

Abstract
Vascular endothelial growth factor (VEGF) is a potent mediator of angiogenesis and vascular permeability, in which c-Src tyrosine kinase plays an essential role. However, the mechanisms by which VEGF stimulates c-Src activation have remained unclear. Here, we demonstrate that vascular endothelial cadherin (VE-cadherin) plays a critical role in regulating c-Src activation in response to VEGF. In vascular endothelial cells, VE-cadherin was basally associated with c-Src and Csk (C-terminal Src kinase), a negative regulator of Src activation. VEGF stimulated Csk release from VE-cadherin by recruiting the protein tyrosine phosphatase SHP2 to VE-cadherin signaling complex, leading to an increase in c-Src activation. Silencing VE-cadherin with small interference RNA significantly reduced VEGF-stimulated c-Src activation. Disrupting the association of VE-cadherin and Csk through the reconstitution of Csk binding-defective mutant of VE-cadherin also diminished Src activation. Moreover, inhibiting SHP2 by small interference RNA and adenovirus-mediated expression of a catalytically inactive mutant of SHP2 attenuated c-Src activation by blocking the disassociation of Csk from VE-cadherin. Furthermore, VE-cadherin and SHP2 differentially regulates VEGF downstream signaling. The inhibition of c-Src, VE-cadherin, and SHP2 diminished VEGF-mediated activation of Akt and endothelial nitric-oxide synthase. In contrast, inhibiting VE-cadherin and SHP2 enhanced ERK1/2 activation in response to VEGF. These findings reveal a novel role for VE-cadherin in modulating c-Src activation in VEGF signaling, thus providing new insights into the importance of VE-cadherin in VEGF signaling and vascular function.
Angiogenesis, the formation of new blood capillaries, is an important component of embryonic vascular development, wound healing, and organ regeneration as well as pathological processes, such as diabetic retinopathies, atherosclerosis, and tumor growth (1–3). Vascular endothelial growth factor (VEGF)2 is essential for many angiogenic processes both in normal and pathological conditions (3). VEGF also plays a critical role in the regulation of vascular permeability (4). VEGF receptors, VEGFR1 (Flt1) and VEGFR2 (mouse Flk1 or human KDR), are restricted in their tissue distribution primarily to endothelial cells. The binding of VEGF to its cognate receptors induces dimerization and subsequent phosphorylation of the receptors, leading to the activation of several intracellular signaling pathways (5–8). In particular, the angiogenic signals triggered by VEGF are mediated through the activation of phospholipase Cγ(PLCγ), protein kinase C, and subsequently the extracellular signal-regulated kinases 1 and 2 (ERK1/2) (5). VEGF also regulates vascular permeability and endothelial cell migration by stimulating the Src family kinases, in particular c-Src, and the phosphatidylinositol 3′-OH-kinase/Akt pathway-dependent endothelial nitric-oxide synthase (eNOS) activation (6, 8–11). The importance of VEGF-induced signaling is demonstrated in that the genetic inactivation of either receptor leads to a complete lack of development of blood vessels in the embryo, and inactivation of VEGFR2 function dramatically impairs the growth of cancer cells in vivo (12–14). However, the molecular mechanisms underlying the differentiated pathways mediated by VEGF are not well defined.
The cytoplasmic tyrosine kinases of the Src family play pivotal roles in mitogenic responses induced by a variety of growth factors, including VEGF (10). Emerging evidence shows an important role of Src kinases in the regulation of angiogenesis and vascular permeability (4). In particular, mice deficient in c-Src showed no vascular permeability response to VEGF and displayed minimal edema and infarction volume following stroke (15, 16). The Src family of tyrosine kinases are basally inactive due to autoinhibition by intramolecular interactions between its Src homology 2 domain and a phosphotyrosine (Tyr-527 in chickens, Tyr-530 in humans) in the C-terminal region and/or between its Src homology 3 domain and a Prorich sequence in the linker region (17). Phosphorylation of c-Src at Tyr-527 is catalyzed by the C-terminal Src kinase (Csk), a cytosolic protein-tyrosine kinase that is regulated by binding to its docking proteins for co-localization with c-Src to exert its inhibitory effect (17). Recent studies suggest that Csk recruitment to its docking proteins, such as paxillin and Cbp/PAG, is controlled by the protein-tyrosine phosphatase SHP2 (18, 19). SHP2 promotes c-Src activation by regulating the phosphorylation of the Csk regulators PAG/Cbp and paxillin, thereby controlling Csk access to Src kinases (17, 20). However, how Src kinase is activated in response to VEGF and whether there is a specific docking protein for Csk to control Src activation remain to be determined.
Vascular endothelial cadherin (VE-cadherin) is the major adhesive protein of the adherens junction and is specific for vascular endothelial cells (21). Deletion or cytosolic truncation of VE-cadherin impairs remodeling and maturation of the vascular network, and on the cellular level, it abolishes transmission of intracellular signal via VEGFR2 (22). VE-cadherin exerts a negative effect on endothelial cell growth by binding VEGFR2 and inhibiting its mitogenic signaling pathways mediated by ERK1/2 activation (22, 23). In addition, VEGF induces tyrosine phosphorylation of VE-cadherin, which has been proposed as a potential mechanism that regulates the stability of cell-cell junctions and vascular permeability (21). Interestingly, the cytoplasmic domain of VE-cadherin contains nine tyrosine residues that represent potential target sites for tyrosine kinases, which may participate in recruiting the signaling proteins that associate with VE-cadherin (24). In particular, it has recently been reported that Csk binds to the phosphorylated Tyr-685 of VE-cadherin. Therefore, it is likely that VE-cadherin plays a pivotal role in VEGF signaling in endothelial cells. In this study, we found that c-Src activation by VEGF was impaired when VE-cadherin expression in endothelial cells was knocked down by small interference RNA (siRNA) and when the Csk binding site in VE-cadherin was mutated. When tyrosine-phosphorylated, VE-cadherin recruited SHP2, which served to disrupt VE-cadherin-Csk complex formation, resulting in the activation of c-Src. Importantly, we also showed that the VE-cadherin/ SHP2/Src signaling module was required for VEGF-mediated Akt and eNOS but not ERK1/2 activation.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
VEGF was from R&D Systems, Inc. (Minneapolis, MN). U73122, LY294002, wortmannin, and 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo{3,4-d}pyrimidine (PP2) were from Calbiochem. Polyclonal antibodies for phospho-Src (Tyr-527), phospho-eNOS (Ser-1179 in bovine eNOS sequence), anti-phospho-Akt (Ser-473), phospho-ERK1/2 (Thr-202/Tyr-204), and Akt antibodies were from Cell Signaling Technologies. Tyrosine phosphorylation antibody 4G10 was from Upstate Biotechnology, Inc. Mouse monoclonal antibodies for eNOS and VE-cadherin were from BD Transduction Laboratories. Polyclonal antibody for phospho-Src (Tyr-416) was from BIOSOURCE. Antibodies for SHP2, c-Src, ERK1, ERK2, Csk, and β-actin were from Santa Cruz Biotechnology.
