

Abstract

A polymeric nanogel has been used to sequester and turn off a lysosomal protein, acid α-glucosidase (GAA). The nanogel contains a β-thiopropionate cross-linker, which endows the nanogel with pH-sensitivity. While encapsulation of the enzyme fully turns off its activity, approximately 75% of the activity is recovered upon reducing the pH to 5.0. The recovered activity is ascribed to pH-induced degradation of the β-thiopropionate cross-linker causing the swelling of the nanogel and ultimately causing the release of the enzyme. We envision that strategies for sequestering protein molecules and releasing them at lysosomal pH might open up new directions for therapeutic treatment of lysosomal storage diseases.
Introduction
Encapsulating a guest molecule stably in one environment and then releasing it in a different environment is one of the hallmarks of supramolecular chemistry.1−5 Many systems have been developed over the years for encapsulating hydrophobic small-molecule guests in molecular cages and amphiphilic assemblies.6−15 Developing such systems for hydrophilic macromolecules, however, is a significant challenge, since there is no chemical distinction between the bulk and the host interior in water-soluble systems.16−23 However, there is a great need for developing encapsulation systems for proteins as guest molecules,24 because imbalance in protein activity is the primary reason for most human pathology.25,26 When an overabundant or overactive protein leads to disease, common therapeutic approaches include small molecules that bind to the active site of the protein and interference RNA molecules that slow down the protein expression.26−28 More recently, supramolecular approaches in which an assembly responds to the presence of excess proteins have also been explored.29−32 On the other hand, when the reduced activity or abundance of a protein causes a pathological condition, the therapeutic options are more limited. Gene delivery approaches are promising, but the safety and efficacy of the delivery vehicles remain as concerns.33−39 An alternative approach is to directly deliver recombinant proteins, which has the advantage of not causing artificial modifications in gene expression.24 Therefore, supramolecular assemblies that can efficiently encapsulate protein molecules and release them in response to a stimulus are of great interest. For example, lysosomal storage diseases27,28 are caused by defective enzyme activity in any one of 50 lysosomal enzymes. The disorders, including Tay-Sachs, Fabry, Gaucher, and Pompe diseases, can sometimes be treated by delivery of recombinant enzyme to replace the missing enzymatic activity.27 Although enzyme replacement therapy is efficacious, it is also very inefficient, with less than 1% of the infused enzyme making it to the target tissues in some treatments.27
Nanoscopic systems involving polymeric molecules and proteins are actively studied as vehicles for protein delivery.24,40−46 A common approach involves covalent conjugation of proteins to polymers using the side chain functional groups or using the initiating/terminating functional group at the chain terminus.47 Noncovalent binding between proteins and polymers has also been investigated.24 Most of these systems use charge complementarity between a polyelectrolyte and the surface charge of the protein as the basis for the formation of the nanoparticle. While this electrostatics-based approach has the advantage of being simple, sterics-based encapsulation has the advantage of providing charge-neutral systems that are often desired for avoiding nonspecific interactions based complexities. In this Article, we report on a pH-responsive and charge-neutral polymer nanogel that stably encapsulates an enzyme at neutral pH and then releases it at low pH using β-thioesters as the stimulus-sensitive functionality in the cross-linker of the nanogel.
Our choice of the cross-linked polymeric nanogels as the host was driven by the fact that these scaffolds have the advantage of being concentration-independent; that is, once formed, the assemblies are stable even at very high dilutions, as they do not require a critical aggregation concentration that is typical for amphiphilic assemblies such as micelles and vesicles.1,2 Similarly, human acid α-glucosidase (GAA) was chosen as the guest enzyme in this study, because GAA is a lysosomal enzyme and is therefore enzymatically active at lysosomal pH (pH ≈ 5), but inactive at neutral pH. Therefore, this enzymatic guest provides a useful readout for the stimulus-sensitive supramolecular chemistry targeted in this work. Defects in GAA cause the lysosomal storage disorder known as Pompe disease, which is clinically treatable by delivery of recombinant enzyme. Finally, we chose β-thioester as the pH-sensitive cross-linking functional group, because: (i) this functional group is stable at neutral pH and is hydrolyzable at lower pH (<pH ≈ 5.3);48,49 (ii) the rate of hydrolysis of the functional group is relatively slow50 and therefore provides an opportunity for a sustained release of the cargo.51,52
Experimental Section
Materials
All the reagents were purchased from commercial sources and used as such without further purification. 1H NMR spectra were recorded on a Bruker DPX-400 MHz NMR spectrometer, and all the spectra were calibrated against tetramethylsilane. Dynamic light scattering (DLS) measurements were carried out on a Malvern Nanozetasizer. Transmission electron microscopy (TEM) images were recorded on a JEOL-2000FX machine operating at an accelerating voltage of 100 kV. Fluorescence emission spectra were recorded on a Photon Technology International Quanta Master fluorometer. Mass spectrometric data were acquired by an electron spray ionization (ESI) technique on a Q-tof-micro quadruple mass spectrometer (Micromass). Absorbance of para-nitophenolate was measured using a plate reader (SpectraMax M5). The enzyme acid alpha glucosidase (GAA) was provided by Genzyme. The GAA was stored as a lyophilized cake at 5 °C until reconstitution with Milli-Q H2O. The reconstituted formulation contained 10 mg/mL GAA, 50 mM sodium phosphate, 4% mannitol, and 0.01% Tween-80 at pH 6.2. Following reconstitution, the solution was stored as frozen aliquots at −80 °C until use.
