

Abstract
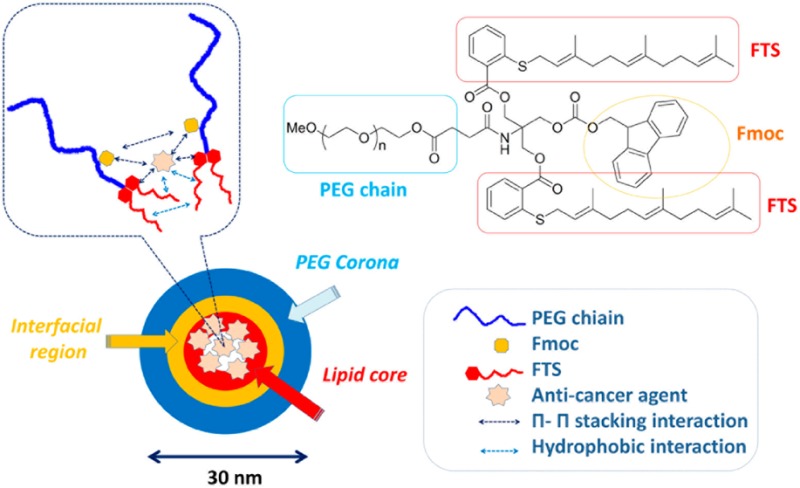
We have developed a dual-function drug carrier, polyethylene glycol (PEG)-derivatized farnesylthiosalicylate (FTS). Here we report that incorporation of a drug-interactive motif (Fmoc) into PEG5k–FTS2 led to further improvement in both drug loading capacity and formulation stability. Doxorubicin (DOX) formulated in PEG5k–Fmoc–FTS2 showed sustained release kinetics slower than those of DOX loaded in PEG5k–FTS2. The maximum tolerated dose of DOX- or paclitaxel (PTX)-loaded PEG5k–Fmoc–FTS2 was significantly higher than that of the free drug. Pharmacokinetics and biodistribution studies showed that DOX/PEG5k–Fmoc–FTS2 mixed micelles were able to retain DOX in the bloodstream for a significant amount of time and efficiently deliver the drug to tumor sites. More importantly, drug (DOX or PTX)-loaded PEG5k–Fmoc–FTS2 led to superior antitumor activity over other treatments including drugs formulated in PEG5k–FTS2 in breast cancer and prostate cancer models. Our improved dual function carrier with a built-in drug-interactive motif represents a simple and effective system for targeted delivery of anticancer agents.
Introduction
Formulations represent an important strategy to improve the therapeutic index of anticancer agents via improvement of their solubility, bioavailability, and pharmacokinetic and biodistribution profiles.1,2 Taxol is an FDA-approved clinical formulation of paclitaxel (PTX) in which a Cremophor EL/ethanol (1:1, v/v) mixture is used to solubilize PTX; however, Taxol can cause hyperactivity reactions, neuropathy, and other serious side effects.3 Polyethylene glycol (PEG)ylated liposomal doxorubicin (DOX) (Doxil) is the first nanoformulation of DOX approved by FDA. Although Doxil has demonstrated decreased cardiotoxicity, it shows limited improvement over free DOX in therapeutic efficacy. In addition, Doxil is associated with other side effects such as hand-foot syndrome.4 Over the last decades, micelles with a nanoscopic supramolecular core–shell structure have gained increasing attention5,6 because of their easy preparation, small sizes, and ability to improve the pharmacokinetics and efficacy of anticancer drugs.7−11 However, most delivery systems involve the use of “inert” materials that do not possess any favorable biological activity. One interesting approach is the development of dual function carriers that have both a delivery function and antitumor activity.1,12−17
We have recently reported a new self-assembling nanomicellar system that is based on PEGylated S-trans, trans-farnesylthiosalicylic acid (FTS).14 FTS, a synthetic farnesylcysteine mimetic, is a potent and especially nontoxic Ras antagonist.18,19 Constitutively active Ras caused by mutation in the Ras family of proto-oncogenes is present in one-third of human cancers.20,21 FTS can inhibit Ras-dependent tumor growth with no adverse toxicity.22 One major mechanism involves affecting membrane interaction of Ras by competing with Ras for binding to Ras-escort proteins and thereby inhibiting its signaling.23 In addition to its antitumor activity in mice and humans, FTS also exhibits anti-inflammatory activity;24,25 however, FTS has poor water solubility and limited oral bioavailability.26 PEGylation was initially designed to improve its solubility. Interestingly, PEG5k–FTS2 conjugate self-assembled to form small-sized micelles (20–30 nm) that were effective in solubilizing other hydrophobic drugs such as PTX. PEG5k–FTS2 differs from most drug carriers in that it shows an antitumor activity that is comparable to that of free FTS.14 Additionally, PTX formulated in PEG5k–FTS2 micelles demonstrated a synergistic antitumor activity that was significantly higher than that of Taxol.14
Most reported micellar systems including PEG5k–FTS2 are designed to load drugs through hydrophobic interactions. While they work well for highly hydrophobic drugs, they only have limited effectiveness in formulating drugs that are either moderately hydrophobic or hydrophilic. The carrier/drug incompatibility will result in not only low drug loading capacity, but also limited stability of the drug-loaded micelles.27 Park’s group has shown that inclusion of less hydrophobic and hydrogen bond-forming “hydrotropic motifs” into the hydrophobic domain of polymeric micelles significantly improved both drug loading capacity and the colloidal stability of drug-loaded micelles.27−29 However, this concept has not been demonstrated in lipidic systems. We recently hypothesized that, incorporated into a surfactant, a drug-interactive motif at an interfacial region will provide an additional carrier/drug interaction mechanism, which could enhance both drug-loading capacity and formulation stability.9,30 Among several motifs screened, 9-fluorenylmethoxycarbony (Fmoc) moiety, a functional group that is routinely used for amino acid protection, was demonstrated to be the most potent drug-interactive group.30 We demonstrated that incorporation of Fmoc motifs into a PEG–lipopeptide conjugate led to a significant improvement in the loading of a number of therapeutic agents of diverse structures.31,32
Considering the significance of Fmoc as a novel “formulation chemophor” or a structural unit capable of interacting with many pharmaceutical agents, we hypothesized that incorporation of Fmoc will further improve the performance of our PEG5k–FTS2 delivery system. We report in this study the development and characterization of a new micellar carrier composed of an FTS-based hydrophobic domain, a PEG hydrophilic segment and an interfacial drug-interactive Fmoc motif (PEG5k–Fmoc–FTS2). Our data showed that inclusion of an Fmoc motif into PEG5k–FTS2 led to a significant improvement in drug loading capacity for both PTX and DOX. More importantly, delivery of PTX or DOX via PEG5k–Fmoc–FTS2 led to superior antitumor activity over other treatments including drugs formulated in PEG5k–FTS2 micelles in breast cancer and prostate cancer models.