Cell Culture
Bovine aortic endothelial cells (BAECs) were purchased from Clonetics (San Diego, CA) and were cultured in medium 199 supplemented with 10% fetal bovine serum (Invitrogen) as described previously (25). Human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical veins and grown in medium 200 with LSGS (Cascade Biologics, Inc.), as described previously (26, 27). Confluent cells cultured in 60-mm dishes were serum-starved for 24 h and exposed to VEGF as indicated. For the inhibitor studies, cells were pretreated with various inhibitors for 30 min in serum-depleted medium.
siRNA and Transfection
To determine the contribution of VE-cadherin and SHP2 in VEGF-stimulated signaling, we treated HUVECs with human VE-cadherin, SHP2, and c-Src siRNA duplex obtained from Dharmacon, Inc. The following siRNAs were used: VE-cadherin siRNA, sense (5′-GGAACCA-GAUGCACAUUGAUU) and antisense (5′-PUCAAUGUGC-AUCUGGUUCCUU); SHP2 siRNA, sense (5′-GAACAUCAC-GGGCAAUUAAUU) and antisense (5′-PUUAAUUGCCCG-UGAUGUUCUU); c-Src siRNA, sense (5′-GAGAACCUGGU-GUGCAAAGUU) and antisense (5′-PCUUUGCACACCAG-GUUCUCUU). The scrambled siRNA control is a nontargeting siRNA pool from Dharmacon, Inc. For transfection of siRNA, HUVECs were seeded into 60-mm dishes for 24 h at about 80% confluence, and then transfection of siRNA was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol, as described previously (27). VEGF stimulation was performed 48 h after siRNA transfection.
Adenovirus Constructs and Infection
Adenoviruses encoding mouse VE-cadherin wild type (WT) and tyrosine 685 replacement mutant (Y685F) were generated from pcDNA3-VE-cadherin-WT and pcDNA3-VE-cadherin-Y685F, which were kindly provided by Dr. Dietmar Vestweber (University of Münster, Münster, Germany) (24), using the ViraPower Adenoviral Expression System (Invitrogen) according to the manufacturer’s protocol. Adenoviruses encoding the catalytically inactive SHP2 mutant with phosphatase domain deletion (Ad-SHP2ΔPTP) and the kinase-inactive c-Src mutant (c-Src-DN) have been described previously (28, 29). Adenoviruses containing β-galactosidase (Ad-LacZ) was used as a control. The infection of endothelial cells with recombinant adenovirus was performed as described previously (30). Briefly, endothelial cells cultured in 60-mm dishes were infected with adenoviruses at the indicated multiplicity of infection (MOI) for 24 h in a growth medium and then treated with VEGF.
Immunoprecipitation and Western Blot Analysis
Cells were harvested in lysis buffer, and the immunoprecipitations were performed according to standard protocols, as described previously (25, 27). The immune complex samples or total cell lysates were resolved on SDS-PAGE according to standard protocols (25, 27). After being transferred to membranes, the samples were immunoblotted with primary antibodies, followed by IRDye infrared secondary antibodies (LI-COR Biosciences). Bands for immunoreactive proteins were visualized by an Odyssey infrared imaging system, and density was quantified by the use of Odyssey software (LI-COR Biotechnology). Results were normalized by arbitrarily setting the densitometry of the control sample to 1.0.
Immunofluorescence
For analysis of the VE-cadherin, Csk, and phospho-Src Tyr-416 localization in monolayer endothelial cells, BAECs or HUVECs were fixed with 3.7% formaldehyde in phosphate-buffered saline after the treatment of VEGF (31). Immunocytochemistry were performed according to a standard protocol, and the images were captured at a magnification of ×60, using a fluorescence microscope (Olympus BX51).
Statistical Analysis
All values are expressed as means ±S.E. The significance of the results was assessed by a paired t test. A p value of <0.05 was considered significant.
RESULTS
VE-cadherin Modulates c-Src Activation in Response to VEGF
To ask whether VEGF-evoked c-Src activation requires VE-cadherin function, we studied endothelial cells in which VE-cadherin expression was silenced acutely by siRNA. Because our siRNAs specifically target on human proteins, all experiments related to siRNA were performed in HUVECs. Freshly isolated HUVECs were transfected with siRNA against human VE-cadherin or with a scrambled nontargeting siRNA control. 48 h after transfection, the cells were stimulated with 20 ng/ml VEGF for 0, 5, 10, and 30 min. Immunoblots revealed a >90% decrease of VE-cadherin expression in VE-cadherin siRNA-transfected cells. VE-cadherin siRNA is specific for targeting VE-cadherin, since expression of β-actin, Akt, and eNOS was unchanged (Fig. 1A). c-Src activation was determined by the status of its autophosphorylation at the active site Tyr-416 residue using specific phospho-Src antibodies. VEGF stimulated increased Src Tyr-416 phosphorylation in control siRNA-treated cells in a time-dependent manner, which is similar to untreated cells (not shown). In contrast, VEGF-evoked c-Src Tyr-416 phosphorylation was decreased markedly in VE-cadherin siRNA-treated cells (Fig. 1, A and B). In addition, silencing VE-cadherin did not significantly affect overall VEGFR2 tyrosine phosphorylation in response to VEGF (Fig. 1C). These results imply that VE-cadherin plays a critical role in c-Src activation by VEGF in ECs.
FIGURE 1. VE-cadherin siRNA inhibited VEGF-induced c-Src activation in endothelial cells.
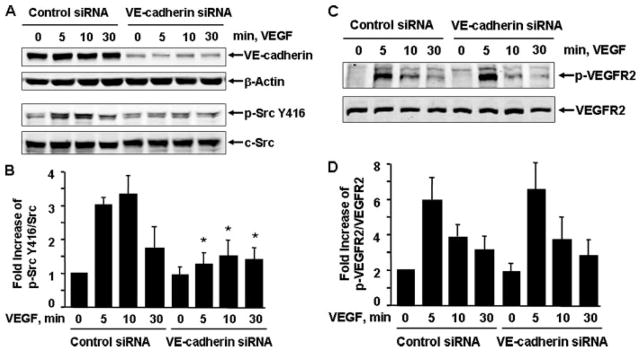
HUVECs were pre-treated with control siRNA or human VE-cadherin siRNA (100 nM) for 48 h and then exposed to VEGF (20 ng/ml) for various times, as indicated. c-Src Tyr-416 phosphorylation in cell lysates was analyzed by Western blot analysis using phosphospecific antibodies (A and B), which recognize c-Src phosphorylated at Tyr-416 (p-Src Y416), and phosphospecific VEGFR2 (p-VEGFR2) (C and D). Total levels of VE-cadherin, β-actin, c-Src, and VEGFR2 were determined by Western blot using their antibodies. Representative immunoblots are shown (A). Quantitative data of Src Tyr-416 phosphorylation and VEGFR2 tyrosine phosphorylation are shown (B) (n = 4).
