

Abstract
Store-operated calcium channels (SOCs) are calcium-selective cation channels that mediate calcium entry in many different cell types. Store-operated calcium entry (SOCE) is involved in various cellular functions. Increasing evidence suggests that impairment of SOCE is responsible for numerous disorders. A previous study demonstrated that YM-58483, a potent SOC inhibitor, strongly attenuates chronic pain by systemic or intrathecal injection and completely blocks the second phase of formalin-induced spontaneous nocifensive behaviour, suggesting a potential role of SOCs in central sensitization. However, the expression of SOCs, their molecular identity and function in spinal cord dorsal horn neurons remain elusive. Here, we demonstrate that SOCs are expressed in dorsal horn neurons. Depletion of calcium stores from the endoplasmic reticulum (ER) induced large sustained calcium entry, which was blocked by SOC inhibitors, but not by voltage-gated calcium channel blockers. Depletion of ER calcium stores activated inward calcium-selective currents, which was reduced by replacing Ca2+ with Ba2+ and reversed by SOC inhibitors. Using the small inhibitory RNA knockdown approach, we identified both STIM1 and STIM2 as important mediators of SOCE and SOC current, and Orai1 as a key component of the Ca2+ release-activated Ca2+ channels in dorsal horn neurons. Knockdown of STIM1, STIM2 or Orai1 decreased resting Ca2+ levels. We also found that activation of neurokinin 1 receptors led to SOCE and activation of SOCs produced an excitatory action in dorsal horn neurons. Our findings reveal that a novel SOC signal is present in dorsal horn neurons and may play an important role in pain transmission.
Introduction
Calcium is an important regulator of many cellular processes and mediates a remarkable variety of cellular functions in many different cell types. In neurons, calcium signalling is crucial for the induction of synaptic plasticity, which contributes to the generation and the maintenance of pain hypersensitivity (Fang et al. 2002; Wei et al. 2006). Impaired Ca2+ homeostasis has been linked to CNS disorders, such as Huntington's disease and Alzheimer's disease (Bezprozvanny & Hayden, 2004; Raza et al. 2007; Wojda et al. 2008). Under conditions of painful peripheral neuropathy and nerve injury, the resting cytoplasmic calcium concentration is increased in dorsal horn neurons (Kawamata & Omote, 1996; Kruglikov et al. 2004), suggesting calcium homeostasis is also impaired under chronic pain conditions. Importantly, decreasing intracellular Ca2+ attenuates pain hypersensitivity induced by paclitaxel and vincristine (Siau & Bennett, 2006). Cytoplasmic calcium signals are generated from intracellular calcium release and external calcium influx from plasma membrane Ca2+-permeable channels. Neurons are endowed with a large repertoire of ion channels, receptors, transporters and plasma membrane Ca2+ ATPase that work together to regulate Ca2+ homeostasis. Although voltage-gated calcium channels (VGCCs) and ligand-gated cation channels were thought to be the primary channels involved in Ca2+ homeostasis in neurons, a growing body of evidence indicates that store-operated calcium channels (SOCs) are also important in mediating Ca2+ influx in cortical, hippocampal and dorsal root ganglion neurons (Berna-Erro et al. 2009; Gemes et al. 2011; Koss et al. 2013).
SOCs are Ca2+-permeable cation channels that can be activated by depletion of endoplasmic reticulum (ER) calcium stores. Store-operated calcium entry (SOCE) is a major mechanism for triggering Ca2+ influx into the non-excitable cell. SOCE underlies sustained Ca2+ signalling, which is required for many calcium-dependent cellular functions, such as enzymatic activity. SOCs comprise two calcium sensors (stromal interaction molecules STIM1 and STIM2) located on the surface of the ER and three structurally related pore-forming subunits (Orai1/2/3), known as Ca2+ release-activated Ca2+ channels (CRAC channels), located in the plasma membrane (Lewis, 2007). SOCs have been extensively studied and the importance of SOCs has been implicated in functions of non-neuronal cells. In the nervous system, SOCE is known to influence neurotransmitter release and synaptic plasticity (Bouron, 2000; Baba et al. 2003) and has been shown to be involved in conditions such as neuropathic pain, ischaemic stroke, Parkinson's disease and Alzheimer's disease (Targos et al. 2005; Berna-Erro et al. 2009; Selvaraj et al. 2009; Gemes et al. 2011). Surprisingly, the nature of SOCE and its molecular identity in neurons is poorly understood. SOCE is present in dorsal root ganglion neurons (Gemes et al. 2011; Gao et al. 2013), hippocampal neurons (Narayanan et al. 2010; Koss et al. 2013) and cortical neurons (Berna-Erro et al. 2009). However, SOCE in spinal cord neurons has not been previously reported and its functional significance in dorsal horn neurons remains elusive.
Recently, we demonstrated that N-[4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl]-4-methyl-1,2,3-thiadiazole-5-carboxamide (YM-58483), a potent SOC channel inhibitor, strongly attenuates chronic pain when administered systemically or intrathecally, and completely blocks the second phase of formalin-induced spontaneous nocifensive behaviour, suggesting that SOCE might be involved in central sensitization, a central mechanism underlying pain hypersensitivity. Here, we demonstrate that SOCs are expressed and mediate SOCE in spinal cord dorsal horn neurons. We identified both STIM1 and STIM2 as important components of SOCs and Orai1 as a key subunit of CRAC channels in dorsal horn neurons. We also found that knockdown of Orai1, STIM1 or STIM2 significantly reduces resting calcium concentration. Furthermore, our results indicate that SOCs are involved in the function of neurokinin 1 (NK1) receptors and activation of SOCs produces excitatory action in dorsal horn neurons. These findings revealed that a novel calcium signal mediated by SOCs, other than voltage- and ligand-gated calcium channels, is present in dorsal horn neurons and may play an important role in pain transmission.
Methods
Animals
All experiments were performed in accordance with the guidelines of the National Institutes of Health, the Committee for Research and Ethical Issues of IASP, and were approved by the Animal Care and Use Committee of Drexel University College of Medicine. Pregnant mice were purchased from Charles River (Wilmington, MA, USA) or Taconic (Hudson, NY, USA) and individually housed in standard cages and maintained on a 12 h light/dark cycle. Neonatal mice from both CD1 and C57BL/6 stains were used for cell cultures and C57BL/6 adult mice were used for slice preparations. Protein levels and functions of SOCs in neurons from C57BL/6 and CD1 mice were not significantly different from each other (data not shown).
