

Summary
Retinoid homeostasis is critical for normal embryonic development. Both the deficiency and excess of these compounds are associated with congenital malformations. Here we demonstrate that SIRT1, the most conserved mammalian NAD+-dependent protein deacetylase, contributes to homeostatic retinoic acid (RA) signaling and modulates mouse embryonic stem cell (mESC) differentiation in part through deacetylation of cellular retinoic acid binding protein II (CRABPII). We show that RA-mediated acetylation of CRABPII at K102 is essential for its nuclear accumulation and subsequent activation of RA signaling. SIRT1 interacts with and deacetylates CRABPII, regulating its subcellular localization. Consequently, SIRT1 deficiency induces hyper-acetylation and nuclear accumulation of CRABPII, enhancing RA signaling and accelerating mESC differentiation in response to RA. Consistently, SIRT1 deficiency is associated with elevated RA signaling and development defects in mice. Our findings reveal a novel molecular mechanism that regulates RA signaling, and highlight the importance of SIRT1 in regulation of ESC pluripotency and embryogenesis.
Keywords: SIRT1, CRABPII, deacetylation, RA signaling, ES cell differentiation
Introduction
Retinoic acid (RA), an active metabolite of vitamin A, is the most potent natural form of vitamin A and plays an important role in a variety of biological processes including vision, reproduction, immune responses, and development (Mark et al., 2009; Soprano et al., 2007). Maintenance of a homeostasis of retinoid is critical for normal embryonic development, as maternal vitamin A-deficiency is associated with a collection of congenital malformations, whereas an excess of vitamin A intake also leads to numerous congenital abnormalities (Mark et al., 2006, 2009; Soprano et al., 2007).
Key regulatory factors that mediate the pleiotropic effects of RA include several classes of RA-binding transcription factors, the retinoic acid receptors RARα, RARβ, and RARγ, the retinoid X receptors RXRα, RXRβ, and RXRγ, and recently the peroxisome proliferator-activated receptor PPARβ/δ (Mark et al., 2009; Noy, 2010). These receptors are members of adopted orphan nuclear receptors that specifically bind with all-trans-RA or 9-cis-RA. Upon ligand binding, they activate a highly dynamic transcriptional network that controls organogenesis, tissue homeostasis, cell proliferation, differentiation, and apoptosis (Chawla et al., 2001; Mark et al., 2009). In addition to the receptors, another class of mediators for the RA signaling includes two isoforms of cellular retinoic acid binding proteins (CRABPs), CRABPI and CRABPII. CRABPI and CRABPII are highly conserved cellular proteins that bind to all-trans-RA with high but varying affinities (Chen et al., 2003). They are expressed in many tissues of the developing embryo in a non-overlapping pattern and exert distinct functions. For example, CRABPI functions to sequester and deliver excess RA for degradation (Boylan and Gudas, 1991, 1992), while CRABPII translocates into the nucleus upon RA binding, channeling RA to the nuclear RARs and activating their transcriptional activity (Delva et al., 1999; Dong et al., 1999; Sessler and Noy, 2005).
SIRT1, a nuclear NAD+-dependent protein deacetylase, is the most conserved member of the sirtuin family of enzymes. SIRT1 is an important cellular metabolic sensor crucially involved in multiple cellular processes, extending mammalian lifespan and affecting diseases related to metabolism and aging (Guarente, 2013; Satoh et al., 2013; Schug and Li, 2011). Numerous mammalian transcription factors important for aging and disease are the deacetylation substrates of SIRT1, including tumor suppressor p53, forkhead transcription factors (FOXOs), nuclear receptor coactivator PGC-1α, histone acetyltransferase p300, and nuclear factor κB (NF-κB) (Guarente, 2011a, b). These actions of SIRT1 link the protein acetylation status to the availability of cellular nutrients, regulating metabolic strategy and cell fate in response to environmental cues (Li, 2013; Schug and Li, 2011).
SIRT1 also plays a critical role in animal development. For example, knocking out SIRT1 systemically in mice on many common genetic backgrounds, such as 129Sv/CD1, 129Sv/C57BL/6, 129SvEv/FVB, and 129SvEV/FVB/Black Swiss, leads to severe developmental defects in multiple tissues, including intrauterine growth retardation, developmental defects of the retina and heart, defective germ cell differentiation, and neonatal lethality (Cheng et al., 2003; McBurney et al., 2003; Wang et al., 2008). In particular, the survival rate of SIRT1 whole body KO mice on a 129SvEv/FVB background is about 1% (Wang et al., 2008). Specifically, SIRT1 has been shown to modulate the neural and glial specification of neural precursors (Kang et al., 2009; Prozorovski et al., 2008), control differentiation of skeletal myoblasts (Fulco et al., 2008; Fulco et al., 2003), and influence spermatogenesis (Coussens et al., 2008). Despite the studies above, however, the molecular mechanisms underlying the important function of SIRT1 in development and stem cell biology are still unclear.
Results
CRABPII is a deacetylation substrate of SIRT1
In search of novel SIRT1 deacetylation substrates, we have identified both CRABPI and CRABPII as hyperacetylated proteins in SIRT1 null MEFs in a global Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-based analysis of Lys acetylation (Figure 1A, Table S1 and (Chen et al., 2012)). Quantification of the SILAC results indicated that the degrees of CRABPs hyper-acetylation in SIRT1 KO MEFs were comparable to those of p53 (Table S1), a well-established SIRT1 deacetylation substrate (Luo et al., 2001; Vaziri et al., 2001).
Figure 1. CRABPII is a deacetylation substrate of SIRT1.
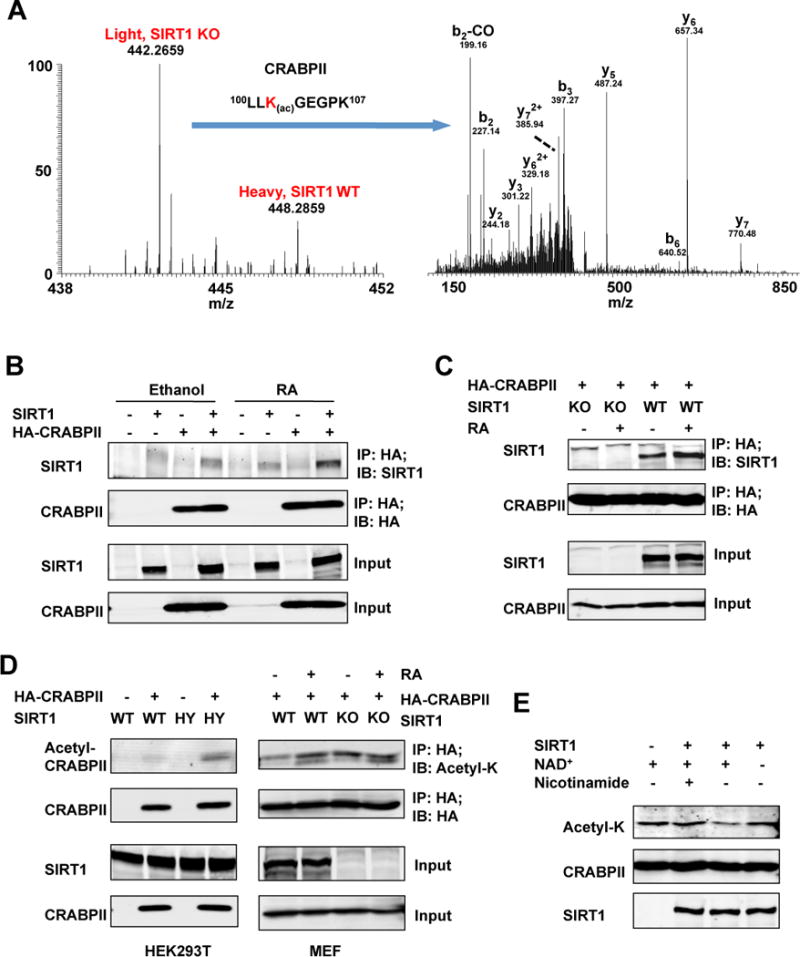
(A) Identification and SILAC quantification of K102 Lys acetylation site on CRABPII. Left panel represents the relative abundances of K102 Lys acetylation sites in SIRT1 KO (light label) and WT (heavy label) MEFs. Right panel shows the MS/MS spectrum that identified “LLK(ac)GEGPK” on CRABPII. b and y ions represent peptide backbone fragmentations containing N- or C- terminus, respectively. Please also see Table S1.
(B) The RA treatment enhances the interaction between HA-CRABPII and SIRT1 in HEK293T cells.
(C) The RA treatment enhances the interaction between CRABPII and endogenous SIRT1 in MEFs.