VE-cadherin Differentially Modulates Akt and ERK1/2 Activation by VEGF
To further determine the role of VE-cadherin in VEGF signaling, we examined the effects of silencing VE-cadherin by siRNA on VEGF-induced activation of ERK1/2, Akt, and eNOS (a substrate of Akt (32)), which have been shown to play pivotal roles in endothelial cell proliferation, survival, migration, and permeability (33). HUVECs were transfected with control or VE-cadherin siRNA; 48 h after transfection, the cells were stimulated with 20 ng/ml VEGF at various times as indicated. Cell lysates were analyzed by immunoblotting with phosphospecific antibodies that recognize their phosphorylation at active sites of Akt Ser-473 (p-Akt), eNOS Ser-1177 (p-eNOS), and ERK1/2 Thr-202/Tyr-204 (p-ERK1/2), respectively. As shown in Fig. 2A, a marked increase in both Akt and eNOS phosphorylation was observed after treatment with VEGF. Cells treated with VE-cadehrin siRNA showed a marked decrease in Akt and eNOS phosphorylation relative to control cells. There was no change in the expression of total Akt and eNOS under any of these conditions (Fig. 2A). Most intriguingly, in contrast to the reduced Akt and eNOS phosphorylation by VE-cadherin siRNA, there was an increase of VEGF-induced ERK1/2 phosphorylation in VE-cadherin siRNA-treated HUVECs compared with control cells (Fig. 2, C and D). Our results suggest that VE-cadherin differentially modulates VEGF signal transduction in endothelial cells.
FIGURE 2. VE-cadherin siRNA inhibited VEGF-induced Akt and eNOS activation but promoted ERK1/2 activation.
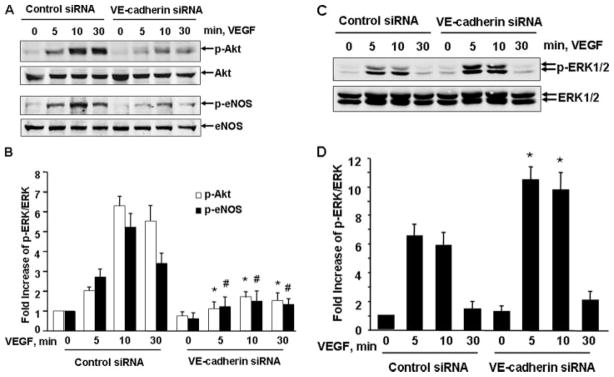
HUVECs were pretreated with control siRNA or human VE-cadherin siRNA (100 nM) for 48 h and then exposed to VEGF (20 ng/ml) for various times, as indicated. The phosphorylation of Akt (Ser-473), eNOS (Ser-1177), and ERK1/2 in cell lysates was determined by Western blot analysis using their phospho-specific antibodies as described under “Experimental Procedures.” Total levels of Akt, eNOS, and ERK1/2 were determined by Western blot using their antibodies. Representative immunoblots (A and C) and quantitative data of protein phosphorylation are shown (B and D) (n = 4).
Notably, the role of VE-cadherin in the regulation of VEGF-stimulated c-Src activation and downstream signaling to Akt, eNOS, and ERK1/2 is specific, because siRNA silencing of platelet-endothelial cell adhesion molecule, another paracellular transmembrane adhesion molecule present in regions of cell-cell contact, had no significant effects on VEGF-mediated Akt, eNOS, and ERK1/2 activation (data not shown).
c-Src Kinase Mediates Activation of Akt and eNOS in Response to VEGF
Src kinases play an import role in signal transduction of various tyrosine kinase receptors. Although it has been suggested that c-Src kinase is involved in VEGF signaling and angiogenesis in endothelial cells, evidence linking Src to Akt/eNOS and ERK1/2 pathways remains to be firmly established (10). Here we exploited the Src kinase-selective inhibitor PP2 to examine the role of c-Src in VEGF signaling. BAECs were pretreated with or without Src kinase inhibitor PP2 at 10 μM for 30 min and then stimulated with 20 ng/ml VEGF for 10 min. The treatment of PP2 inhibited VEGF-induced Akt and eNOS activation. In contrast, inhibiting Src kinases by PP2 enhanced ERK1/2 activation (Fig. 3A). Similar results were observed in HUVECs treated with PP2 (data not shown). Our data suggest differential roles of c-Src in VEGF-mediated Akt/eNOS and ERK1/2 pathways.
FIGURE 3. Src kinase is required for mediating VEGF-induced Akt and eNOS activation.
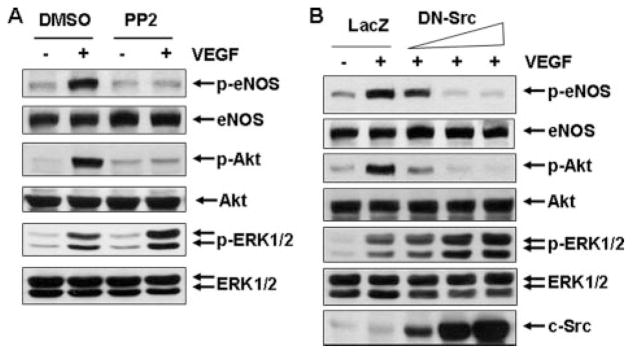
A, BAECs were pretreated with Src kinase inhibitor PP2 (10 μM) and then stimulated with VEGF (20 ng/ml) for 10 min. DMSO, Me2SO. B, HUVECs were infected with adenovirus encoding LacZ (control, 300 MOI) and adenovirus encoding c-Src-DN at the different doses (30, 100, and 300 MOI) for 24 h and then exposed to VEGF for 10 min. The phosphorylation of Akt, eNOS, and ERK1/2 in cell lysates was determined by Western blot analysis as described under “Experimental Procedures.” Total levels of Akt, eNOS, ERK1/2, and c-Src were determined by Western blot using their antibodies. Representative immunoblots were shown (n = 3).