Cell culture and slice preparation
Primary cultures of spinal cord superficial dorsal horn neurons were prepared from neonatal (postnatal day 1 (P1) or P2) mice as previously described (Hu & Gereau, 2003). Briefly, neonatal mice were decapitated after inducing hypothermia on ice. A laminectomy was performed and the spinal cord was carefully removed. The superficial dorsal horn was dissected with a surgical blade cut in approximately lamina III. The superficial dorsal horn strips were incubated for 30 min at 37°C in Hanks balance salt solution (HBSS; Invitrogen, Carlsbad, CA, USA) (in mm: 137 NaCl, 5.4 KCl, 0.4 KH2PO4, 1 CaCl2, 0.5 MgCl2, 0.4 MgSO4, 4.2 NaHCO3, 0.3 Na2HPO4 and 5.6 glucose) containing papain (15 U ml−1; Worthington Biochemical, Lakewood, NJ, USA), rinsed three times with HBSS, and placed in culture medium containing Neurobasal A (Invitrogen), fetal calf serum (2%; Invitrogen), heat-inactivated horse serum (2%; Invitrogen), l-glutamax-1 (0.2 mm; Invitrogen) and B-27 (2%; Invitrogen). The strips were mechanically dissociated by gently triturating with a pipette. The resulting cells (88% neurons, stained with a neuron marker NeuN) were plated onto poly-d-lysine- and collagen-coated coverslips or plates. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 for 2–4 days. For slice preparation, 7- to 10-week-old C57BL/6 mice were anaesthetized with inhalation of an overdose of isoflurane. After decapitation, the vertebral column and surrounding tissue were isolated and immersed in ice-cold oxygenated sucrose artificial cerebrospinal fluid (ACSF) containing the following (in mm): 190 sucrose, 2.5 KCl, 26 NaHCO3, 6 MgCl2, 1.25 NaH2PO4, 0.5 CaCl2, 5 ethyl pyruvate, 3 myo-inositol, 1 sodium l-ascorbate and 12 glucose. The lumbar enlargement of the spinal cord was dissected out and glued onto the cutting platform with the adhesive Loctite 404 (Loctite, Rocky Hill, CT, USA). Transverse spinal cord slices (300 μm) were cut in sucrose ACSF with a Vibratome 3000 (Vibratome, St Louis, MO, USA). Slices were maintained at room temperature in ACSF containing (in mm) 115 NaCl, 5 KCl, 26 NaHCO3, 1 MgCl2, 1.25 NaH2PO4, 1 CaCl2, 3 myo-inositol and 12 glucose under continuous oxygenation and were allowed to recover for 1 h before electrophysiological recording.
Transfection
For knocking down STIM1 and STIM2, neurons were cultured on glass coverslips (for calcium imaging and electrophysiology experiments) and plates (for Western blot analysis) for 24 h, and then transfected with small inhibitory RNA (siRNA) targeting STIM1 (Dharmacon, Lafayette, CO, USA), siRNA targeting STIM2 (Dharmacon) or Scramble siRNA (Dharmacon) using X-tremeGENE HP DNA Transfection Reagent (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer's instructions. For knocking down Orai proteins, acutely isolated neurons were electroporated using a mouse neuron nucleofector kit according to the manufacturer's instructions (Lonza Group, Basel, Switzerland) as described by Motiani et al. (2010). Briefly, acutely dissociated neurons were transfected with 12 μg of siRNA targeting Orai1 (Life Technologies, Grand Island, NY, USA), siRNA targeting Orai2 (Dharmacon), siRNA targeting Orai3 (Dharmacon) or Scramble siRNA (Life Technologies for Orai1; Dharmacon for Orai2 and Orai3) per 3 × 106 cells. For electrophysiological recordings, neurons were co-transfected with green fluorescent protein (GFP)-containing plasmid and targeting siRNA or scramble siRNA. After 16 h, transfection medium was removed and neurons were fed with fresh culture medium. Calcium imaging, patch-clamp recordings and Western blot analysis were performed 48–72 h after transfection.
Real-time PCR analysis of mRNA expression
Total RNA was extracted from neonatal and adult spinal cords and acutely dissociated neurons using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA). RNA concentration was determined by optical density at 260 nm (using an optical density 260-unit equivalent to 40 μg ml−1). Total RNA was reverse transcribed into cDNA for each sample using a Fermentas maxima first-strand cDNA synthesis kit (Thermo Scientific, Rockford, IL, USA) following the manufacturer's instructions. Primers specific for mouse STIM1 (Mm00486423_m1), STIM2 (Mm01223103_m1), Orai1 (Mm00774349_m1), Orai2 (Mm04214089_s1), Orai3 (Mm01612888_m1) and GAPDH were purchased from Applied Biosystems (Foster City, CA, USA). Real-time quantitative PCR was performed in a 7900HT fast real-time PCR System (Applied Biosystems). Amplification was with the following conditions: 5 min of initial denaturation at 96°C, then 35 cycles of 96°C for 30 s, 55°C for 30 s and 72°C for 1.5 min. The threshold cycle for each gene was determined and analysed using Relative Quantitation software (Applied Biosystems). The relative expression of the target genes was calculated using the 2(–Delta Delta CT, 2−ΔΔCT) method (Livak & Schmittgen, 2001). Messenger RNA (mRNA) levels of STIM1, STIM2, Orai1, Orai2 and Orai3 were normalized to the housekeeping gene GAPDH.