Experimental Section
Materials and Reagents
Paclitaxel (98%) was purchased from AK Scientific Inc. (CA, USA). Doxorubicin (>99%) was purchased from LC Laboratories (MA, USA). 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD) was purchased from Invitrogen (NY, USA). N-hydroxysuccinimide (NHS) and dicyclohexylcarbodiimide (DCC) were purchased from Alfa Aesar (MA, USA). 4-(Dimethylamino) pridine (DMAP) was purchased from Calbiochem–Novabiochem Corporation (CA, USA). FTS was synthesized and purified following a published literature.33
Synthesis of PEG5k–Fmoc–FTS2 Conjugate
PEG5k–Fmoc–FTS2 was synthesized via solution condensation reactions from PEG methyl ether (mPEG–OH, Mw = 5000 Da) (mPEG5K–OH) (Scheme 1). MPEG5K–OH was reacted with succinate anhydride (5 equiv) in CH2Cl2 overnight, and DMAP (5 equiv) was used as a catalyst. The PEG derivative was precipitated with 10 volumes of cold ether and washed with ether twice. Excess DMAP was removed by additional washes with cold ethanol (Yield = 91%). The carboxy-terminated mPEG (mPEG5K–COOH) was then reacted with tris(hydroxymethyl)aminomethane (Tris) in the presence of NHS (3 equiv) and DCC (3 equiv) in CH2Cl2 for 1 day, followed by a similar purification step as described above (Yield = 92%). The two hydroxyl groups in the PEG-derivatized Tris were blocked by forming acetonide using p-toluenesulfonic acid (TsOH) as a catalyst in acetone. Then, the Fmoc group was coupled to the remaining OH of Tris via reaction with 9-fluorenylmethoxycarbonyl chloride (Fmoc–Cl) (2 equiv) and triethylamine (3 equiv) in CH2Cl2 overnight. PEGylated molecules were similarly purified as described above, and the acetonide group was removed by treatment with 1% TsOH in CH2Cl2 (Yield = 50%, two steps). Finally, FTS (4 equiv) was coupled onto the PEGylated molecules with DCC (4 equiv) and DMAP (0.4 equiv) as the coupling reagents. The reaction mixture was filtered and precipitated with ether and ethanol twice and was concentrated under vacuum (Yield = 85%). The powder was then dissolved in water and filtered through a filter with a pore size of 0.2 μm. The final product was obtained by lyophilizing the filtrate. PEG5k–FTS2 was synthesized following our reported method.14
Scheme 1. Synthesis Scheme of PEG5k–Fmoc–FTS2 Conjugate.

Preparation and Characterization of PTX- and DOX-Loaded Micelles
PTX (10 mM in chloroform) and PEG5k–Fmoc–FTS2 conjugate (10 mM in chloroform) were mixed with various carrier/drug ratios. After chloroform was removed, a thin film of drug/carrier mixture was formed. PTX-loaded micelles were formed by adding Dulbecco’s phosphate-buffered saline (DPBS) to hydrate the thin film followed by gentle votexing. To load DOX into the PEG5k–Fmoc–FTS2 micelle, DOX·HCl was first treated with triethylamine (3 equiv) in a mixture of chloroform (CHCl3)/methanol (MeOH) (1:1, v/v) to remove HCl from DOX·HCl. DOX-loaded PEG5k–Fmoc–FTS2 micelles were then similarly prepared as described above. The PTX loading efficiency was quantified by high performance liquid chromatography (HPLC) as described before.14 The DOX loading efficiency was examined by Waters Alliance 2695 Separations Module combined with Waters 2475 Fluorescence Detector (excitation, 490 nm; emission, 590 nm; gain, 3; sensitivity (FUFS), 10 000). Hibar 250–4 LiChrosorb RP-8 (5 μm) column (sorbent lot no. L59040432) was used, and the mobile phase consisted of acetonitrile/water (52.5:47.5, v/v) with 2.5 mM CH3COONH4 and 0.05% (v/v) CH3COOH. The flow rate of the mobile phase was 1 mL/min, and running time was 12 min. Drug loading capacity (DLC) and drug loading efficiency (DLE) were calculated from the following equations:
The mean diameter, morphology, and size distribution of PEG5k–Fmoc–FTS2 micelles were assessed by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The critical micelle concentration (CMC) of PEG5k–Fmoc–FTS2 micelles was determined using pyrene as a fluorescence probe.34 The in vitro kinetics of DOX release from PEG5k–Fmoc–FTS2 micelles was examined by a dialysis method.35 The hemolytic effect of PEG5k–Fmoc–FTS2 micelles was examined as described.16
Cell Culture and Animals
4T1.2 (mouse metastatic breast cancer cell line), MCF-7 (human breast carcinoma cell line), and A549 (human lung adenocarcinoma epithelial cell line) were obtained from ATCC (VA, USA). HCT116 (human colon carcinoma cell line) was kindly provided by Dr. Lin Zhang (University of Pittsburgh Cancer Institute). All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 5% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37 °C in a humidified 5% CO2 atmosphere. Female BALB/c mice, 4–6 weeks in age, and CD-1 mice, 4–6 weeks in age, were purchased from Charles River (Davis, CA). Male nude mice, 6–8 weeks in age, were purchased from Harlan (Livermore, CA). All animals were housed under pathogen-free conditions according to Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines. All animal-related experiments were performed in full compliance with institutional guidelines and approved by the Animal Use and Care Administrative Advisory Committee at the University of Pittsburgh.
In Vitro Cytotoxicity Study
The cytotoxicity of drugs (PTX and DOX) formulated in PEG5k–Fmoc–FTS2 micelles was assessed with several cancer cell lines and compared to Taxol and free DOX by MTT assay.14 The cytotoxicity of free PEG5k–FTS2, PEG5k–Fmoc–FTS2, and FTS was also examined.