Synthesis of Nanogel
We have used monomer and cross-linker in the molar ratios of 95:5. Monomer M and cross-linker C were taken in a vial and diluted with 100 μL of initiator, 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (I) solution (5.5 mg/mL) prepared with phosphate buffered saline (PBS) buffer (10 mM, pH = 7.4). The vial was then vortexed for 2 min to make it a homogeneous mixture. Inverse microemulsion (prepared separately) consisted of 5 mL of heptane and Brij L4 surfactant (0.60 g). The microemulsion was added to the vial containing M, C, I and was then subjected to vortex (5 min) followed by sonication (5 min). The reaction mixture was purged with argon gas for 10 min to remove oxygen. Finally, the reaction vessel was placed inside a UV chamber and exposed to UV light with mild stirring for 20 min. After the polymerization, it was diluted with measured volume of PBS buffer (pH = 7.4) followed by addition of ∼2 mL of n-butanol. It was centrifuged for 15 min at 2943g to remove all the surfactants and organic solvent. This was repeated twice to make sure all of the surfactants were removed. The resulting aqueous solution was dialyzed (MWCO 7000 Da) against PBS buffer (pH = 7.4) for 24 h at 5 °C while water was changed every 6 h.
Synthesis of GAA Loaded Nanogel
For the synthesis of acid degradable nanogel following inverse microemulsion polymerization process, we have used monomer (150 mg, 0.604 mmol) and cross-linker (18 mg, 0.06 mmol) in the molar ratio of 95:5. Monomer M and cross-linker C were taken in a vial and diluted with 100 μL of initiator (I) solution (5.5 mg/mL) prepared with PBS buffer (10 mmol, pH = 7.4). Then, 2 mg of protein (acid α-glucosidase) was added to the mixture. The vial was then vortexed for 2 min to make it a homogeneous mixture. In separate vial inverse microemulsion was prepared using 5 mL of heptanes and 0.60 g of Brij L4 surfactant. The microemulsion was added to the vial containing M, C, I and protein and was then subjected to vortex (5 min) followed by sonication (5 min). The reaction mixture was purged with argon gas for 10 min to remove oxygen. Finally, the reaction vessel was placed inside UV Chamber and exposed to UV light with mild stirring for 20 min. After the polymerization, it was diluted with measured volume of PBS buffer (pH = 7.4) followed by addition of ∼2 mL of n-butanol. It was centrifuged for 15 min at 2943g to remove all the surfactants and organic solvent. This was repeated twice to make sure all of the surfactants were removed. The resulting aqueous solution was dialyzed (MWCO 7000 Da) against PBS buffer (pH = 7.4) for 24 h at 5 °C while water was changed in every 6 h.
Synthesis of GAA Loaded Control Nanogel
The control nanogel was synthesized using a similar procedure, except we used a control cross-linker (CC) lacking the β-thiopropionate functional group. The monomer to cross-linker molar ratio was maintained at 95:5.
Dynamic Light Scattering Study
For the DLS measurements, the concentration of the nanogel was 1 mg/mL. The solution was filtered using a hydrophilic membrane (pore size 0.450 μm) before the experiment was performed.
Transmission Electron Microscopy Study
For the TEM measurements, the nanogel solution was prepared in 1 mg/mL concentration. One drop of the sample was drop-casted on a carbon coated Cu grid. About 3 min after the deposition, the grid was tapped with filter paper to remove surface water. Finally, it was dried in air for another 6 h before images were taken.
