

Abstract
The skin is the front line of defense against insult and injury and contains many epidermal and immune elements that comprise the skin-associated lymphoid tissue (SALT). The reaction of these components to injury allows an effective cutaneous response to restore homeostasis. Psoriasis vulgaris is the best-understood and most accessible human disease that is mediated by T cells and dendritic cells. Inflammatory myeloid dendritic cells release IL-23 and IL-12 to activate IL-17-producing T cells, Th1 cells, and Th22 cells to produce abundant psoriatic cytokines IL-17, IFN-γ, TNF, and IL-22. These cytokines mediate effects on keratinocytes to amplify psoriatic inflammation. Therapeutic studies with anticytokine antibodies have shown the importance of the key cytokines IL-23, TNF, and IL-17 in this process. We discuss the genetic background of psoriasis and its relationship to immune function, specifically genetic mutations, key PSORS loci, single nucleotide polymorphisms, and the skin transcriptome. The association between comorbidities and psoriasis is reviewed by correlating the skin transcriptome and serum proteins. Psoriasis-related cytokine-response pathways are considered in the context of the transcriptome of different mouse models. This approach offers a model for other inflammatory skin and autoimmune diseases.
Keywords: healthy skin, keratinocyte, T cells, dendritic cells, DCs, skin-associated lymphoid tissue, molecular studies, mouse models
Introduction
Psoriasis vulgaris is an inflammatory skin disease mediated by the cells and molecules of both the innate and adaptive immune systems, but with key responses of normal skin cells to associated products. In many ways, the immune pathways that become activated in psoriasis represent amplifications of background immune circuits that exist as constitutive or inducible pathways in normal human skin. These include epidermal keratinocytes as key participants in innate immunity, which can induce and switch classes of T cells that are recruited to the skin. Because skin must be considered as an immune-competent tissue with a dedicated T cell population, we introduce this review with a description of relevant immune properties of healthy human skin.
Healthy Skin
Structure of Skin
Skin consists of three major tissue segments. The epidermis is composed mainly of keratinocytes, but with an integral population of dendritic antigen-presenting cells termed Langerhans cells (LCs). The dermis is largely composed of collagenous connective tissue with blood vessels, but it also contains numerous other cell types including immune cells and has several associated appendages such as hair follicles, sweat glands, and sebaceous glands. The adipose tissue or subcutis makes up the third layer.
As illustrated in Figure 1a, the epidermis is formed by keratinocytes that undergo a progressive differentiation program (homeostatic growth) in which proliferative cells in the basal layer differentiate into spinous and then granular keratinocytes. Granular layer keratinocytes express several molecules associated with innate immunity, such as antimicrobial peptides (AMPs) including S100A7 (psoriasin), S100A8 (calgranulin A), S100A9 (calgranulin B), lipocalin 2, β-defensin, and cathelicidin (CAMP/LL-37) (1, 2). With further differentiation, granular keratinocytes transition to corneocytes that have lost their nuclei, but they gain a cross-linked protein membrane structure termed the cornified envelope, between which many layers of neutral lipids are deposited. Effectively, the cornified layer creates a physical barrier to outward water loss as well as to inward bacterial penetration.
Figure 1.

Components of healthy and inflamed skin. (a) The epidermis is formed by slowly differentiating keratinocytes. In granular layer keratinocytes, antimicrobial peptides (AMPs) may be stored, including S100A7, S100A8, S100A9, β-defensins, cathelicidin (CAMP/LL-37), and lipocalin 2 (LCN2). The nucleus is lost as granular keratinocytes transition to corneocytes, and a cross-linked protein membrane structure termed the cornified envelope is formed, between which many layers of neutral lipids are deposited. This produces an effective water-impermeable barrier. The epidermis contains Langerhans cells (LCs) that are immature antigen-presenting cells, and the dermis contains resident myeloid dendritic cells (DCs). Although there are nonrecirculating cutaneous lymphocyte antigen (CLA)+ resident memory T cells (Trm cells) in the skin, keratinocytes constitutively synthesize CCL27 (CTACK), which is the major chemokine that attracts CCR10+ CLA+ skin-homing T cells into noninflamed skin for immune surveillance. These components maintain steady-state cutaneous immunity or, effectively, a state of tolerance. (b) The epidermis can also participate in innate or adaptive immune responses to triggers such as injury, infection, or cytokine stimulation. Keratinocytes may ① proliferate in response to cytokines such as IL-22 to accelerate loss of surface keratinocytes and eliminate pathogens, ② increase synthesis of innate effector molecules such as AMPs, and ③ direct migration of new T cell subsets and other immune effector cells into the skin through production of chemokines. (Additional abbreviations used in figure: IFN-α, interferon-α; TNF, tumor necrosis factor; TSLP, thymic stromal lymphopoietin; T17, IL-17-producing CD4+ and CD8+ T cells; Th, T helper cells.)
Skin-Associated Lymphoid Tissue
For many years, normal human skin has been recognized as a specialized lymphoid tissue, and the cellular immune elements have been termed SALT (skin-associated lymphoid tissue), first described by Streilein in 1983 (3–6). Healthy skin is in an effective state of immune tolerance. Normal human skin contains a large population of cutaneous lymphocyte antigen (CLA)+ effector memory T cells termed resident-memory T cells (Trm), that are believed to be nonrecirculating T cells that mediate protective immunity in the skin. Nearly all these CD3+ T cells bear the skin migratory receptor CLA, which enables binding to E-selectin expressed on cutaneous blood vessels, and are CD45RO+ CCR4+ T cells (7). Calculations of the number of T cells in healthy skin suggest that there are 1 × 106 T cells/cm2 of normal skin and 2 × 1010 T cells in the entire skin surface, nearly twice the number in the circulation (8).
Keratinocytes constitutively synthesize CCL27 (CTACK), which is the major chemokine that attracts CCR10+ T cells into noninflamed skin for immune surveillance (9, 10). Approximately 10% of T cells in the peripheral circulation of adults have become differentiated for skin homing/protective immunity through expression of CLA, and these represent a mixture of central memory and effector memory cells that respond to different sets of chemokines. Hence, healthy skin contains abundant resident T cells and the capacity to recruit additional recirculating T cells.
Figure 1b diagrams how the epidermis may participate in innate or adaptive immune responses, either by directing migration of new T cell subsets into the skin through production of cytokines or by increased synthesis of innate effector molecules directly in keratinocytes (5). For example, epidermal injury can trigger high-level production of CCL20 in keratinocytes (11), and this chemokine then has the ability to attract CD11c+ myeloid dendritic cells (DCs) into the dermis as well as CCR6+ IL-17-producing T cells into sites of injury. Alternatively, infection may activate innate immune pathways leading to production of TNF or IFN-α in the skin. These cytokines then have the ability to induce a somewhat differing array of chemokines in epidermal keratinocytes, which would control recruitment of specific leukocyte effector populations. For example, TNF serves to induce CCL20, which leads to myeloid DCs and T17 recruitment (IL-17-producing CD4+ and CD8+ T cells) as well as to neutrophil recruitment by keratinocyte-derived CXCL chemokines. IFN-γ can induce CXCL10 and CXCL11 in keratinocytes, leading to recruitment of Th1 cells, while also increasing synthesis of Mx-1 and other antiviral gene products in keratinocytes (12). Activated keratinocytes can also synthesize thymic stromal lymphopoietin (TSLP), which is considered a likely initiator of Th2-centered immune responsesin the skin (13).
Whether the keratinocytes are capable of initiating inflammation or are simply responders to the cytokine milieu is the subject of much debate. Keratinocytes can upregulate molecules such as HLA-DR and may thus present antigens to T cells; however, they cannot act as true antigen-presenting cells as they cannot deliver the signal 2 that activates antigen-specific T cells. Overall, the epidermis responds to and integrates different danger signals, and can orchestrate defined innate and adaptive immune responses.
Keratinocytes Amplify Specific Cytokine Signals from T Cells
Activation of skin-resident T cells could lead to the production of IFN-γ, IL-17, or IL-22, depending on the nature of the activation stimulus or antigen. This can lead to induction of cytokine-specific chemokines, which then amplify specific effector responses. In this manner, Th1 activation, leading to increased production of IFN-γ, induces synthesis of chemokines (CXCL9, CXCL10, and CXCL11) that can recruit more Th1 cells. Likewise, activation of IL-17-producing T cells, leading to IL-17 release, activates CCL20, CXCL1, CXCL2, and CXCL8/IL-8 synthesis, leading to recruitment of more IL-17-producing T cells and neutrophils into the skin. IL-17 is also a strong inducer for synthesis of AMPs in keratinocytes (12). Activation of Th22 cells results in increased production of IL-22, which can induce keratinocyte hyperplasia (14). This causes a switch into an epidermal regenerative growth pathway (with increased synthesis of S100 proteins and other AMPs), and it causes faster growth of keratinocytes, which leads to accelerated loss of surface keratinocytes and elimination of pathogens (Figure 1b).
Langerhans Cells in Healthy Skin
The epidermis contains LCs that are immature antigen-presenting cells (Figure 2a). If these LCs become activated by antigensor cytokines, they can migrate out of the epidermis and carry antigens to draining lymph nodes to activate T cell responses. The changing views on the role of LCs were recently thoroughly reviewed by Romani and colleagues (15). Initially, LCs were considered to be nerve endings, or “effete” melanocytes; then studies in the 1980s led to an appreciation that they can produce an effective immune response. LCs in human skin preferentially activate Th2 and Th22 cells, based on ex vivo functional analyses (16). In a series of elegant experiments coculturing epidermal CD1a+ LCs and T cells from healthy skin of the same donor, Seneschal et al. (17) showed that LCs could induce are gulatory T cell (Treg) phenotype (CD3+CD4+CD25hiFoxp3+). However, when the LCs were pulsed with increasing doses of Candida albicans, more effector T cells and fewer Tregs were induced. This supports the concept that the type of immune response induced by the LCs depends upon their surrounding environment.
Figure 2.
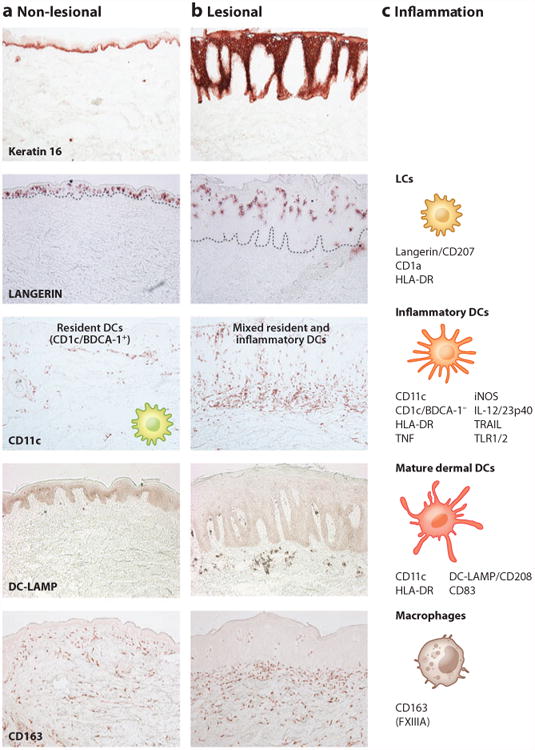
Immune cellular components of normal-appearing and inflamed psoriatic skin. Representative immunohistochemistry of (a) normal-appearing, nonlesional skin of psoriasis patients and (b) lesional psoriasis skin is shown, as is (c) a diagram of immune cellular components and their surface receptors during inflammation. Keratin 16 stains basal epidermis in nonlesional skin but stains full thickness epidermis in psoriasis. Langerin/CD207+ Langerhans cells (LCs) are found scattered in the lower epidermis in nonlesional skin and are found higher up in the thicker epidermis of psoriasis lesions. LCs are also identified by CD1a and HLA-DR. CD11c+ BDCA-1+ resident myeloid dendritic cells (DCs) are found in the upper dermis in nonlesional skin. During psoriatic inflammation, numbers of CD11c+ inflammatory DCs that are CD1c/BDCA-1− increase in the epidermis and dermis. These cells also express HLA-DR, TNF, iNOS, IL-12/23p40, TRAIL, and TLR1/2. Resident DCs are stable in number between nonlesional and lesional skin and are both CD11c+ and BDCA-1+. However, resident myeloid dermal DCs become more mature in psoriasis, as evidenced by expression of DC-LAMP/CD208. CD163+ macrophages are scattered throughout the dermis in nonlesional skin and increased approximately twofold in lesional skin. These CD163 + cells are also FXIIIA+, although CD163 is preferred as a marker. All images are 10 × magnification. (Additional abbreviations used in figure: BDCA, blood dendritic cell antigen; DC-LAMP, DC-lysosome-associated membrane protein; iNOS, inducible nitric oxide synthase; TLR, Toll-like receptor; TNF, tumor necrosis factor; TRAIL, tumor necrosis factor–related apoptosis-inducing ligand.)