Maximum Tolerated Dose Studies
Groups of four female CD-1 mice were administered intravenously with free DOX (5, 10, 15, 20 mg DOX/kg), DOX-loaded PEG5k–Fmoc–FTS2 micelles (5, 10, 15, 20, 25, 30 mg DOX/kg), Taxol (15, 20, 25 mg PTX/kg), or PTX-loaded PEG5k–Fmoc–FTS2 micelles (50, 75, 100, 120, 140 mg PTX/kg). Changes in body weight and mouse survival were monitored daily for 2 weeks. The maximum tolerated dose (MTD) was defined as the maximal dose that causes neither greater than 15% of body weight loss nor mouse mortality within 2 weeks after administration.16
Near-Infrared Fluorescence Optical Imaging
Nude mice bearing bilateral s.c. PC-3 xenografts were intravenously injected with 200 μL of DiD-loaded PEG5k–Fmoc–FTS2 at a concentration of 0.4 mg/mL. At indicated times (0.5 h, 6 h, 24 h, 48 h, 72 h, and 96 h), the mice were scanned using a Carestream Molecular Imaging System, in Vivo Multispectral FX PRO, with the excitation at 630 nm and the emission at 700 nm using a 30 s exposure time. The mice were anesthetized by isoflurane inhalation before each imaging. After 96 h, the mice were euthanized by CO2 overdose. The tumor and major organs were excised for ex vivo imaging.
Plasma Pharmacokinetics and Tissue Distribution
DOX·HCl and DOX-loaded PEG5k–Fmoc–FTS2 micelles were injected into female BALB/c mice via tail vein at a dose of 5 mg DOX/kg. The blood samples were collected in heparinized tubes at different time points (3 min, 8 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, and 12 h) postinjection. The blood was centrifuged (2500 rpm, 10 min), and plasma was collected for the analysis. DOX in plasma was extracted by extraction buffer (10% Triton X-100, deionized water, and isopropanol at a volumetric ratio of 1:2:15).9 DOX was examined by HPLC using fluorescence detection. Noncompartmental pharmacokinetic analysis was done by WinNonlin. For the tissue biodistribution study, DOX·HCl and DOX-loaded PEG5k–Fmoc–FTS2 were injected into female BALB/c mice bearing 4T1.2 breast tumor at a dose of 5 mg DOX/kg. At 1 day postinjection, tumor tissues and major organs were collected from the mice. The tissues were weighed and homogenized using Homogenizer PowerGen 500 (Fisher Scientific). The tissue solutions were mixed with the extraction buffer, and DOX was extracted overnight at −20 °C. The solutions were centrifuged (2500 rpm, 10 min), and the supernatant was used for HPLC measurement. The concentrations of DOX in tissues were determined based on the standard curve of DOX in blood.
In Vivo Therapeutic Study
Two tumor models (a syngeneic murine breast cancer model (4T1.2) and a human prostate cancer (PC-3) xenograft model) were used to assess the therapeutic activity of PTX or DOX formulated in PEG5k–Fmoc–FTS2 micelles. The breast cancer model was established by inoculation of 4T1.2 cells (1 × 105) in 200 μL of PBS at the right flank of female BALB/c mice. Treatments were started when tumors achieved a volume of ∼50 mm3, and this day was designated as day one. Then tumor-bearing mice were randomly divided into six groups (n = 5) and administered intravenously with PBS (control), PEG5k–Fmoc–FTS2 micelles, Taxol (10 mg PTX/kg), PTX-loaded PEG5k–Fmoc–FTS2 micelles (10, 20 mg PTX/kg), and PTX-loaded PEG5k–FTS2 (10 mg PTX/kg), respectively, on days 1, 3, 5, 8, 11, and 14. Free PEG5k–Fmoc–FTS2 micelles were given at the equivalent dosage of the carrier in the group of PTX-loaded PEG5k–Fmoc–FTS2 micelles (10 mg PTX/kg). The therapeutic effect of DOX-loaded PEG5k–Fmoc–FTS2 micelles (5, 10 mg DOX/kg) was similarly evaluated in 4T1.2 tumor model. Controls include PBS, PEG5k–Fmoc–FTS2 micelles, DOX·HCl (5 mg DOX/kg), liposomal DOX (5 mg DOX/kg), and DOX-loaded PEG5k–FTS2 (5 mg DOX/kg). Tumor sizes were measured with digital caliper three times per week and calculated as V = (L × W2)/2; L = the longest diameter (mm); W = the shortest diameter (mm). Each group was compared by relative tumor volume (RTV) (where RTV equals the tumor volume divided by the initial tumor volume before treatment). Mice were sacrificed when the tumors developed ulceration or reached 2000 mm3. The body weights of all mice from different groups were monitored every 3 days. The antitumor activity of PTX-loaded PEG5k–Fmoc–FTS2 micelles was further evaluated in a human prostate cancer xenograft model, PC-3. Different groups were similarly treated as described above on days 1, 3, 6, 9, and 12. Tumor size and body weight were monitored as described above.
Statistical Analysis
Data are presented as mean ± standard deviation (SD). Statistical analysis was performed by Student’s t test for comparison of two groups, and comparisons for multiple groups were made with one-way analysis of variance (ANOVA), followed by Newman–Keuls test if the overall p-value is <0.05. In all statistical analyses, the threshold of significance was defined as P < 0.05.
Results and Discussion
Preparation and Characterization of Drug-Loaded PEG5k–Fmoc–FTS2 Micelles
PEG5k–Fmoc–FTS2 conjugate, containing one Fmoc motif and two molecules of FTS coupled to one molecule of PEG via a labile ester linkage, was synthesized via solution condensation reactions (Scheme 1and Figure 1). 1H NMR spectrum of PEG5k–Fmoc–FTS2 conjugate is shown in Figure S1 of the Supporting Information, with signals at 3.63 ppm and 7–8 ppm attributable to the methylene protons of PEG and benzene ring protons of Fmoc motif, respectively. Carbon chain and benzene ring signals of FTS were located at 1.5–2.2 ppm and 7–8 ppm, respectively. The molecular weight of the PEG5k–Fmoc–FTS2 conjugate, measured by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry, is close to the theoretical value (6105) (Figure S2, Supporting Information), which indicates the successful synthesis of the PEG5k–Fmoc–FTS2 conjugate.
Figure 1.
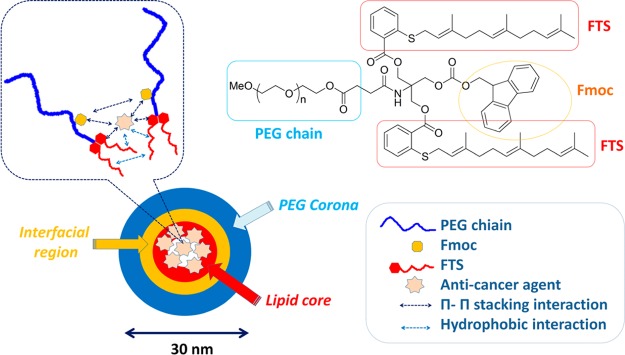
Chemical structure of PEG5k–Fmoc–FTS2 and the postulated model of carrier/drug interaction.