Photoluminescence Study
Fluorescence spectroscopy was performed on a Photon Technology International Quanta Master fluorometer. GAA samples were diluted to 1 μM in 10 mM sodium acetate or 10 mM PBS buffer at pH 5.0 or 7.4, respectively. Tryptophans were excited at 295 nm (slit width 1 nm), and emission was monitored from 305 to 400 nm (slit width 3 nm) at 20 °C. Five scans were performed on each sample and averaged. A blank buffer spectrum was collected for each buffering system and subtracted from the results prior to analysis. Data collection was handled by using Felix32 software.
Bicinconinic Acid (BCA) Assay for Quantification of the Protein
A calibration curve was generated for known concentrations of the protein using a BCA assay kit. Absorbance (at 562 nm) of the unknown protein solution was measured in a plate reader. The absorbance value of the unknown protein solution was fitted in the calibration curve to derive the protein concentration.
GAA Activity Assay
GAA activity was measured using para-nitrophenol-α-d-glucopyranoside as a substrate in 100 mM citrate/100 mM sodium phosphate buffer at pH 5.0. The enzyme was incubated with the prewarmed substrate at 37 °C in a Bio-Rad C1000 thermocycler. Aliquots were then quenched in 200 mM borate buffer at pH 9.0 in a clear-bottom 96-well plate. para-Nitrophenolate product formation was then measured by absorbance of 400 nm light on a Molecular Devices Spectra Max M5 instrument at 25 °C. Final product concentrations were corrected for dilution factor and converted to molar concentrations using the known extinction coefficient (18 200 M–1 cm–1).
Cell Viability
The in vitro cellular viability of the nanogels and the degraded nanogels was evaluated on healthy 293T and MDA-MB-231 breast cancer cell lines. The cells were cultured in T75 cell culture flasks using Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 (DMEM/F12) with 10% fetal bovine serum (FBS) supplement. The cells were seeded at 10 000 cells/well/200 μL in a 96-well plate and allowed to grow for 24 h under incubation at 37 °C and 5% CO2. These cells were then treated with nanogels of different concentrations and were incubated for another 24 h. Cell viability was measured using the Alamar Blue assay with each data point measured in triplicate. Fluorescence measurements were made using the plate reader SpectraMax M5 by setting the excitation wavelength at 560 nm and monitoring emission at 590 nm on a black well plate.
Results and Discussion
The structures of the monomers and cross-linker used in this study are shown in Scheme 1. The monomer is based on a charge neutral tetraethylene glycol, as these monomers typically render the systems biocompatible and obviate the electrostatic-based nonspecific interactions between the enzymatic guest and the host assembly. The β-thioester based cross-linker also contains oligoethylene glycol units to endow them with the water solubility needed for convenient incorporation of the cross-linking monomer in the aqueous phase during the nanogel synthesis using inverse emulsion polymerization.53−55 The water-soluble monomers and cross-linkers, combined with the nanogel synthesis in a water/oil emulsion polymerization, also provides an opportunity for the in situ encapsulation of the water-soluble enzymatic guest in a water-soluble nanocontainer.
Scheme 1.

The inverse emulsion was formed using heptane as the continuous phase and Brij L4 as the surfactant. The polymerization between the monomer and the cross-linker was initiated within the dispersed aqueous phase of the water/oil emulsion using 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone as the photoinitiator. The reaction vessel containing the reaction mixture was exposed to UV light with mild stirring for 20 min. After the polymerization, the surfactants were removed by addition of n-butanol and PBS buffer, followed by centrifugation (see Scheme 1 for stepwise protein encapsulation and release). The resultant aqueous solution was dialyzed against PBS buffer for 24 h at 5 °C to obtain the protein-encapsulated nanogel.
The nanogels were characterized by 1H NMR spectroscopy (Figure 1a). First, there were no signals from the acrylate protons in the 1H NMR spectrum of the NG, indicating complete conversion of the acrylate monomer. Second, the polymer contains both the TEG-acrylate monomer and the β-thioester cross-linker incorporated into the nanogel. Note that the signal at 2.6–2.8 ppm corresponds to the β-protons of the β-thioester. From the integration, the percentage of cross-linker was found to correspond to the feed-ratio of the monomers at ∼5% (see the calculation and NMR in the Supporting Information). The cross-linkers that are buried within the interiors of the nanogel are discernible in the NMR, also supporting the low cross-link density of ∼5%. To examine the size of the nanogel particle, we carried out DLS experiments, which suggest an average hydrodynamic diameter (Dh) of about 16 nm for the nanogels (Figure 1b). Although DLS is a clearer indication of the nanogel sizes in solution, we also show support for the size data using TEM (Figure 1c). The 10–20 nm sizes observed in TEM are in agreement with the DLS results.
Figure 1.