Myeloid Dendritic Cells in Healthy Skin
A resident population of DCs in the dermis (Figure 2a) also contributes to the ability of skin DCs to activate T cells after encountering antigens. In the late 1980s, dermal immune cells were first characterized using a marker to the clotting factor Factor XIIIa (FXIIIA) (18, 19). FXIIIA+ cells were large, “fluffy” cells with dendritic morphology scattered throughout the dermis, and they were called dermal DCs or dermal dendrocytes. However, in recent years, newer antibodies identifying these dendritic dermal cell populations have become available, including the αx integrin CD11c, which identifies interstitial DCs (20). When healthy dermis was stained with CD11c, a different pattern of positive cells was observed in the upper dermis, which contrasted with the FXIIIA+ cells scattered throughout the dermis (21). Two-color immunofluorescence showed that CD11c and FXIIIA indeed identified two distinct populations of cells.
Given that CD11c can identify all myeloid cells with varying levels of expression, a more specific cutaneous DC marker was sought. A panel of blood dendritic cell antigen (BDCA) antibodies was developed (22), with BDCA-1 (CD1c) and BDCA-3 (CD141) identifying distinct circulating myeloid DC subsets. In healthy skin, these two antibodies identify two subpopulations of CD11c+ DCs, with the major subpopulation consisting of BDCA-1+ cells (21). FACS-sorted BDCA-1+ cells from healthy dermis were able to induce minor allogeneic T cell proliferation, but if the DCs were first cultured with DC-maturing cytokines (a cocktail of IL-1β, IL-6, TNF, and PGE2), the T cell proliferation was much more robust. This suggests that BDCA-1+ DCs in steady state are immature, but their responses can be augmented and become more effective when matured.
BDCA-3+ cells were increased twofold after acute narrow-band UV radiation (NB-UVB), suggesting they may have a role in acute response to injury (23). BDCA-3+ cells, the minor CD11c+ DC subset in healthy skin, are capable of cross-presentation (24). Briefly, under normal circumstances, an exogenous antigen (such as bacteria) is taken up by DCs, processed internally, and complexed with MHC class II molecules for presentation with other costimulatory molecules to CD4+ T cells. In contrast, endogenous antigens (such as viral particles) are processed with MHC class I for presentation to CD8+ cytotoxic T cells. During cross-presentation, endogenous antigens are processed for additional CD4+ T cell responses, which may be highly desirable for generating effective and long-lived immune responses (25). Although initial studies suggested that circulating BDCA-3+ DCs were the most efficient cross-presenting subset of DCs (24), more recent studies have shown that both BDCA-1+ and BDCA-3+ DCs from human tonsils have this capability (26). This reinforces the concept that plasticity is a general feature of DCs and that they can behave as the conditions require, rather than having a predefined T cell stimulatory capacity.
However, not all investigators use the same DC markers, and it can be quite difficult to interpret and compare studies. The choice of antibody clones to identify cell subsets and methods of obtaining cells from skin for studies may influence results. Some investigators use CD14 and CD1a to identify two subsets of dermal DCs in healthy skin (27–29). CD14 identifies a minor subset of DCs that appear to drive T cell help for B cell and plasma cell responses. However, dermal CD14+ cells may also express BDCA-1 and BDCA-3 (30). The CD1a+ dermal myeloid DC subset is also BDCA-1+ (30), but as CD1a is also an epidermal LC marker, BDCA-1 may be more suitable for the dermal subset as it shows more selectivity here.
Macrophages in Healthy Skin
While searching for an improved marker for the FXIIIA+ cells, we found that CD163 is a much more robust marker of this population (Figure 2a) (21). CD163 is a myeloid cell hemoglobin/haptoglobin scavenger receptor that binds TWEAK (31). Ligation of this marker produces heme oxygenase and IL-10. Cutaneous CD163+ cells have electron microscopic features of macrophages and ingest tattoo pigment (unlike BDCA-1+ cells), supporting a phagocytic role for these cells (21). Cutaneous macrophages can also produce many mediators and cytokines (32). Functional studies with cutaneous macrophages are challenging given that healthy live macrophages can be quite difficult to extract from skin. However, they were not effective stimulators of T cells in an allogeneic mixed leukocyte reaction (21). Although many investigators have used CD68 as a macrophage marker, it is not as specific as CD163 for identifying cutaneous macrophages (32). Hence, the main populations of myeloid cells in the dermis are CD11c+ DCs—both the BDCA-1+ and BDCA-3+ subsets—and CD163+ macrophages.
Psoriasis
Clinical and Histological Features of Psoriasis
Psoriasis is a common skin disease affecting 1–3% of the North American population. Classic psoriasis, called large plaque psoriasis or psoriasis vulgaris, is the most common type. It can be fairly easily diagnosed as characteristic red colored plaques with well-defined borders and silvery-white dry scale, located on elbows, knees, and scalp and in the lumbosacral area (Figure 3a), although it can be more extensive (Figure 3b). Other less common types of psoriasis also occur, such as guttate, inverse, pustular, erythrodermic, palmo-plantar, and drug-associated psoriasis (33–35).
Figure 3.
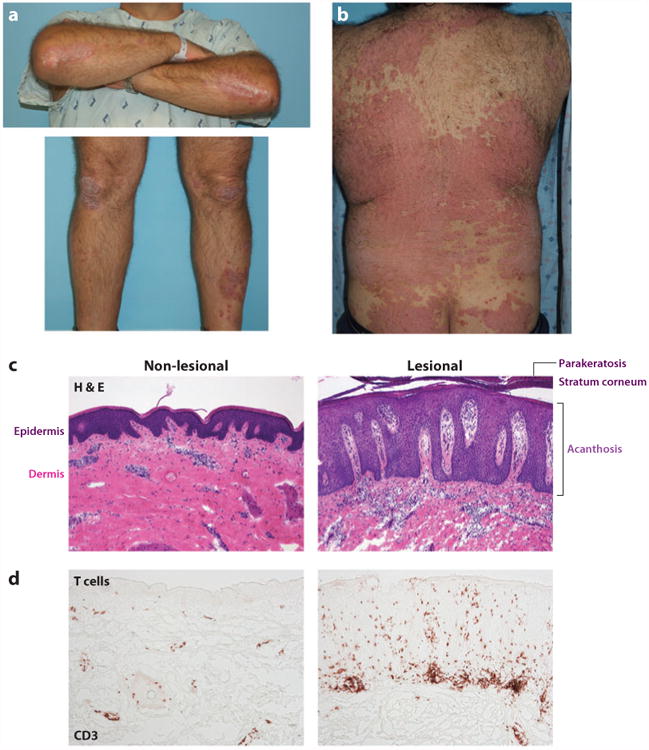
Clinical and histological features of psoriasis. (a) Clinical appearance of chronic psoriasis vulgaris, showing well-defined erythematous scaly plaques of psoriasis on elbows and knees. (b) Back showing more extensive psoriasis lesions. (c) Histology of nonlesional and lesional skin biopsy at the same magnification, with hematoxylin and eosin stain (H&E) where cellular nuclei stain blue. The epidermis is seen as a dark layer due to keratinocyte nuclei and forms an undulating border with the pink dermis below. Nuclei of resident structural and immune cells are seen in the dermis. Lesional psoriasis skin shows a greatly thickened epidermis (acanthosis) with elongations into the dermis (rete ridges). Retention of nuclei (parakeratosis) can be seen in the thickened stratum corneum. There is a dramatic increase in the number of cells in the dermis, composed predominantly of DCs and T cells. (d) Increased numbers of CD3+ T cells are seen in lesional psoriasis skin, often forming lymphoid-like clusters with DCs. All images 10 × magnification.
The amount of psoriasis covering the body can be measured roughly as a percentage of body area, using the palm to represent 1% of the body. In approximately one-third of patients, more than 10% of the body is covered, and this is termed moderate to severe psoriasis. Clinical disease can also be assessed by a trained health-care practitioner, using the Psoriasis Activity and Severity Index (PASI) score. This tool ranks severity and area of erythema (redness), induration (thickness), and desquamation (scale) of the plaques in different body sections, with 72 as the maximal score. A baseline PASI score is assigned (for inclusion in clinical trials a PASI of 12 or greater is often required), the score is then reevaluated at various time points, and the improvement is calculated. Most clinical studies consider that an improvement of 75% from baseline is required for the treatment to be considered successful (reported as PASI75), although a PASI50 can also be very meaningful for an individual patient.
The classic histological features of psoriasis can help explain the clinical appearance, demonstrated by hematoxylin and eosin stain (Figure 3c) (36). The epidermis is greatly thickened (acanthosis) as the keratinocytes move through the epidermis over 4–5 days, a tenfold acceleration. As the normal process of differentiation cannot occur, there is a loss of the normal granular layer, thickened stratum corneum (hyperkeratosis), and retention of nuclei in the upper layers and stratum corneum (parakeratosis). There is increased keratin 16 staining throughout the epidermis (Figure 2b), and neutrophils collect in the epidermis and stratum corneum (Kogoj pustules and Munro's microabscesses). In the dermis, there are abundant mononuclear cells, predominantly myeloid cells (Figure 2b,c) and T cells (Figure 3d). The erythema of psoriasis lesions is due to a greater number of dilated dermal blood vessels.
Initiation Phase of Psoriasis
Psoriasis can be triggered by many factors, including injury and trauma (termed the Koebner effect), infection, medications, and the topical biological response modifier imiquimod (a TLR7 agonist) (Figure 4a). Murine studies have shown that topical imiquimod may induce psoriasiform skin inflammation, mediated by the IL-23/IL-17 axis and activated DCs (37). Whereas most studies have focused on the maintenance phase of psoriasis because of the difficulty of obtaining samples to study initiation, Gilliet and coworkers have developed a mechanistic model to explain the early stages of disease, shown in Figure 4a (38–40). Injury to the skin causes cell death and the production of the AMP LL37 by keratinocytes. DNA/LL37 complexes bind to intracellular TLR9 in plasmacytoid dendritic cells (pDCs), which causes activation and production of type I interferons IFN-α and -β. LL37/RNA complexes can activate plasmacytoid DCs through TLR7, and myeloid DCs can be activated by this complex through TLR8. Hence, myeloid DCs can be activated by the LL37/RNA complex as well as by type 1 interferons, driving T cell activation and the production of cytokines found in psoriasis. Extracellular DNAhas recently been shown in the epidermis in association with neutrophil extracellular traps (NETs) (41), supporting this model of psoriasis initiation.