Western blot analysis
Mice were killed via overdose isoflurane anaesthesia. The lumbar section of the spinal cord was dissected and homogenized using a Dounce homogenizer in an ice-cold radio immunoprecipitation assay (RIPA) buffer containing 50 mm Tris HCl, 150 mm NaCl, 0.2 mm EDTA, 1% Triton X-100, 2% sodium dodecyl sulfate, 1% deoxycholate, 0.1 mm phenylmethanesulfonyl fluoride and protease inhibitor cocktails (Thermo Fisher Scientific). For cultures, neurons were washed in PBS and lysed in RIPA buffer. The lysed tissues and neurons were sonicated at a constant intensity of 2.5 for 10 s, and centrifuged at 18,000 × g (4°C) for 5 min. The concentrations of total protein were determined using a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific) following the manufacturer's instructions. Protein samples were heated at 95°C for 5 min, electrophoresed in 10% SDS polyacrylamide gel, and transferred onto PVDF membranes (Millipore, Billerica, MA, USA). The blots were blocked with 5% skimmed milk in Tris-buffered saline–Tween 0.1% for 1 h at room temperature and probed with rabbit anti-STIM1 (1:1000, Cell Signaling, Danvers, MA, USA), anti-STIM2 (1:4000, ProSci, Poway, CA, USA), anti-Orai1 (1:500, ProSci), anti-Orai2 (1:500, ProSci), anti-Orai3 (1:500, ProSci) and anti-GAPDH (1:10,000, ProSci) primary antibodies at 4°C overnight. The blots were washed and incubated for 1 h at room temperature with the horseradish peroxidase-conjugated secondary antibody (1:10,000, Cell Signaling), then developed with enhanced chemiluminescence (Millipore). The densitometry of protein bands was quantified using Image J software (NIH, Bethesda, MD, USA).
Calcium imaging
The changes in intracellular Ca2+ concentration ([Ca2+]i) were examined using fura-2-based microfluorimetry and imaging analysis as previously described (Nicolai et al. 2010). Neurons were loaded with 4 μm fura-2AM (Life Technologies) for 30 min at room temperature in HBSS, washed, and further incubated in normal bath solution (Tyrode's) containing (in mm) 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes and 5.6 glucose for an additional 20 min. Coverslips were mounted in a small laminar-flow perfusion chamber (Model RC-25, Warner Instruments, Hamden, CT, USA) and continuously perfused at 5–7 ml min−1 with Tyrode's solution. Images were acquired at 3 s intervals at room temperature (20–22°C) using an Olympus inverted microscope equipped with a CCD camera (Hamamatsu ORCA-03G, Tokyo, Japan). The fluorescence images were recorded and analysed using the software MetaFluor 7.7.9 (Molecular Devices, Sunnyvale, CA, USA). The fluorescence ratio was determined as the fluorescence intensities excited at 340 and 380 nm with background subtraction. For measuring the resting calcium level, the free [Ca2+]i was calculated by the formula [Ca2+]i = Kd*β* (R – Rmin)/(Rmax – R), where β = (I380 max)/(I380 min). Rmin, Rmax and β were determined by in situ calibration, as described previously (Fuchs et al. 2005), and 224 nm was used as the dissociation constant Kd (Grynkiewicz et al. 1985). Only one recording was made from each coverslip.
Electrophysiological recording
Standard whole-cell recordings were made at room temperature using an EPC 10 amplifier and PatchMaster software (HEKA Elektronik, Lambrecht, Germany) as previously described (Hu & Gereau, 2003). Electrode resistances were 3–5 MΩ with series resistances of 6–15 MΩ, which was not compensated for the current amplitudes in this study (≤100 pA, voltage errors < 2 mV). Only neurons with a resting membrane potential more hyperpolarized than −50 mV were used. For recording SOC currents, three standard protocols were used: a gap-free protocol in which the membrane voltage was held at −80 mV without depolarization or hyperpolarization voltage pulses applied; a ramp protocol in which the membrane voltage was held at 0 mV, and a voltage ramp was applied from −100 mV up to +100 mV every 10 s; and a step protocol in which neurons were held at 0 mV, and a step hyperpolarizing voltage pulse to −100 mV was applied every 5 s. The normal bath solution was Tyrode's solution. For recording SOC currents in cultured neurons, the bath solution contained (in mm) 135 N-methyl-d-glucamine (NMDG), 10 CaCl2, 2 MgCl2, 10 Hepes, 0.0005 tetrodotoxin (TTX) and 5.6 glucose. For recording SOC currents in neurons from slices, the bath solution was modified ACSF containing (in mm) 115 NMDG, 26 NaHCO3, 2 CaCl2, 2 CsCl, 1 MgCl2, 1.25 NaH2PO4, 0.0005 TTX, 0.005 mibefradil and 12 glucose. The electrode solution contained (in mm) 125 CsMeSO4, 8 MgCl2, 10 BAPTA, 10 Hepes, 3 Na2ATP and 0.3 Na2GTP, pH 7.4. Only one neuron was recorded from each coverslip or slice.
Drug application
YM-58483, TTX, substance P (SP) and (3aR,7aR)-octahydro-2-[1-imino-2-(2-methoxyphenyl)ethyl]-7,7-diphenyl-4H-isoindol (RP 67580) were purchased from Tocris (Minneapolis, MN, USA). ω-Conotoxin MVIIC was purchased from Alomone Labs (Jerusalem, Israel). Cyclopiazonic acid (CPA), thapsigargin (TG), nimodipine, mibefradil, gadolinium chloride, 2-aminoethyl diphenyl borate (2-APB) and BAPTA were purchased from Sigma (St Louis, MO, USA). They were dissolved in Milli-Q water or dimethyl sulfoxide (DMSO) as stock solutions and further diluted to final concentrations in 0.1% DMSO.
Data analysis
Off-line evaluation was done using PatchMaster and Origin 8.1 (OriginLab, Northampton, MA, USA). Data are expressed as original traces or as mean ± standard error of the mean (SEM). Treatment effects were statistically analyxed with a one-way analysis of variance (ANOVA). When ANOVA showed a significant difference, pairwise comparisons between means were performed by the post hoc Bonferroni method. Paired or two-sample Student's t tests were used when comparisons were restricted to two means. Error probabilities of P < 0.05 were considered statistically significant. The statistical software Origin 8.1 was used to perform all statistical analyses.