(a) 1H NMR spectra of monomer (M), cross-linker (C), and nanogel without protein (NG). Asterisk (*) indicates solvent peak, and chemical shift values are shown in ppm on the X-axis. (b) DLS spectrum of the nanogel (PDI = 0.45). (c) HRTEM image of the nanogel.
To investigate the de-cross-linking phenomenon in the nanogel due to hydrolytic cleavage of the β-thiopropionate linker in acidic conditions, (Figure2a), we treated the nanogel solution with acetate buffer, where the pH of the solution was maintained at 5.0. The variations in the nanogel were monitored by assessing size change using DLS over time (Figure 2b). Interestingly, the average size of the nanogel increased over time (from ∼18 to ∼60 nm) at pH 5.0, which is attributed to de-cross-linking-induced swelling.55 The larger size particle formation (see autocorrelation function for the NG in the Supporting Information) is attributed to acid-induced swelling which eventually leads to interparticle fusion. To further verify the size change, we have analyzed dried sample of de-cross-linked nanogel (aliquot taken after 48 h) using TEM, which revealed the presence of spherical particles in the range of 40–70 nm, along with a few even larger particles (more than 100 nm in size) (Figure 3b). The larger particles that are likely to occur due to interparticle fusion are observed in DLS also, when investigated using volume-based assessment of the scattering data (Figure 3a). Overall, the TEM data corroborates the DLS data. To confirm whether de-cross-linking induced swelling occurs selectively in acidic pH, we monitored time variable DLS (Figure 2c) of the nanogel at pH 7.4 and observed even after 7 days there was no change in the size of the nanogel. This suggests that swelling of the nanogel is indeed due to pH variation.
Figure 2.

(a) Acid induced hydrolysis of the β-thiopropionate functional group. (b) DLS profile (PDI = 0.45, 0.35, 0.36, 0.40, 0.39) of the nanogel at different time intervals at pH 5.0. (c) DLS profile (PDI = 0.45, 0.41, 0.41, 0.40) of the nanogel at different time intervals at pH 7.4.
Figure 3.

(a) DLS profile (PDI = 0.457) of the nanogel after 48 h incubation at pH 5. (b) TEM image of the nanogel after 48 h incubation at pH 5; temperature = 25 °C.
Next, we were interested in understanding the possibility of releasing the encapsulated cargo in response to pH change. To this end, GAA was encapsulated within the nanogel network using the same procedure described above (see Experimental Section). We used phosphate buffer solution of GAA (10 mg/mL) in the inverse microemulsion polymerization to generate the protein-encapsulated nanogel. To evaluate the concentration of GAA in the nanogel solution, we performed a BCA assay (a standard assay for determination of protein concentration in an unknown solution; for detailed information, see the Experimental Section and Figure S1 in the Supporting Information), which revealed 28 μg of protein per milligram of nanogel. The encapsulation efficiency of the nanogel was found to be ∼90%. The high encapsulation efficiency may be due to the monomer, cross-linker, initiator, and protein all being hydrophilic and therefore partitioning in the aqueous phase of the inverse micelle formed by Brij L4 surfactant prior to polymerization.
The activity of the encapsulated GAA, relative to the free GAA, was evaluated next. We hypothesized that the enzyme would be less available to the substrate, when encapsulated, and therefore would have a lower activity. It follows then that the activity will be recovered when exposing the nanogel to lower pH, as the enzyme will be released in response to the β-thioester cleavage based de-cross-linking. The enzymatic activity was measured using para-nitrophenol-α-d-glucopyranoside as substrate, the GAA-assisted cleavage of the α-1,4-linkage of which releases the chromophore p-nitrophenolate (pNP) (Figure 4a). At pH 7.4, the nanogel:enzyme complex did not exhibit any enzymatic activity. Note however that the enzymatic activity of GAA itself at pH 7.4 is undetectable (Figure S2, Supporting Information). To activate the GAA from the nanogel, we reduced the pH of the solution to 5.0 using acetate buffer. Indeed, the activity of GAA dramatically increased (Figure 4b). To calibrate the percentage of activity that was recovered after 48 h of incubation at pH 5.0, the enzymatic activity of the GAA from the nanogel was compared with that of the free enzyme at the same concentration. Approximately 75% of the enzymatic activity was recovered upon exposing the nanogel to lower pH for 48 h (75% in respect to activity of native protein at 48 h time point, but 62% when activity of the native protein at 0 min time point is taken into account). This recovery is presumably due to the de-cross-linking of the nanogel leading to accessibility of the enzyme to substrate (Figures S3 and S4, Supporting Information).
Figure 4.