Figure 4.

Pathways for initiation and maintenance of psoriasis. (a) Early disease: Imiquimod (IMQ), a TLR7 agonist, can activate plasmacytoid dendritic cells (pDCs) to produce interferons (IFN). LL37, a peptide derived from cathelicidin, may have an important role in the initiation of psoriasis lesions via this pathway. LL37 released from keratinocytes (KCs) can bind to nucleic acids to activate pDCs to release IFN-α/β. LL37/RNA complexes can also activate resident myeloid DCs to produce IL-12 and IL-23, key psoriatic cytokines. (b) Chronic disease: The major pathogenic pathway in psoriasis occurs when (I) mature dermal DCs and inflammatory myeloid DCs produce cytokines such as IL-23 and IL-12. (II) These cytokines activate T17 (Th17 and Tc17), Th1, and Th22 cells to contribute to the cytokine milieu and further act on keratinocytes. (III) As outlined in Figure 1, keratinocytes can produce chemokines and antimicrobial peptides (AMPs) to (IV) augment cutaneous immune responses.
Studies That Established Psoriasis as a T Cell-Mediated Disease
For many years, there was a debate about whether the primary process in psoriasis involved hyperplastic keratinocytes with secondary immune activation or vice versa. In part, this debate was fueled by a lack of knowledge regarding therapeutic mechanisms of commonly used agents. On the one hand, corticosteroids and some immunosuppressants could be used to treat psoriasis, but on the other hand, systemic agents such as methotrexate were viewed as keratinocyte-directed agents. The first specific indication that the immune system could be playing a more integral role came with the clinical trial targeting T cells with the DAB389IL-2 agent, a fusion protein also called denileukin diftitox or Ontak®, that causes apoptosis in activated T cells expressing functional IL-2 receptors (42). Table 1 lists this and other immune treatments that have been used in psoriasis. This study showed that specific depletion of activated T cells in psoriasis lesions could cause clinical and histological disease resolution.
Table 1. Key immune-targeted biologics tested in psoriasisa.
| Target | Agent | Drug | Trade name |
|---|---|---|---|
| IL-2R | DAB389IL-2 FP | Denileukin diftitox | Ontak |
| B7 (CD80/CD86) | CTLA-4-Ig FP | Abatacept | Orencia |
| CD2 | LFA-3-Ig FP | Alefaceptb | Amevive |
| CD11a | Anti-CD11a mAb | Efalizumab | Raptiva |
| TNF | Anti-TNF mAb | Infliximabb, adalimumabb | Remicade, Humira |
| TNF | TNFR-Ig FP | Etanerceptb | Enbrel |
| IL-12/-23 | Anti-IL-12/-23p40 mAb | Ustekinumabb, briakinumab | Stelara, Ozespa |
| IL-23 | Anti-IL-23p19 mAb | LY2525623 SCH 900222, CNTO 1959, AMG 139 | |
| IL-17A | Anti-IL-17A mAb | Secukinumab, ixekizumab | |
| IL-17R | Anti-IL-17RA mAb | Brodalumab |
Abbreviations: CTLA, cytotoxic T lymphocyte antigen; FP, fusion protein; LFA, lymphocyte function-associated antigen; mAb, monoclonal antibody; TNF, tumor necrosis factor.
Current FDA-approved biologics for plaque psoriasis.
Hence, this study set up the general hypothesis that psoriasis is a disease mediated by activated T cells that are present in focal skin regions (plaques of disease). This view has been solidified and refined by the availability of a series of immune-targeted drugs that have been tested in psoriasis patients and by the ability to study immune cell subsets and molecular pathways in diseased tissue through biopsies of skin. CTLA-4-Ig (abatacept) was used to block B7-mediated costimulation to T cells (43). At high doses (above 10 mg/kg), consistent improvements in psoriasis were detected that correlated with a decrease of DC and T cell subsets from diseased skin regions. Hence, this study was the first to show that disease activity could be restrained by a specific T cell antagonist that did not deplete T cells as its primary mechanism of action.
Subsequently, two biologics targeted primarily at T cell activation pathways became FDA approved therapeutics for psoriasis. One of these agents was an LFA-3-Ig fusion protein(alefacept), which blocks CD2-mediated T cell activation. With this agent, strong clearing of psoriasis lesions was seen in patients in which the drug induced large decreases in T cells and DC populations in the skin (44). Effector memory T cells were often depleted in the peripheral circulation of patients treated with alefacept (45), providing additional evidence for pathogenic actions of activated T cells that infiltrate skin lesions of psoriasis vulgaris.
Another T cell–targeted biologic used for psoriasis was a monoclonal antibody to the integrin CD11a (efalizumab), although it is no longer FDA-approved. T cells selectively use the integrin CD11a/CD18 (LFA-1) for migration to peripheral tissues and as a part of T cell costimulation. Efalizumab blocks T cell migration and activation responses in psoriasis patients, again without inducing T cell cytotoxicity as a primary mechanism. Strong improvements in psoriasis lesions were seen in patients that had accompanying reductions in T cell and DC subsets that infiltrate skin lesions (46). During studies of the mechanism of action of efalizumab, an inflammatory DC population was discovered in psoriasis lesions called TNF- and iNOS-producing dendritic cells, or TIP-DCs (46), as discussed in more detail in the next section. Subsequently, the role of different T cell subsets in psoriasis, including Th1 (IFN-γ), Th17 (IL-17), and Th22 (IL-22), has been dissected through the testing of a range of cytokine antagonists (47), listed in Table 1.
A General Model of Immune Circuits in Chronic Skin Disease
Figure 4b shows a current pathogenic model for the involvement of T cell and DC subsets in sustaining disease activity in psoriasis plaques. Activation and differentiation of T cell subsets are supported by IL-12 and IL-23, which appear to be produced mainly from myeloid DC subsets in the skin. Psoriasis lesions contain T cells that discretely produce IFN-γ, IL-17, and IL-22, with initial labeling of these cells as Th1, Th17, and Th22, respectively. There are also CD8+ T cell populations that make the same range of cytokines, so these have been termed Tc1, Tc17, and Tc22, respectively. More recently, γδ T cells have been found to be IL-17-producing cells in psoriasis, so we have adopted the more general term T17 to encompass IL-17-producing lymphocyte subsets in the skin. Keratinocytes respond to cytokines from each of these subsets by upregulating mRNAs for a range of inflammatory products. Broadly, the induced keratinocyte products have the ability to feedback on immune cells in the skin so that chronic T cell activation persists. Chemokines made by keratinocytes are proposed to be important for continuing inward migration of leukocyte subsets that have relatively short life spans, e.g., neutrophils and myeloid DCs.
Chronic disease activity may also be supported by mature (DC-LAMP+) DCs that form cellular clusters with T cells in the dermis, and this structure can be considered as a form of induced SALT (iSALT) or tertiary lymphoid tissue (4, 48, 49). In the dermis of psoriasis lesions, some elements of lymph node structure are contained in dermal aggregates, such as CCR7+ T cells and DCs, along with local production of CCL19, which is a CCR7 agonist and an inducer of lymphoid structure (50). Psoriasis could also result from failure to turn off inflammation, which is perpetuated by this cutaneous tertiary lymphoid tissue.
From testing and mechanistic studies with evolving IL-17 antagonists (Table 1), the view of psoriasis as an IL-23/T17-centered inflammatory disease is evolving. These studies have shown that blockade of IL-17A or the IL-17 receptor A subunit can reverse clinical, histologic, and molecular features of psoriasis in approximately 80% of psoriasis patients given higher levels of the antagonists (51–54). The strong effect of IL-17 antagonists raises a question about pathogenic function(s) of Th1 and Th22 cell subsets in chronic disease, although there are clear molecular pathways in psoriasis that can be traced to individual cytokines of each T cell class.
Myeloid Dendritic Cells in Psoriasis
The earliest indication that myeloid DCs may be important in psoriasis were experiments by Nestle et al. (55) showing that psoriasis lesion-derived dermal DCs stimulated a T cell response with production of IL-2 and IFN-γ. It is now appreciated that myeloid DCs are key proximal cells in the pathogenic psoriatic pathway (Figure 4b). CD11c+ DC cell counts were increased in psoriasis lesions and reduced with all successful treatments studied (alefacept, efalizumab, etanercept, infliximab, NB-UVB). However, in psoriasis lesions, there were many CD11c+ cells that did not costain BDCA-1+ or BDCA-3+, and were subsequently termed inflammatory myeloid DCs (56). We consider TIP-DCs (46, 57) to be a subset of the inflammatory DCs because not all CD11c+ cells express TNF and iNOS. BDCA-1+ DCs expressed markers of DC maturity, such as CD208 (DC-LAMP) and CD205 (DEC-205), in contrast to BDCA-1- DCs, which showed increased CD209 (DC-SIGN) (Figure 2b).
Interestingly, these two populations of CD11c+ myeloid DCs (BDCA-1+ and BDCA-1−) could both stimulate T cells robustly in an allogeneic mixed lymphocyte reaction and similarly induce allogeneic T cells to produce IFN-γ and IL-17. To further characterize these cells, the transcriptome of the FACS-sorted cells was determined by comparing gene expression of BDCA-1+ versus BDCA-1− DCs. Several new markers that define the inflammatory DCs were discovered with this approach, including TRAIL and TLR1 and TLR2 (58). Myeloid DCs also produce IL-20 in psoriasis lesions (59), and this could be a driver of epidermal hyperplasia. Myeloid cells expressing 6-sulfo-LacNAc (slan) have also been proposed to be inflammatory DC precursors in psoriasis, driving strong Th17 and Th1 responses (60).
Although the above markers describe inflammatory DCs in psoriasis, a recent paper defined CD11c+HLA-DRhi myeloid subsets in ascites fluid from cancer patients differently, using BDCA-1 to define a subset to inflammatory DCs and CD16 to define inflammatory macrophages (61). We have previously shown, by two-color immunofluorescence and flow cytometry, that cutaneous DCs are CD16+ (32, 56), so CD16 does not appear to define two discrete inflammatory myeloid subsets in the skin. Additional studies are required, and the DCs may have different markers in specific organs or across various disease states.
T Cells in Psoriasis
Many investigators have now demonstrated that psoriasis lesions contain increased numbers of T cells (62) (Figure 3d). In the next steps of the central pathogenic pathway in psoriasis after DC activation, IL-23 is required for expansion and survival of T cells that produce IL-17. IL-23 is composed of two chains, the unique p19 chain and the p40 chain shared with IL-12. In situ in psoriasis lesions, abundant IL-23 is available from DCs and macrophages (32, 62). Most psoriasis-associated T cells are CD3+ CD2+ CD45RO+ CLA+ with a subset having activation markers CD25, HLA-DR, and CD27 (63, 64). Additionally, there was a skewed Th1 cell polarization profile with a resultant increase in production of IFN-γ and TNF-α (65). These Th1 cells migrate into psoriatic lesions by T cell chemokines such as CXCL9, CXCL10, and CXCL11, which are produced by myeloid cells and keratinocytes. In contrast to T cells of healthy skin, psoriasis lesional T cells produce abundant IL-17 when activated ex vivo by anti-CD3/CD28 or PMA/ionomycin (66).