(D) SIRT1 deficiency increases acetylation levels of CRABPII in HEK293T cells (left panel), and the RA treatment induces the acetylation of CRABPII in WT but not SIRT1 KO MEFs (right panel).
(E) SIRT1 deacetylates CRABPII in vitro. Affinity purified acetylated HA-CRABPII proteins were incubated with or without recombinant SIRT1 protein in buffers containing indicated chemicals as described in Experimental Procedures.
To confirm the SILAC results and to test whether CRABPI and CRABPII are bona fide SIRT1 deacetylation substrates, we first examined whether SIRT1 could physically interact with CRABPs. As shown in Figure S1A, HA-CRABPII but not HA-CRABPI was co-immunoprecipitated with SIRT1 under normal culture conditions in HEK293T cells (IP-HA; IB-SIRT1), indicating that SIRT1 specifically interacts with CRABPII but not CRABPI. Moreover, consistent with previous observations that CRABPII functions to shuttle RA from the cytosol to the nucleus (Delva et al., 1999; Dong et al., 1999; Sessler and Noy, 2005), whereas SIRT1 is predominately a nuclear protein, the interaction between HA-CRABPII and overexpressed SIRT1 (Figure 1B) or endogenous SIRT1 (Figure 1C) was increased upon the RA treatment. Therefore, CRABPII but not CRABPI functions as a physiological interacting partner of SIRT1.
Next, we investigated whether hyper-acetylation of CRABPII in SIRT1 deficient MEFs is due to loss of SIRT1 deacetylase activity. As shown in Figure 1D, left panels, the acetylated HA-CRABPII levels were higher in cells co-expressing a catalytically inactive SIRT1 mutant protein (H355Y, HY) than in cells expressing the wild-type (WT) SIRT1 protein. Furthermore, the acetylation levels of HA-CRABPII were induced upon the RA treatment in WT MEFs, but not further in SIRT1 KO MEFs (Figure 1D, right panels), suggesting that SIRT1 is the prime protein deacetylase for CRABPII in MEFs.
Finally, to confirm that SIRT1 could directly deacetylate CRABPII, we carried out in vitro deacetylation assays with affinity purified acetyl-HA-CRABPII protein from SIRT1 KO MEFs and recombinant human SIRT1 protein. As shown in Figure 1E, acetyl-CRABPII protein was deacetylated by SIRT1 in a NAD+-dependent manner, and treatment with nicotinamide, a SIRT1 inhibitor, completely abolished the action of SIRT1 on CRABPII. Taken together, these data indicate that CRABPII is a deacetylation substrate of SIRT1 in cells and in vitro.
Loss of SIRT1 increases the nuclear accumulation of CRABPII and enhances RA signaling in cultured cells
CRABPII is a cellular RA carrier that translocates from the cytosol into the nucleus upon RA binding to activate the nuclear RA receptors (Delva et al., 1999; Dong et al., 1999; Sessler and Noy, 2005). The observation that CRABPII is hyper-acetylated upon the RA treatment (Figure 1D) suggests the acetylation modification of this protein is associated with its nuclear translocation. To test this possibility, we analyzed the subcellular localizations of HA-CRABPII in WT and SIRT1 KO MEFs by immuno-fluorescent staining (Figure 2A and Figure S1B). In WT MEFs, HA-CRABPII was predominately in the cytoplasm after switching from a normal culture medium to a medium containing RA-free charcoal-stripped FBS (Ethanol). Treatment with RA in this medium led to translocation of cytosolic HA-CRABPII into the nucleus in most cells (more than 80%), and washing out RA with the charcoal-stripped medium (Washout) cycled HA-CRABPII back to the cytosol. In SIRT1 KO MEFs, on the other hand, HA-CRABPII proteins in the majority of cells (more than 90%) were largely in the nucleus regardless of RA in the culture medium. These observations indicate that RA- or SIRT1 deficiency- induced hyper-acetylation of CRABPII enhances its nuclear localization.
Figure 2. The acetylation status at K102 of CRABPII affects its subcellular localization and subsequent RAR activation in cultured cells.
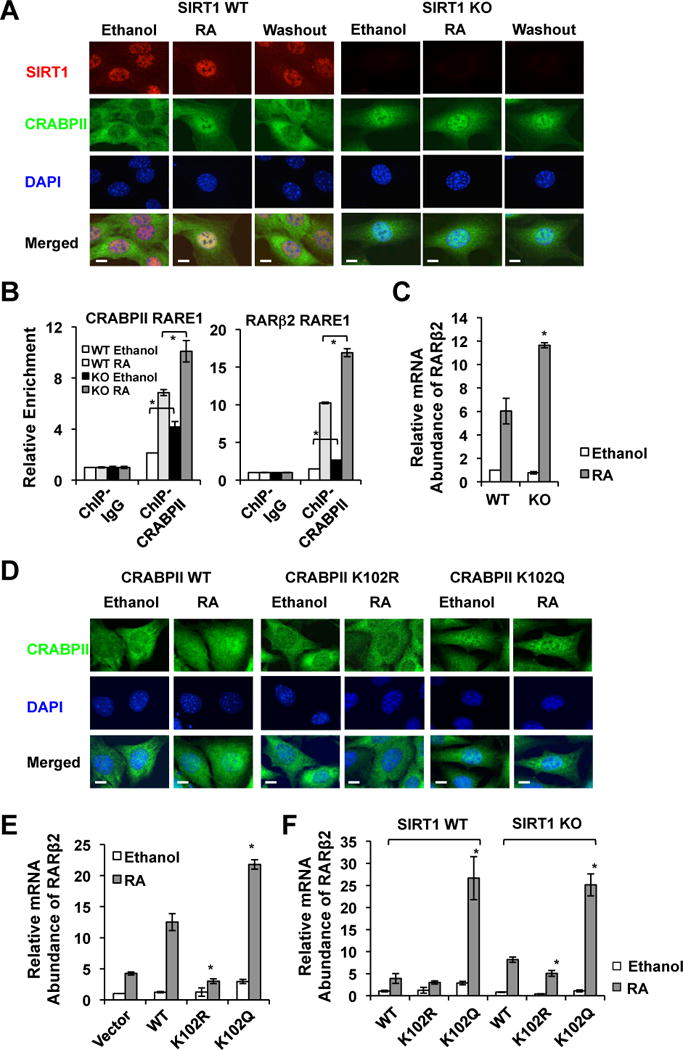
(A) SIRT1 deficiency induces the nuclear accumulation of CRABPII regardless of RA in MEFs. Bar, 10 μm.
(B) SIRT1 deficient MEFs have increased association of CRABPII with RARE on the promoters of RA target genes. *p<0.05, values are represented as mean ± SEM.
(C) SIRT1 deficient MEFs have increased expression levels of RARβ2 after the RA treatment. *p<0.05, values are represented as mean ± SEM.
(D) Acetylation of K102 results in increased nuclear accumulation of CRABPII. MEFs stably expressing indicated proteins were subjected for indirect immunofluorescence detection of the CRABPII location as described in Experimental Procedures. Bar, 10 μm.
(E) The CRABPII K102R mutant is defective in promoting RARβ2 expression, whereas the K102Q mutant enhances the RA-induced expression of RARβ2. *p<0.05, values are represented as mean ± SEM.
(F) The CRABPII K102R mutant is less active, whereas the K102Q mutant is more active independently of SIRT1. *p<0.05, values are represented as mean ± SEM. Please also see Figure S1.
In line with the increased nuclear accumulation of CRABPII, SIRT1 KO MEFs had enhanced association of CRABPII with retinoic acid response elements (RAREs) on RA target promoters (i.e. RARβ2 and CRABPII, two classical RA target genes) even in the absence of RA, as revealed by a chromatin immuno-precipitation (ChIP) assay (Figure 2B, Ethanol). The RA treatment recruited additional CRABPII to the indicated RAREs, but SIRT1 KO MEFs displayed an increased recruitment compared to WT MEFs (Figure 2B, RA). Accordingly, SIRT1 KO MEFs had an increased expression level of RARβ2 after the RA treatment (Figure 2C). In summary, our data indicate that SIRT1 deficiency-induced CRABPII hyper-acetylation increases its nuclear accumulation and chromatin association, thereby enhancing subsequent RA signaling in cells.