Similar to the PEG5k–FTS2 micellar system, the PEG5k–Fmoc–FTS2 conjugate readily formed micelles in aqueous solution (Figure 2). DLS measurements showed that PEG5k–Fmoc–FTS2 micelles had hydrodynamic sizes around 20 nm at the concentration of 20 mg/mL (Figure 2A). TEM showed spherical particles with a uniform size distribution (Figure 2B). PEG5k–Fmoc–FTS2 micelles were highly effective in solubilizing various anticancer drugs such as PTX, DOX (Tables S1 and S2, Supporting Information), and many others (data not shown). The size, DLC, DLE, and colloidal stability of drug-loaded PEG5k–Fmoc–FTS2 micelles were then examined and compared to those of the counterparts without an Fmoc motif. In general, incorporation of DOX or PTX led to an increase in the particle sizes for both micellar systems; however, the sizes gradually became close to the original size with the increase in the carrier/drug molar ratio. For DOX, a minimal carrier/drug molar ratio of 0.5:1 was needed to formulate the drug in PEG5k–FTS2 micelles (Table S1, Supporting Information). In contrast, DOX could be loaded into PEG5k–Fmoc–FTS2 micelles at a carrier/drug molar ratio as low as 0.1:1. The DLC for DOX/PEG5k–Fmoc–FTS2 mixed micelles at this carrier/drug ratio is 32.8%, which represents a 3.2-fold increase compared to the PEG5k–FTS2 formulation. In addition, DOX/PEG5k–Fmoc–FTS2 mixed micelles were significantly more stable than the counterparts without an Fmoc motif under all carrier/drug ratios examined (Table S1, Supporting Information). Similarly, incorporation of an Fmoc motif significantly improved the performance of PEG5k–FTS2 micelles in formulating PTX (Table S2, Supporting Information). The DLC for PTX/PEG5k–Fmoc–FTS2 mixed micelles was 12.1%, which is a 2.7-fold increase compared to the counterpart without an Fmoc. The morphology and size uniformity were largely retained following loading of PTX (Figure 2C,D) or DOX (Figures 2E,F) at a respective molar ratio of 2.5:1 and 1:1. The improvements in drug-loading capacity and formulation stability are likely due to an enhanced drug/carrier interaction. Fmoc group contains a bulky, fused fluorenylmethyl ring structure capable of providing strong hydrophobic interaction and forming π–π stacking with compounds that carry aromatic moieties (Figure 1, unpublished data). Thus, in addition to hydrophobic interaction, PEG5k–Fmoc–FTS2 can further interact with PTX or DOX through π–π stacking, which leads to improved carrier/drug compatibility.
Figure 2.

Size distribution and morphology of (A, B) free drug PEG5k–Fmoc–FTS2 micelles, (C, D) PTX-loaded PEG5k–Fmoc–FTS2 micelles, and (E, F) DOX-loaded PEG5k–Fmoc–FTS2 micelles measured by DLS and TEM, respectively. Drug concentration in micelle was kept at 1 mg/mL. Blank micelle concentration was 20 mg/mL.
The CMC of PEG5k–Fmoc–FTS2 micelles was examined using pyrene as a fluorescence probe and was found to be 0.2 μM (Figure S3, Supporting Information), which is lower than that of PEG5k–FTS2 micelles (0.68 μM).14. This is likely due to the fact that Fmoc can not only enhance the carrier/drug interaction, but also facilitate the interaction among the carrier molecules themselves. The reduced CMC shall improve the stability of PEG5k–Fmoc–FTS2 micelles upon dilution in vivo.
In Vitro Drug Release Study
The profile of DOX release from PEG5k–Fmoc–FTS2 micelles was examined by a dialysis method and compared to that of DOX-loaded PEG5k–FTS2 micelles. For the initial 8 h, about 50.7% of DOX was released from PEG5k–FTS2 formulation, while only 38.3% of DOX was released from DOX-loaded PEG5k–Fmoc–FTS2 micelles (Figure 3). Overall, DOX formulated in PEG5k–Fmoc–FTS2 micelles exhibited a slower relative rate of DOX release compared to the counterpart without an Fmoc motif.
Figure 3.
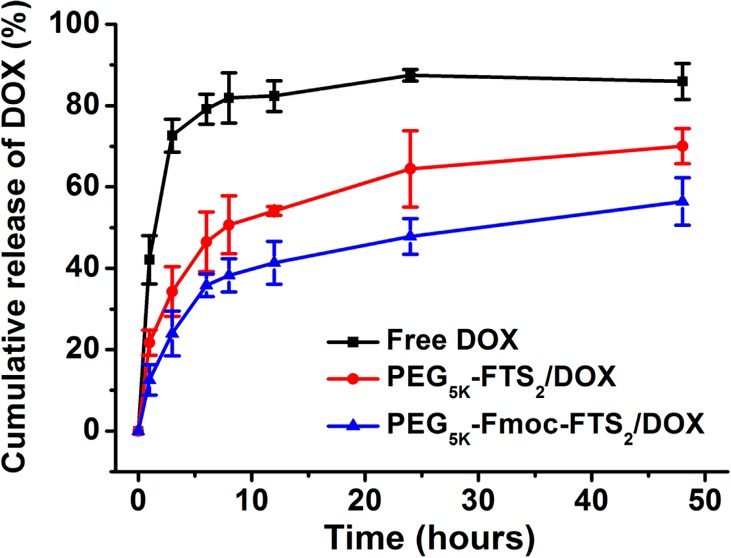
Cumulative DOX release profile from free DOX, DOX-loaded PEG5k–FTS2, and PEG5k–Fmoc–FTS2 micelles. DPBS (pH = 7.4) containing 0.5% (w/v) Tween 80 was used as the release medium. Values reported are the means ± SD for triplicate samples.
Hemolytic Effect of Micelles
Figure S4 of the Supporting Information shows the hemolytic activities of drug-free PEG5k–Fmoc–FTS2 micelles and polyethylenimine (PEI), a cationic polymer known to have significant hemolytic effect.38 PEI induced hemolysis in a dose-dependent manner. In contrast, PEG5k–Fmoc–FTS2 micelles showed only negligible levels of hemolytic activity at the same experimental concentrations, which suggests a safe profile of PEG5k–Fmoc–FTS2 micelles.
In Vitro Cytotoxicity of Free and Drug-Loaded Micelles
The cytotoxicity of drug-free PEG5k–Fmoc–FTS2 and PEG5k–FTS2 micelles was examined in 4T1.2 and HCT-116 tumor cells and compared to free FTS (Figure 4A,B). Free FTS inhibited the tumor cell growth in a concentration-dependent manner. The two FTS conjugates were comparable but slightly less active than free FTS in cytotoxicity toward both 4T1.2 and HCT-116 tumor cells (Figures 4A,B). The cytotoxicity of PEG5k–Fmoc–FTS2 is unlikely to be attributed to the surface activity since PEG–Fmoc–FTS2 micelles showed minimal hemolytic activity at the concentrations that were much higher than those used in the cytotoxicity study (Figure S4, Supporting Information). It is likely that the cytotoxicity of PEG–FTS comes from the released FTS following intracellular delivery. This is supported by the observation that a PEG5k–FTS2 conjugate with a relatively labile ester linkage is more active than a similar conjugate with a relatively stable amide linkage.14
Figure 4.