To further determine the role of c-Src in VEGF signaling, we examined the effects of an adenovirus encoding for a kinase-inactive c-Src on VEGF signaling. The dominant negative function of this kinase-inactive c-Src mutant (c-Src-DN) has been characterized previously (29). Infection of c-Src-DN at 30, 100, and 300 MOI in HUVECs increased c-Src expression in a dose-dependent manner (Fig. 3B). Consistent with the PP2 results, overexpression of c-Src-DN dose-dependently inhibited Akt and eNOS activation in response to VEGF. However, VEGF-induced ERK1/2 activation was increased by overexpression of a c-Src-DN in HUVECs. Thus, our findings imply that c-Src has a positive role in VEGF-induced activation of Akt and eNOS but a negative role in ERK1/2 activation.
SHP2 Is Essential for Mediating VE-cadherin to c-Src Activation in Response to VEGF
To define the molecular mechanisms by which VE-cadherin modulates VEGF activation of c-Src, we studied the potential signal molecules associated with VE-cadherin, focusing on SHP2 because of its involvement in Src activation in other systems (17). BAECs were treated with VEGF for 0, 5, 10, and 30 min, and cell lysates were subjected to immunoprecipitation with VE-cadherin antibody. As shown in Fig. 4A, the association of SHP2 and VE-cadherin was increased by VEGF stimulation in a time-dependent manner. We also analyzed the tyrosine phosphorylation of SHP2. We found that VE-cadherin-associated SHP2 was tyrosine-phosphorylated in response to VEGF, which was detected by immmunoblotting with anti-phosphotyrosine antibodies (4G10) after VE-cadherin immunoprecipitation (Fig. 4A). These results suggest that SHP2 associates with VE-cadherin in endothelial cells in response to VEGF.
FIGURE 4. SHP2 is essential for mediating VEGF-induced c-Src activation and its downstream signaling.

A, BAECs were stimulated with VEGF (20 ng/ml) for the indicated times. The cell lysates were subjected to immunoprecipitation (IP) with VE-cadherin antibodies or mouse IgG as a control followed by immunoblotting with 4G10, SHP2, and VE-cadherin. Representative immunoblots were shown (n = 3). B–D, HUVECs were pretreated with control siRNA and human SHP2 siRNA (150 nM) for 48 h and then exposed to VEGF for 10 min. The phosphorylation of Src Tyr-416, Akt, eNOS, and ERK1/2 in cell lysates was determined by Western blot analysis as described under “Experimental Procedures.” Total levels of SHP2, β-actin, c-Src, Akt, eNOS, and ERK1/2 were determined by Western blot using their antibodies. Representative immunoblots (B and C) and quantitative data of protein phosphorylation were shown (D) (n = 4).
To determine the role of SHP2 in VEGF-evoked Src activation in endothelial cells, HUVECs were transfected with control siRNA or human SHP2 siRNA and then stimulated with VEGF for 10 min. SHP2 siRNA specifically knocked down expression of SHP2 in endothelial cells, but not β-actin, compared with control siRNA-transfected HUVECs (Fig. 4B). VEGF-evoked c-Src Tyr-416 phosphorylation was decreased markedly in SHP2 knockdown cells compared with control cells (Fig. 4B). Thus, our data indicate that in VEGF signaling, SHP2 promotes c-Src activation. Consistent with the critical role of c-Src in VEGF signal transduction, silencing SHP2 by siRNA also inhibited Akt and eNOS activation. In contrast, VEGF-induced ERK1/2 activation was enhanced (Fig. 4C). These results suggest that VE-cadherin, SHP2, and c-Src may lie on similar pathways in VEGF signaling.
Next, we sought to determine whether the catalytic activity of SHP2 is required for VEGF-induced c-Src activation by infecting endothelial cells with either adenoviruses encoding β-galactosidase (Ad-LacZ, as a control) or adenoviruses expressing a PTP catalytic domain deletion mutant of SHP2 (Ad-SHP2ΔPTP) (28). The dominant negative effect of this mutant has been demonstrated previously (28). SHP2ΔPTP was overexpressed in cells, as confirmed by immunoblotting for the expression of the truncated SHP2 mutant (Fig. 5A). We observed that VEGF appropriately stimulated Src Tyr-416 phosphorylation in Ad-LacZ-infected BAECs (Fig. 5A). In contrast, Src Tyr-416 phosphorylation by VEGF is dramatically inhibited in cells infected with Ad-SHP2ΔPTP (Fig. 5, A and B). Concomitantly, VEGF-induced activation of Akt and eNOS were impaired in the cells infected with Ad-SHP2ΔPTP, whereas VEGF-induced ERK1/2 activation was enhanced by Ad-SHP2ΔPTP expression (Fig. 5C). Similar data were obtained in HUVECs infected with Ad-SHP2ΔPTP (not shown). These data revealed that the catalytic activity of SHP-2 is required for VEGF-induced c-Src activation and its downstream signaling to Akt and eNOS in endothelial cells.
FIGURE 5. SHP2 activity is involved in mediating VEGF-induced c-Src activation and its downstream signaling.

BAECs were infected with Ad-LacZ (control) or Ad-SHP2ΔPTP for 24 h and then exposed to VEGF (20 ng/ml) for 10 min. The phosphorylation of Src Tyr-416, Akt, eNOS, and ERK1/2 in cell lysates was determined by Western blot analysis as described under “Experimental Procedures.” Total levels of SHP2ΔPTP, c-Src, Akt, eNOS, and ERK1/2 were determined by Western blot using their antibodies. Representative immunoblots (A and C) and quantitative data of protein phosphorylation were shown (B) (n = 5).
SHP2 Regulates VEGF-induced Src Activation by Negatively Controlling Csk Recruitment to VE-cadherin
Csk is a negative regulator of c-Src activation (17). To ask whether Csk is involved in the regulation of c-Src activation via VE-cadherin and SHP2 by VEGF, we examined the potential interaction of Csk and VE-cadherin in response to VEGF with or without functional SHP2. BAECs were infected with Ad-LacZ or Ad-SHP2ΔPTP and stimulated with VEGF for 10 min. The cell lysates were subjected to immunoprecipitation with anti-VE-cadherin antibody or control mouse IgG followed by immunoblotting with anti-Csk antibody. Csk was found to be constitutively associated with VE-cadherin (Fig. 6A), consistent with the relatively low level of c-Src activation under basal conditions (Fig. 6, A and B). Intriguingly, VEGF substantially reduced the amount of Csk that was associated with VE-cadherin. Infection of Ad-SHP2ΔPTP blocked such a decrease of VE-cadherin-associated Csk (Fig. 6, A and B). Of note, the amount of c-Src associated with VE-cadherin was unaltered regardless of either VEGF stimulation or Ad-SHP2ΔPTP infection (Fig. 6A). In addition, Ad-SHP2ΔPTP infection did not change the interaction of VE-cadherin and VEGFR2 in endothelial cells in response to VEGF (Fig. 6C). No Csk, Src, and VE-cadherin were immunoprecipitated when control mouse IgG was used, showing the specificity of protein-protein interactions by co-immunoprecipitation and Western blotting. Taken together, these results demonstrate that SHP2 activity plays a critical role in the regulation of VE-cadherin and Csk dynamic interactions and c-Src activation in response to VEGF.