Results
SOCs are expressed in the spinal cord
Our previous study demonstrated that YM-58483, a potent SOC inhibitor, produces a strong central analgesic effect in chronic pain conditions (Gao et al. 2013), suggesting that SOCs may play a role in pain hypersensitivity. Thus, we investigated whether SOCs are present in spinal cord dorsal horn neurons. To examine the expression of SOCs in the spinal cord, we first performed Taqman RT-PCR in spinal cord tissues from neonatal and adult mice, and acutely dissociated dorsal horn neurons from neonatal mice. We found that the mRNA of SOC family was expressed in both spinal tissue and acutely dissociated neonatal dorsal horn neurons (88% purity). The expression pattern was similar among neonatal and adult mice, but the mRNA level of STIM2 was about twofold greater than that of STIM1 (Fig.1A). Orai1 expression was twofold greater than Orai2 in neurons, opposite of the expression in tissue. To validate the protein expression of SOCs in the spinal cord, Western blot analysis was conducted with comparison of the cortex and lymph nodes, which are known to express SOCs (Wissenbach et al. 2007; Ma et al. 2010). To determine the specificity of antibodies against SOC proteins, blots were incubated in blocking solution containing the specific antibody neutralized with an excess of the immunizing peptide (corresponding to the epitope recognized by the antibody). We observed a single band at 85 kDa with the STIM1 antibody, two bands at 105 and 115 kDa with the STIM2 antibody, two main bands at 45 and 55 kDa with the Orai1 antibody, one main band at 28 kDa with the Orai2 antibody and one band at 33 kDa with the Orai3 antibody (Fig.1B). However, in the presence of the neutralized antibody, STIM1, STIM2 and Orai3 staining was absent; Orai1 and Orai2 staining in corresponding molecular weights was dramatically reduced (data not shown). Expression levels of SOCs in the spinal cord were comparable to those in the cortex and lymph nodes (Fig.1B).
Figure 1. Store-operated calcium channels (SOCs) are expressed in spinal cord dorsal horn neurons.
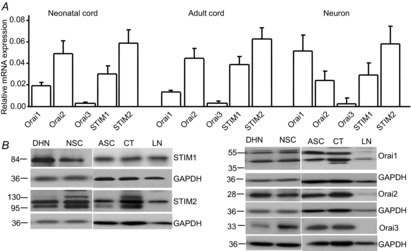
A, mRNA levels of STIM1, STIM2, Orai1, Orai2 and Orai3 in neonatal and adult spinal cord tissue and acutely dissociated dorsal horn neurons by quantitative PCR (normalized to GAPDH). Values represent mean ± SEM, n = 4 samples each. B, protein levels of STIM1, STIM2, Orai1, Orai2 and Orai3 in dorsal horn neurons (DHN), neonatal spinal cord (NSC), adult spinal cord (ASC), the cortex (CT) and lymph nodes (LN) by Western blot analysis.
To further confirm the protein expression of SOC family members in spinal cord dorsal neurons, we used an RNA interference gene silencing approach to knock down SOC proteins. Neurons were transfected against STIM1, STIM2 or control siRNA using X-tremeGENE HP DNA transfection reagent. The siRNA concentration was determined based on our pilot study (two concentrations of non-targeting (control) or targeting siRNA were tested). After 48 h of transfection, total mRNA and protein were extracted from neurons. The specific siRNA against STIM1 at a concentration of 1 μg ml−1 dramatically reduced STIM1 mRNA and protein levels (Fig.2A and B), and had no effect on STIM2 expression. Similarly, the specific STIM2 siRNA (1 μg ml−1) drastically decreased STIM2 mRNA and protein levels at both bands (Fig.2A and B), but had no effect on STIM1 expression. Knocking down Orai proteins was challenging in neurons and did not work with lipofectamine 2000 or X-tremeGENE HP DNA Transfection Reagent. Luckily, Orai proteins were successfully knocked down by electroporation. Neurons transfected with siRNAs targeting Orai1, Orai2 or Orai3 (12 μg siRNA per 3 × 106 neurons) showed a dramatic reduction of their own mRNA and protein levels at the corresponding band(s) (Fig.2C and D), and showed no interference with each other (data not shown). These results demonstrate that the SOC family is expressed in spinal cord dorsal horn neurons.
Figure 2. Expression of the SOC family is significantly decreased by transfection of target-specific siRNAs in dorsal horn neurons.
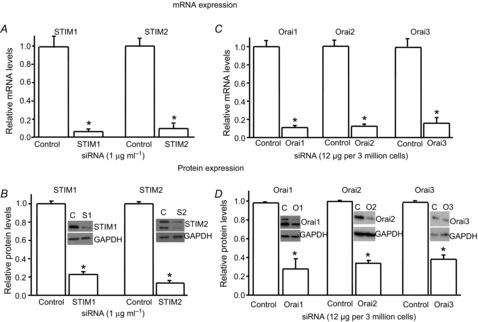
A and B, effects of specific siRNAs against STIM1 or STIM2 on mRNA levels (A) and protein levels (B) of STIM1 and STIM2, respectively (n = 4–5 samples). C and D, effects of specific siRNAs against Orai1, Orai2 or Orai3 on mRNA levels (C) and protein levels (D) of Orai1, Orai2 and Orai3, respectively (n = 4 samples each). The mRNA and protein levels were normalized to control (non-target siRNA). Values represent mean ± SEM; *P < 0.05, compared with control by Student's t test.
Depletion of calcium stores induces SOCE in dorsal horn neurons
Given the expression of SOCs in dorsal horn neurons, we next investigated whether SOCs are functional in dorsal horn neurons. For this, we performed calcium imaging recordings in living cells. Cultured neurons were loaded with a fura-2 calcium dye in HBSS. Under the microscope, neurons were easily distinguishable from glia: they appeared phase bright, and had small, smooth, rounded somata and visible processes (confirmed by staining with MAP2b, a neuronal marker). To further determine neurons in our cultured cells, we applied 60 mm KCl for 30 s, which induced a fast and transient calcium response (Δ ratio >0.2) in most cells (glial cells had no or small KCl responses). The KCl-induced calcium response was completely blocked by 200 μm CdCl2 (data not shown). When neurons were pretreated with the Ca2+-free Tyrode's solution, 1 μm TG or 30 μm CPA transiently elevated intracellular Ca2+; the subsequent addition of 2 mm CaCl2 caused sustained responses with different amplitudes in almost every neurons (Fig.3A, B and D), while CaCl2 addition induced only small calcium responses (Δ ratio around 0.12) in the absence of TG or CPA (Fig.3C and D). Both TG and CPA induced robust sustained calcium responses (Δ ratio > 0.2) in a majority of neurons. However, a small population of neurons (30 of 260) had little TG- or CPA-induced calcium responses (Δ ratio ≤0.2). To compare Ca2+ entry induced by activation of VGCCs to SOCE in individual neurons, application of 30, 60 or 90 mm KCl for 1 min induced transient calcium responses in most cells. The Ca2+ responses induced by 60 and 90 mm KCl were indistinguishable. TG (1 or 2 μm) evoked sustained calcium responses (Fig.3E). The Ca2+ responses induced by 1 and 2 μm TG were similar. TG-induced Ca2+ entry was comparable to Ca2+ entry mediated by VGCCs (Fig.3F). These data reveal a new calcium signal other than VGCCs in dorsal horn neurons.