(a) GAA activity assay after 48 h incubation of the GAA loaded nanogel at pH 5.0 and pH 7.4: monitoring absorbance at 400 nm of the pNP produced due to cleavage of the α-1,4-linkages of para-nitrophenol-α-d-glucopyranoside substrate. (b) Comparison of the end point activity; protein concentration in each case = 0.173 μM; temperature = 37 °C. (c) Time variable DLS profile (PDI = 0.354, 0.44, 0.40, 0.45) of native GAA and GAA loaded in the nanogel. (d) TEM image of GAA encapsulated NG; inset shows zoomed NG.
We then examined DLS (Figure 4c) and fluorescence (Figures 5 and S5) of the GAA-loaded nanogel before and after treatment in acidic conditions (pH ≈ 5) over 48 h. The DLS profile showed that the average hydrodynamic diameter of the GAA loaded nanogel at pH 7.4 is ∼18 nm, which corroborates well with the TEM results (Figure 4d). However, the DLS profile of this nanogel changed after low pH treatment (see autocorrelation function in the Supporting Information). We observed a bimodal distribution (Figure 4c) after the GAA-loaded nanogel was exposed to acidic pH; two distinct average hydrodynamic diameters of 24 and 90 nm were observed. We interpret the two radii as corresponding to the released protein and to the swollen de-cross-linked nanogel, respectively. The increase in the size of the released GAA might be attributed to the aggregation of some protein over 48 h at room temperature. To confirm this, we incubated native GAA at pH 5.0 for 48 h and found a similar increase in the size (Figure 4c). This aggregation phenomenon might be the cause of ∼25% loss of the enzymatic activity of the released GAA. In another control experiment, we found that there was no change in the hydrodynamic diameter of the GAA-loaded nanogel at pH 7.4 even after 10 days (Figure S6, Supporting Information). These results support the notion that the enzyme becomes more accessible for the substrate upon de-cross-linking in response to reduced pH. The DLS provides the supporting evidence that there might be a pH-induced de-cross-linking and accessibility of the enzyme.
Figure 5.

(a) Normalized emission spectra of the GAA loaded in the nanogel (black line) and native GAA (red line) at pH 7.4. (b) Normalized emission spectra of GAA loaded in the nanogel (black line) and GAA released from nanogel after 48 h incubation at pH 5.0 (red line). After loading of the GAA inside the nanogel, the spectrum becomes blue-shifted because of the hydrophobic environment compared to bulk solvent. After releasing from the nanogel, spectrum becomes red-shifted because of the polar environment of the bulk water. Tryptophan excitation wavelength = 295 nm; temperature = 25 °C.
A remaining question however is whether the protein is indeed fully released from the nanogel network or the swelling of the de-cross-linking nanogel causes the substrate to diffuse inside the nanogel. To address this, we examined the time-dependent increase in the enzymatic activity (Figure S13, Supporting Information). It is clear that the enzymatic activity reaches a high activity even after 5 min of incubation at low pH and remains at that activity up to about 24 h. Interestingly, however, there is a marked increase in the activity between 24 and 48 h. We attribute these findings to suggest that at initial time scales that the de-cross-linking reaction simply causes the nanogel to swell to provide substrate access to the enzyme. The activity increase at longer time scales is attributed to the release of the enzyme from the nanogel.
To further test that (a) all the proteins used initially have encapsulated within the nanogel network and (b) the β-thiopropionate linker is responsible for the release of the encapsulated protein molecules, we designed and synthesized a control nanogel using a cross-linker that lacks β-thiopropionate functional group (Figure 6a). In this case, the enzyme is not expected to be released from the nanogel at low pH. The structure of the control cross-linker is very similar to the pH-sensitive cross-linker, except the sulfur atom is replaced by a methylene unit in the cross-linker. The absence of the β-thiopropionate functionality removes the pH trigger from the nanogel.52 Encapsulation of GAA in this control nanogel was achieved using a method similar to the one above (see Experimental Section for details). The control nanogel’s encapsulation efficiency was ∼50%, as assessed by the BCA assay. The lower encapsulation efficiency is likely due to the reduced hydrophilicity of the cross-linker (CC).56 Nonetheless, the control nanogel/GAA complex provides the opportunity to test our encapsulation and pH-induced de-cross-linking/release hypotheses.
Figure 6.