FIGURE 6. SHP2 regulates c-Src activation by regulating the dissociation of Csk from VE-cadherin in response to VEGF.
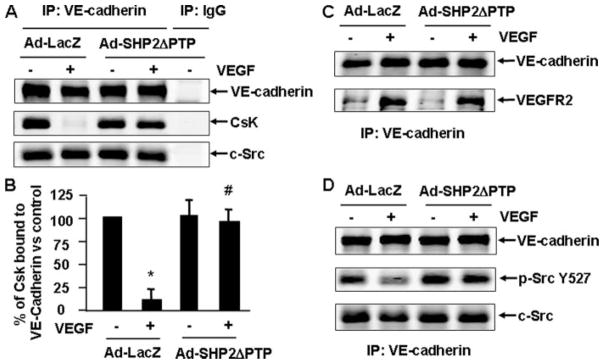
BAECs were infected with Ad-LacZ (control) or Ad-SHP2ΔPTP for 24 h and then exposed to VEGF (20 ng/ml) for 10 min. The cell lysates were subjected to immunoprecipitation (IP) with VE-cadherin antibodies or mouse IgG as a control followed with immunoblotting with Csk (A), VE-cadherin (A–D), c-Src (A and D), VEGFR2 (C), and phospho-Src Tyr-527 (D). Representative immunoblots (A, C, and D) and quantitative data for the association of Csk and VE-cadherin were shown (B) (n = 6).
VEGF Reduces Phosphorylation of Tyr-527 in VE-cadehrin-associated c-Src
Csk-catalyzed phosphorylation of Tyr-527 on c-Src is known to negatively regulate c-Src activity. To determine whether VEGF reduces c-Src Tyr-527 phosphorylation, we analyzed c-Src Tyr-527 phosphorylation in VEGF-treated endothelial cell lysates by immunoblotting with an antibody specifically to phosphorylated c-Src Tyr-527. A VEGF-induced change in c-Src Tyr-527 phosphorylation was not detectable in total cell lysates (data not shown), indicating that VEGF either did not induce Tyr-527 dephosphorylation or that the change was too small to be detected in the total c-Src population. Next we asked whether VE-cadherin-associated c-Src was dephosphorylated at Src Tyr-527 in response to VEGF and, if so, whether SHP2 is involved. ECs were treated with or without VEGF after infection of Ad-LacZ and Ad-SHP2ΔPTP, and the cell lysates were immunoprecipitated with VE-cadherin antibody followed by immunoblotting with phospho-Src Tyr-527 antibody. Interestingly, VEGF-induced c-Src Tyr-527 dephosphorylation was detected in VE-cadherin immunoprecipitates, and this change in c-Src Tyr-527 phosphorylation was greatly inhibited by overexpression of Ad-SHP2ΔPTP (Fig. 6D). These results indicate that the VE-cadherin-bound c-Src represents a major fraction of c-Src where Tyr-527 dephosphorylation occurs in VEGF-stimulated cells. Moreover, SHP2-mediated the disassociation of Csk from VE-cadherin is essential for the reduction of c-Src Tyr-527 phosphorylation associated with VE-cadherin in VEGF-stimulated cells.
SHP2 Modulates Csk and Activated Src in the Areas of Cell-Cell Contacts
To further investigate the turnover of VE-cadherin and Csk association in response to VEGF and the essential role of SHP2 in this process, the localization of VE-cadherin and Csk was examined in the EC monolayer by immunofluorescence microscopy in HUVECs infected with Ad-LacZ and Ad-SHP2ΔPTP in either the presence or absence of VEGF. In all conditions, VE-cadherin was localized to the areas of cell-cell contacts (Fig. 7, middle). In Ad-LacZ-infected cells without VEGF treatment, there was some Csk localized in the areas of cell-cell contacts (Fig. 7, right). After VEGF treatment for 10 min, very little Csk was found in the areas of cell-cell contacts, but VE-cadherin remained at the areas of cell-cell contacts (Fig. 7). In contrast, in HUVECs infected with Ad-SHP2ΔPTP, we observed an accumulation of Csk in the areas of cell-cell contacts regardless of VEGF treatment, suggesting that the inhibition of SHP2 activity blocked the dislocation of Csk from the areas of cell-cell contacts. Similar data were observed when BAECs were used for these experiments (data not shown). Taken together, our findings suggest that SHP2 plays an important role in modulating Csk localization in the areas of cell-cell contacts in response to VEGF. However, in the absence of VEGF stimulation, there is no apparent difference in Csk accumulation at cell-cell contacts in control versus Ad-SHP2delPTP-infected cells, which is consistent with the immunoprecipitation results (Fig. 6A) that show that a similar amount of Csk was associated with VE-cadherin in control versus Ad-SHP2delPTP-infected cells.
FIGURE 7. SHP2 regulates Csk dissociation from VE-cadherin in living endothelial cells in response to VEGF.

HUVECs were infected with Ad-LacZ (control) or Ad-SHP2ΔPTP for 24 h and then exposed to VEGF (20 ng/ml) for 10 min. The cells were fixed, and Csk and VE-cadherin distribution in cells was analyzed by immunocytochemistry with Csk and VE-cadherin antibodies and by fluorescence microscopy as described under “Experimental Procedures.” Representative images (magnification, ×60) were from four independent experiments.
To demonstrate c-Src activation and its localization in ECs, phosphospecific Src Tyr-416 antibodies were used to identify activated c-Src in HUVEC monolayer. In response to VEGF, the signal of c-Src Tyr-416 phosphorylation was observed in the areas of cell-cell contacts (Fig. 8). However, the amount of phosphorylated c-Src Tyr-416 in the areas of cell-cell contacts was greatly decreased by treating the cells with Ad-SHP2ΔPTP (Fig. 8), indicating that catalytic activity of SHP2 is required for c-Src activation in the areas of cell-cell contacts. Similar results were obtained when endogenous SHP2 was knocked down by siRNA (data not shown). We noted that there is a strong signal of phosphorylated c-Src Tyr-416 in the nucleus, which may be related to nuclear c-Src activation because there is some evidence that c-Src is localized to the nucleus and is active (34). Combined with biochemistry analysis (Figs. 5 and 6), these results suggest that the loss of Csk association with VE-cadherin is mediated by SHP2, which leads to c-Src activation in the areas of cell-cell contacts in ECs in response to VEGF.