The acetylation status of CRABPII on K102 directly modulates its subcellular localization and subsequent RAR activation
CRABPII was hyper-acetylated at five lysine sites in SIRT1 KO MEFs (Table S1). To identify the acetylation site(s) that is responsible for the enhanced nuclear localization of CRABPII and RA signaling, we generated five single acetylation deficient CRABPII mutants (K to R mutation) by site-direct mutagenesis. In WT MEFs, only the K102R mutant appeared to have an impaired ability to translocate into the nucleus after the RA treatment (Figure S1C, Figure 2D and S1B, the K102R panels). More importantly, a CRABPII mutant that mimics the acetylation of K102, K102Q, increased its nuclear accumulation independently of RA (Figure 2D and S1B, the K102Q panels), indicating that acetylation of K102 is not only necessary but also sufficient to build up the nuclear CRABPII levels.
Consistent with above observations (Figure 2D and S1B), when stably expressed in MEFs (Figure S1D), the K102R mutant displayed a blunted ability to promote the expression of RARβ2, whereas the K102Q mutant enhanced the RA-induced expression of this gene (Figure 2E). Moreover, the K102R mutant was less able to induce RARβ2 expression in both WT and SIRT1 KO MEFs compared to WT CRAPBII, and in contrast, the K102Q mutant increased the mRNA abundance of RARβ2 independently of cellular SIRT1 levels (Figure 2F), further supporting the notion that the acetylation status of K102 directly determines its ability to activate RAR. Collectively, our data indicate that K102 acetylation of CRABPII is an essential element in the RA-induced nuclear localization of CRABPII and subsequent transactivation of RARs. Through deacetylation of K102 on CRABPII, SIRT1 plays a critical role in regulation of the cellular RA signaling.
SIRT1 deficiency accelerates RA-induced mouse embryonic stem cell (mESC) differentiation
RA is essential for a variety of biological processes, including growth and differentiation. In embryonic stem cells (ESCs), depending on the timing of the RA treatment as well as the addition of other factors, RA can induce their differentiation into a large number of different cell types (Mark et al., 2009; Soprano et al., 2007). To further assess the impact of SIRT1 deficiency-induced hyper-acetylation of CRABPII on the RA signaling and its physiological functions, we investigated RA-induced differentiation of WT and SIRT1 KO mESCs (Figure 3). Consistent with our observations in HEK293T and MEFs, SIRT1 interacted with and deacetylated CRABPII but not CRABPI in mESCs (Figure 3A and Figure S2A). Nuclear accumulation of CRABPII was also enhanced in SIRT1 KO mESCs (Figure 3B and Figure S2B). When cultured in a defined serum-free ES cell maintenance medium, ESGRO medium, SIRT1 KO mESCs displayed a reduced staining intensity for the activity of Alkaline Phosphatase (AP, Figure S2C), a marker of undifferentiated ESCs, indicating that SIRT1 KO mESCs are more differentiated than WT mESCs. Consistent with previous reports (Calvanese et al., 2010; Saunders et al., 2010), WT mESCs gradually reduced their cellular SIRT1 mRNA levels when treated with 0.2 μM of RA in a differentiating M10 medium (Figure 3C). The cellular mRNA levels of RARβ2 and CRABPII, on the other hand, were dramatically induced within 24-hour of the treatment, then gradually went down. This induction was significantly elevated and extended in SIRT1 KO mESCs (Figure 3D), indicating an increase in RA response. In contrast to CRABPII, the mRNA levels of CRABPI were severely reduced in SIRT1 KO mESCs even in the basal medium (Figure 3D), suggesting that SIRT1 modulates CRABPII and CRABPI via distinct mechanisms.
Figure 3. SIRT1 KO mESCs are hypersensitive to RA-induced differentiation.
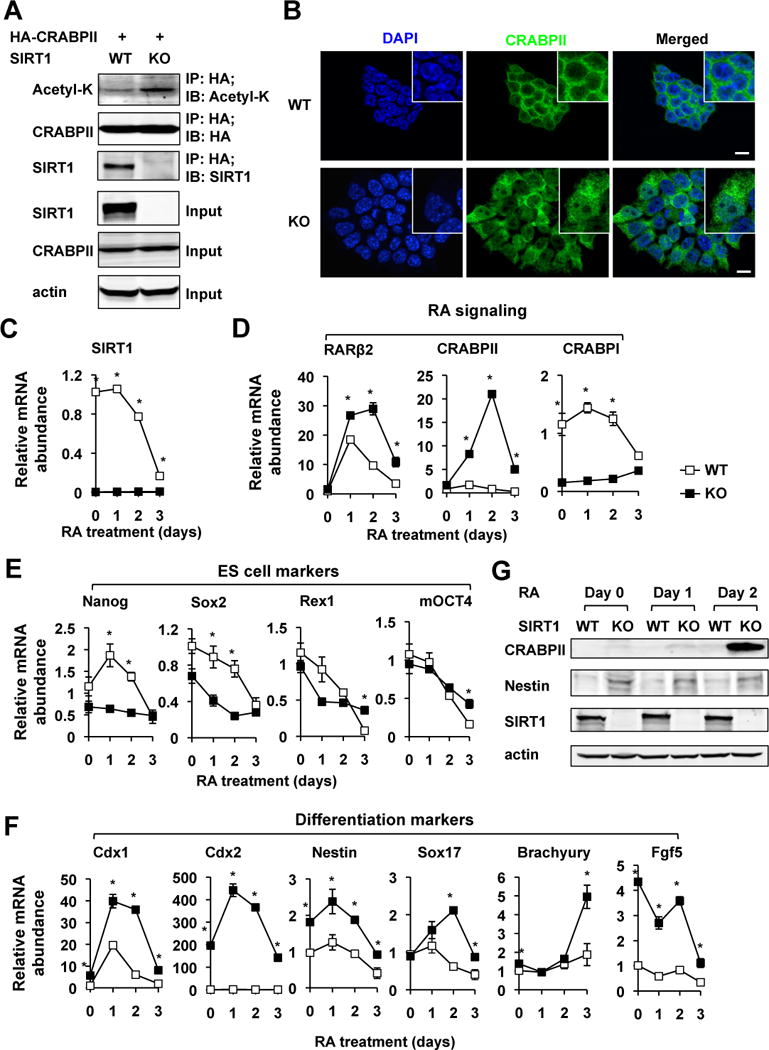
(A) SIRT1 interacts with CRABPII and SIRT1 deficiency increases the acetylation levels of CRABPII in mESCs.
(B) SIRT1 KO mESCs have increased accumulation of CRABPII in the nucleus. Bar, 10 μm.
(C) The expression of SIRT1 is reduced during RA-induced differentiation of mESCs. WT and SIRT1 KO mESCs were treated with 0.2 μM of RA in the M10 medium for indicated days. *p<0.05, values are represented as mean ± SEM.
(D) SIRT1 KO mESCs have an increased induction of RARβ2 and CRABPII, while reduced levels of CRABPI during RA-induced differentiation. *p<0.05, values are represented as mean ± SEM.
(E) Deletion of SIRT1 in mESCs reduces expression levels of several ES cell markers. *p<0.05, values are represented as mean ± SEM.
(F) SIRT1 KO mESCs have increased expression of differentiation markers in response to RA. *p<0.05, values are represented as mean ± SEM.
(G) SIRT1 KO mESCs have increased induction of CRABPII protein but reduced levels of Nestin protein in response to RA treatment. The protein levels of CRABPII and Nestin were analyzed by immuno-blotting.
Please also see Figure S2.
In line with the increased differentiation morphology, SIRT1 deficient mESCs had decreased expression levels of a couple of ES cell markers, particularly Nanog and Sox2 (Figure 3E), while the levels of a number of differentiation markers, including Cdx1 and Cdx2, two members of the caudal-like Cdx gene family encoding homeodomain transcription factors that are essential for Hox gene expression and vertebral anteroposterior patterning, were significantly increased at different times during the RA treatment in SIRT1 KO mESCs (Figure 3F). Cdx1 is a direct target of RA (Allan et al., 2001; Houle et al., 2000; Houle et al., 2003). The elevated expression of CRABPII and Nestin proteins in SIRT1 KO mES cells was further confirmed by immuno-blotting (Figure 3G).
To confirm our observation that deletion of SIRT1 leads to increased RA-induced mESC differentiation, we generated ES cell lines in which SIRT1 was stably knocked down by shRNA (Figure 4A). Consistent with our data in Figure 3, SIRT1 shRNA mESCs (sh-SIRT1) displayed a hyper-differentiation morphology compared to control shRNA mESCs (sh-Control), including flattening of the ES cell colonies and reduction of AP staining intensity when cultured in the ESGRO medium (Figure 4B and Figure S2D). They also had significantly increased expression levels of several differentiation markers in the same medium (Figure 4C). When cultured in the M10 medium followed by a low dose RA treatment (20 nM), sh-SIRT1 mESCs have accelerated appearance of differentiation morphology (Figure S2E), along with elevated induction of RA target genes RARβ2, CRABPII and Cdx1, reduced levels of ES cell markers Nanog and Sox2, and increased expression of differentiation markers Cdx2 and Nestin (Figure 4D). Altogether, these observations demonstrate that SIRT1 deficiency accelerates RA-induced mESC differentiation.