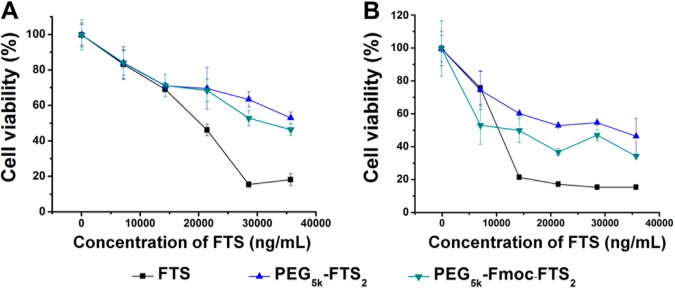
Cytotoxicity of drug-free PEG5k–FTS2 and PEG5k–Fmoc–FTS2 micelles compared to FTS against (A) 4T1.2 mouse breast cancer cell line and (B) HCT-116 human colon carcinoma cell line.
Figure 5, panels A–C show the cytotoxicity of Taxol and PTX formulated in PEG5k–Fmoc–FTS2 micelles in several cancer cell lines. Taxol inhibited the tumor cell growth in a concentration-dependent manner. Delivery of PTX via PEG5k–Fmoc–FTS2 micelles led to a significant increase in the cytotoxicity at low concentrations. We also tested the cytotoxicity of DOX formulated in PEG5k–Fmoc–FTS2 micelles and compared to free DOX·HCl. Similarly, DOX-loaded PEG5k–Fmoc–FTS2 micelles showed more potent cytotoxicity than did free DOX at the low concentration range (Figure 5D–F). When PTX or DOX was formulated into the PEG5k–Fmoc–FTS2 micelles, they showed higher levels of cytotoxicity to MCF-7, A549, and HCT-116 cancer cells compared to Taxol formulation or free DOX (Figure 5A–F), which suggests that PTX or DOX can be more effectively delivered into tumor cells by PEG5k–Fmoc–FTS2 micelles, which will result in enhanced tumor cell killing.
Figure 5.

Anticancer effect of Taxol, drug-free, and PTX-loaded PEG5k–Fmoc–FTS2 micelles was performed on (A) MCF-7 human breast carcinoma cell line, (B) A549 human lung adenocarcinoma epithelial cell line, and (C) HCT-116 human colon carcinoma cell line. Tumor cell killing of free DOX, drug-free, and DOX-loaded PEG5k–Fmoc–FTS2 micelles was performed in (D) MCF-7 human breast carcinoma cell line, (E) A549 human lung adenocarcinoma epithelial cell line, and (F) HCT-116 human colon carcinoma cell line. Cells were treated for 72 h, and cytotoxicity was determined by MTT assay. Values reported are the means ± SD for triplicate samples.
Maximum Tolerated Dose Studies
Groups of four female CD-1 mice were administered intravenously a single dose of either Taxol (15–25 mg PTX/kg) or PTX-loaded PEG5k–Fmoc–FTS2 micelles (50–140 mg PTX/kg). Taxol was well tolerated at the dose of 15 mg PTX/kg (Table S3, Supporting Information); however, an increase of the PTX dosage to 20 mg/kg resulted in the death of one out of four treated mice (Table S3, Supporting Information). For the mice treated with PTX-loaded PEG5k–Fmoc–FTS2 micelles, there was neither significant weight loss nor noticeable changes in normal activity at a PTX dosage as high as 140 mg/kg (Table S3, Supporting Information). Therefore, the maximum tolerated dose (MTD) for PTX/PEG5k–Fmoc–FTS2 is greater than 140 mg/kg, which is significantly higher than that of Taxol (15–20 mg/kg). Figure S5 of the Supporting Information shows the results of the MTD study for DOX-loaded PEG5k–Fmoc–FTS2 micelles. There were no obvious body weight loss or other toxicity signs in the mice treated with DOX-loaded PEG5k–Fmoc–FTS2 micelles at the doses of 5–15 mg DOX/kg within 2 weeks. Only a moderate body weight loss (<10%) was found in mice treated with 20 mg/kg of DOX/PEG5k–Fmoc–FTS2 mixed micelles on day four, which was recovered on day five. In contrast, although free DOX was well tolerated at the dose of 10 mg/kg, it caused a significant decrease (>15%) in the body weight and eventually the death of all treated mice at a dose of 15 mg DOX/kg (Table S3, Supporting Information). Thus, the MTDs of DOX-loaded PEG5k–Fmoc–FTS2 micelles and free DOX are about 20 mg/kg and 10 mg/kg, respectively. The improved safety profile of the PEG5k–Fmoc–FTS2 formulation is likely due to a slower rate of drug release from the micelles before they reach the tumor site, which leads to reduced drug uptake by normal tissues.
Near-Infrared Fluorescence Optical Imaging
Biodistribution and tumor-targeting efficiency of PEG5k–Fmoc–FTS2 micelles were tested in a human prostate cancer xenograft model (PC-3). DiD, a hydrophobic near-infrared fluorescence (NIRF) dye with high penetration, low tissue absorption, and scattering, was loaded into the PEG5k–Fmoc–FTS2 micelles for tissue imaging. Figure 6, panel A shows that DiD-loaded PEG5k–Fmoc–FTS2 micelles were able to accumulate at the tumor as early as 6 h postinjection, and these micelles retained in the tumor at 96 h after injection. No obvious tumor accumulation was observed in the mice treated with free DiD dye.7 After the last imaging at 96 h postinjection, tumors and major organs were excised for ex vivo imaging. As shown in Figure 6, panels B and C, significantly higher levels of signal were observed in the tumor tissues compared to normal organs, except the lung. The very small size of our micellar system may contribute significantly to the effective tumor localization (Figure 6A). It has been reported that the size of the particles must be within sub-100 nm range for them to efficiently reach the poorly vascularized tumors.36 Such small sizes are also critical to enable the particles effective in deep penetration into the tumor tissues, especially the tumors with a tough tangle of collagen such as pancreatic and some breast cancers.37 Interestingly, little fluorescence signal was observed in the liver and spleen, the two major internal organs that are involved in the nonspecific clearance of nanoparticles. The low signal in the liver may be due to a combination of (a) very low uptake of the particles because of their small sizes and (b) rapid metabolism of the particles in the liver.
