Wong C, Berk BC. Arterioscler Thromb Vasc Biol. 2004;24:1186–1191. doi: 10.1161/01.ATV.0000130664.51010.28. [DOI] [PubMed] [Google Scholar]
- 34.Cunnick JM, Dorsey JF, Munoz-Antonia T, Mei L, Wu J. J Biol Chem. 2000;275:13842–13848. doi: 10.1074/jbc.275.18.13842. [DOI] [PubMed] [Google Scholar]
- 35.Tai LK, Okuda M, Abe J, Yan C, Berk BC. Arterioscler Thromb Vasc Biol. 2002;22:1790–1796. doi: 10.1161/01.atv.0000034475.40227.40. [DOI] [PubMed] [Google Scholar]
- 36.Jin ZG, Melaragno MG, Liao DF, Yan C, Haendeler J, Suh YA, Lambeth JD, Berk BC. Circ Res. 2000;87:789–796. doi: 10.1161/01.res.87.9.789. [DOI] [PubMed] [Google Scholar]
- 37.Ingham RJ, Holgado-Madruga M, Siu C, Wong AJ, Gold MR. J Biol Chem. 1998;273:30630–30637. doi: 10.1074/jbc.273.46.30630. [DOI] [PubMed] [Google Scholar]
- 38.Dayanir V, Meyer RD, Lashkari K, Rahimi N. J Biol Chem. 2001;276:17686–17692. doi: 10.1074/jbc.M009128200. [DOI] [PubMed] [Google Scholar]
- 39.Claesson-Welsh L. Biochem Soc Trans. 2003;31:20–24. doi: 10.1042/bst0310020. [DOI] [PubMed] [Google Scholar]
- 40.Zachary I. Biochem Soc Trans. 2003;31:1171–1177. doi: 10.1042/bst0311171. [DOI] [PubMed] [Google Scholar]
- 41.Papapetropoulos A, Garcia CG, Madri JA, Sessa WC. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsao PS, Buitrago R, Chan JR, Cooke JP. Circulation. 1996;94:1682–1689. doi: 10.1161/01.cir.94.7.1682. [DOI] [PubMed] [Google Scholar]