Figure 4. SIRT1 silencing induces differentiation of mESCs.
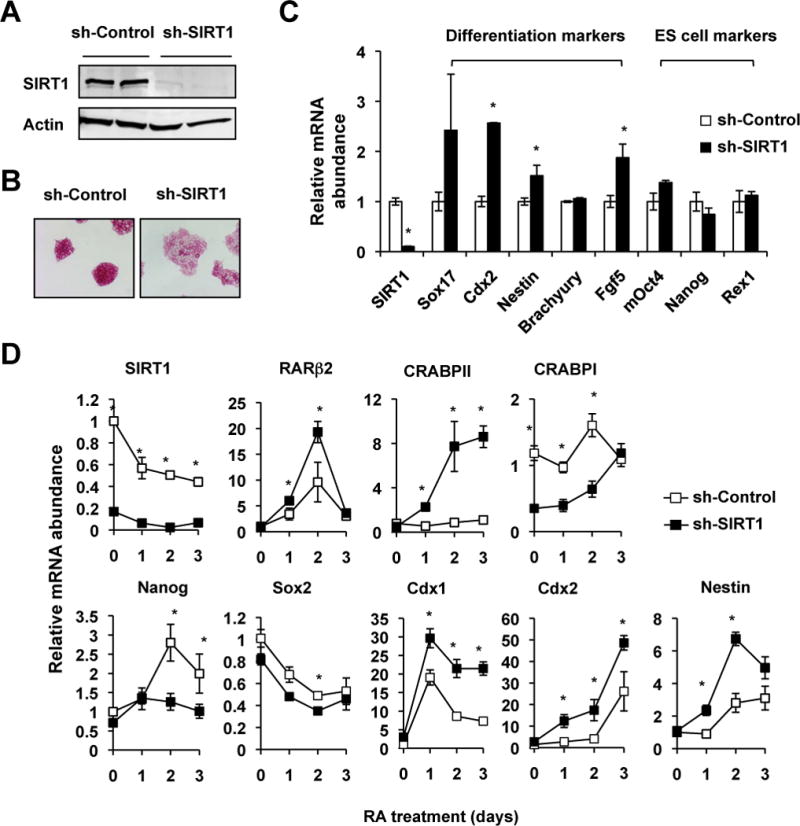
(A) SIRT1 protein levels in control (sh-Control) and SIRT1 (sh-SIRT1) shRNA E14 mES cells.
(B) Knocking-down SIRT1 induces differentiation of mES cells in the ESGRO medium. Sh-Control and sh-SIRT1 mESCs were stained for the AP activities.
(C) Knocking-down SIRT1 induces the expression of differentiation markers without alteration of stem cell markers in the ESGRO medium (n=3 independent stably clones, *p<0.05, values are represented as mean ± SEM).
(D) SIRT1 shRNA cells have increased induction of RA target genes and differentiation makers, yet decreased levels of ES markers after the RA treatment days (n=3 independent stably clones, *p<0.05, values are represented as mean ± SEM).
Please also see Figure S2.
To further explore the transcriptional networks that had been altered by SIRT1 deficiency in mESCs, we determined the transcriptomes of sh-Control and sh-SIRT1 mESCs in the M10 medium treated with vehicle ethanol or with 20 nM of RA for 2 days by microarrays. As shown in Figure 5A, knocking down SIRT1 in mESCs significantly altered the expression levels of 846 gene probes by more than 1.5-fold in basal culture conditions (ethanol). Two-day treatment of RA significantly changed the expression of 4,745 and 6,201 gene probes by more than 1.5-fold in sh-Control and sh-SIRT1 mESCs, respectively. Among which 3,461 were common to both cells (Figure 5B). In support of our observations that SIRT1 deficient mESCs were more sensitive to the RA treatment (Figure 3 and Figure 4), the majority of affected genes were significantly more induced or repressed by RA in sh-SIRT1 mESCs than in sh-Control mESCs (Figure 5C, boxed genes). Further Ingenuity Pathway Analysis (IPA) of 1,468 significantly changed gene probes in RA treated samples (Table S2) showed that sh-SIRT1 and sh-Control mESCs displayed distinct transcriptional responses to RA. Compared to sh-Control mESCs, pathways involved in maintenance of ESC pluripotency were significantly enriched in the downregulated gene list, whereas pathways that mediate ESC differentiation and growth were highly enriched in the upregulated gene list in the sh-SIRT1 mESCs, (Figure 5D, Figure S3, Table S3). This observation confirms that SIRT1 deficiency in mESCs reduces the pluripotency while increasing the sensitivity to RA-induced differentiation. Interestingly, comparisons of transcriptomes between knockdown of SIRT1 and deficiency of known factors critical for ESC pluripotency/differentiation revealed that SIRT1 correlated positively with Prdm14, a core transcription factor required for the maintenance and reacquisition of ESC pluripotency (Chia et al., 2010; Tsuneyoshi et al., 2008) (Figure 5E), further supporting the notion that SIRT1 is crucial for maintenance of ES cell pluripotency.
Figure 5. Knocking-down SIRT1 promotes RA-induced differentiation of mESCs.
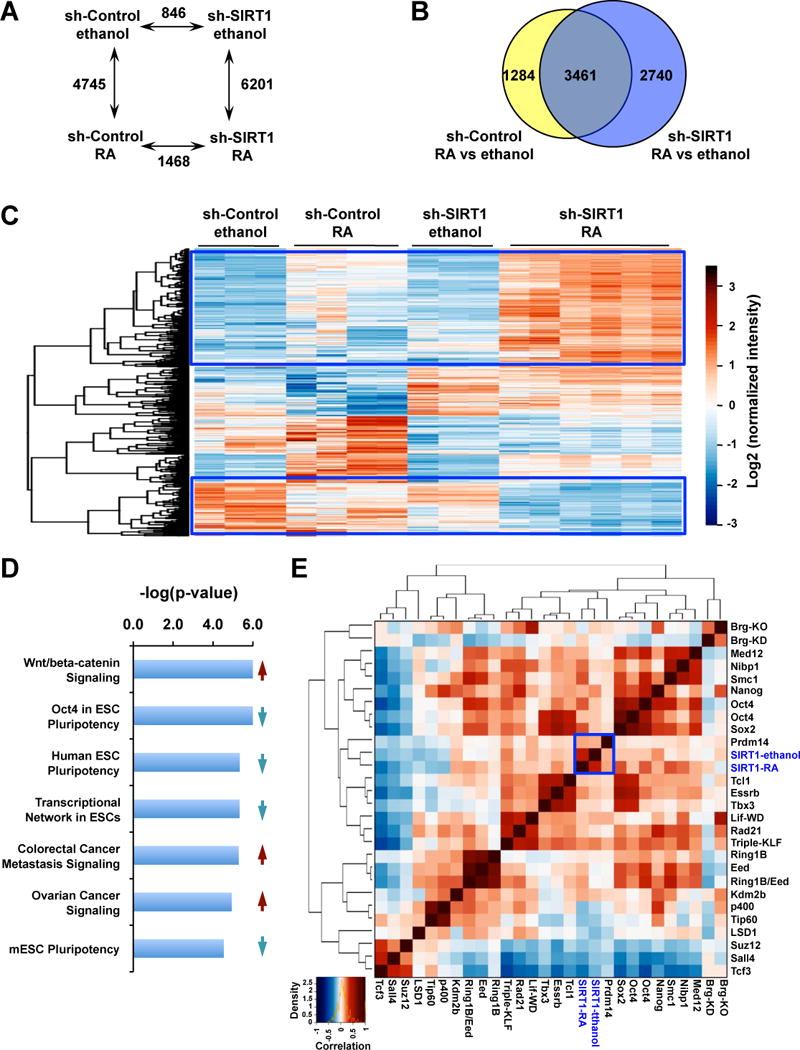
(A) The numbers of differentially expressed gene probes between sh-Control and sh-SIRT1 mESCs with and without treatment of RA. The mRNAs were analyzed by the mouse whole genome microarrays as described in Experimental Procedures (n=3–6, adjusted p<0.05, cutoff fold changes: 1.5).
(B) Venn-diagram representation of the subset of genes that were significantly altered by more than 1.5 fold upon RA treatment in sh-Control and sh-SIRT1 mESCs.
(C) sh-SIRT1 mESCs display an enhanced response to RA. The mRNA levels of 1,468 differentially expressed gene probes in RA treated sh-Control and sh-SIRT1 mESCs in panel A were presented by heatmaps.