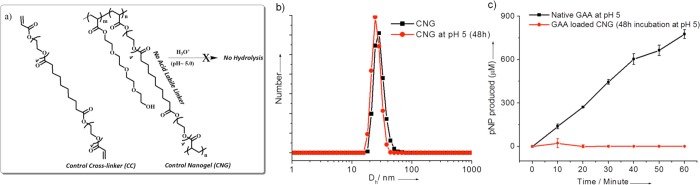
(a) Structure of the control cross-linker and control nanogel (CNG). (b) DLS profile (PDI = 0.45, 0.60) of the control nanogel after 48h incubation at pH 5.0. (c) GAA activity assay profile of control nanogel; protein concentration = 0.173 μM; temperature = 37 °C.
The time-dependent size change of the control nanogel was assessed by DLS at pH 5.0. Even after 48 h, no significant change in the hydrodynamic diameter of the control nanogel was observed, suggesting that there is no de-cross-linking mediated swelling (Figure 6b). This result was also reflected in the protein activity assay. Even after 48 h at pH 5.0, there was no significant enzymatic activity of the GAA in the control nanogel complex (Figure 6c). These observations suggest the following: (i) The protein encapsulation in the nanogel turns off the enzymatic activity. (ii) GAA encapsulation through inverse emulsion polymerization places the enzyme inside the nanogel. If these were not present inside the nanogel, the unencapsulated enzyme would be active in the control nanogel experiments. (iii) The β-thiopropionate linker is indeed responsible for the pH-sensitive de-cross-linking/swelling and enzymatic activation.
Finally, since the nanogels are based on oligoethylene glycol units as the surface functional groups and cross-linking moiety, we hypothesized that these nanogels might not be cytotoxic. To test this, we carried out in vitro cell viability assay using an Alamar blue assay with 293T and MDA-MB-231 cell lines (Figure 7). The cells were incubated with varying concentration of the nanogel solution for 24 h at 37 °C. The nanogels exhibit >80% cell viability for both 293T and MDA-MB-231 cell lines in the entire concentration range. In addition to testing the cytotoxicity of the nanogels, it is also critical to investigate whether the degradation products of the nanogels are cytotoxic. Therefore, the cell viability of the degraded nanogel was also investigated at various concentrations. These too showed concentration-independent cell viability for both 293T and MDA-MB 231 cell lines (Figure S7, Supporting Information). These results are promising first steps to ultimately utilize these nanogels for in vivo applications.
Figure 7.

In vitro cytoxicities of nanogel on 293T cell line (black) and MDA-MB-231 cell line (red).
Conclusions
In summary, we have shown that (i) an enzyme can be conveniently encapsulated into a polymeric nanogel synthesized through the inverse microemulsion method; (ii) the enzymatic activity is turned off when encapsulated within the nanogel; (iii) when the nanogel contains an acid labile cross-linker, the nanogel can be de-cross-linked in response to pH changes; and (iv) the pH-induced de-cross-linking event turns the enzymatic activity back on. This demonstration of protein encapsulation and pH-induced release using polymeric nanogels has clear biological implications. The protein encapsulated in this study, GAA, is used in massive doses in recombinant enzyme replacement therapy to treat Pompe disease patients. Since, the enzyme is inactive at pH 7.4 and is active at acidic lysosomal pH, the method of reversibly turning off the enzymatic activity has biomedical implications for the delivery of proteins to lysosomes. From an even broader perspective, the nanomaterials platform of protein encapsulation and stimulus-sensitive release can be utilized in several biological applications.
Acknowledgments
Support from the National Science Foundation (CHE-1307118) and the NIGMS (GM 062255 to S.T.) and NIDDK (DK 76877 to S.C.G.) is gratefully acknowledged. We also acknowledge the infrastructure support provided by the Center for Hierarchical Manufacturing at UMass Amherst. We also thank Garrett Loomis for helping in the synthesis of monomer.