Studies have shown that some psoriasis lesional T cells are oligoclonal; for example, there is an increase in Vβ13.1+ cells (67). T cells recognizing both streptococcal M protein and keratin can be found in the peripheral blood of psoriasis patients (68, 69, 70, 71). The antigen most often implicated is streptococcal antigen, and the proposed mechanism is that streptococcus-specific T cells cross-react with cutaneous antigens or proteins such as keratins, an example of molecular mimicry (71).
There are numerous T cell phenotypes with specific transcription factors, surface markers, and cytokine profiles (72). CD4 and CD8 T cells producing IL-17 have been identified in psoriasis lesions (73, 74). Until recently, it was assumed that lesional IL-17-producing T cells expressed the αβ T cell receptor (TCR). Hence, it was somewhat surprising when recent studies characterized many IL-17-producing T cells in human and murine psoriasis skin as γδ T cells (reviewed in 75). Psoriasis dermal suspensions and lesions showed significantly more CD3+ T cells expressing γδ TCR than is the case in healthy control skin (76). γδ T cells in peripheral blood have been characterized as Vγ9Vδ2, CLA+, and CCR6+, and they are able to produce IL-17A. They can activate keratinocytes via TNF and IFN-γ (77). In psoriasis, these cells accumulated in lesions but apparently were reduced in the circulation. Because Th17 and Tc17 are normally used to designate conventional (αβTCR+) T cells, we have used the term T17 to be broadly inclusive of all IL-17-producing T cell subsets (78). Regardless of the TCR subclass in psoriasis lesions, as mentioned above, targeting IL-17 as a therapeutic strategy appears to be the most successful treatment to date.
Tregs are a heterogeneous group of cells that maintain antigen-specific self-tolerance and are one mechanism in the arsenal of the immune system to prevent tissue damage due to inflammation. Some naturally occurring circulating Tregs can be identified as CD4+, CD25 (IL-2R)hi, Foxp3 (forkhead/winged helix transcription factor 3)+, and CD127 (IL-7R)− (72). Tregs use diverse mechanisms to maintain immune tolerance, including release of inhibitory cytokines, induction of apoptosis, and inhibition of IL-2 secretion (79). Buckner (80) recently reviewed how a healthy immune state is a consequence of effector T cells being held in check by Tregs, leading to a balanced immune status. However, unrestrained effector T cell effects leading to autoimmunity could be a result of decreased Treg numbers, function, or resistance to Treg effects (80). Some studies have shown Tregs to be dysfunctional in psoriasis, with decreased suppressive capacity (81), suggesting that psoriasis may result from the inability to suppress auto-inflammation. In fact, the function of skin-derived Tregs has not yet been examined, and further studies are needed to evaluate their contribution and how they can be harnessed for therapeutic benefit.
NK and NKT Cells in Psoriasis
Natural killer (NK) cells are a specialized subset of CD56+CD16+ cells with the ability to kill cancer and virally infected cells in a non-MHC-dependent manner (82). However, NK cells may play a role in psoriasis by releasing cytokines such as IFN-γ, TNF, and IL-22. NKT cells are a heterogeneous group of innate cells that share some features of both NK cells and T cells (83). There are three subsets, and they may also play a role in psoriasis by releasing cytokines such as IFN-γ. CD1d, an invariant stimulator of NKT cells, is abundantly expressed in psoriatic epidermis (84). In general, however, the roles of these immune cell subsets in psoriasis are not fully understood.
Keratinocytes in Psoriasis
How exactly do the cytokines found in psoriasis contribute to the psoriatic histological phenotype? To dissect the effect of cytokines on the epidermis, our group and others have treated cultured keratinocytes with cytokines to determine the specific keratinocyte gene set for each cytokine. One of the earliest of these transcriptional profiling experiments was treatment of epidermal keratinocytes with TNF, showing that this cytokine regulated gene expression for immune and inflammatory responses and also tissue remodeling, cell motility, cell cycle, and apoptosis (85). In a similar experimental design, IL-22 was shown to promote innate immunity, partially via upregulation of gene expression for keratinocyte AMPs (86). The main cell type expressing IL-17R in psoriasis is keratinocytes; however, IL-17 regulates a surprisingly small list of only 35–40 genes in human keratinocytes (12). Now, we and others have curated gene sets for keratinocyte responses to cytokines found in psoriasis, alone and in combinations, including IL-17, TNF, IFN-γ, IL-1, IFN-α, and IL-22 (12, 87–89). Many of these gene sets are enriched in the psoriatic transcriptome (90). Collectively, the cytokines IL-17, IFN-γ, IL-22, and TNF can cause keratinocyte proliferation as well as chemokine, cytokine, and AMP production (Figure 1). This becomes a self-amplifying loop, where these products act back on the DCs, T cells, and neutrophils to perpetuate the cutaneous inflammatory process.
The enrichment of the keratinocyte IL-17 gene set in the psoriasis transcriptome, as well as the superior success of anti-IL-17 treatments, suggests that IL-17 is a key cytokine in the formation of the psoriatic phenotype. Many of the genes induced by this cytokine in keratinocytes are indeed highly expressed in psoriasis lesions, such as chemokines and AMPs (Figures 1 and 4b). However, it is perhaps surprising that targeting a cytokine so distal in the pathogenic pathway, with such a small number of responsive genes, is so critical. One possible explanation for this is that IL-17 can stabilize chemokine mRNA, such as CXCL1 (91). Furthermore, synergistic effects likely exist between IL-17 and TNF, given that a combination of the two cytokines induces greater changes in gene expression than either alone (89). TNF-α regulates gene expression through NF-κB, and although IL-17 may also activate NF-κB, its major signaling pathway is through CCAAT/enhancer binding protein (C/EBP) transcription (92, 93). IL-17 induces IL-19 and IL-36γ in psoriasis lesions, which may then lead to proliferative responses in keratinocytes. IL-19, IL-20, and IL-22 have similar trophic effects on the epidermis (94), and transgenic models have shown psoriasis-related pathologies in mouse skin for IL-20, IL-22, and IL-36 cytokines (reviewed in 95).
Investigators are reevaluating the contributions to this disease of other immune cells found in psoriasis lesions, such as neutrophils (41). There are abundant neutrophil chemokines, such as CXCL1, CXCL2, and CXCL8/IL-8, and neutrophils in psoriasis collect predominantly in the epidermis. Neutrophils are a first line of defense against an immune attack of any type, possessing many intracellular AMPs that can be released by a process called NETosis. NETs are web-like extracellular structures containing protein-covered chromatin. NETs have been identified in psoriasis by staining for nucleic acid with DAPI and neutrophil elastase (96, 97). NETs have been implicated in causing organ damage associated with autoimmune diseases (98). These studies support the potential role of AMPs and nucleic acids in the initiation of psoriasis, as discussed above. Neutrophils were also positive for IL-17 (96), suggesting another potential role for neutrophils in psoriasis. However, neutrophils are inconsistently found in chronic psoriasis lesions and are absent from some mouse models of psoriasis.
Genetic Background of Psoriasis and Its Relationship to Immune Function
Investigators have long appreciated the genetic nature of psoriasis. The concordance rate of psoriasis is approximately 70% in monogenic twins and 20% in dizygotic twins, depending on the study and the population (99). Approximately three billion base pairs exist in the human genome, and only 3–5% of these sequences code for proteins. A disease-causing mutation is usually quite rare (<1%) and is commonly found in a coding or regulatory region. Psoriasis is a complex disease with over 30 single nucleotide polymorphisms (SNPs) contributing to disease risk, but two gene mutations have recently been found that can independently induce psoriasis (IL36RN and CARD14), and these genes have an effect on both the skin and the immune system.
CARD14 Mutations
Eighteen years ago, PSORS2 was identified on chromosome 17q in a large family with typical large plaque psoriasis. Recently, through NexGen sequencing of patients with familial psoriasis, a gain-of-function mutation in the Caspase Recruitment Domain-Containing Protein 14 (CARD14) gene was found at this site, which segregated with psoriasis (100, 101). A de novo mutation in CARD14 was concurrently discovered in a pediatric patient with a severe clinical presentation of psoriasis, without a family history. The CARD14 gene region was resequenced in many patients and controls (>6,000 cases and >4,000 controls), and numerous additional CARD14 missense mutations were found (100). CARD14 mRNA was found to be elevated 2.7-fold in the psoriasis transcriptome (101), and a CARD14 SNP was also recently discovered (102). CARD14 protein was expressed in the epidermis and dermis of psoriasis plaques of a patient with this mutation as well as in classic psoriasis. How might CARD14 mutations cause psoriasis? CARD proteins are involved in scaffold formation for inflammasome activation, and wild-type CARD14 activates Bcl10 and NF-κB. Mutations in the CARD14 gene lead to altered CARD14 protein and in association with an inflammatory trigger may induce increased activation of NF-κB, leading to transcription of many genes including key chemokines upregulated in psoriasis such as CCL20, CXCL8/IL-8, and IL-36γ/IL-1F9. These chemokines recruit additional cells such as neutrophils, DCs, and T cells that then produce their own inflammatory mediators. All of these events contribute to the vicious cycle of inflammation and acanthosis seen in psoriasis.
IL36RN Mutations
Mutations in IL36RN were first described in 2009 in two families with severe pustular psoriasis (103, 104). This gene, also called IL-1F5, encodes for an anti-inflammatory protein IL-36Ra, which is a natural antagonist of IL-1F9. Hence, a mutation in this gene leads to an altered protein with decreased effect and to unopposed IL-1F9 effects of NF-κB and MAPK activation through IL-1Rrp2 and IL-1RacP. Peripheral blood mononuclear cells from an affected patient compared with a healthy volunteer showed increased production of cytokines downstream of NF-κB (IL-1α, IL-6, CXCL8/IL-8, TNF). These two studies point to a loss of function in IL36RN as the genetic basis for generalized pustular psoriasis. These monogenic forms suggest that psoriasis is a phenotype that may occur as a result of different pathologies, and perhaps psoriasis with mutations can be separated out from those forms of psoriasis without genetic mutations.
Other Psoriasis Susceptibility (PSORS) Loci
The human genome is inherited in blocks, with linkage disequilibrium of genes in close proximity being inherited together. At least 12 major psoriasis susceptibility (PSORS) loci have now been identified, originally by linkage disequilibrium in family-based studies. Often, several candidate genes at each PSORS locus may be contributing to the disease. In the future, the gene at each locus will ultimately be identified by deep sequencing at each chromosomal position.
The first psoriasis-associated susceptibility locus (PSORS1) at chromosomal position 6p21.3 was identified by genome-wide linkage scans on the background of the serological association between psoriasis and HLA-Cw6 (99, 105). The PSORS1 locus has the highest odds ratio (OR) of any PSORS loci, of approximately 3.0. In fact, this region has the greatest impact on psoriasis heritability, a major association upheld even when 10 susceptibility loci were considered as a group (106). The exact identity of the PSORS1 gene is controversial due to extensive disequilibrium across the region, with approximately 10 genes in the 300 kb candidate region that are genetically inseparable. PSORS1 lies within the class I region of the MHC, and there is consensus that HLA-C is the most likely PSORS1 gene (105). The exact functional implications of alleles at this locus are not yet fully known, although there appear to be allele-specific differences in HLA-C expression and regulation by cytokines in psoriasis (107). Given our understanding that HLA class I molecules are important for antigen presentation to CD8+ T cells, this locus links the genetics of psoriasis with the immunological basis of the disease.