Figure 3. Depletion of endoplasmic reticulum Ca2+ stores by thapsigargin (TG) and cyclopiazonic acid (CPA) induces calcium responses in cultured dorsal horn neurons.
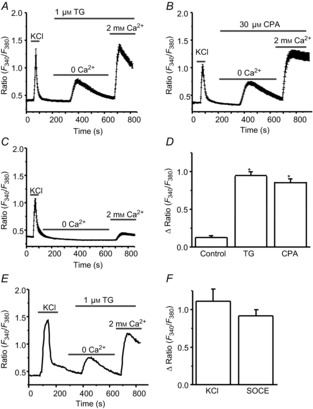
A, TG-induced calcium entry (n = 53 neurons). B, CPA-induced calcium entry (n = 57 neurons). C, addition of 2 mm Ca2+-induced calcium entry in the absence of TG or CPA (n = 39 neurons). D, summary of TG- and CPA-induced calcium influx. E, KCl- and TG-induced calcium entry (n = 19). F, summary of KCl- and TG-induced calcium influx. Values represent mean ± SEM; *P < 0.05, compared with control by one-way ANOVA.
To characterize the pharmacological properties of the TG- or CPA-induced calcium entry, we tested the effect of YM-58483, 2-APB and GdCl3, well-defined SOC inhibitors in non-excitable cells (Ishikawa et al. 2003; Putney, 2010), on TG- or CPA-induced calcium responses in dorsal horn neurons. We bath-applied 1 μm YM-58483, 30 μm 2-APB or 1 μm GdCl3 and measured the changes in the peak amplitude of calcium responses induced by 1 μm TG. Pretreatments with GdCl3 and YM-58483 did not alter calcium release from the ER stores. However, both GdCl3 and YM-58483 strongly prevented TG-induced calcium influx (Fig.4A and B). 2-APB significantly reduced TG-induced calcium influx, but also decreased calcium release in these neurons (Fig.4A–D), suggesting TG-induced calcium entry is SOCE.
Figure 4. TG-induced calcium entry is inhibited by SOC inhibitors.
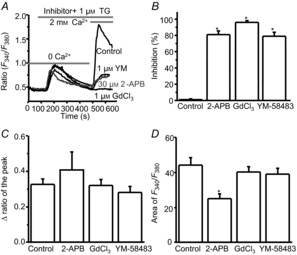
A, representative TG-induced calcium responses recorded in neurons pretreated with 2-APB, YM-58483 or GdCl3; B, summary of the effects of 2-APB (n = 32 neurons), YM-58483 (n = 41 neurons) and GdCl3 (n = 45 neurons) on TG-induced calcium entry. C and D, summary of the effects of 2-APB, YM-58483 and GdCl3 on TG-induced calcium release presented as the peak amplitude (C) or as area (D). Values represent mean ± SEM; *P < 0.05, compared with control by one-way ANOVA.
To examine whether these inhibitors can reverse SOCE, we used CPA to deplete calcium stores as it produced a more sustained calcium response. Neurons were pretreated with CPA in the Ca2+-free Tyrode's solution for more than 5 min, and the subsequent addition of 2 mm CaCl2 induced sustained responses with limited reductions over 7–8 min. Consistent with the results from pretreatment, GdCl3 completely reversed CPA-induced SOCE at 1 μm concentration (Fig.5A). YM-58483 markedly attenuated SOCE in a concentration-dependent manner with an IC50 value of about 0.67 μm (Fig.5B). Interestingly, 2-APB slightly potentiated SOCE initially, and then inhibited SOCE at both concentrations (Fig.5C). These results suggest that depletion of calcium stores activates SOCE.
Figure 5. CPA-induced calcium entry is attenuated by SOC inhibitors.

A, effect of GdCl3 on CPA-induced calcium influx. Left, representative examples; right, summary of the effect of GdCl3 (n = 15 neurons). B, effect of YM-58483 on CPA-induced calcium influx. Left, representative examples; right, summary of the effect of YM-58483 (n = 18 neurons). C, effect of 2-APB on CPA-induced calcium influx. Left, representative examples; right, summary of the effect of 2-APB (n = 11 neurons). Values represent mean ± SEM; *P < 0.05, compared with control by one-way ANOVA.
Spinal cord dorsal horn neurons express a variety of VGCCs including T, L, N and P/Q types (Heinke et al. 2004). While GdCl3 and 2-APB are not highly selective for SOCs, YM-58483 has been reported to be a potent and selective inhibitor of SOCs in lymphocytes (Ishikawa et al. 2003). To rule out the possibility that YM-58483-induced reduction of SOCE may be mediated by blocking VGCCs in dorsal horn neurons, we tested the effect of YM-58483 on VGCCs in dorsal horn neurons. Pretreatment with 3 μm YM-58483, which drastically inhibited SOCE, did not alter the amplitude of 60 mm KCl-induced calcium response (Fig.6A), suggesting that the YM-58483-induced inhibition of SOCE is not mediated by interaction with VGCCs. To further examine the impact of voltage-sensitive Ca2+ entry on SOCE, we used specific calcium channel blockers mibefradil (a T-type channel blocker), nimodipine (an L-type channel blocker) and ω-conotoxin MVIlC (an N-, P/Q-type channel blocker) as pharmacological tools as a non-selective calcium channel blocker, CdCl2, also blocks SOCE (McElroy et al. 2008). To avoid potential drug–drug interactions, these blockers were bath applied individually. Their concentrations were selected based on previous reports (Martin et al. 2000; McDonough et al. 2002; Wojda & Kuznicki, 2013). Pretreatment of neurons with 5 μm mibefradil, 5 μm nimodipine or 1 μm ω-conotoxin MVIlC did not affect the average peak of TG-induced calcium entry (Fig.6B and C), while they blocked 60 mm KCl-induced calcium responses by 56 ± 4% (n = 32), 65 ± 3% (n = 36) or 30 ± 4% (n = 35), respectively. Taken together, these results demonstrate that VGCCs were not involved in SOCE.
Figure 6. Store-operated calcium entry (SOCE) is independent of voltage-gated calcium channels (VGCCs).