Supporting Information Available
Additonal synthesis details and figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Evans D. F.; Wennerstrom H.. The Colloidal Domain, 2nd ed.;Wiley-VCH:New York, 1999. [Google Scholar]
- Holmberg K.; Jönsson B.; Kronberg B.; Lindman B.. Surfactants and Polymers in Aqueous Solution, 2nd ed.; JohnWiley & Sons, Ltd: New York, 2003. [Google Scholar]
- Saito G.; Swanson J. A.; Lee K.-D. Adv. Drug Delivery Rev. 2003, 55, 199–215. [DOI] [PubMed] [Google Scholar]
- Roy D.; Cambre J. N.; Sumerlin B. S. Prog. Polym. Sci. 2010, 35, 278–301. [Google Scholar]
- Guo D. S.; Liu Y. Acc. Chem. Res. 2014, 47, 1925–1934. [DOI] [PubMed] [Google Scholar]
- Sawada T.; Yoshizawa M.; Sato S.; Fujita M. Nat.Chem. 2009, 1, 53–56. [DOI] [PubMed] [Google Scholar]
- Liu M. J.; Kono K.; Fréchet J. M. J. J. Controlled Release 2000, 65, 121–131. [DOI] [PubMed] [Google Scholar]
- Ronson T. K.; Giri C.; Beyeh N. K.; Minkkinen A.; Topić F.; Holstein J. J.; Rissanen K.; Nitschke J. R. Chem.—Eur. J. 2013, 19, 3374–3382. [DOI] [PubMed] [Google Scholar]
- Zhong S.; Cui H. G.; Chen Z. Y.; Wooley K. L.; Pochan D. J. Soft Matter 2008, 4, 90–93. [DOI] [PubMed] [Google Scholar]
- Bae Y.; Kataoka K. Adv. Drug Delivery Rev. 2009, 61, 768–784. [DOI] [PubMed] [Google Scholar]
- Ono K.; Yoshizawa M.; Kato T.; Watanabe K.; Fujita M. Angew. Chem., Int. Ed. 2007, 46, 1803–1806. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Kumazawa K.; Fujita M. J. Am. Chem. Soc. 2005, 127, 13456–13457. [DOI] [PubMed] [Google Scholar]
- Formina N.; McFearin C.; Sermsakdi M.; Edigin O.; Almutairi A. J. Am. Chem. Soc. 2010, 132, 9540–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko R.; Ventura J.; Zhuang J.; Thayumanavan S. Adv. Drug Delivery Rev. 2012, 64, 836–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J.-H.; Chacko R.; Jiwpanich S.; Bickerton S.; Babu R. P.; Thayumanavan S. J. Am. Chem. Soc. 2010, 132, 17227–17235. [DOI] [PubMed] [Google Scholar]
- Mahmoud E. A.; Sankaranarayanan J.; Morachis J. M.; Kim G.; Almutairi A. Bioconjugate Chem. 2011, 22, 1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin V. P. Nat. Rev. Drug Discovery 2005, 4, 145–160. [DOI] [PubMed] [Google Scholar]
- Abu Lila A. S.; Ishida T.; Kiwada H. Expert Opin. Drug Delivery 2009, 6, 1297–1309. [DOI] [PubMed] [Google Scholar]
- Eliaz R. E.; Nir S.; Marty C.; Szoka F. C. Cancer Res. 2004, 64, 711–718. [DOI] [PubMed] [Google Scholar]
- Haag R.; Kratz F. Angew. Chem., Int. Ed. 2006, 45, 1198–1215. [DOI] [PubMed] [Google Scholar]
- Murthy N.; Xu M.; Schuck S.; Kunisawa J.; Shastri N.; Fréchet J. M. J. Proc. Natl. Acad.Sci. U. S. A. 2003, 100, 4995–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramonov S.; Bachelder E.; Beaudette T.; Standley S.; Lee C.; Dashe J.; Fréchet J. M. J. Bioconjugate Chem. 2008, 19, 911–919. [DOI] [PubMed] [Google Scholar]
- Kabanov A. V.; Vinogradov S. V. Angew. Chem., Int. Ed. 2009, 48, 5418–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z.; Biswas A.; Zhao M.; Tnag Y. Chem. Soc. Rev. 2011, 40, 3638–3655. [DOI] [PubMed] [Google Scholar]
- Walsh G. Nat. Biotechnol. 2010, 28, 917–924. [DOI] [PubMed] [Google Scholar]
- Leader B.; Baca Q. J.; Golan D. E. Nat. Rev. Drug Discovery 2008, 7, 21–39. [DOI] [PubMed] [Google Scholar]
- Desnick R. J.; Schuchman E. H. Annu. Rev. Genomics Hum. Genet. 2012, 13, 307–335. [DOI] [PubMed] [Google Scholar]
- Parenti G.; Pignata C.; Vajro P.; Salerno M. Int. J. Mol. Med. 2013, 31, 11–20. [DOI] [PubMed] [Google Scholar]
- Savariar E. N.; Ghosh S.; Gonzalez D. C.; Thayumanavan S. J. Am. Chem. Soc. 2008, 130, 5416–5417. [DOI] [PubMed] [Google Scholar]