Single Nucleotide Polymorphisms
SNPs are defined as substitutions of one base pair for another in >1% of the population, and are most commonly found in noncoding regions of the genome. Genome-wide association studies (GWAS) aim to identify SNPs in DNA associated with a clinically defined disease (phenotype) by comparing the allele frequency of each SNP between a group of individuals with disease (cases) versus participants without disease (controls). The International HapMap Project identified the majority of common SNPs, which can be evaluated by GWAS using a SNP array. For each SNP, the disease locus could lie anywhere within an inherited block, and the significance of each SNP may reflect several potential genes that are in linkage disequilibrium.
Significant SNPs have been identified in psoriasis by GWAS, especially several psoriasis SNPs in the IL-23/IL-17 axis (108). A particular SNP in the IL23 locus (rs11209026G>A) may have functional significance, with a gradient of IL-17 responses to T cell stimulation by IL-23 in both healthy volunteers and psoriasis patients (109). Continuing to study cellular responsiveness as it relates to genotype is a critical stepping-stone on the pathway toward a deeper understanding of psoriasis.
In a recent meta-GWAS of psoriasis patients and controls, 21 SNPs were confirmed, and 15 SNPs were newly identified (102). Notable genes associated with each SNP in a 500 kb region around each SNP were described in that manuscript. Supplemental Table S1 lists the top GO Biological Processes associated with these genes (follow the Supplemental Material link from the Annual Reviews home page at http://www.annualreviews.org). Although the top pathways were keratinization and keratinization differentiation, many are associated with immunological processes, including T cell and NK cell proliferation, cytokine responses, regulation of Th17 and Th1 cells, JAK-STAT cascade, and leukocyte adhesion. These pathways have all been implicated in psoriasis pathogenesis and suggest functional significance of the genes that may be regulated by these SNPs.
Integration of Genetics and the Transcriptome of Psoriasis
To learn the potential functional implications of the SNPs uncovered in this meta-GWAS (102), we evaluated whether positional candidate genes around each SNP had altered gene expression in psoriasis. Many studies have examined the transcriptome of psoriasis, which is defined as differentially expressed genes (DEGs) between lesional and nonlesional skin. Recently, our group conducted a meta-analysis-derived (MAD) transcriptome of all the published studies using HGU133 Plus 2.0 chips, called MAD3 transcriptome, defining a robust list of psoriasis DEGs (110). Figure 5 shows a Manhattan-type plot depicting the gene expression fold change on the MAD3 transcriptome ordered by their position on the genome (in gray shades). Genes located +/− 500 kb around each of 36 psoriasis SNPs identified in the meta-GWAS (102) are highlighted in colors (by chromosome). The subset of these positional genes that were DEGs in the MAD3 transcriptome are represented as dots of darker shades and listed on the right-hand side (colored by chromosome). The notable genes IL23R, IL12B, IL23A, and IL4 were added because there were SNPs in these genes, and although they were not detected by microarray, they were determined by RT-PCR or FACS to be differentially regulated (90, 111). Thus, many genetic susceptibility loci also contain genes with strong upregulation of mRNA products. It is interesting and logical that the Th2 cytokine IL-4 is the only gene downregulated [log2(FCH) = −1.45, p = 0.034].
Figure 5.
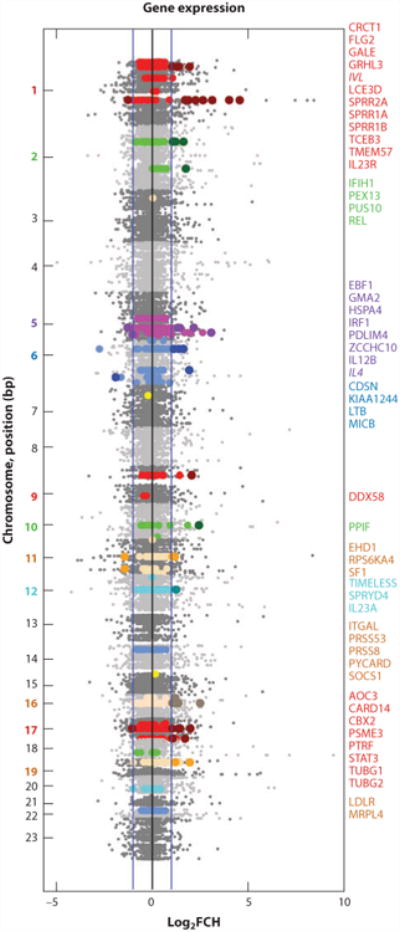
SNPs and gene expression in psoriasis lesions. Manhattan-type plot depicting (in gray shades) the magnitude of dysregulation of the HGU133 Plus 2.0 genes on the MAD3 psoriasis transcriptome by their base pair (bp) position on the genome (110). Blue lines on the x-axis represent a twofold change in the log2 scale. In a recent meta-GWAS, 36 SNPs were identified as psoriasis susceptibility loci (21 known loci, 15 new loci) (102). Genes that are located +/− 500 kb around each SNP are highlighted in colors (by chromosome). Dots in darker shades are for those genes differentially regulated in the MAD3 transcriptome, with the corresponding gene name listed on right, colored by chromosome.
It thus seems that many of the genetic risk loci for psoriasis map to immune pathways that become activated in the disease and are likely drivers of the psoriasis phenotype. For example, as discussed above, IL-23R, IL-12B, and IL-23A are components of the IL-23-IL-17 axis. REL is part of the NF-κB complex, which may be important as a TNF- and IL-17-dependent transcription factor in psoriasis (112), and STAT3 is required for Th17 differentiation (113). SOCS1 is a member of the suppressor of cytokine signaling family of proteins, with a role in T17 differentiation. MICB encodes the MHC class I chain–related gene B, which further supports the potential importance of genes at the PSORS1 locus. There were a number of IFN-related genes, including (a) DDX58, the RIG-I innate antiviral receptor that recognizes double-stranded RNA and regulates IFN production (102); (b) IFIH1, which encodes the interferon-induced helicase c domain-containing protein; and (c) IRF1, the IFN-regulatory factor 1. Several SNP-DEGs are involved in keratinization, such as FLG2, LCE3D, and CDSN, supporting the potential for genetic influence in epidermal processes during psoriatic inflammation as well.
Comorbidities in Psoriasis
Investigators have had a long-standing appreciation of the relationship between psoriasis and nail dystrophy, psoriatic arthritis, depression, and cancer (squamous cell carcinoma and lymphoma) (33). However, more recently, epidemiological studies have uncovered an association between psoriasis and other systemic diseases (114–116). Psoriasis-associated comorbidities also include cardiovascular risk/acute myocardial infarction, diabetes, and metabolic syndrome (defined as central obesity plus two of the following: increased triglycerides, decreased HDL cholesterol, increased blood pressure, and increased fasting plasma glucose). These comorbidities appear to be correlated with psoriasis severity. Evaluation of patients with psoriasis now needs to include consideration of these associated comorbidities and their response to treatment.
The causal relationship between psoriasis and systemic comorbidities is not fully understood, but shared genetic risks, common environmental factors, or inflammatory pathways may provide the links (117, 118). Insights into the skin disease and systemic inflammation may come from evaluating skin and circulating proteins concurrently. Recently, our group published a study of the skin transcriptome and serum protein measurements (using a 92-protein multiplex Luminex-based panel) in 85 patients with moderate to severe psoriasis, compared with serum proteomics in a cohort of healthy volunteers (119). Figure 6 summarizes the data for a subset of these serum proteins and their encoding genes.
Figure 6.

Serum proteomics and lesional transcriptomics in psoriasis patients. Recently, our group compared serum proteins of psoriasis patients and healthy volunteers, with the accompanying skin psoriasis transcriptome [DEGs for lesional (LS) versus nonlesional (NL) skin] (119). This figure depicts a subset of these serum proteins (right side of y-axis) and their encoding genes (mRNA; left side of y-axis). Yellow lines represent a twofold change (FCH) in the log2 scale (x-axis). Green dots represent the protein dysregulation between psoriasis patients (PS) and healthy volunteers, with a more sensitive assay for IL-17. Red dots represent the log2FCH (LS versus NL) detected by microarray in skin for genes encoding each protein. Dark blue dots represent differential expression by RT-PCR in the same cohort of patients, and light blue dots represent differential expression by RT-PCR in a separate patient cohort (90). An asterisk indicates results where protein determination was below the limit of detection for at least one group, so fold change may be biased. Results are grouped as (I) upregulated serum protein and skin mRNA; (II) increased serum protein without expression in skin, which could indicate production of mediators in tissues outside of the skin; (III) decreased serum protein, which could indicated consumption of the serum proteins; (IV) abundant gene expression with little protein; and (V) decreased tissue gene expression with minimal or low serum protein.
The data in Figure 6 support a model for systemic inflammation in which a subset of inflammatory products is produced at high levels in psoriasis skin lesions and may then diffuse into the systemic circulation. Other tissues beyond the skin could have inflammation induced by exposure to high levels of inflammatory cytokines or other mediators (117). Many interesting immune molecules were significantly elevated both in the circulation and as lesional mRNA, including IL-17, IL-1ra, and TNF-α. The chemokines CCL5, CCL2, and CCL4 were also elevated. Numerous immune molecules were elevated in the circulation but without elevation in corresponding skin mRNA, including IL-16, IL-18, and CD40 ligand. PAI1 is a product of inflamed endothelial cells (120), that could be induced by cytokines increased in the blood. Leptin, the hormone associated with obesity, was 1.5 log2(FCH) times higher in psoriasis patients versus healthy volunteers, and higher in psoriasis patients with BMI >30 (mean 24 ng/ml) versus psoriasis patients with BMI <30 (mean 7 ng/ml) (119). C-reactive protein was elevated in the circulation and is a general indicator of inflammation. Some immune molecules were decreased in the peripheral circulation, such as IL-1α and IGF1, possibly indicating consumption. Overall, given that many of the pathogenic pathways of psoriasis are well understood, this type of data analysis could offer some mechanistic insights into the relationship between skin and systemic inflammation.
Murine Models of Psoriasis
Over the past decades, murine models of psoriasis have been developed as tools for understanding the pathogenesis of this disease and also as preclinical models. In 2007, Gudjonsson et al. (121) reviewed the current mouse models, classifying them as xenografts, allografts, transgenics, targeted mutations, and spontaneous models, and ranked features of human psoriasis in each model. Wagner et al. (122) provided a comprehensive update in 2010, covering 27 models. Transcriptomic profiling of five of the most representative models (K14-amphiregulin, K5-Stat3C, K5-Tie2, K5-TGF-β1, and Imiquimod) were compared to each other and to human psoriasis (123). At a global level, there were strong and statistically significant similarities between gene expression patterns in human psoriasis and each of these mouse models, in particular gene expression patterns associated with epidermal development and keratinization. However, marked differences also existed in immune-associated gene expression across the models. The dermal injection of IL-23 into mice is a novel murine model of psoriasis (124, 125), although transcriptional profiling of this model has not yet been published. (See Note Added in Proof.)