Figure 6.

(A) In vivo and (B) ex vivo NIRF imaging of DiD-loaded PEG5k–Fmoc–FTS2 micelles in prostate cancer PC-3 xenograft-bearing mice. (C) Quantitative fluorescence intensities of tumors and major organs from ex vivo images.
Plasma Pharmacokinetics and Tissue Distribution
Free DOX·HCl and DOX-loaded PEG5k–Fmoc–FTS2 micelles were injected to tumor-free mice at a dose of 5 mg/kg, and the DOX concentration in plasma was measured at different time points. At this dose, the initial plasma concentration of the carrier is ∼0.6 μM, which is significantly higher than its CMC (0.2 μM). Figure 7, panel A shows the percentage of injected dose of DOX in the blood over time following intravenous administration. The pharmacokinetic parameters were calculated based on a noncompartment model and are summarized in Table 1. The T1/2 of DOX in DOX-loaded PEG5k–Fmoc–FTS2 micelles was 6.67-fold higher than that of free DOX. Furthermore, the plasma AUC0-inf of DOX for the micellar DOX was almost 19-fold higher than that of free DOX (64.8 μg × h/mL vs 3.43 μg × h/mL). In contrast, the Vd of DOX for micellar DOX was significantly lower than that of free DOX (2.58 L/kg vs 7.34 L/kg), which suggests prolonged blood circulation of DOX-loaded PEG5k–Fmoc–FTS2 micelles.
Figure 7.
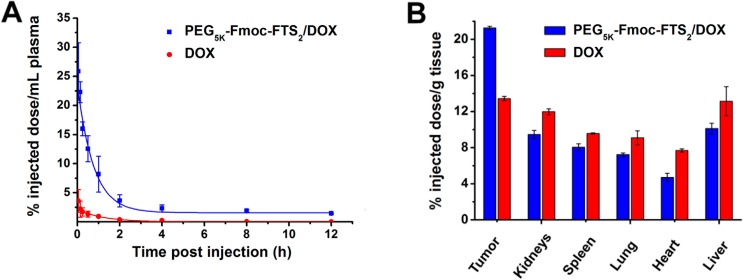
(A) Blood retention kinetics of DOX·HCl and DOX-loaded PEG5k–Fmoc–FTS2 micelles in mice. DOX·HCl and DOX- loaded PEG5k–Fmoc–FTS2 micelles were injected into female BALB/c mice via tail vein at a dose of 5 mg DOX/kg. (B) Tissue distribution of DOX 1 day following the injection. DOX·HCl and DOX-loaded PEG5k–Fmoc–FTS2 were injected into female BALB/c mice bearing 4T1.2 breast tumor at the dose of 5 mg DOX/kg, respectively. Values are means ± SEM.
Table 1. Pharmacokinetic Variables of Free DOX and DOX-Loaded PEG5k–Fmoc–FTS2 Micelles.
| groups | T1/2 (h) | AUC0-inf (μg × h/mL) | Cmax (μg/mL) | CL (L/h/kg) | Vd (L/kg) |
|---|---|---|---|---|---|
| DOX | 1.74 | 3.43 | 3.59 | 2.91 | 7.34 |
| PEG5k–Fmoc–FTS2/DOX | 11.60 | 64.80 | 25.84 | 0.15 | 2.58 |
The tissue distribution of DOX·HCl and DOX-loaded PEG5k–Fmoc–FTS2 micelles was also investigated in female BALB/c mice bearing 4T1.2 breast tumor. Free DOX·HCl and DOX-loaded PEG5k–Fmoc–FTS2 micelles were injected at the same DOX dose of 5 mg/kg. At 24 h postinjection, major organs and tumors were excised for DOX determination. As shown in Figure 7, panel B, there was about two-fold increase in the tumor uptake of DOX for DOX-loaded PEG5k–Fmoc–FTS2 micelles compared to free DOX. The enhanced DOX accumulation is likely due to the very small size of the DOX-loaded micelles and their excellent stability. In addition, DOX-loaded PEG5k–Fmoc–FTS2 micelles were associated with a reduced DOX accumulation in normal organs such as the heart compared to free DOX (Figure 7B). These findings showed that DOX-loaded PEG5k–Fmoc–FTS2 can not only increase the tumor–target efficacy of DOX, but also decrease DOX-associated cardiotoxicity.
In Vivo Therapeutic Study
The antitumor activity of PTX- and DOX-loaded PEG5k–Fmoc–FTS2 micelles was first investigated in a syngeneic murine breast cancer model (4T1.2). As shown in Figure 8, panel A, free PEG5k–Fmoc–FTS2 micelles alone showed no effects in inhibiting the tumor growth at the dose used. Taxol formulation showed a modest effect in inhibiting the tumor growth at a dose of 10 mg PTX/kg. In contrast, PTX formulated in PEG5k–Fmoc–FTS2 micelles showed a much more pronounced antitumor activity at the same dosage. It is also apparent that PTX/PEG5k–Fmoc–FTS2 mixed micelles were more active than was the counterpart without an Fmoc motif (P < 0.05). By increasing the PTX dosage to 20 mg/kg, a further improvement in the therapeutic effect resulted.
Figure 8.

(A) Enhanced antitumor activity of PTX formulated in PEG5k–Fmoc–FTS2 micelles in a syngeneic murine breast cancer model (4T1.2). (B) Changes of body weight in mice that received different treatments; P < 0.01 (10 mg PTX/kg PEG5k–Fmoc–FTS2 vs Taxol), P < 0.05 (10 mg PTX/kg PEG5k–Fmoc–FTS2 vs 10 mg PTX/kg PEG5k–FTS2). (C) Enhanced antitumor activity of DOX formulated in PEG5k–Fmoc–FTS2 micelles in a syngeneic murine breast cancer model (4T1.2) compared to DOX·HCl and DOX-loaded liposome. (D) Changes of body weight in mice that received different treatments; P < 0.01 (5 mg DOX/kg PEG5k–Fmoc–FTS2 vs DOX·HCl), P < 0.01 (5 mg DOX/kg PEG5k–Fmoc–FTS2 vs 5 mg DOX/kg liposome).
Figure 8, panel C shows the result of therapy study on DOX/PEG5k–Fmoc–FTS2 mixed micelles in the 4T1.2 tumor model. Both DOX/PEG5k–FTS2 and DOX/PEG5k–Fmoc–FTS2 were significantly more active than were free DOX or liposomal DOX in inhibiting the tumor growth. There was also a trend of improvement in antitumor activity for DOX/PEG5k–Fmoc–FTS2 compared to DOX/PEG5k–FTS2, although it is not statistically significant (P = 0.16). No significant changes were noticed in body weight in all treatment groups compared to the PBS control group (Figure 8B,D).