- Takaoka Y.; Sakamoto T.; Tsukiji S.; Narazaki M.; Matsuda T.; Tochio H.; Shirakawa M.; Hamachi I. Nat. Chem. 2009, 1, 557–561. [DOI] [PubMed] [Google Scholar]
- Azagarsamy M. A.; Yesilyurt V.; Thayumanavan S. J. Am. Chem. Soc. 2010, 132, 4550–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizusawa K.; Ishida Y.; Takaoka Y.; Masayoshi M.; Tsukiji S.; Hamachi I. J. Am. Chem. Soc. 2010, 132, 7291–7293. [DOI] [PubMed] [Google Scholar]
- Mastrobattista E.; Van der Aa M.; Hennink W. E.; Crommelin D. J. Nat. Rev. Drug Discovery 2006, 5, 115–121. [DOI] [PubMed] [Google Scholar]
- Pack D. W.; Hoffman A. S.; Pun S.; Stayton P. S. Nat. Rev. Drug Discovery 2005, 4, 581–593. [DOI] [PubMed] [Google Scholar]
- Ogris M.; Brunner S.; Schemler S.; Kircheis R.; Wagner E. Gene Ther. 1999, 6, 595–605. [DOI] [PubMed] [Google Scholar]
- Wattiaux R.; Laurent N.; Coninck S. W.-D.; Jadot M. Adv. Drug Delivery Rev. 2000, 41, 201–208. [DOI] [PubMed] [Google Scholar]
- Hunter A. C. Adv. Drug Delivery Rev. 2006, 58, 1523–1531. [DOI] [PubMed] [Google Scholar]
- Tang M. X.; Redemann C. T.; Szoka F. C. Bioconjugate Chem. 1996, 7, 703–714. [DOI] [PubMed] [Google Scholar]
- Boussif O.; LezoualcIh F.; Zanta M. A.; Mergny M. D.; Scherman D.; Demeneix B.; Behr J. P. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla R. I.; Gao Y.; Li X.; Serpe M. J. J. Mater. Chem. B 2014, 2, 2444–2451. [DOI] [PubMed] [Google Scholar]
- Smith M. H.; Lyon L. A. Macromolecules 2011, 44, 8154–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermonden T.; Censi R.; Hennink W. E. Chem. Rev. 2012, 112, 2853–2888. [DOI] [PubMed] [Google Scholar]
- Nochi T.; Yuki Y.; Takahashi H.; Sawada S-i.; Mejima M.; Kohda T.; Harada N.; Kong G.; Sato A.; Kataoka N.; Tokuhara D.; Kurokawa S.; Takahashi Y.; Tsukada H.; Kozaki S.; Akiyoshi K.; Kiyono H. Nat. Mater. 2010, 9, 572–578. [DOI] [PubMed] [Google Scholar]
- Yan M.; Du J.; Gu Z.; Liang M.; Hu Y.; Zhang W.; Priceman S.; Wu L.; Zhou Z. H.; Liu Z.; Segura T.; Tang Y.; Lu Y. Nat. Nanotechnol. 2010, 5, 48–53. [DOI] [PubMed] [Google Scholar]
- Azagarsamy M. A.; Anseth K. S. Angew. Chem., Int. Ed. 2013, 52, 13803–13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J.; Anderson S. M.; Du J.; Yan M.; Wang J.; Shen M.; Lu Y.; Segura T. Adv. Mater. 2011, 23, 4549–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Toro D.; Thayumanavan S. Eur. Polym. J. 2013, 49, 2906–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi M.; Sasaki S.; Nagasaki Y.; Kataoka K. Biomacromolecules 2003, 4, 1426–1432. [DOI] [PubMed] [Google Scholar]
- Oishi M.; Nagasaki Y.; Itaka K.; Nishiyama N.; Kataoka K. J. Am. Chem. Soc. 2005, 127, 1624–1625. [DOI] [PubMed] [Google Scholar]
- Schoenmakers R. G.; Wetering P.; van de Elbert D. L.; Hubbell J. A. J. Controlled Release 2004, 95, 291–300. [DOI] [PubMed] [Google Scholar]
- Dan K.; Ghosh S. Angew. Chem., Int. Ed. 2013, 52, 7300–7305. [DOI] [PubMed] [Google Scholar]
- Dan K.; Pan R.; Ghosh S. Langmuir 2011, 27, 612–617. [DOI] [PubMed] [Google Scholar]
- Schillemans J. P.; Flesch F. M.; Hennink W. E.; van Nostrum C. F. Macromolecules 2006, 39, 5885–5890. [Google Scholar]
- Lawrence G M. J.; Rees D. Adv. Drug Delivery Rev. 2000, 45, 89–121. [DOI] [PubMed] [Google Scholar]
- Azagarsamy M. A.; Alge D. L.; Radhakrishnan S. J.; Tibbitt M. W.; Anseth K. S. Biomacromolecules 2012, 13, 2219–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton J.; Kawamoto S.; Fujimoto K.; Kawaguchi H.; Capek I. Polym. Int. 2000, 49, 358–366. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.