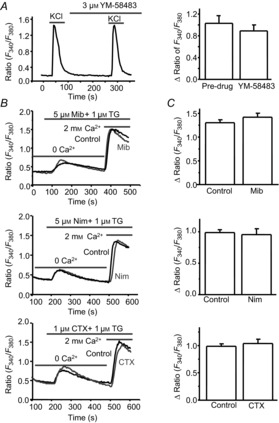
A, effect of YM-58483 on KCl-induced calcium response (n = 15 neurons). Left, representative 60 mm KCl-induced calcium responses; right, summary of the effect of YM-58483. B, representative TG-induced calcium responses recorded in neurons pretreated with mibefradil (Mib), nimodipine (Nim) or ω-conotoxin MVIIC (CTX). C, summary of the effects of mibefradil (n = 50 neurons), nimodipine (n = 45 neurons) and ω-conotoxin MVIIC (n = 32 neurons) on TG-induced SOCE. Values represent mean ± SEM; *P < 0.05, compared with control by Student's t test.
Depletion of the ER calcium stores activates SOC currents in dorsal horn neurons
Despite the very small conductance of SOCs, SOC-mediated inward calcium currents have been successfully recorded in native excitable cells (Gemes et al. 2011; Spinelli et al. 2012). We then investigated whether depletion of the ER calcium stores activates membrane conductance in dorsal horn neurons. BAPTA-induced membrane conductance changes were recorded using the standard method for measuring SOC currents as described previously (Bird et al. 2008). Neurons were selected first based on morphology and confirmed by firing an action potential in response to a current injection. To avoid interference of voltage-gated ion channels, neurons were held at −80 mV (around Cl− current reversal potential in our recording conditions) with a gap-free recording protocol (no depolarization stimulus). Intracellular application of 10 mm BAPTA developed inward currents 1 min after break-in in Tyrode's solution. These currents were decreased by replacing 10 mm Na+ with 10 mm Cs+ and completely diminished by further removing Ca2+ (Fig.7A). Similarly, using a voltage step protocol (hyperpolarized to −100 mV from 0 mV holding potential), the inward current was decreased by 10 mm Cs+, and almost eliminated by removing Ca2+ from the external solution (Fig.7B), suggesting that BAPTA activates a Cs+-insensitive calcium current, which was attenuated by 1 μm GdCl3 and by 3 μm YM-58483 (Fig.7C and D). However, these currents were only observed in one-third of neurons within 5 min after break-in. To facilitate depletion of ER calcium stores, 1 μm TG was bath applied after 1–2 min break-in. Depletion of ER calcium stores-induced calcium currents were recorded by replacing Na+ and K+ with NMDG+ and increasing Ca2+ concentration from 2 to 10 mm in the bath solution. When neurons were held at −80 mV with the gap-free protocol, depletion of ER calcium stores induced sustained calcium currents with various amplitudes from 5 to 125 pA in 82 of 95 neurons (Fig.8A), which did not reverse immediately by removing TG from the bath solution. To determine the reversal potential of these currents, a ramp protocol (from −100 to +100 mV) was used with a holding potential of 0 mV. A small basal current was recorded with the CsMeSO4-based internal solution. Depletion of ER calcium stores induced a greater inward current than an outward current (Fig.8B). The net SOC currents were obtained by subtracting the basal current (before SOC current induction) from total currents (after SOC current induction). The reversal potential of SOC currents was 55.8 ± 5.4 mV (n = 6).
Figure 7. Depletion of Ca2+ stores by BAPTA induces SOC currents in dorsal horn neurons.
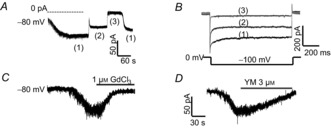
A and B, effects of replacing 10 mm Na+ with 10 mm Cs+ or removing Ca2+ on BAPTA-induced currents recorded with a gap-free protocol (A) and a step protocol (B). (1) normal Tyrode's solution; (2) replacing 10 mm NaCl with CsCl; (3) removing Ca2+ from (2). C and D, BAPTA-induced currents were attenuated by GdCl3 (C) or YM-58483 (D).
Figure 8. BAPTA/TG-induced SOC currents are mediated by CRAC channels in dorsal horn neurons.
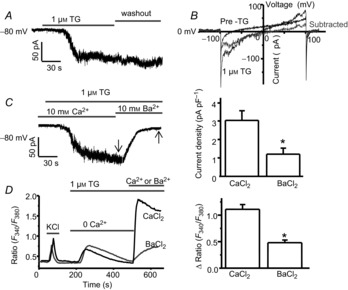
A and B, BAPTA/TG-induced calcium currents recorded with the gap-free protocol (A) and with a ramp protocol (B). C, effect of replacing 10 mm Ca2+ with 10 mm Ba2+ on BAPTA/TG-induced current recorded with the gap-free protocol. Left, a representative BAPTA/TG-induced current recorded in 10 mm Ca2+ or 10 mm Ba2+; right, summary of BAPTA/TG-induced Ca2+ current and Ba2+ current (n = 9 neurons). D, effect of replacing 2 mm Ca2+ with 2 mm Ba2+ on TG-induced SOCE. Left, representative TG-induced Ca2+ or Ba2+ entry; right, summary of TG-induced Ca2+ entry (n = 52 neurons) and Ba2+ entry (n = 40 neurons). Arrows indicate where current amplitudes were measured. Values represent mean ± SEM; *P < 0.05, compared by Student's t test.
A distinctive feature of CRAC channels is that they conduct Ca2+ better than Ba2+ in non-excitable cells (Hoth, 1995; Bakowski & Parekh, 2007). To determine whether these currents can be carried by Ba2+, we replaced 10 mm Ca2+ by 10 mm Ba2+. The currents were markedly reduced by replacing Ca2+ with Ba2+ (Fig.8C). To confirm this result, we performed calcium imaging recordings. Consistent with the finding from patch-clamp recordings, TG-induced responses were dramatically decreased by replacing Ca2+ with Ba2+ (Fig.8D), suggesting that depletion of ER calcium stores activates CRAC channels.