The in vivo therapeutic activity of PTX formulated in PEG5k–Fmoc–FTS2 micelles was further evaluated in a human prostate cancer xenograft model (PC-3) (Figure 9A). The PC-3 model has a relatively slower growth rate than does the 4T1.2 tumor model, and tumor growth was more effectively controlled by the different treatments in the PC-3 model. Nonetheless, PTX/PEG5k–Fmoc–FTS2 mixed micelles were significantly more effective than either Taxol or PTX/PEG5k–FTS2 mixed micelles in inhibiting the tumor growth at the same dose of 10 mg PTX/kg. Figure 9, panel C shows representative images of nude mice bearing PC-3 tumors treated with PBS and various PTX formulations by day 17 after initial treatment. By day 17, the RTV for PTX/PEG5k–Fmoc–FTS2 mixed micelles was 1.71, while the RTVs for mice treated with Taxol and PTX/PEG5k–FTS2 mixed micelles were 5.2 and 2.9, respectively. An increase of the dose of PTX to 20 mg PTX/kg led to a further improvement in antitumor activity; one out of the five mice in this group became tumor-free after day 38 without further treatment. No weight loss was observed in mice treated with all PTX formulations (Figure 9B), while consistent weight loss was shown in mice treated with PBS or the carrier alone after day nine (Figure 9B).
Figure 9.

(A) Enhanced antitumor activity of PTX formulated in PEG5k–Fmoc–FTS2 micelles in a human prostate cancer xenograft model (PC-3). (B) Changes of body weight in mice that received different treatments; P < 0.01 (10 mg PTX/kg PEG5k–Fmoc–FTS2 vs Taxol), P < 0.01 (10 mg PTX/kg PEG5k–Fmoc–FTS2 vs 10 mg PTX/kg PEG5k–FTS2). (C) Photograph shows representative images of nude mice bearing PC-3 tumors treated with PBS and different PTX formulations by day 17 after initial treatment.
In vivo therapy study demonstrated that significant therapeutic effect can be achieved with minimal toxicity using our PEG5k–Fmoc–FTS2 micellar system (Figures 8 and 9) in both prostate and breast cancer models. The superior antitumor efficacy along with the minimal toxicity of the improved system could be ascribed to the very small size of the micelles and their improved pharmacokinetic profile, which led to effective tumor targeting and reduced nonspecific uptake by normal tissues.
Conclusions
We have developed in this study a simple and well characterized nanomicellar system that consists of an FTS-based hydrophobic domain, a PEG hydrophilic segment, and a drug-interactive Fmoc motif. Both drug loading capacity and formulation stability were significantly improved by inclusion of a drug-interactive Fmoc motif. In contrast to many existing micellar systems that have no favorable biological activity, our PEG5k–Fmoc–FTS2 conjugate well retained the biological activity of FTS. In addition to its antitumor activity, PEG5k–Fmoc–FTS2 synergized with codelivered anticancer agents in inhibiting the cell growth. Pharmacokinetics and biodistribution studies showed that DOX-loaded PEG5k–Fmoc–FTS2 micelles were able to retain DOX in the bloodstream for a prolonged period of time and were highly effective in targeted delivery of DOX to tumors. More importantly, PTX- or DOX-loaded PEG5k–Fmoc–FTS2 micelles led to a superior antitumor activity over other treatments including drugs formulated in PEG5k–FTS2 micelles in both breast cancer and prostate cancer models. PEG5k–Fmoc–FTS2 may represent a promising micellar system for effective delivery of anticancer agents to tumors.
Acknowledgments
This work was supported by NIH Grants No. RO1CA173887, No. R01GM102989, and No. R21CA173887.
Supporting Information Available
Additional figures and tables as discussed in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
¶ X.Z. and Y.H. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zhang X.; Huang Y.; Li S. Nanomicellar carriers for targeted delivery of anticancer agents. Ther. Delivery 2014, 5, 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serajuddin A. T. Salt formation to improve drug solubility. Adv. Drug Delivery Rev. 2007, 59, 603–16. [DOI] [PubMed] [Google Scholar]
- Weiss R. B.; Donehower R. C.; Wiernik P. H.; Ohnuma T.; Gralla R. J.; Trump D. L.; Baker J. R. Jr.; Van Echo D. A.; Von Hoff D. D.; Leyland-Jones B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263–8. [DOI] [PubMed] [Google Scholar]
- El-Rayes B. F.; Ibrahim D.; Shields A. F.; LoRusso P. M.; Zalupski M. M.; Philip P. A. Phase I study of liposomal doxorubicin (Doxil) and cyclophosphamide in solid tumors. Invest. New Drugs 2005, 23, 57–62. [DOI] [PubMed] [Google Scholar]
- Torchilin V. P. Micellar nanocarriers: Pharmaceutical perspectives. Pharm. Res. 2007, 24, 1–16. [DOI] [PubMed] [Google Scholar]
- Lukyanov A. N.; Torchilin V. P. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv. Drug Delivery Rev. 2004, 56, 1273–89. [DOI] [PubMed] [Google Scholar]
- Xiao K.; Luo J.; Li Y.; Lee J. S.; Fung G.; Lam K. S. PEG-oligocholic acid telodendrimer micelles for the targeted delivery of doxorubicin to B-cell lymphoma. J. Controlled Release 2011, 155, 272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K.; Li Y.; Lee J. S.; Gonik A. M.; Dong T.; Fung G.; Sanchez E.; Xing L.; Cheng H. R.; Luo J.; Lam K. S. “OA02” peptide facilitates the precise targeting of paclitaxel-loaded micellar nanoparticles to ovarian cancer in vivo. Cancer Res. 2012, 72, 2100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Y.; Kim S.; Tyler J. Y.; Park K.; Cheng J. X. Blood-stable, tumor-adaptable disulfide bonded mPEG-(Cys)4-PDLLA micelles for chemotherapy. Biomaterials 2013, 34, 552–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan K.; Zhang X.; Tang X.; Yu J.; Liu S.; Wang D.; Li Y.; Huang J. Fabrication of cationic nanomicelle from chitosan-graft-polycaprolactone as the carrier of 7-ethyl-10-hydroxy-camptothecin. Colloids Surf., B 2010, 76, 475–82. [DOI] [PubMed] [Google Scholar]