None of the mouse models fully represents the set of cellular changes and molecular features of psoriasis, but they contain sub-elements of this disease that differ across the various strategies used to create a murine inflammatory or hyperplastic epidermal phenotype. To visualize how psoriasis-related cytokine-response pathways are reflected in different mouse models, a graphic representation of cytokine pathway expression is shown in Figure 7. In this figure, normalized enrichment scores (NES) from Gene Set Enrichment Analysis (126) have been created for gene sets that are induced in cultured human cells or reconstructed human epidermis by the cytokines IL-17, IL-22, IFN-γ, IFN-α, TNF, IL-1, IL-4, and IL-13, either alone or in combinations that produced additive or synergistic effects in psoriasis lesions (12, 90, 127–129). High expression of that pathway in a target tissue produces a NES of greater than 2, whereas low expression of that pathway would be a NES of 1 or less. Results are also coded for level of significance by color, with dark red being most significant [false discovery rate (FDR) < 0.0001]. The actual representations of the cytokine pathways in lesions of psoriasis vulgaris are shown in columns a and b using the MAD3 transcriptome (lesional versus nonlesional skin; Figure 7a) (110) and lesional versus normal skin (Figure 7b) (130) as the reference data sets. This shows that psoriasis vulgaris has a high enrichment of gene sets related to TNF, IL-1, IL-17, IL-22, and IFNs but a poor signal of genes induced by Th2 cytokines (IL-4, IL-13).
Figure 7.

Enrichment of cytokine-related inflammatory pathways (gene sets) in human psoriasis transcriptomes and in five mouse models of psoriasis. Normalized enrichment scores (NES) and false discovery rate (FDR) are shown for gene sets regulated in keratinocytes (KC), monocytes, immature dendritic cells (iDCs), fibroblasts, and reconstituted human epidermis (RHE) by several psoriatic inflammatory cytokines. An asterisk indicates cytokine pathways described in Reference 129. (a,b) Expression of these cytokine pathways in human psoriasis vulgaris is shown for lesional (LS) versus nonlesional (NL) skin in the MAD3 transcriptome (a) (110) and for lesional versus normal (N) skin (b) (130). (c–h) Cytokine enrichment in the five mouse models (123) is illustrated as follows: K14-amphiregulin (AREG) ear skin (c) and tail skin (d), K5-Stat3C (Stat-3) (e), K5-Tie2 (Tie-2) (f), K5-TGF-β1 (TGF-β) (g), and Imiquimod (h). (See Note Added in Proof.)
Data generated from gene-array analysis of murine psoriasis-related models are shown in columns c-h. Overall, the highest overlap of cytokine pathways with psoriasis vulgaris is seen in the model in which amphiregulin is overexpressed by a transgene. In particular, the representation of IL-17 and TNF pathways is particularly strong, but the NES is still lower than in the human disease. The STAT3, Tie2, and TGF-β transgene models, along with the imiquimod treatment model, represent models with expression of some inflammatory pathways that are present in psoriasis vulgaris, but with decreasing fidelity to the range of pathways that are expressed in the human disease. Note that STAT3, Tie2, and TGF-β models all have higher expression of IL-13 and/or IL-4 pathways than is seen in psoriasis vulgaris. These might therefore be better models for cytokine expression in intrinsic atopic dermatitis, where IL-17 is expressed in skin lesions along with Th2 cytokines (131).
The most interesting aspect of this range of inflammatory models is that direct, high-level activation of keratinocytes via an autocrine growth factor (amphiregulin) (132) has the ability to induce cytokine-related gene circuits that most closely resemble psoriasis vulgaris. Hence, these data cement the concept that activated keratinocytes, whether via transgenes, endogenous mutations (CARD14), damage/injury, or exposure to specific T cell–produced cytokines, can induce and sustain a chronic inflammatory response that is highly aligned with psoriasis. Overall, the transgenic models provide interesting insights into means by which inflammatory skin disease phenotypes might be induced or regulated. However, fixed overexpression of transgenes is not well suited to preclinical testing of therapeutics that act through collapsing complex inducible/repressible pathways.
One interesting model that meets nearly all human psoriasis features is the xenotransplantation of nonlesional skin onto AGR mice (that lack B and T cells and IFN-γ receptors) (133, 134). In this model, there is spontaneous conversion of grafted nonlesional skin to lesional skin over weeks. The lesions are characterized by acanthosis, loss of granular layer, parakeratosis, dermal and epidermal infiltrates of T cells, and TNF-producing DCs. The model's limitation is the difficulty of obtaining large grafts of nonlesional psoriasis skin, and therefore the numbers of mice that can be grafted are relatively small. This model has been used for preclinical studies, showing the therapeutic benefit of TNF blockade, and the critical role of T cells moving into the epidermis via VLA-1 in the pathogenesis of psoriasis (135).
Conclusions and Perspective
Psoriasis vulgaris is the best-understood and most accessible human disease that is mediated by T cells and DCs. The ability to measure cellular and molecular inflammatory pathways in diseased human tissue, combined with the availability of specific immune antagonists to relevant disease-related pathways, has permitted a dissection of pathogenic circuits to a much larger extent than in less accessible diseases such as rheumatoid arthritis, Crohn's disease, or multiple sclerosis. Existing and emerging therapeutic immune antagonists are highly aligned with elements of genetic risk, most especially the common p40 subunit of IL-12/-23, the p19 subunit that defines IL-23, and the IL-23 receptor. These genes control development of Th17 cells and other IL-17-producing T cells, so the emerging IL-17 antagonists are also highly related to core genetic elements of disease risk and development. The risk genes also identify numerous genes of TNF/NF-κB signaling that have a major interplay with IL-17 immunity and keratinocyte or DC responses to innate immune signals. The progressive refinement of immune-targeted therapeutics, which has been enabled by the scientific studies of psoriasis in humans, has now resulted in the ability to control most clinical disease signs and symptoms in approximately 80% of patients treated with antagonists to IL-23 or IL-17. Future goals should include developing strategies of treatment that do not require continuous, long-term immune suppression, i.e., strategies to restore immune regulation or tolerance in this disease, and to derive a better understanding of the specific antigenic triggers that may induce and sustain T cell activity in focal skin lesions. Hopefully, this approach taken to psoriasis pathogenic dissection and treatment will be a model for the approach to other inflammatory diseases of the skin and to inflammatory diseases in other tissues that are less accessible to direct analysis by tissue biopsy.
Note Added in Proof
In a recent publication by our group, the IL-23-induced mouse transcriptome showed the greatest fidelity overall to human psoriasis. This close relationship was seen when (a) cytokine gene sets were analyzed in a manner similar to Figure 7 in this review and (b) the IL-23-induced murine upregulated genes were compared to the human psoriasis transcriptome (136).
Supplementary Material
Acknowledgments
Our psoriasis studies are supported in part by grant #UL1 TR000043 from the National Center for Advancing Translational Sciences and from the National Institutes of Health (NIH) Clinical and Translational Science Award program. M.A.L. is supported by NIH 1R01AR060222. We thank Dr. Wittkowski for helpful discussions. We are grateful to Dr. Gudjonsson, Dr. Swindell, Dr. Elder, Dr. Brodmerkel, and Dr. Li for generous and expeditious sharing of their data.
Footnotes
Disclosure Statement: The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review. J.G.K. has been a consultant or received research support from companies developing therapeutics for psoriasis, including Amgen, Boehringer, Centocor/Janssen, Merck, Pfizer, Idera, Astellas, and Japan Tobacco.
Contributor Information
Michelle A. Lowes, Email: lowesm@rockefeller.edu.
Mayte Suárez-Fariñas, Email: farinam@rockefeller.edu.
James G. Krueger, Email: jgk@rockefeller.edu.
Literature Cited
- 1.Morizane S, Gallo RL. Antimicrobial peptides in the pathogenesis of psoriasis. J Dermatol. 2012;39:225–30. doi: 10.1111/j.1346-8138.2011.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorensen OE, Cowland JB, Theilgaard-Monch K, Liu L, Ganz T, Borregaard N. Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J Immunol. 2003;170:5583–89. doi: 10.4049/jimmunol.170.11.5583. [DOI] [PubMed] [Google Scholar]
- 3.Streilein JW. Skin-associated lymphoid tissues (SALT): origins and functions. J Investig Dermatol. 1983;80(Suppl):12s–16s. doi: 10.1111/1523-1747.ep12536743. [DOI] [PubMed] [Google Scholar]
- 4.Egawa G, Kabashima K. Skin as a peripheral lymphoid organ: revisiting the concept of skin-associated lymphoid tissues. J Investig Dermatol. 2011;131:2178–85. doi: 10.1038/jid.2011.198. [DOI] [PubMed] [Google Scholar]
- 5.Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9:679–91. doi: 10.1038/nri2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Meglio P, Perera GK, Nestle FO. The multitasking organ: recent insights into skin immune function. Immunity. 2011;35:857–69. doi: 10.1016/j.immuni.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Clark RA. Skin-resident T cells: the ups and downs of on site immunity. J Investig Dermatol. 2010;130:362–70. doi: 10.1038/jid.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, Kupper TS. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. 2006;176:4431–39. doi: 10.4049/jimmunol.176.7.4431. [DOI] [PubMed] [Google Scholar]
- 9.Homey B, Alenius H, Muller A, Soto H, Bowman EP, et al. CCL27-CCR10 interactions regulate T cell-mediated skin inflammation. Nat Med. 2002;8:157–65. doi: 10.1038/nm0202-157. [DOI] [PubMed] [Google Scholar]
- 10.Morales J, Homey B, Vicari AP, Hudak S, Oldham E, et al. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proc Natl Acad Sci USA. 1999;96:14470–75. doi: 10.1073/pnas.96.25.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kennedy-Crispin M, Billick E, Mitsui H, Gulati N, Fujita H, et al. Human keratinocytes' response to injury upregulates CCL20 and other genes linking innate and adaptive immunity. J Investig Dermatol. 2012;132:105–13. doi: 10.1038/jid.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suarez-Farinas M, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159:1086–91. doi: 10.1111/j.1365-2133.2008.08769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takai T. TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol Int. 2012;61:3–17. doi: 10.2332/allergolint.11-RAI-0395. [DOI] [PubMed] [Google Scholar]
- 14.Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Investig. 2009;119:3573–85. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romani N, Brunner PM, Stingl G. Changing views of the role of Langerhans cells. J Investig Dermatol. 2012;132:872–81. doi: 10.1038/jid.2011.437. [DOI] [PubMed] [Google Scholar]
- 16.Fujita H, Nograles KE, Kikuchi T, Gonzalez J, Carucci JA, Krueger JG. Human Langerhans cells induce distinct IL-22-producing CD4+ T cells lacking IL-17 production. Proc Natl Acad Sci USA. 2009;106:21795–800. doi: 10.1073/pnas.0911472106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36:873–84. doi: 10.1016/j.immuni.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cerio R, Griffiths CE, Cooper KD, Nickoloff BJ, Headington JT. Characterization of factor XIIIa positive dermal dendritic cells in normal and inflamed skin. Br J Dermatol. 1989;121:421–31. doi: 10.1111/j.1365-2133.1989.tb15509.x. [DOI] [PubMed] [Google Scholar]