To determine whether depletion of the calcium stores induces SOC currents in lamina I/II neurons from adult mouse spinal cord slices, we performed SOC current recordings in spinal cord slices from adult mice using a gap-free recording protocol. Neurons were held at −80 mV, and BAPTA/TG-induced currents were recorded in the modified ACSF solution (NMDG+ based) with a gap-free protocol (no synaptic currents were observed under this condition). BAPTA/TG application activated inward currents in 59 of 67 dorsal horn neurons. The current amplitude ranged from 6 to 111 pA. Bath application of 30 μm 2-APB slightly enhanced this inward current transiently, and then drastically inhibited it (Fig.9A). GdCl3 (1 μm) reversed BAPTA/TG-induced calcium currents (Fig.9B). BAPTA/TG-induced SOC currents were also dramatically diminished by 3 μm YM-58483 (Fig.9C). The effects of SOCE inhibitors on SOC currents in slices were similar to what we observed in cultured neurons (Fig.9D–F). These results confirm that SOCs are functionally expressed in adult dorsal horn neurons.
Figure 9. SOCs are functional in lamina I/II neurons from spinal cord slices of adult mice.

A–C, representative BAPTA/TG-induced SOC currents recorded in neurons from slices and from cultures, which are reduced by 2-APB (A), GdCl3 (B) or YM-58483 (C). D–F, summary of inhibition of BAPTA/TG-induced SOC currents by 2-APB (n = 7–9 neurons), GdCl3 (n = 8–11 neurons) or YM-58483 (n = 9–12 neurons). Arrows indicate where current amplitudes were measured. Values represent mean ± SEM; *P < 0.05, compared with control (pre-drug, Pre) by paired Student's t test.
Knockdown of STIM1, STIM2 or Orai1 attenuates SOCE and reduces resting calcium level
As demonstrated above, SOCs are functionally expressed in dorsal horn neurons. It is well known that STIM1 and Orai1 are major determinants of SOCE in non-excitable cells. To identify the molecular identity of SOCs in dorsal horn neurons, the RNA interference gene silencing approach was used to knock down individual SOC proteins. We first determined whether STIM proteins mediate SOCE in dorsal horn neurons. Neurons were transfected with 1 μg ml−1 siRNAs as described above. After 48 h of transfection, calcium imaging was performed in neurons. TG (1 μm)-induced SOCE was markedly reduced by STIM1 knockdown, significantly decreased by STIM2 knockdown, and further diminished by knockdown of both STIM1 and STIM2 (Fig.10A). We next investigated whether Orai members are involved in SOCE. Neurons were transfected with siRNAs targeting Orai1, Orai2 or Orai3 as described above. Knockdown of Orai1 almost abolished SOCE. However, knockdown of Orai2 or Orai3 had no effect on SOCE (Fig.10B). These results indicated that Orai1 is necessary, STIM1 is critical and STIM2 is important for SOCE in dorsal horn neurons.
Figure 10. Orai1 is required, and STIM1 and STIM2 are important for SOCE and resting calcium homeostasis in dorsal horn neurons.
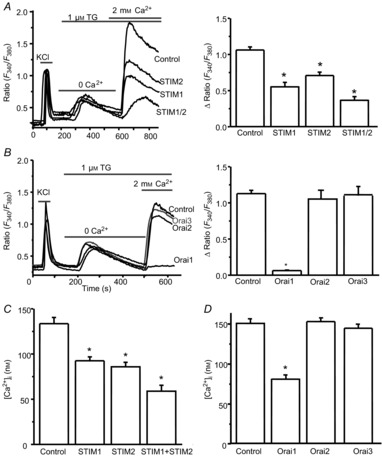
A, effects of specific siRNAs against STIMs on TG-induced SOCE. Left, representative examples of TG-induced calcium responses recorded in neurons transfected with control siRNA or targeting siRNAs against STIMs; right, summary of effects of control siRNA (n = 78 neurons), STIM1 siRNA (n = 77 neurons), STIM2 siRNA (n = 69 neurons) and mixed siRNAs against STIM1 and STIM2 (n = 32 neurons), respectively. B, effects of specific siRNAs against Orai members on TG-induced SOCE. Left, representative examples of TG-induced calcium responses recorded in neurons transfected with control siRNA or targeting siRNAs against Orai members; right, summary of the effects of control siRNA (n = 112 neurons), Orai1 siRNA (n = 76 neurons), Orai2 siRNA (n = 40 neurons) or Orai3 siRNA (n = 42 neurons). C, effects of specific siRNAs against STIMs on the resting calcium concentration measured in neurons transfected with control siRNA (n = 99 neurons), STIM1 siRNA (n = 99 neurons), STIM2 siRNA (n = 67 neurons) or mixed siRNAs against STIM1 and STIM2 (n = 45). D, effects of specific siRNAs against Orai members on the resting calcium concentration measured in neurons transfected with control siRNA (n = 329 neurons), Orai1 siRNA (n = 109 neurons), Orai2 siRNA (n = 114 neurons) or Orai3 siRNA (n = 98 neurons). Values represent mean ± SEM, *P < 0.05, compared with control by one-way ANOVA.
STIM2 has been identified as a regulator of basal Ca2+ levels in cell lines and cortical neurons (Brandman et al. 2007; Berna-Erro et al. 2009). We tested whether STIM proteins regulate the basal calcium level in dorsal horn neurons. Calcium imaging was performed in cultured dorsal horn neurons. Neurons were transfected with siRNA targeting STIM1, STIM2 or non-targeting siRNA. In the normal Tyrode's solution (2 mm Ca2+), the resting calcium concentration of dorsal horn neurons was about 150 nm. Knockdown of STIM2 reduced the basal cytosolic calcium concentration by 35%. Surprisingly, knockdown of STIM1 also led to a marked decrease in the basal cytosolic calcium level in dorsal horn neurons by 30% (Fig.10C). We were interested in determining whether the CRAC channels Orai1, Orai2 or Orai3 regulate the basal calcium level. Neurons were transfected with specific siRNAs targeting Orai1, Orai2, Orai3 or non-targeting siRNA. Knockdown of Orai1 decreased basal Ca2+ by 47%, while knockdown of Orai2 or Orai3 had no effect (Fig.10D). These findings indicate that STIM1, STIM2 and Orai1 are important regulators of basal cytosolic calcium homeostasis.
To confirm the data obtained from calcium imaging recordings and the involvement of STIM1, STIM2 and Orai1 in mediating SOC currents, whole-cell patch-clamp recordings were performed. Neurons were co-transfected with a GFP-expressing plasmid and STIM1, STIM2, Orai1 or control siRNAs. BAPTA/TG-induced SOC currents were recorded in GFP-positive neurons. Neurons were held at −80 mV using a gap-free recording protocol. SOC currents were drastically reduced by knockdown of STIM1, STIM2 or Orai1 (Fig.11A and B). These results confirmed that STIM1, STIM2 and Orai1 are major components of SOCs that mediate the SOC current in dorsal horn neurons.