(D) Pathways involved in the maintenance of ESC pluripotency are enriched in the downregulated gene list of sh-SIRT1 mESCs (blue down-arrow), whereas pathways that mediate ESC differentiation and growth were highly enriched in the upregulated gene list of sh-SIRT1 mESCs (red up-arrow). 1,468 significantly changed gene probes in RA treated sh-SIRT1 and sh-Control samples were analyzed by IPA.
(E) A correlation map of transcriptomes between knockdown of SIRT1 and deficiency of other known factors that are critical for ESC pluripotency/differentiation.
SIRT1 deficiency accelerates RA-induced mESC differentiation in part through CRABPII
To test whether SIRT1 modulates RA-induced mESC differentiation in part through deacetylation of CRABPII, we silenced CRABPII in sh-Control and sh-SIRT1 mESCs by siRNA (Figure 6A, CRABPII panel, and Figure S4A, CRABPII siRNA). As shown in Figure 6A, knocking-down CRABPII partially rescued the elevation of a number of RA targets and ES differentiation markers, RARβ, Cdx1, and Nestin, in sh-SIRT1 mESCs. By contrast, the expression of a stem cell marker Nanog (Figure 6A), as well as many other differentiation makers that are not direct targets of RA (not-shown), was not significantly affected by CRABPII silencing, indicating that CRABPII specifically regulates a subset of ES differentiation markers.
Figure 6. The acetylation status of CRABPII directly influences mESC differentiation in response to RA.
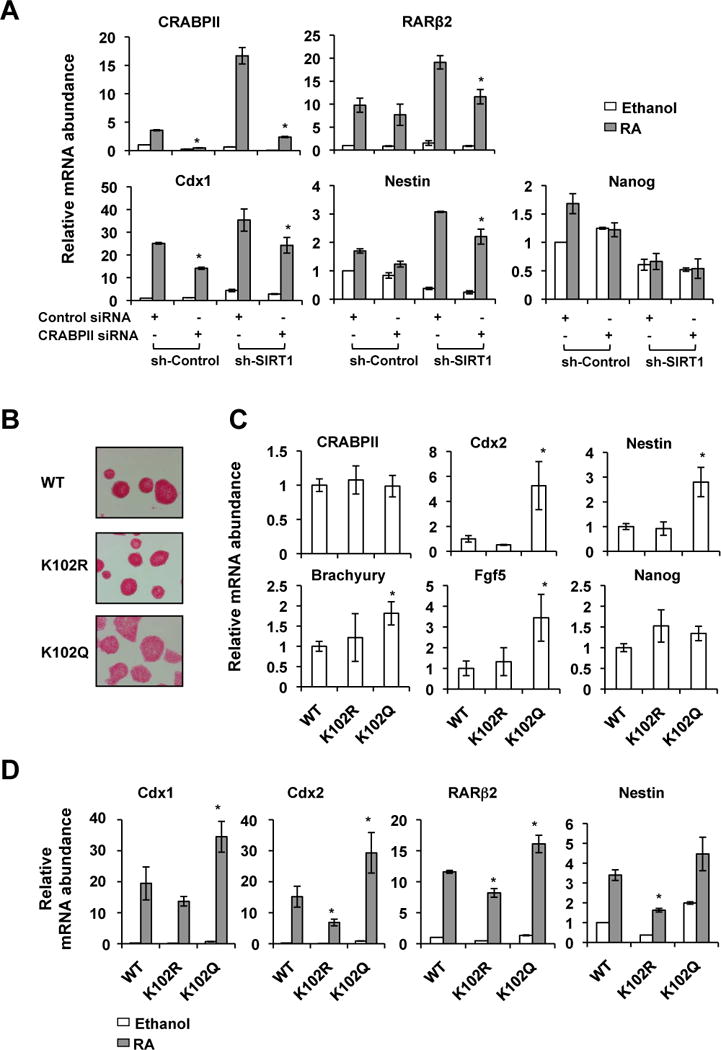
(A) Silencing CRABPII partially rescues the elevation of a number of ES differentiation markers in sh-SIRT1 mESCs. Sh-Control and sh-SIRT1 mESCs were transfected with siRNA specific against CRABPII. They were then treated with ethanol or 20 nM RA for two days (n=3 independent stably clones, *p<0.05, values are represented as mean ± SEM).
(B) The CRABPII K102Q mutant has increased ability to induce mESC differentiation morphologically. mESCs stably expressing indicated CRABPII proteins were cultured in the ESGRO medium.
(C) The CRABPII K102Q mutant induces expression of differentiation makers in mESCs in the ESGRO medium (n=4–7 independent stably clones with comparably CRABPII expression levels for each protein, *p<0.05, values are represented as mean ± SEM).
(D) The CRABPII K102Q mutant enhances RA-induced expression of ES cell differentiation makers, whereas the K102R mutant represses their induction. WT, K102R, and K102Q mutant CRABPII were stably expressed in mESCs in which endogenous CRABPII was silenced by siRNAs against its 3′-UTR. Cells were then treated with ethanol or 20 nM RA for two days. *p<0.05, values are represented as mean ± SEM.
Please also see Figure S4.
To further confirm that SIRT1 deficiency induced CRABPII hyper-acetylation is indeed able to accelerate mESC differentiation, we stably expressed WT, K102R, and K102Q mutant CRABPII in mESCs in which endogenous CRABPII was silenced by siRNAs against its 3′-UTR (Figure S4, CRABPII 3′UTR siRNA). As shown in Figure 6B and Figure S4C, expression of the acetylation mimic of CRABPII, K102Q, induced differentiation morphology of mESCs in the ESGRO medium. Consistently, the expression levels of many ES differentiation makers were significantly induced by the K102Q mutant (Figure 6C). Moreover, when treated with RA in the M10 medium, mESCs expressing the K102Q mutant elevated the levels of several ES cell differentiation markers, whereas mESCs expressing the K102R mutant had reduced expression of these markers compared with mESCs expressing WT CRABPII (Figure 6D). Taken together, our findings demonstrate that through modulation of the acetylation status and subcellular localization of CRABPII, SIRT1 is critically involved in repression of the RA signaling and maintenance of ES cell pluripotency.
SIRT1 deficiency induced developmental defects are associated with elevated RA signaling in mice
To further assess the importance of SIRT1 deficiency induced CRABPII/RAR activation and enhanced ES cell differentiation in animal development, we investigated whether the reported developmental defects in SIRT1 KO mice are associated with altered RA signaling at different developmental stages. Consistent with previous reports (Cheng et al., 2003; McBurney et al., 2003; Wang et al., 2008), whole body SIRT1 KO mice displayed intrauterine growth retardation (Figure S5A), developmental defects of the retina at various developmental stages (not shown), and neonatal lethality (Table S4) on the C57BL/6 background. Further investigation revealed that the endochondral ossification of vertebrates was delayed in SIRT1 KO mice compared to WT littermates, as indicated by reduced Alizarin Red staining (Figure 7A and 7B). This delay was associated with increased expression of RARβ proteins in both osteoblasts (Figure 7C) and chondrocytes (Figure 7D) (IgG negative controls, Figure S5B). The elevation in RA signaling had also been observed in other tissues of SIRT1 KO mice, including heart (Figure 7E) and intestine (Figure 7F) of E16.5 SIRT1 KO embryos on the C57BL/6 background, and testis of adult SIRT1 KO mice on a mixed 129SVJ/CD1 background (Figure S5C). SIRT1 deficient heart and testis have been reported to have various developmental abnormalities (Cheng et al., 2003; McBurney et al., 2003). To directly test whether SIRT1 deficiency has any impacts on animal’s response to RA, we gave Flox controls and SIRT1 liver-specific KO (SIRT1 LKO) mice a single dose of RA via intragastric gavage after feeding with a vitamin A deficient diet for 8 weeks to clean the background RA signaling. In agreement with our observations in cultured cells, deletion of hepatic SIRT1 led to increased mRNA levels of a number of RA target genes in the mouse liver after the RA dosing (Figure S5D). Collectively, our observations indicate that a number of SIRT1 deficiency induced developmental defects are associated with enhanced RA signaling in mice, and that deletion of SIRT1 increases the transcriptional responses to RA in vivo.
Figure 7. SIRT1 deficiency induced developmental defects are associated with elevated retinoic acid signaling in C57BL/6 mice.