FIGURE 8. SHP2 regulates VEGF-induced c-Src activation at the areas of cell-cell contacts in living endothelial cells.

HUVECs were infected with Ad-LacZ (control) or Ad-SHP2ΔPTP for 24 h and then exposed to VEGF (20 ng/ml) for 10 min. The cells were fixed and c-Src Tyr-416 phosphorylation and VE-cadherin distribution in cells was analyzed by immunocytochemistry with phospho-specific c-Src Tyr-416 antibody and VE-cadherin antibody and by fluorescence microscopy as described under “Experimental Procedures.” Representative images (magnification, ×60) were from four independent experiments.
Csk Binding Site Tyr-685 in VE-cadherin Is Critical for Mediating c-Src Activation in Response to VEGF
It has recently been reported that Csk binds to the phosphorylated tyrosine 685 of VE-cadherin (24). To further determine the molecular mechanisms underlying the involvement of VE-cadherin/SHP2 in VEGF-stimulated c-Src activation and downstream signaling, we generated adenoviruses encoding mouse VE-cadherin-WT and Y685F (24). The dosages of these adenoviruses were tested in HUVECs. 200 MOI of adenoviruses encoding VE-cadherin-WT and Y685F produce an equivalent level of expression compared with endogenous human VE-cadherin (Fig. 9, A and B). As noted, increasing expression of exogenous VE-cadherin also decreased the level of endogenous VE-cadherin, suggesting that VE-cadherin expression in endothelial cells may be tightly controlled. Since human VE-cadherin siRNA does not target on mouse VE-cadherin, we reconstituted mouse VE-cadherin-WT and Y685F with adenovirus infection to an equivalent level of endogenous VE-cadherin in HUVECs after endogenous VE-cadherin was silenced by siRNA (Fig. 9C). Csk was associated with mouse VE-cadherin-WT but with Y685F in the absence of VEGF treatment (Fig. 9D), which agreed with the previous report (24). Consistent with the results shown in Fig. 6A, VEGF stimulated Csk dissociation with mouse VE-cadherin-WT. Interestingly, VE-cadherin-WT, but not Y685F mutant, mediated VEGF-stimulated c-Src activation (Fig. 9, E and F). Compared with that in VE-cadherin-WT reconstituted cells, VEGF-induced Akt and eNOS activation was also impaired in VE-cadherin-Y685F reconstituted cells, whereas ERK1/2 activation was slightly enhanced in these cells (Fig. 9, G and H). These results further imply that Csk and VE-cadherin association is critical for VE-cadehrin/SHP2 to modulate Src activation and downstream signaling in response to VEGF.
FIGURE 9. Csk binding site Tyr-685 in VE-cadherin is critical for mediating c-Src activation in response to VEGF.

A and B, HUVECs were infected with adenoviruses encoding mouse VE-cadherin-WT or Y685F at different doses. The endogenous human VE-cadherin and exogenous mouse VE-cadherin were analyzed by immunoblotting. C–H, HUVECs were transfected with VE-cadherin siRNA and then reconstituted with adenoviruses carrying VE-cadherin-WT or Y685F (200 MOI), followed by the exposure of the cells to VEGF (20 ng/ml) for 10 min. The cell lysates were analyzed by immunoblotting with VE-cadherin and eNOS (C) or subjected to immunoprecipitation (IP) with VE-cadherin antibodies or mouse IgG as a control, followed by immunoblotting with Csk and VE-cadherin (D), or analyzed by immunoblotting with the antibodies of phospho-Src Tyr-416 and c-Src (E and F), with the antibodies of phospho-Akt, phospho-eNOS (G), and phospho-ERK1/2 (H). Representative immunoblots and quantitative data for c-Src activation (F) were shown (n = 3).
DISCUSSION
The major findings of the present study are that VE-cadherin mediates VEGF-induced c-Src activation through SHP2 to control Csk recruitment, resulting in the differential regulation of Akt/eNOS and ERK pathways (Fig. 10). We found that VEGF stimulated Csk dissociation with VE-cadherin, resulting in the activation of c-Src. SHP2 regulates c-Src activation through the control of Csk/VE-cadherin dynamic interactions in response to VEGF. Furthermore, we found that VE-cadherin- and SHP2-dependent activation of c-Src is important for VEGF signal transduction, which positively regulates Akt and eNOS activity but negatively regulates ERK1/2 activity (Fig. 10). Therefore, our results demonstrate a novel role of VE-cadherin in modulating c-Src activation and downstream signaling of VEGF.
FIGURE 10. Model for a role of VE-cadherin for VEGF-induced c-Src activation and downstream signaling.

In resting cells, Csk binds to VE-cadherin through constitutive phosphotyrosine Tyr-685-dependent interaction. c-Src is also associated with VE-cadherin. Such proximate localization of Csk and c-Src in cell adherens junction areas allows Csk to effectively phosphorylate c-Src Tyr-527 and inhibit c-Src activation. When VEGFR2 is activated by VEGF, the VEGF receptor tyrosine kinase phosphorylates VE-cadherin, which permits VE-cadherin to recruit SHP2. The VE-cadherin-bound SHP2 induces release of Csk from VE-cadherin through potentially dephosphorylating VE-cadherin on Csk binding site Tyr-685. Consequently, the c-Src Tyr-527 phosphorylation level decreases, and the c-Src Tyr-416 phosphorylation level increases. The VE-cadherin/SHP2/c-Src module mediates the VEGF-induced phosphatidylinositol 3′-OH-kinase-dependent Akt/eNOS pathway but negatively regulates PLCγ/protein kinase C-dependent ERK activation.
VE-cadherin is a key adherens junction molecule, and it functions to maintain adherens junction stability and to suppress apoptosis of endothelial cells by forming a complex with VEGFR2 and members of the armadillo family of cytoplasmic proteins, including β-catenin, β-catenin, and p120-catenin (21). However, the mechanisms by which VE-cadherin regulates VEGF signaling via VEGFR2 have not been fully understood. In this study, we found that VE-cadherin modulates VEGF-induced c-Src and Akt/eNOS activation. Specifically, we showed that disrupting VE-cadherin function by siRNA knockdown inhibited c-Src Tyr-416 phosphorylation in ECs in response to VEGF. Furthermore, VEGF-mediated Akt and eNOS activation were reduced by siRNA silencing of VE-cadherin expression and by inhibiting c-Src activation with the pharmacological inhibitor PP2 and adenovirus c-Src-DN. Together, these results suggest a critical role of VE-cadherin in regulation of c-Src activation and Akt/eNOS activation in response to VEGF.