- 19.Nestle FO, Zheng XG, Thompson CB, Turka LA, Nickoloff BJ. Characterization of dermal dendritic cells obtained from normal human skin reveals phenotypic and functionally distinctive subsets. J Immunol. 1993;151:6535–45. [PubMed] [Google Scholar]
- 20.Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. 2001;106:259–62. doi: 10.1016/s0092-8674(01)00456-1. [DOI] [PubMed] [Google Scholar]
- 21.Zaba LC, Fuentes-Duculan J, Steinman RM, Krueger JG, Lowes MA. Normal human dermis contains distinct populations of CD11c+BDCA-1+ dendritic cells and CD163+FXIIIA+ macrophages. J Clin Investig. 2007;117:2517–25. doi: 10.1172/JCI32282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, et al. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J Immunol. 2000;165:6037–46. doi: 10.4049/jimmunol.165.11.6037. [DOI] [PubMed] [Google Scholar]
- 23.Kennedy Crispin M, Fuentes-Duculan J, Gulati N, Johnson-Huang LM, Lentini T, et al. Gene profiling of narrowband UVB-induced skin injury defines cellular and molecular innate immune responses. J Investig Dermatol. 2013;133:692–701. doi: 10.1038/jid.2012.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207:1247–60. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–69. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 26.Segura E, Durand M, Amigorena S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med. 2013;210:1035–47. doi: 10.1084/jem.20121103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angel CE, George E, Brooks AE, Ostrovsky LL, Brown TL, Dunbar PR. Cutting edge: CD1a+ antigen-presenting cells in human dermis respond rapidly to CCR7 ligands. J Immunol. 2006;176:5730–34. doi: 10.4049/jimmunol.176.10.5730. [DOI] [PubMed] [Google Scholar]
- 28.Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29:497–510. doi: 10.1016/j.immuni.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haniffa M, Ginhoux F, Wang XN, Bigley V, Abel M, et al. Differential rates of replacement of human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation. J Exp Med. 2009;206:371–85. doi: 10.1084/jem.20081633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, Amigorena S. Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med. 2012;209:653–60. doi: 10.1084/jem.20111457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fabriek BO, Dijkstra CD, Van den Berg TK. The macrophage scavenger receptor CD163. Immunobiology. 2005;210:153–60. doi: 10.1016/j.imbio.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Fuentes-Duculan J, Suárez-Fariñas M, Zaba LC, Nograles KE, Pierson KC, et al. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J Investig Dermatol. 2010;130:2412–22. doi: 10.1038/jid.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- 34.Lebwohl M. Psoriasis. Lancet. 2003;361:1197–204. doi: 10.1016/S0140-6736(03)12954-6. [DOI] [PubMed] [Google Scholar]
- 35.Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol Mech Dis. 2012;7:385–422. doi: 10.1146/annurev-pathol-011811-132448. [DOI] [PubMed] [Google Scholar]
- 36.Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–73. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 37.Van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182:5836–45. doi: 10.4049/jimmunol.0802999. [DOI] [PubMed] [Google Scholar]
- 38.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–69. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 39.Gilliet M, Lande R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr Opin Immunol. 2008;20:401–7. doi: 10.1016/j.coi.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206:1983–94. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar V, Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol. 2010;10:1325–34. doi: 10.1016/j.intimp.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 42.Gottlieb SL, Gilleaudeau P, Johnson R, Estes L, Woodworth TG, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med. 1995;1:442–47. doi: 10.1038/nm0595-442. [DOI] [PubMed] [Google Scholar]
- 43.Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Investig. 1999;103:1243–52. doi: 10.1172/JCI5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chamian F, Lowes MA, Lin SL, Lee E, Kikuchi T, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes inpsoriasis vulgaris. Proc Natl Acad Sci USA. 2005;102:2075–80. doi: 10.1073/pnas.0409569102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chamian F, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, et al. Alefacept (anti-CD2) causes a selective reduction in circulating effector memory T cells (Tem) and relative preservation of central memory T cells (Tcm) in psoriasis. J Transl Med. 2007;5:27. doi: 10.1186/1479-5876-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, et al. Increase in TNF-α and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a) Proc Natl Acad Sci USA. 2005;102:19057–62. doi: 10.1073/pnas.0509736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nograles KE, Krueger JG. Anti-cytokine therapies for psoriasis. Exp Cell Res. 2011;317:1293–300. doi: 10.1016/j.yexcr.2011.01.024. [DOI] [PubMed] [Google Scholar]
- 48.Lew W, Bowcock AM, Krueger JG. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and ‘Type 1’ inflammatory gene expression. Trends Immunol. 2004;25:295–305. doi: 10.1016/j.it.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 49.Johnson-Huang LM, Suarez-Farinas M, Pierson KC, Fuentes-Duculan J, Cueto I, et al. A single intradermal injection of IFN-γ induces an inflammatory state in both non-lesional psoriatic and healthy skin. J Investig Dermatol. 2012;132:1177–87. doi: 10.1038/jid.2011.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitsui H, Suarez-Farinas M, Belkin DA, Levenkova N, Fuentes-Duculan J, et al. Combined use of laser capture microdissection and cDNA microarray analysis identifies locally expressed disease-related genes in focal regions of psoriasis vulgaris skin lesions. J Investig Dermatol. 2012;132:1615–26. doi: 10.1038/jid.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krueger JG, Fretzin S, Suarez-Farinas M, Haslett PA, Phipps KM, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. 2012;130:145–54. doi: 10.1016/j.jaci.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papp KA, Reid C, Foley P, Sinclair R, Salinger DH, et al. Anti-IL-17 receptor antibody AMG 827 leads to rapid clinical response in subjects with moderate to severe psoriasis: results from a phase I, randomized, placebo-controlled trial. J Investig Dermatol. 2012;132:2466–69. doi: 10.1038/jid.2012.163. [DOI] [PubMed] [Google Scholar]
- 53.Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366:1181–89. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 54.Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190–99. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- 55.Nestle FO, Turka LA, Nickoloff BJ. Characterization of dermal dendritic cells in psoriasis. Autostimulation of T lymphocytes and induction of Th1 type cytokines. J Clin Investig. 1994;94:202–9. doi: 10.1172/JCI117308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, Abello MV, Novitskaya I, et al. Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Investig Dermatol. 2009;129:79–88. doi: 10.1038/jid.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 58.Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, Johnson-Huang LM, Nograles KE, et al. Identification of TNF-related apoptosis-inducing ligand and other molecules that distinguish inflammatory from resident dendritic cells in patients with psoriasis. J Allergy Clin Immunol. 2010;125:1261–68. e9. doi: 10.1016/j.jaci.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang F, Lee E, Lowes MA, Haider AS, Fuentes-Duculan J, et al. Prominent production of IL-20 by CD68+/CD11c+ myeloid-derived cells in psoriasis: gene regulation and cellular effects. J Investig Dermatol. 2006;126:1590–99. doi: 10.1038/sj.jid.5700310. [DOI] [PubMed] [Google Scholar]
- 60.Hansel A, Gunther C, Ingwersen J, Starke J, Schmitz M, et al. Human slan (6-sulfo LacNAc) dendritic cells are inflammatory dermal dendritic cells in psoriasis and drive strong TH17/TH1 T-cell responses. J Allergy Clin Immunol. 2011;127:787–94. e9. doi: 10.1016/j.jaci.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 61.Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. 2013;38:336–48. doi: 10.1016/j.immuni.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 62.Bos JD, Hulsebosch HJ, Krieg SR, Bakker PM, Cormane RH. Immunocompetent cells in psoriasis. In situ immunophenotyping by monoclonal antibodies. Arch Dermatol Res. 1983;275:181–89. doi: 10.1007/BF00510050. [DOI] [PubMed] [Google Scholar]
- 63.Bos JD, De Rie MA. The pathogenesis of psoriasis: immunological facts and speculations. Immunol Today. 1999;20:40–46. doi: 10.1016/s0167-5699(98)01381-4. [DOI] [PubMed] [Google Scholar]
- 64.Ferenczi K, Burack L, Pope M, Krueger JG, Austin LM. CD69, HLA-DR and the IL-2R identify persistently activated T cells in psoriasis vulgaris lesional skin: blood and skin comparisons by flow cytometry. J Autoimmun. 2000;14:63–78. doi: 10.1006/jaut.1999.0343. [DOI] [PubMed] [Google Scholar]
- 65.Austin LM, Ozawa M, Kikuchi T, Walters IB, Krueger JG. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-γ, interleukin-2, and tumor necrosis factor-α, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Investig Dermatol. 1999;113:752–59. doi: 10.1046/j.1523-1747.1999.00749.x. [DOI] [PubMed] [Google Scholar]
- 66.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Investig Dermatol. 2008;128:1207–11. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 67.Chang JC, Smith LR, Froning KJ, Kurland HH, Schwabe BJ, et al. Persistence of T-cell clones in psoriatic lesions. Arch Dermatol. 1997;133:703–8. [PubMed] [Google Scholar]
- 68.Prinz JC, Grob B, Vollmer S, Trommler P, Strobel I, et al. T cell clones from psoriasis skin lesions can promote keratinocyte proliferation in vitro via secreted products. Eur J Immunol. 1994;24:593–98. doi: 10.1002/eji.1830240315. [DOI] [PubMed] [Google Scholar]
- 69.Kim SM, Bhonsle L, Besgen P, Nickel J, Backes A, et al. Analysis of the paired TCR α- and β- chains of single human T cells. PLoS ONE. 2012;7:e37338. doi: 10.1371/journal.pone.0037338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diluvio L, Vollmer S, Besgen P, Ellwart JW, Chimenti S, Prinz JC. Identical TCR β-chain rearrangements in streptococcal angina and skin lesions of patients with psoriasis vulgaris. J Immunol. 2006;176:7104–11. doi: 10.4049/jimmunol.176.11.7104. [DOI] [PubMed] [Google Scholar]
- 71.Valdimarsson H, Thorleifsdottir RH, Sigurdardottir SL, Gudjonsson JE, Johnston A. Psoriasis—as an autoimmune disease caused by molecular mimicry. Trends Immunol. 2009;30:494–501. doi: 10.1016/j.it.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 72.Palmer MT, Weaver CT. Autoimmunity: increasing suspects in the CD4+ T cell lineup. Nat Immunol. 2010;11:36–40. doi: 10.1038/ni.1802. [DOI] [PubMed] [Google Scholar]
- 73.Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, et al. Induction of IL-17+ T cell trafficking and development by IFN-γ: mechanism and pathological relevance in psoriasis. J Immunol. 2008;181:4733–41. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ortega C, Fernandez AS, Carrillo JM, Romero P, Molina IJ, et al. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. 2009;86:435–43. doi: 10.1189/JLB.0109046. [DOI] [PubMed] [Google Scholar]