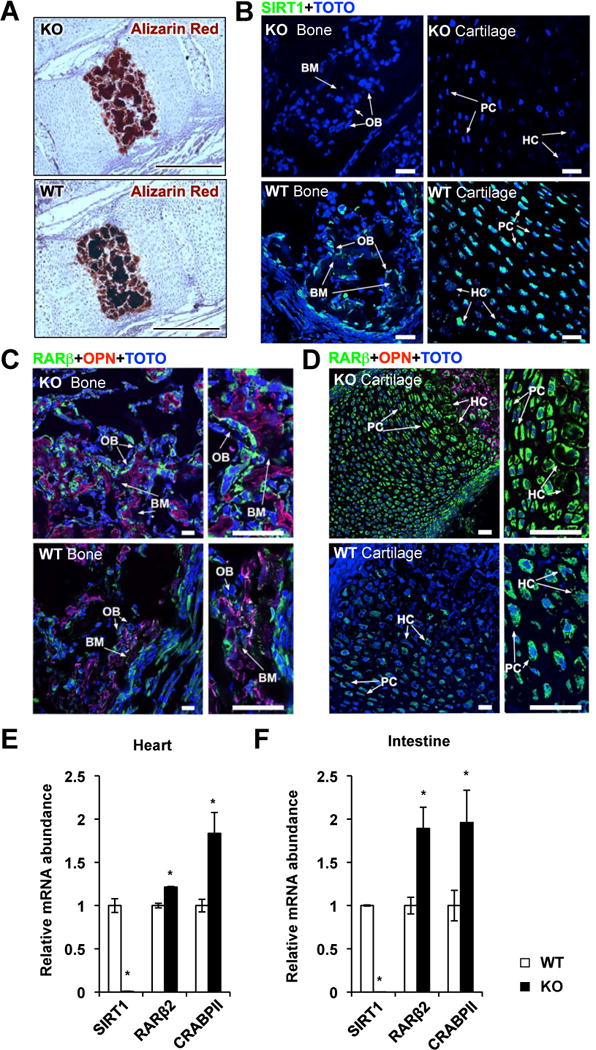
(A) SIRT1 KO mice have decreased endochondral mineralization. Vertebral sections from WT and SIRT1 KO newborn pups (P0.5) were stained with Alizarin Red for mineralization. Images were taken from sections in parallel to the liver. Bar, 100 μm.
(B) Loss of SIRT1 protein in bone and cartilage of SIRT1 KO newborn pups. Bar, 10 μm.
(C) Increased expression of RARβ in SIRT1 KO osteoblasts. Images were taken from the mineralization (bone matrix: BM) parts of long bones during endochondral ossification process. The bone mineralization parts were identified with the osteoblast cells (OB) middle stage maturation marker osteopontin (OPN). Bar, 10 μm.
(D) Increased expression of RARβ in SIRT1 KO chondrocytes, in regions of proliferative cartilage (PC) and hypertrophic cartilage (HC). Images were taken from the cartilage parts of long bones during endochondral ossification process. The PC and HC regions were identified by the cell nuclear morphological feature of proliferative chondrocytes and hypertrophic chondrocytes. Bar, 10 μm. (E–F) E16.5 SIRT1 KO embryos have elevated expression levels of RARβ2 and CRABPII in heart (E) and intestine (F). n=5, *p<0.05, values are represented as mean ± SEM.
Discussion
As a critical cellular metabolic sensor, SIRT1 has been well established as a master regulator of metabolism (Guarente, 2011a, b; Li, 2013). However, in spite of severe developmental defects in SIRT1 KO mice (Cheng et al., 2003; McBurney et al., 2003; Wang et al., 2008), the roles of SIRT1 in animal development remain undefined. In our present study, we showed that SIRT1 is able to regulate embryonic stem cell pluripotency/differentiation and animal development in part through CRABPII and RA signaling. We identified CRABPII as a new SIRT1 deacetylation substrate and provided evidence that SIRT1 controls the nuclear-cytoplasmic distribution of RA by modulating the acetylation status of CRABPII. SIRT1 deficiency led to hyper-acetylation and nuclear accumulation of CRABPII, resulting in increased RA signaling. This elevated transcriptional response to RA further reduced the pluripotency of mESC at the ground state and enhanced their differentiation upon the RA treatment, which finally resulted in various developmental defects in mice. Our findings uncover a novel mechanism that mediates SIRT1’s action in ES cell biology, and underscore a critical role of SIRT1 in transcriptional regulation of embryogenesis and animal development.
Several lines of evidence suggest a link between SIRT1 and RA signaling during animal development. For instance, the developmental defects observed in SIRT1 KO mice (Figure S5A, (Cheng et al., 2003; McBurney et al., 2003)) closely resemble the developmental defects caused by altered vitamin A metabolism and RA signaling (Ghyselinck et al., 1998; Grondona et al., 1996; Kastner et al., 1996; MacLean et al., 2007; Sucov et al., 1994). SIRT1 has also been shown to function as a co-repressor in inhibition of the RAR-mediated neuronal differentiation of P19 cells (Kang et al., 2009; Yu et al., 2012). Here we showed that by direct deacetylation of CRABPII, SIRT1 limits nuclear RAs that are available to their receptors and represses RA-induced ESC differentiation. This action of SIRT1 appears to link to a number of SIRT1 deficiency-induced developmental defects, particularly skeletal development. Vitamin A and RA signaling have been shown to exert profound influences on skeletal development (Weston et al., 2003). For example, inhibition of RA signaling is an active step in bone morphogenetic protein 4 (BMP4) mediated skeletogenesis (Hoffman et al., 2006). CRABPII null mice have minor limb defects (Fawcett et al., 1995), and its expression is dramatically induced in mouse models of degenerative joint disease (Welch et al., 2009). Therefore, it will be interesting to see whether SIRT1 KO mice are sensitive to these joint diseases. In addition, given the broad tissue expression pattern of CRABPII in developing embryos, this previously uncharacterized regulatory mechanism may have a general role in embryogenesis, particularly under the vitamin A excessive condition.
It is of importance to note that the penetrance and severity of developmental defects caused by SIRT1 deletion in mice are dependent on the genetic background (Cheng et al., 2003; McBurney et al., 2003; Wang et al., 2008; Satoh et al., 2010). In particular, SIRT1 KO mice appear to develop normally on the FVB background (Satoh et al., 2010). Although the detailed mechanisms underlying this interesting phenomenon remain to be defined, one plausible explanation is mice have distinct vitamin A metabolism and RA signaling on different genetic backgrounds. In line with this possibility, the RAR activation pathway is among the top differential pathways in various brain regions of C57BL/6 mice compared to FVB mice (Figure S6). Additional analyses will be needed to further test this possibility. Nevertheless, our findings demonstrate that enhanced RA signaling is associated with several developmental defects in SIRT1 KO mice on the C57BL/6 background. Our studies further suggest that reduction of the cellular RA signaling might ameliorate a number of SIRT1 deficiency-induced developmental defects. Therefore, it will be of great interest to test whether deletion of CRABPII or feeding of vitamin A deficient diets will rescue certain developmental abnormalities and increase the survival rate of SIRT1 KO mice.
RA-induced nuclear translocation of CRABPII has been proposed to be a multi-step process (Budhu and Noy, 2002; Majumdar et al., 2011; Sessler and Noy, 2005). In this model, CRABPII is associated with endoplasmic reticulum (ER) in the absence of RA. Binding of RA triggers some conformational rearrangements on ER-associated CRABPII, resulting in its release from ER and exposure of three-dimensional nuclear localization signal (NLS). The RA-bound holo-CRABPII is then recognized by importin α and transported into the nucleus (Budhu and Noy, 2002; Sessler and Noy, 2005). A recent study further proposed that SUMOylation of K102 is critical for release of ER-associated holo-CRABPII (Majumdar et al., 2011). However, several observations cannot fit well into this model. For example, although mutation of CRABPII K102 into R, which eliminates both SUMOylation and acetylation, appears to disrupt the nuclear translocation, the K102R mutant retains a normal ability to interact with importin α after the RA treatment in cells (Majumdar et al., 2011). Moreover, post-translational modifications of CRABPII are not required for the formation of the CRABPII/RAR complex (Dong et al., 1999). More importantly, we showed that hyper-acetylated CRABPII and the CRABPII K102 acetylation mimic (the K102Q mutant), two modified CRABPII proteins that cannot be further SUMOylated at the K102 site, are still able to accumulate in the nucleus (Figure 2A and 3A), strongly suggesting that CRABPII K102 SUMOylation is not required for its nuclear accumulation. Taking these observations together, we propose here that neither SUMOylation nor acetylation of CRABPII have significant impacts on its RA binding, nuclear translocation, and subsequent interaction with RAR. Instead, deacetylation of CRABPII is required to recycle CRABPII back to the cytosol and terminate cellular RA signaling upon RA removal (Figure S7A). It appears that acetylation of CRABPII occurs in the nucleus after holo-CRABPII docks on the RAR/RXR complex, possibly by nuclear acetyltransferases recruited by the RA-bound active RAR/RXR heterodimer. This modification stalls CRABPII on the RAR target promoters, prolonging their state of active transcription. Deacetylation of CRABPII by SIRT1, particularly at the K102 site, is then required to recycle CRABPII back into the cytosol and switch off the active state (Figure S7A). Interestingly, SIRT1 appears to remain on the RAREs after RA removal (Figure S7B), probably functioning as a co-repressor to maintain the repressive state of the promoter. In our model, both K102R and K102Q mutants are able to translocate into the nucleus by RA. However, the acetylation-deficient K102R mutant will be swiftly removed from the nucleus after its short interaction with the RAR/RXR heterodimer, whereas the K102Q mutant will be trapped on the active RAR target promoters due to its structural and/or sequential resemblance to the acetylated CRABPII. Additional biochemical and structural studies with WT, K102R and K102Q CRABPII proteins are needed to investigate how the acetylation status of CRABPII affects its chromatin association as well as its interaction with RAR and the nuclear export machinery.