Following VEGF stimulation, phosphorylation of the c-Src activation site (Tyr-416) increases. Normally, this is accompanied by decreased phosphorylation of the c-Src inactivation site (Tyr-527); however, this residue was hyperphosphorylated basally, and we were unable to detect Tyr-527 dephosphorylation in VEGF-stimulated cells. Because Tyr-527 dephosphorylation results in c-Src activation, even a low level of dephospho-Tyr-527 should be physiologically relevant (17). Indeed, Tyr-527 dephosphorylation of c-Src was detected in a fraction of c-Src that associated with the VE-cadherin complex in response to VEGF stimulation (Fig. 6D). Moreover, a significant amount of c-Src Tyr-416 phosphorylation was observed in the areas of cell-cell contacts where VE-cadherin is located in the endothelial cell monolayer in response to VEGF (Fig. 8). These data suggest that c-Src-associated VE-cadherin complex represents an important fraction of c-Src activation in response to VEGF.
Phosphorylation of c-Src Tyr-527 is catalyzed by Csk, which is a cytosolic protein-tyrosine kinase that is regulated by binding to its docking proteins for co-localization with Src to exert its inhibitory effect (17). Csk binds to paxillin, resulting in c-Src inactivation in focal adhesions in fibroblast and epithelial cells (20). In this study, we found that Csk was associated with VE-cadherin basally in HUVECs and BAECs, which is consistent with a recent report showing that both Csk and VE-cadherin can be co-precipitated from cell lysates of transfected COS7 cells and endothelial cell line bEND.3 cells (24). Importantly, our results suggest that Csk was involved in VEGF-induced c-Src activation. In particular, VEGF stimulated a marked reduction in the amount of Csk bound to VE-cadherin. This correlated with a decrease in c-Src Tyr-527 phosphorylation and an increase of c-Src Tyr-416 phosphorylation in endothelial cells. The dynamic interactions of Csk with VE-cadherin were also observed in the monolayer of endothelial cells in response to VEGF. The studies with the reconstitution of mouse VE-cadherin-WT and Y685F in human VE-cadherin siRNA-treated cells further demonstrate that the interaction of Csk and VE-cadherin is critical for c-Src activation in response to VEGF.
Moreover, we showed that SHP2 regulates of c-Src activation through control of the Csk/VE-cadherin dynamic interactions in response to VEGF. siRNA knockdown of SHP2 expression in endothelial cells inhibited VEGF-induced c-Src Tyr-416 phosphorylation, and infection of Ad-SHP2ΔPTP, which encodes a catalytically inactive SHP2 mutant, attenuated c-Src Tyr-416 phosphorylation by VEGF. In agreement with our results, SHP2 has been shown to mediate Src activation in epithelial growth factor signaling in fibroblasts (17) and epithelial cells (20). Inhibiting SHP2 function with either SHP2 siRNA or Ad-SHP2ΔPTP also blocked VEGF-mediated Akt/eNOS activation, suggesting that VE-cadherin, SHP-2, and c-Src act in the same pathway during VEGF signaling. Although the exact mechanisms involved in regulation of c-Src activation by SHP2 are not clear, our studies with VE-cadherin-WT and Y685F imply that SHP2 dissociates Csk from VE-cadherin through dephosphorylation of VE-cadherin at the Tyr-685 site to remove the inhibition of Csk on c-Src in the complex. In keeping with this notion, both Csk and c-Src bind to VE-cadherin in endothelial cells in the basal condition. Simultaneous binding of c-Src and Csk to VE-cadherin could constitute a mechanism for Src kinase inhibition (17). We also found that VEGF stimulated the dissociation of Csk but not c-Src from VE-cadherin, concomitant with an increase of c-Src Tyr-416 phosphorylation. The inhibition of SHP2 function by overexpression of the SHP2ΔPTP mutant prevented VEGF-induced dissociation of Csk from VE-cadherin and VE-cadherin-associated c-Src Tyr-527 dephosphorylation. Taken together, these observations support the model in which SHP2 indirectly activates c-Src through targeting on VE-cadherin and Csk. Although it is clear for the direct interaction of Csk and VE-cadherin, the nature of SHP2 association with VE-cadherin is unknown. It is possible that SHP2 may directly interact with VE-cadherin or alternatively bind to VE-cadherin-associated protein(s), such as β-catenin and p120-catenin. It has been reported that VEGF stimulates tyrosine phosphorylation of VE-cadherin, β-catenin, and p120-catenins in endothelial cells (35), and ligand-binding blots using a SHP2-glutathione S-transferase fusion peptide in vitro established that SHP2 associates selectively with β-catenin but not p120-catenin in VE-cadherin complexes (36). Further studies are needed to determine molecular basis underlying the involvement of SHP2 in VE-cadherin signaling complex.
In contrast to the crucial role of VE-cadherin and SHP2 in positive regulation of phosphatidylinositol 3′-OH-kinase-dependent Akt and eNOS activation in VEGF signaling through modulating c-Src kinase activity, we found that both VE-cadherin and SHP2 negatively regulate ERK1/2 activation in endothelial cells in response to VEGF. It has been demonstrated that VEGF-mediated ERK1/2 activation is PLCγ/protein kinase C-dependent via VEGFR2 activation, which plays a key role in VEGF-induced endothelial cell proliferation and angiogenesis (5, 7, 37). In this study, we found that knockdown of VE-cadherin by siRNA enhanced ERK1/2 activation by VEGF. ERK1/2 have many substrates, such as p90 ribosomal S6 kinase, c-Myc, and Elk-1, which are important in regulation of gene expression and cell proliferation. Thus, our observations are consistent with the results derived from mouse VE-cadherin-deficient endothelial cells in which VEGF-induced ERK1/2 activation and endothelial cell proliferation are increased compared with VE-cadherin-reconstituted mouse endothelial cells (38). Unexpectedly, we observed that VEGF-induced ERK1/2 activation was also elevated in endothelial cells treated with SHP2 siRNA or Ad-SHP2ΔPTP compared with control cells. These results are in contrast to the positive role of SHP-2 in the regulation of ERK1/2 in several other growth factor signaling pathways, including epidermal growth factor, hepatocyte growth factor, and fibroblast growth factor in epithelial cells and fibroblasts through a Ras-dependent pathway (28, 39–42). The mechanisms by which SHP2 negatively regulates VEGF-induced ERK1/2 activation remain to be determined. It is possible that SHP2 may target at PLCγ to decrease its tyrosine phosphorylation, resulting in a reduction of ERK1/2 activation, since a PLCγ/protein kinase C-dependent but not Ras-dependent pathway mediates VEGF-induced ERK1/2 activation in endothelial cells. Together, our results suggest that the role of SHP2 for ERK1/2 signaling may differ based on the cell type and stimulus.