- 75.Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol. 2012;9:302–9. doi: 10.1038/cmi.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cai Y, Shen X, Ding C, Qi C, Li K, et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laggner U, Di Meglio P, Perera GK, Hundhausen C, Lacy KE, et al. Identification of a novel proinflammatory human skin-homing Vγ9Vδ2 T cell subset with a potential role in psoriasis. J Immunol. 2011;187:2783–93. doi: 10.4049/jimmunol.1100804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013;34:174–81. doi: 10.1016/j.it.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goodman WA, Cooper KD, McCormick TS. Regulation generation: the suppressive functions of human regulatory T cells. Crit Rev Immunol. 2012;32:65–79. doi: 10.1615/critrevimmunol.v32.i1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buckner JH. Mechanisms of impaired regulation by CD4+CD25+FOXP3+ regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849–59. doi: 10.1038/nri2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, et al. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol. 2005;174:164–73. doi: 10.4049/jimmunol.174.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dunphy S, Gardiner CM. NK cells and psoriasis. J Biomed Biotechnol. 2011;2011:248317. doi: 10.1155/2011/248317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Simoni Y, Diana J, Ghazarian L, Beaudoin L, Lehuen A. Therapeutic manipulation of natural killer (NK) T cells in autoimmunity: Are we close to reality? Clin Exp Immunol. 2013;171:8–19. doi: 10.1111/j.1365-2249.2012.04625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bonish B, Jullien D, Dutronc Y, Huang BB, Modlin R, et al. Overexpression of CD1d by keratinocytes in psoriasis and CD1d-dependent IFN-γ production by NK-T cells. J Immunol. 2000;165:4076–85. doi: 10.4049/jimmunol.165.7.4076. [DOI] [PubMed] [Google Scholar]
- 85.Banno T, Gazel A, Blumenberg M. Effects of tumor necrosis factor-α (TNF α) in epidermal keratinocytes revealed using global transcriptional profiling. J Biol Chem. 2004;279:32633–42. doi: 10.1074/jbc.M400642200. [DOI] [PubMed] [Google Scholar]
- 86.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–54. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 87.Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Investig Dermatol. 2009;129:2175–83. doi: 10.1038/jid.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blumenberg M. Skinomics: transcriptional profiling in dermatology and skin biology. Curr Genomics. 2012;13:363–68. doi: 10.2174/138920212801619241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M, Nograles KE, Tian S, et al. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Investig Dermatol. 2011;131:677–87. doi: 10.1038/jid.2010.340. [DOI] [PubMed] [Google Scholar]
- 90.Suárez-Fariñas M, Lowes MA, Zaba LC, Krueger JG. Evaluation of the psoriasis transcriptome across different studies by gene set enrichment analysis (GSEA) PLoS ONE. 2010;5:e10247. doi: 10.1371/journal.pone.0010247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Datta S, Novotny M, Pavicic PG, Jr, Zhao C, Herjan T, et al. IL-17 regulates CXCL1 mRNA stability via an AUUUA/tristetraprolin-independent sequence. J Immunol. 2010;184:1484–91. doi: 10.4049/jimmunol.0902423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, et al. Functional cooperation between interleukin-17 and tumor necrosis factor-ex is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–67. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- 93.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–48. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 94.Sa SM, Valdez PA, Wu J, Jung K, Zhong F, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. 2007;178:2229–40. doi: 10.4049/jimmunol.178.4.2229. [DOI] [PubMed] [Google Scholar]
- 95.Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013;34:174–81. doi: 10.1016/j.it.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. 2011;187:490–500. doi: 10.4049/jimmunol.1100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aubert P, Suarez-Farinas M, Mitsui H, Johnson-Huang LM, Harden JL, et al. Homeostatic tissue responses in skin biopsies from NOMID patients with constitutive overproduction of IL-1β. PLoS ONE. 2012;7:e49408. doi: 10.1371/journal.pone.0049408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Knight JS, Carmona-Rivera C, Kaplan MJ. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front Immunol. 2012;3:380. doi: 10.3389/fimmu.2012.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bowcock AM. The genetics of psoriasis and autoimmunity. Annu Rev Genomics Hum Genet. 2005;6:93–122. doi: 10.1146/annurev.genom.6.080604.162324. [DOI] [PubMed] [Google Scholar]
- 100.Jordan CT, Cao L, Roberson ED, Duan S, Helms CA, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-κB, in psoriasis. Am J Hum Genet. 2012;90:796–808. doi: 10.1016/j.ajhg.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jordan CT, Cao L, Roberson ED, Pierson KC, Yang CF, et al. PSORS2 is due to mutations in CARD14. Am J Hum Genet. 2012;90:784–95. doi: 10.1016/j.ajhg.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44:1341–48. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365:620–28. doi: 10.1056/NEJMoa1013068. [DOI] [PubMed] [Google Scholar]
- 104.Onoufriadis A, Simpson MA, Pink AE, DiMeglio P, Smith CH, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet. 2011;89:432–37. doi: 10.1016/j.ajhg.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Elder JT. PSORS1: linking genetics and immunology. J Investig Dermatol. 2006;126:1205–6. doi: 10.1038/sj.jid.5700357. [DOI] [PubMed] [Google Scholar]
- 106.Chen H, Poon A, Yeung C, Helms C, Pons J, et al. A genetic risk score combining ten psoriasis risk loci improves disease prediction. PLoS ONE. 2011;6:e19454. doi: 10.1371/journal.pone.0019454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hundhausen C, Bertoni A, Mak RK, Botti E, Di Meglio P, et al. Allele-specific cytokine responses at the HLA-C locus: implications for psoriasis. J Investig Dermatol. 2012;132:635–41. doi: 10.1038/jid.2011.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Duffin KC, Krueger GG. Genetic variations in cytokines and cytokine receptors associated with psoriasis found by genome-wide association. J Investig Dermatol. 2009;129:827–33. doi: 10.1038/jid.2008.308. [DOI] [PubMed] [Google Scholar]
- 109.Di Meglio P, Di Cesare A, Laggner U, Chu CC, Napolitano L, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE. 2011;6:e17160. doi: 10.1371/journal.pone.0017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tian S, Krueger JG, Li K, Jabbari A, Brodmerkel C, et al. Meta-analysis derived (MAD) transcriptome of psoriasis defines the “core” pathogenesis of disease. PLoS ONE. 2012;7:e44274. doi: 10.1371/journal.pone.0044274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tonel G, Conrad C, Laggner U, Di Meglio P, Grys K, et al. Cutting edge: A critical functional role for IL-23 in psoriasis. J Immunol. 2010;185:5688–91. doi: 10.4049/jimmunol.1001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lizzul PF, Aphale A, Malaviya R, Sun Y, Masud S, et al. Differential expression of phosphorylated NF-κB/RelA in normal and psoriatic epidermis and downregulation of NF-κB in response to treatment with etanercept. J Investig Dermatol. 2005;124:1275–83. doi: 10.1111/j.0022-202X.2005.23735.x. [DOI] [PubMed] [Google Scholar]
- 113.Camporeale A, Poli V. IL-6, IL-17 and STAT3: a holy trinity in auto-immunity? Front Biosci. 2012;17:2306–26. doi: 10.2741/4054. [DOI] [PubMed] [Google Scholar]
- 114.Azfar RS, Gelfand JM. Psoriasis and metabolic disease: epidemiology and pathophysiology. Curr Opin Rheumatol. 2008;20:416–22. doi: 10.1097/BOR.0b013e3283031c99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kimball AB, Gladman D, Gelfand JM, Gordon K, Horn EJ, et al. National Psoriasis Foundation clinical consensus on psoriasis comorbidities and recommendations for screening. J Am Acad Dermatol. 2008;58:1031–42. doi: 10.1016/j.jaad.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mehta NN, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Patients with severe psoriasis are at increased risk of cardiovascular mortality: cohort study using the General Practice Research Database. Eur Heart J. 2010;31:1000–6. doi: 10.1093/eurheartj/ehp567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Davidovici BB, Sattar N, Prinz JC, Puig L, Emery P, et al. Psoriasis and systemic inflammatory diseases: potential mechanistic links between skin disease and co-morbid conditions. J Investig Dermatol. 2010;130:1785–96. doi: 10.1038/jid.2010.103. [DOI] [PubMed] [Google Scholar]
- 118.Boehncke WH, Boehncke S, Tobin AM, Kirby B. The ‘psoriatic march’: a concept of how severe psoriasis may drive cardiovascular comorbidity. Exp Dermatol. 2011;20:303–7. doi: 10.1111/j.1600-0625.2011.01261.x. [DOI] [PubMed] [Google Scholar]
- 119.Suarez-Farinas M, Li K, Fuentes-Duculan J, Hayden K, Brodmerkel C, Krueger JG. Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate-to-severe psoriasis. J Investig Dermatol. 2012;132:2552–64. doi: 10.1038/jid.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chrobak I, Lenna S, Stawski L, Trojanowska M. Interferon-γ promotes vascular remodeling in human microvascular endothelial cells by upregulating endothelin (ET)-1 and transforming growth factor (TGF) β2. J Cell Physiol. 2013;228:1774–83. doi: 10.1002/jcp.24337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gudjonsson JE, Johnston A, Dyson M, Valdimarsson H, Elder JT. Mouse models of psoriasis. J Investig Dermatol. 2007;127:1292–308. doi: 10.1038/sj.jid.5700807. [DOI] [PubMed] [Google Scholar]
- 122.Wagner EF, Schonthaler HB, Guinea-Viniegra J, Tschachler E. Psoriasis: what we have learned from mouse models. Nat Rev Rheumatol. 2010;6:704–14. doi: 10.1038/nrrheum.2010.157. [DOI] [PubMed] [Google Scholar]
- 123.Swindell WR, Johnston A, Carbajal S, Han G, Wohn C, et al. Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PLoS ONE. 2011;6:e18266. doi: 10.1371/journal.pone.0018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hedrick MN, Lonsdorf AS, Shirakawa AK, Richard Lee CC, Liao F, et al. CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J Clin Investig. 2009;119:2317–29. doi: 10.1172/JCI37378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jiang W, Zhu FG, Bhagat L, Yu D, Tang JX, et al. A Toll-like receptor 7, 8, and 9 antagonist inhibits Th1 and Th17 responses and inflammasome activation in a model of IL-23-induced psoriasis. J Investig Dermatol. 2013;133:1777–84. doi: 10.1038/jid.2013.57. [DOI] [PubMed] [Google Scholar]
- 126.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mee JB, Johnson CM, Morar N, Burslem F, Groves RW. The psoriatic transcriptome closely resembles that induced by interleukin-1 in cultured keratinocytes: dominance of innate immune responses in psoriasis. Am J Pathol. 2007;171:32–42. doi: 10.2353/ajpath.2007.061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, Nograles KE, Guttman-Yassky E, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022–30. e395. doi: 10.1016/j.jaci.2009.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Swindell WR, Xing X, Stuart PE, Chen CS, Aphale A, et al. Heterogeneity of inflammatory and cytokine networks in chronic plaque psoriasis. PLoS ONE. 2012;7:e34594. doi: 10.1371/journal.pone.0034594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gudjonsson JE, Aphale A, Grachtchouk M, Ding J, Nair RP, et al. Lack of evidence for activation of the hedgehog pathway in psoriasis. J Investig Dermatol. 2009;129:635–40. doi: 10.1038/jid.2008.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Suarez-Farinas M, Dhingra N, Gittler J, Shemer A, Cardinale I, et al. Intrinsic atopic dermatitis shows similar T2 and higher T17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol. 2013;132:361–70. doi: 10.1016/j.jaci.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cook PW, Piepkorn M, Clegg CH, Plowman GD, DeMay JM, et al. Transgenic expression of the human amphiregulin gene induces a psoriasis-like phenotype. J Clin Investig. 1997;100:2286–94. doi: 10.1172/JCI119766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-α. J Exp Med. 2004;199:731–36. doi: 10.1084/jem.20031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nestle FO, Nickoloff BJ. From classical mouse models of psoriasis to a spontaneous xenograft model featuring use of AGR mice. Ernst Schering Res Found Workshop. 2005;50:203–12. doi: 10.1007/3-540-26811-1_11. [DOI] [PubMed] [Google Scholar]
- 135.Conrad C, Boyman O, Tonel G, Tun-Kyi A, Laggner U, et al. α1 β1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med. 2007;13:836–42. doi: 10.1038/nm1605. [DOI] [PubMed] [Google Scholar]
- 136.Suárez-Fariñas M, Arbeit R, Jiang W, Ortenzio FS, Sullivan T, Krueger JG. Suppression of molecular inflammatory pathways by Toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS ONE. 2013;8:e84634. doi: 10.1371/journal.pone.0084634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.