In summary, we have shown that through deacetylation of CRABPII, SIRT1 plays a vital role in the regulation of cellular RA signaling and mESCs pluripotency. Since SIRT1 is a key metabolic sensor that is hypersensitive to the cellular metabolic/redox status and environmental signals, this newly characterized SIRT1/CRABPII/RAR signaling cascade will provide a new avenue to study gene-environment interactions that affect animal development.
Experimental Procedures
Animal studies
Whole body SIRT1 KO mice (SIRT1 KO) on the mixed 129SVJ/CD1 background (McBurney et al., 2003) were backcrossed into the C57BL/6 background for 8 generations to generate SIRT1 KO mice on over 99% C57BL/6 background. Embryos/newborn pups from heterozygous breeding pairs were analyzed at indicated developmental stages for developmental defects. Liver-specific SIRT1 knockout mice (SIRT1 LKO) and their age-matched littermate Flox controls have been reported before (Purushotham et al., 2009). 5-month old male Flox and SIRT1 LKO mice were fed with a vitamin A deficient diet for 8 weeks, and then given a dose of 5mg/kg all-trans retinoic acid (Sigma) via intragastric gavage. Livers were harvested 24 hours later. All animal experiments were conducted in accordance with guidelines of NIEHS/NIH Animal Care and Use Committee.
Cell culture
E14 mESC line was from ATCC, and WT and SIRT1 KO mESCs have been described (McBurney et al., 2003). All stem cells were maintained in the ESGRO Complete Clonal Grade Medium (ESGRO medium, Millipore). To induce the differentiation of mESCs with RA, mESCs were first cultured on gelatin-coated plates in the M10 medium. Sh-Control and sh-SIRT1 mESCs were treated with ethanol or 20 nM RA in the M10 medium for indicated times. WT and SIRT1 KO mESCs were treated with 0.2 μM RA.
Immuno-fluorescence assays
Immuno-fluorescence (IF) assays in MEFs and mouse tissues were preformed as described (Grant et al., 2013; Guo et al., 2010). WT and SIRT1 KO MEFs stably expressing HA-CRABPII were treated with ethanol or 0.1 μM RA for 6 hours before analysis. Images were taken by a Zeiss LSM 710 confocal microscope and quantified using ImageJ and MetaMorph Offline Version 7.8.6.0 (Sunnyvale).
Immunoprecipitation assay
To investigate the interaction between SIRT1 and HA-CRABP proteins in HEK293T cells, HEK293T cells transfected with indicated expressing constructs were treated with ethanol or 20 nM RA for 24 hours, cells were then lyzed in the NP40 buffer. The whole-cell extracts were immunoprecipitated with monoclonal anti-HA antibody-conjugated agarose beads (Santa Cruz biotech). To analyze the interaction between endogenous SIRT1 and HA-CRABPII, WT and SIRT1 KO MEFs stably expressing HA-CRABPII were treated with ethanol or 0.1 μM RA for 6 hours. Cell extracts were then immunoprecipitated by anti-HA Agarose beads, and then immunoblotted using antibodies against SIRT1 (Sigma).
Chromatin immunoprecipitation (ChIP) analysis
Chromatin immunoprecipitation (ChIP) analysis was performed essentially as described by the manufacturer (Millipore) with some modifications.
Protein Acetylation Analysis
The hyper-acetylation sites of endogenous CRABPI and CRABPII proteins in SIRT1 KO MEFs were identified with a SILAC-based lys-acetylomic method (Chen et al., 2012). To analyze the acetylation levels of CRABPII in cells, HEK293T cells transfected with constructs expressing HA-CRABPII or WT or HY mutant of SIRT1, and WT and SIRT1 KO MEFs stably expressing HA-CRABPII, were treated with ethanol or 0.1 μM RA for 6 hours. HA-CRABPII was then immunopurified from cell extracts and the acetylation levels were analyzed with anti-acetyl-lysine polyclonal antibodies (Chemicon). To test whether SIRT1 deacetylates CRABPII in vitro, acetylated HA-CRABPII protein was immuno-purified with anti-HA antibodies from TSA treated SIRT1 KO MEFs, then incubated with 2 units of purified recombinant human SIRT1 protein with or without 200 μM NAD+ or 5 mM Nicotinamide as indicated in the deacetylation buffer for 1 hour at 37 °C.
Microarray Analysis
Total RNAs from sh-Control and sh-SIRT1 mESCs treated with or without RA for 2-day were isolated using the Qiagen RNeasy mini-kit, and gene expression profiles were analyzed using the Agilent Mouse whole genome arrays (Agilent Technologies) following the Agilent 1-color microarray- based gene expression analysis protocol.
Accession Number
The Gene Expression Omnibus accession number for the data reported in this paper is GSE59140.
Statistical analysis
Values are expressed as mean ± standard error of mean (SEM) from at least three independent experiments or biological replicate, unless otherwise indicated in the figure legend. Significant differences between the means were analyzed by the two-tailed, unpaired, non-parametric Mann-Whitney test, and differences were considered significant at p< 0.05.
Supplementary Material
Highlights.
CRABPII is a novel deacetylation substrate of SIRT1.
CRABPII K102 acetylation increases its nuclear accumulation and RAR activation
SIRT1 deficiency increases CRABPII acetylation and enhances RA signaling.
Loss of SIRT1 accelerates RA-induced mESC differentiation in part through CRABPII.