- Bae Y.; Kataoka K. Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers. Adv. Drug Delivery Rev. 2009, 61, 768–84. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Liu K.; Huang Y.; Xu J.; Li J.; Ma X.; Li S. Reduction-sensitive dual functional nanomicelles for improved delivery of paclitaxel. Bioconjugate Chem. 2014, 25, 1689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Zhang X.; Lu J.; Huang Y.; Li J.; Li S. Targeted delivery of curcumin to tumors via PEG-derivatized FTS-based micellar system. AAPS J. 2014, 16, 600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Lu J.; Huang Y.; Zhao W.; Chen Y.; Li J.; Gao X.; Venkataramanan R.; Sun M.; Stolz D. B.; Zhang L.; Li S. PEG-farnesylthiosalicylate conjugate as a nanomicellar carrier for delivery of paclitaxel. Bioconjugate Chem. 2013, 24, 464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Huang Y.; Zhao W.; Chen Y.; Li J.; Gao X.; Venkataramanan R.; Li S. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Mol. Pharmaceutics 2013, 10, 2880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Huang Y.; Zhao W.; Marquez R. T.; Meng X.; Li J.; Gao X.; Venkataramanan R.; Wang Z.; Li S. PEG-derivatized embelin as a nanomicellar carrier for delivery of paclitaxel to breast and prostate cancers. Biomaterials 2013, 34, 1591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Huang Y.; Zhao W.; Chen Y.; Zhang P.; Li J.; Venkataramanan R.; Li S. PEG-farnesyl thiosalicylic acid telodendrimer micelles as an improved formulation for targeted delivery of paclitaxel. Mol. Pharmaceutics 2014, 11, 2807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloog Y.; Cox A. D. RAS inhibitors: Potential for cancer therapeutics. Mol. Med. Today 2000, 6, 398–402. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [DOI] [PubMed] [Google Scholar]
- Asher W. M.; Parvin S.; Virgillo R. W.; Haber K. Echographic evaluation of splenic injury after blunt trauma. Radiology 1976, 118, 411–5. [DOI] [PubMed] [Google Scholar]
- Cox A. D.; Der C. J. Ras family signaling: Therapeutic targeting. Cancer Biol. Ther. 2002, 1, 599–606. [DOI] [PubMed] [Google Scholar]
- Niv H.; Gutman O.; Henis Y. I.; Kloog Y. Membrane interactions of a constitutively active GFP-Ki-Ras 4B and their role in signaling. Evidence from lateral mobility studies. J. Biol. Chem. 1999, 274, 1606–13. [DOI] [PubMed] [Google Scholar]
- Rotblat B.; Ehrlich M.; Haklai R.; Kloog Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: A potential treatment for cancer. Methods Enzymol. 2008, 439, 467–89. [DOI] [PubMed] [Google Scholar]
- Mor A.; Aizman E.; Kloog Y. Celecoxib enhances the anti-inflammatory effects of farnesylthiosalicylic acid on T cells independent of prostaglandin E(2) production. Inflammation 2012, 35, 1706–14. [DOI] [PubMed] [Google Scholar]
- Mor A.; Aizman E.; Chapman J.; Kloog Y. Immunomodulatory properties of farnesoids: The new steroids?. Curr. Med. Chem. 2013, 20, 1218–24. [DOI] [PubMed] [Google Scholar]
- Kraitzer A.; Kloog Y.; Haklai R.; Zilberman M. Composite fiber structures with antiproliferative agents exhibit advantageous drug delivery and cell growth inhibition in vitro. J. Pharm. Sci. 2011, 100, 133–49. [DOI] [PubMed] [Google Scholar]
- Huh K. M.; Lee S. C.; Cho Y. W.; Lee J.; Jeong J. H.; Park K. Hydrotropic polymer micelle system for delivery of paclitaxel. J. Controlled Release 2005, 101, 59–68. [DOI] [PubMed] [Google Scholar]
- Kim J. Y.; Kim S.; Pinal R.; Park K. Hydrotropic polymer micelles as versatile vehicles for delivery of poorly water-soluble drugs. J. Controlled Release 2011, 152, 13–20. [DOI] [PubMed] [Google Scholar]
- Saravanakumar G.; Min K. H.; Min D. S.; Kim A. Y.; Lee C. M.; Cho Y. W.; Lee S. C.; Kim K.; Jeong S. Y.; Park K.; Park J. H.; Kwon I. C. Hydrotropic oligomer-conjugated glycol chitosan as a carrier of paclitaxel: Synthesis, characterization, and in vivo biodistribution. J. Controlled Release 2009, 140, 210–7. [DOI] [PubMed] [Google Scholar]
- Gao X.; Huang Y.; Makhov A. M.; Epperly M.; Lu J.; Grab S.; Zhang P.; Rohan L.; Xie X. Q.; Wipf P.; Greenberger J.; Li S. Nanoassembly of surfactants with interfacial drug-interactive motifs as tailor-designed drug carriers. Mol. Pharmaceutics 2013, 10, 187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Lu J.; Huang Y.; Zhao W.; Zhang Y.; Zhang X.; Li J.; Venkataramanan R.; Gao X.; Li S. Design and evaluation of a PEGylated lipopeptide equipped with drug-interactive motifs as an improved drug carrier. AAPS J. 2014, 16, 114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Huang Y.; Liu H.; Marquez R. T.; Lu J.; Zhao W.; Zhang X.; Gao X.; Li J.; Venkataramanan R.; Xu L.; Li S. A PEG-Fmoc conjugate as a nanocarrier for paclitaxel. Biomaterials 2014, 35, 7146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano D.; Ben-Baruch G.; Marom M.; Egozi Y.; Haklai R.; Kloog Y. Farnesyl derivatives of rigid carboxylic acids-inhibitors of Ras-dependent cell growth. J. Med. Chem. 1995, 38, 1267–72. [DOI] [PubMed] [Google Scholar]
- La S. B.; Okano T.; Kataoka K. Preparation and characterization of the micelle-forming polymeric drug indomethacin-incorporated poly(ethylene oxide)-poly(beta-benzyl l-aspartate) block copolymer micelles. J. Pharm. Sci. 1996, 85, 85–90. [DOI] [PubMed] [Google Scholar]
- Xiong X. B.; Huang Y.; Lu W. L.; Zhang X.; Zhang H.; Nagai T.; Zhang Q. Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J. Controlled Release 2005, 107, 262–75. [DOI] [PubMed] [Google Scholar]
- Cabral H.; Matsumoto Y.; Mizuno K.; Chen Q.; Murakami M.; Kimura M.; Terada Y.; Kano M. R.; Miyazono K.; Uesaka M.; Nishiyama N.; Kataoka K. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–23. [DOI] [PubMed] [Google Scholar]
- Bourzac K. Nanotechnology: Carrying drugs. Nature 2012, 491, S58–60. [DOI] [PubMed] [Google Scholar]
- Reul R.; Nguyen J.; Kissel T. Amine-modified hyperbranched polyesters as non-toxic, biodegradable gene delivery systems. Biomaterials 2009, 30, 5815–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.