In summary, our findings demonstrate a novel role of VE-cadherin in modulating c-Src activation through SHP2 and Csk in response to VEGF. Moreover, our data imply that VE-cadehrin/Csk/SHP2/Src signal module differentially regulates Akt/ eNOS and ERK1/2 pathways in VEGF signaling (Fig. 10). Since the activation of c-Src and its downstream signaling play a critical role in mediating VEGF-induced angiogenesis and vascular permeability (8), further elucidation of the molecular mechanisms underlying the involvement of VE-cadherin in VEGF signal transduction may provide new therapeutic strategies for angiogenesis and vascular permeability-related cardiovascular disease, cancer, and diabetes.
Acknowledgments
We are grateful to Dr. Dietmar Vestweber for the plasmids of mouse VE-cadherin-WT and Y685F mutant. We thank Chelsea Wong for the isolation of HUVECs and other technical assistance.
Footnotes
This work was supported by National Institutes of Health Grants RO1 HL080611 (to Z. G. J.) and R01 AR046524-01 (to A. M. B.), American Diabetes Association Thomas R. Lee Award 1-06-CD-13 (to Z. G. J.) and, American Heart Association Grant-in-aid 0755916T (to Z. G. J.).
The abbreviations used are: VEGF, vascular endothelial growth factor; PLCγ, phospholipase γ; ERK, extracellular signal-regulated kinase; eNOS, endothelial nitric-oxide synthase; VE-cadherin, vascular endothelial cadherin; siRNA, small interference RNA; PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo{3,4-d}pyrimidine; BAEC, bovine aortic endothelial cell; HUVEC, human umbilical vein endothelial cell; WT, wild type; MOI, multiplicity of infection; EC, endothelial cell.
References
- 1.Folkman J. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N, Gerber HP, LeCouter J. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 4.Weis SM, Cheresh DA. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi T, Yamaguchi S, Chida K, Shibuya M. EMBO J. 2001;20:2768–2778. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zachary I. Biochem Soc Trans. 2003;31:1171–1177. doi: 10.1042/bst0311171. [DOI] [PubMed] [Google Scholar]
- 7.Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M. Proc Natl Acad Sci U S A. 2005;102:1076–1081. doi: 10.1073/pnas.0404984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. Nat Rev. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 9.Radisavljevic Z, Avraham H, Avraham S. J Biol Chem. 2000;275:20770–20774. doi: 10.1074/jbc.M002448200. [DOI] [PubMed] [Google Scholar]
- 10.Schlessinger J. Cell. 2000;100:293–296. doi: 10.1016/s0092-8674(00)80664-9. [DOI] [PubMed] [Google Scholar]
- 11.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Proc Natl Acad Sci U S A. 2001;98:2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 13.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 14.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 15.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Mol Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 16.Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, Cheresh DA. J Cell Biol. 2002;157:149–160. doi: 10.1083/jcb.200109079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, Luo J, Thompson JA, Schraven BL, Philips MR, Neel BG. Mol Cell. 2004;13:341–355. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- 18.Chan RJ, Feng GS. Blood. 2007;109:862–867. doi: 10.1182/blood-2006-07-028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neel BG, Gu H, Pao L. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 20.Ren Y, Meng S, Mei L, Zhao ZJ, Jove R, Wu J. J Biol Chem. 2004;279:8497–8505. doi: 10.1074/jbc.M312575200. [DOI] [PubMed] [Google Scholar]
- 21.Dejana E, Spagnuolo R, Bazzoni G. Thromb Haemost. 2001;86:308–315. [PubMed] [Google Scholar]
- 22.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 23.Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N. Proc Natl Acad Sci U S A. 2002;99:9462–9467. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumeister U, Funke R, Ebnet K, Vorschmitt H, Koch S, Vestweber D. EMBO J. 2005;24:1686–1695. doi: 10.1038/sj.emboj.7600647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Circ Res. 2003;93:354–363. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 26.Jin ZG, Wong C, Wu J, Berk BC. J Biol Chem. 2005;280:12305–12309. doi: 10.1074/jbc.M500294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong C, Jin ZG. J Biol Chem. 2005;280:33262–33269. doi: 10.1074/jbc.M503198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kontaridis MI, Liu X, Zhang L, Bennett AM. Mol Cell Biol. 2002;22:3875–3891. doi: 10.1128/MCB.22.11.3875-3891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okuda M, Takahashi M, Suero J, Murry CE, Traub O, Kawakatsu H, Berk BC. J Biol Chem. 1999;274:26803–26809. doi: 10.1074/jbc.274.38.26803. [DOI] [PubMed] [Google Scholar]
- 30.Lungu AO, Jin ZG, Yamawaki H, Tanimoto T, Wong C, Berk BC. J Biol Chem. 2004;279:48794–48800. doi: 10.1074/jbc.M313897200. [DOI] [PubMed] [Google Scholar]
- 31.Yamaoka-Tojo M, Tojo T, Kim HW, Hilenski L, Patrushev NA, Zhang L, Fukai T, Ushio-Fukai M. Arterioscler Thromb Vasc Biol. 2006;26:1991–1997. doi: 10.1161/01.ATV.0000231524.14873.e7. [DOI] [PubMed] [Google Scholar]
- 32.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sessa WC. Mem Inst Oswaldo Cruz. 2005;100(Suppl 1):15–18. doi: 10.1590/s0074-02762005000900004. [DOI] [PubMed] [Google Scholar]
- 34.Liebl EC, Martin GS. Oncogene. 1992;7:2417–2428. [PubMed] [Google Scholar]
- 35.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. J Cell Sci. 1998;111:1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 36.Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ. J Biol Chem. 2000;275:5983–5986. doi: 10.1074/jbc.275.8.5983. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi T, Ueno H, Shibuya M. Oncogene. 1999;18:2221–2230. doi: 10.1038/sj.onc.1202527. [DOI] [PubMed] [Google Scholar]
- 38.Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO, Dejana E. J Cell Biol. 2003;161:793–804. doi: 10.1083/jcb.200209019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi ZQ, Lu W, Feng GS. J Biol Chem. 1998;273:4904–4908. doi: 10.1074/jbc.273.9.4904. [DOI] [PubMed] [Google Scholar]
- 40.Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. Mol Cell Biol. 2000;20:8513–8525. doi: 10.1128/mcb.20.22.8513-8525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cunnick JM, Dorsey JF, Munoz-Antonia T, Mei L, Wu J. J Biol Chem. 2000;275:13842–13848. doi: 10.1074/jbc.275.18.13842. [DOI] [PubMed] [Google Scholar]
- 42.Shi ZQ, Yu DH, Park M, Marshall M, Feng GS. Mol Cell Biol. 2000;20:1526–1536. doi: 10.1128/mcb.20.5.1526-1536.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]