Acknowledgments
We thank Drs. Paul Wade and Raja Jothi, and members of the Li laboratory for critical reading of the manuscript. We would also like to thank Dr. Sue Edelstein from the NIEHS Photography & Graphics Service Center for the cartoon graph of Figure S7A and the Graphic Abstract; Ms Julie Foley from the NIEHS Cellular & Molecular Pathology Branch for histological analyses of mouse embryos; and Mr. C. Jeff Tucker and Dr. Agnes Janoshazi from NIEHS Fluorescence Microscopy and Imaging Center for quantification of confocal images. This research was supported by the Intramural Research Program of National Institute of Environmental Health Sciences of the NIH to X.L. (Z01 ES102205), and by NIH grants to Y. Z. (GM105933 and CA160036). S. T. was supported by a pre-doctoral fellowship from Shanghai Jiao Tong University and by a research grant to G. Huang (New Drug Discovery Project, 2012ZX09506-001-005).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan D, Houle M, Bouchard N, Meyer BI, Gruss P, Lohnes D. RARgamma and Cdx1 interactions in vertebral patterning. Dev Biol. 2001;240:46–60. doi: 10.1006/dbio.2001.0455. [DOI] [PubMed] [Google Scholar]
- Boylan JF, Gudas LJ. Overexpression of the cellular retinoic acid binding protein-I (CRABP-I) results in a reduction in differentiation-specific gene expression in F9 teratocarcinoma cells. J Cell Biol. 1991;112:965–979. doi: 10.1083/jcb.112.5.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan JF, Gudas LJ. The level of CRABP-I expression influences the amounts and types of all-trans-retinoic acid metabolites in F9 teratocarcinoma stem cells. J Biol Chem. 1992;267:21486–21491. [PubMed] [Google Scholar]
- Budhu AS, Noy N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol Cell Biol. 2002;22:2632–2641. doi: 10.1128/MCB.22.8.2632-2641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvanese V, Lara E, Suarez-Alvarez B, Abu Dawud R, Vazquez-Chantada M, Martinez-Chantar ML, Embade N, Lopez-Nieva P, Horrillo A, Hmadcha A, et al. Sirtuin 1 regulation of developmental genes during differentiation of stem cells. Proc Natl Acad Sci U S A. 2010;107:13736–13741. doi: 10.1073/pnas.1001399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- Chen AC, Yu K, Lane MA, Gudas LJ. Homozygous deletion of the CRABPI gene in AB1 embryonic stem cells results in increased CRABPII gene expression and decreased intracellular retinoic acid concentration. Arch Biochem Biophys. 2003;411:159–173. doi: 10.1016/s0003-9861(02)00732-4. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhao W, Yang JS, Cheng Z, Luo H, Lu Z, Tan M, Gu W, Zhao Y. Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol Cell Proteomics. 2012;11:1048–1062. doi: 10.1074/mcp.M112.019547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003 doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia NY, Chan YS, Feng B, Lu X, Orlov YL, Moreau D, Kumar P, Yang L, Jiang J, Lau MS, et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature. 2010;468:316–320. doi: 10.1038/nature09531. [DOI] [PubMed] [Google Scholar]
- Coussens M, Maresh JG, Yanagimachi R, Maeda G, Allsopp R. Sirt1 deficiency attenuates spermatogenesis and germ cell function. PLoS One. 2008;3:e1571. doi: 10.1371/journal.pone.0001571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delva L, Bastie JN, Rochette-Egly C, Kraiba R, Balitrand N, Despouy G, Chambon P, Chomienne C. Physical and functional interactions between cellular retinoic acid binding protein II and the retinoic acid-dependent nuclear complex. Mol Cell Biol. 1999;19:7158–7167. doi: 10.1128/mcb.19.10.7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Ruuska SE, Levinthal DJ, Noy N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J Biol Chem. 1999;274:23695–23698. doi: 10.1074/jbc.274.34.23695. [DOI] [PubMed] [Google Scholar]
- Fawcett D, Pasceri P, Fraser R, Colbert M, Rossant J, Giguere V. Postaxial polydactyly in forelimbs of CRABP-II mutant mice. Development. 1995;121:671–679. doi: 10.1242/dev.121.3.671. [DOI] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- Ghyselinck NB, Wendling O, Messaddeq N, Dierich A, Lampron C, Decimo D, Viville S, Chambon P, Mark M. Contribution of retinoic acid receptor beta isoforms to the formation of the conotruncal septum of the embryonic heart. Dev Biol. 1998;198:303–318. [PubMed] [Google Scholar]
- Grant P, Ahlemeyer B, Karnati S, Berg T, Stelzig I, Nenicu A, Kuchelmeister K, Crane DI, Baumgart-Vogt E. The biogenesis protein PEX14 is an optimal marker for the identification and localization of peroxisomes in different cell types, tissues, and species in morphological studies. Histochem Cell Biol. 2013;140:423–442. doi: 10.1007/s00418-013-1133-6. [DOI] [PubMed] [Google Scholar]
- Grondona JM, Kastner P, Gansmuller A, Decimo D, Chambon P, Mark M. Retinal dysplasia and degeneration in RARbeta2/RARgamma2 compound mutant mice. Development. 1996;122:2173–2188. doi: 10.1242/dev.122.7.2173. [DOI] [PubMed] [Google Scholar]
- Guarente L. Sirtuins, aging, and metabolism. Cold Spring Harb Symp Quant Biol. 2011a;76:81–90. doi: 10.1101/sqb.2011.76.010629. [DOI] [PubMed] [Google Scholar]
- Guarente L. The logic linking protein acetylation and metabolism. Cell Metab. 2011b;14:151–153. doi: 10.1016/j.cmet.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27:2072–2085. doi: 10.1101/gad.227439.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285:13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Garcha K, Karamboulas K, Cowan MF, Drysdale LM, Horton WA, Underhill TM. BMP action in skeletogenesis involves attenuation of retinoid signaling. J Cell Biol. 2006;174:101–113. doi: 10.1083/jcb.200604150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle M, Prinos P, Iulianella A, Bouchard N, Lohnes D. Retinoic acid regulation of Cdx1: an indirect mechanism for retinoids and vertebral specification. Mol Cell Biol. 2000;20:6579–6586. doi: 10.1128/mcb.20.17.6579-6586.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle M, Sylvestre JR, Lohnes D. Retinoic acid regulates a subset of Cdx1 function in vivo. Development. 2003;130:6555–6567. doi: 10.1242/dev.00889. [DOI] [PubMed] [Google Scholar]
- Kang MR, Lee SW, Um E, Kang HT, Hwang ES, Kim EJ, Um SJ. Reciprocal roles of SIRT1 and SKIP in the regulation of RAR activity: implication in the retinoic acid-induced neuronal differentiation of P19 cells. Nucleic Acids Res. 2009;38:822–831. doi: 10.1093/nar/gkp1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner P, Mark M, Leid M, Gansmuller A, Chin W, Grondona JM, Decimo D, Krezel W, Dierich A, Chambon P. Abnormal spermatogenesis in RXR beta mutant mice. Genes Dev. 1996;10:80–92. doi: 10.1101/gad.10.1.80. [DOI] [PubMed] [Google Scholar]
- Li X. SIRT1 and energy metabolism. Acta Biochim Biophys Sin (Shanghai) 2013;45:51–60. doi: 10.1093/abbs/gms108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- MacLean G, Li H, Metzger D, Chambon P, Petkovich M. Apoptotic extinction of germ cells in testes of Cyp26b1 knockout mice. Endocrinology. 2007;148:4560–4567. doi: 10.1210/en.2007-0492. [DOI] [PubMed] [Google Scholar]
- Majumdar A, Petrescu AD, Xiong Y, Noy N. Nuclear translocation of cellular retinoic acid-binding protein II is regulated by retinoic acid-controlled SUMOylation. J Biol Chem. 2011;286:42749–42757. doi: 10.1074/jbc.M111.293464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark M, Ghyselinck NB, Chambon P. Function receptors: of retinoid nuclear lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol. 2006;46:451–480. doi: 10.1146/annurev.pharmtox.46.120604.141156. [DOI] [PubMed] [Google Scholar]
- Mark M, Ghyselinck NB, Chambon P. Function of retinoic acid receptors during embryonic development. Nucl Recept Signal. 2009;7:e002. doi: 10.1621/nrs.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy N. Between death and survival: retinoic acid in regulation of apoptosis. Annu Rev Nutr. 2010;30:201–217. doi: 10.1146/annurev.nutr.28.061807.155509. [DOI] [PubMed] [Google Scholar]
- Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schroter F, Ninnemann O, Siegert E, Bendix I, Brustle O, Nitsch R, et al. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat Cell Biol. 2008;10:385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Brace CS, Ben-Josef G, West T, Wozniak DF, Holtzman DM, Herzog ED, Imai S. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J Neurosci. 2010;30:10220–10232. doi: 10.1523/JNEUROSCI.1385-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, Imai S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18:416–430. doi: 10.1016/j.cmet.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, Sharma AD, Tawney J, Nakagawa M, Okita K, Yamanaka S, Willenbring H, Verdin E. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging (Albany NY) 2010;2:415–431. doi: 10.18632/aging.100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Annals of Medicine. 2011;43:198–211. doi: 10.3109/07853890.2010.547211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessler RJ, Noy N. A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol Cell. 2005;18:343–353. doi: 10.1016/j.molcel.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Soprano DR, Teets BW, Soprano KJ. Role of retinoic acid in the differentiation of embryonal carcinoma and embryonic stem cells. Vitam Horm. 2007;75:69–95. doi: 10.1016/S0083-6729(06)75003-8. [DOI] [PubMed] [Google Scholar]
- Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994;8:1007–1018. doi: 10.1101/gad.8.9.1007. [DOI] [PubMed] [Google Scholar]
- Tsuneyoshi N, Sumi T, Onda H, Nojima H, Nakatsuji N, Suemori H. PRDM14 suppresses expression of differentiation marker genes in human embryonic stem cells. Biochem Biophys Res Commun. 2008;367:899–905. doi: 10.1016/j.bbrc.2007.12.189. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch ID, Cowan MF, Beier F, Underhill TM. The retinoic acid binding protein CRABP2 is increased in murine models of degenerative joint disease. Arthritis Res Ther. 2009;11:R14. doi: 10.1186/ar2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston AD, Hoffman LM, Underhill TM. Revisiting the role of retinoid signaling in skeletal development. Birth Defects Res C Embryo Today. 2003;69:156–173. doi: 10.1002/bdrc.10010. [DOI] [PubMed] [Google Scholar]
- Yu S, Levi L, Siegel R, Noy N. Retinoic acid induces neurogenesis by activating both retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) J Biol Chem. 2012;287:42195–42205. doi: 10.1074/jbc.M112.410381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.