Figure 11. SOC currents are mediated by STIM1, STIM2 and Orai1.
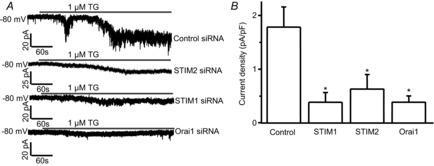
Effects of specific siRNAs against STIM1, STIM2 or Orai1 on BAPTA/TG-induced calcium currents. A, representative BAPTA/TG-induced calcium currents recorded with the gap-free protocol in neurons transfected with control siRNA, targeting siRNAs. B, summary of the effects of control siRNA (n = 25 neurons), STIM1 siRNAs (n = 16 neurons), STIM2 siRNA (n = 18 neurons) and Orai1 siRNA (n = 18 neurons), respectively. Values represent mean ± SEM; *P < 0.05, compared with control by one-way ANOVA.
Activation of SOCs induces membrane depolarization in dorsal horn neurons
To understand the functional significance of SOCs in dorsal horn neurons, we examine the overall effect of depletion of Ca2+ stores on neuronal intrinsic excitability. Current-clamp recordings were performed and normal Tyrode's solution was used as the bath solution. The resting membrane potentials of recorded neurons were around −50 to −60 mV. As shown in Fig.8B, SOC currents are greater at more hypolarized potentials. To obtain a consistent and robust effect, membrane potentials in all recorded neurons were maintained at a more hypolarized potentials (around −80 mV) by injection of negative currents (−22 ± 3 pA). BAPTA/TG produced significant membrane depolarization in 19 of 22 recorded neurons. In some neurons, BAPTA/TG evoked action potentials. This effect was almost completely abolished by 0.3 μm GdCl3 or 3 μm YM-58483 (Fig.12). These results indicate that activation of SOCs produces an excitatory action in spinal cord dorsal horn neurons.
Figure 12. Activation of SOCs induces membrane depolarization in dorsal horn neurons.
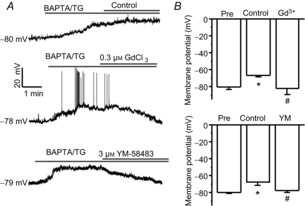
A, representative examples of BAPTA/TG-induced depolarization treated with control, 0.3 μm GdCl3 or 3 μm YM-58483. B, summary of the effects of GdCl3 or YM-58483 on BAPTA/TG-induced changes in membrane potential. Values represent mean ± SEM; n = 5–9 neurons each; *P < 0.05 compared with pre (before TG application), #P < 0.05 compared with control by one-way ANOVA.
Activation of NK1 receptors leads to SOCE in dorsal horn neurons
Our previous study demonstrated that inhibition of SOCE by YM-58483 attenuates pain hypersensitivity (Gao et al. 2013). We investigated whether SOCs are involved in pain transmission. SP plays an important role in pain processing through the activation of NK1 receptors in dorsal horn neurons. To test whether SP can induce SOCE in these neurons, bath application of 100 nm SP induced a transient calcium response in the absence of Ca2+; addition of 2 mm Ca2+ caused a robust calcium influx (Fig.13A). To determine whether this effect is mediated by NK1 receptors, we tested the effect of RP 67580, a potent and selective NK1 receptor antagonist (Garret et al. 1991), on SP-induced calcium entry. Neurons were pretreated with 1 μm RP 67580; SP-induced Ca2+ release and Ca2+ influx were completely blocked by RP 67580 (Fig.13B and C). To determine whether this Ca2+ entry is through SOCs, we pretreated neurons with 3 μm YM-58483, and found that SP-induced Ca2+ release was not affected by YM-58483, although Ca2+ influx was blocked by YM-58483 (Fig.13D and F). To further confirm that this effect was mediated by Orai1, neurons were transfected with specific Orai1 siRNA. Knockdown of Orai1 also abolished SP-induced Ca2+ entry, but had no effect on Ca2+ release (Fig.13E and F). These results suggested that SOCs are involved in the function of NK1 receptors.
Figure 13. Activation of NK1 receptors results in SOCE in dorsal horn neurons.
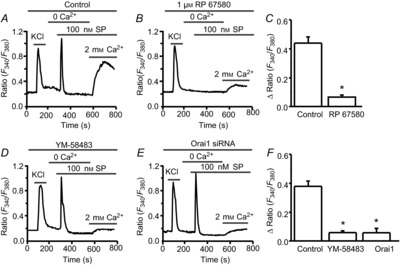
A and B, representative examples of substance P (SP)-induced Ca2+ responses in the absence (A), or in the presence of 1 μm RP 67580 (B). C, summary of the effect of RP 67580 on SP-induced SOCE. D and E, representative examples of SP-induced Ca2+ responses in the presence of YM-58483 (D), or after knockdown of Orai1 (E). F, summary of the effects of 3 μm YM-58483 and knockdown of Orai1 on SP-induced SOCE. Values represent mean ± SEM; n = 42–54 neurons each; *P < 0.05, compared with control by Student's t test (C) or one-way ANOVA (F).
Discussion
The major findings of this study are the demonstration that SOCE, as a new calcium signal, is present in dorsal horn neurons and that SOCs play a role in the physiological function of dorsal horn neurons. This is the first study identifying STIM1, STIM2 and Orai1 as major players mediating SOCE and SOC current in dorsal horn neurons.
Our RT-PCR results demonstrated that the SOC family is expressed in the spinal cord and dorsal horn neurons. STIM2 level is greater than STIM1, consistent with previous reports that STIM2 is the dominant isoform in the brain (Berna-Erro et al. 2009; Skibinska-Kijek et al. 2009). Western blot analysis with the specific STIM1 antibody shows a single band at 85 kDa, demonstrating STIM1 protein expression. We observed two bands of STIM2 at 105 and 115 kDa in Western blots, which agrees with a previous study showing that STIM2 has two differential levels of phosphorylation isoforms at 105 and 115 kDa (Williams et al. 2001). We also found that Orai1 protein is present as two molecular weights at 45 and 55 kDa. It has been shown that Orai1 has two isoforms: a longer form (Orai1α) of 33 kDa and a shorter form (Orai1β) of 23 kDa (Fukushima et al. 2012). These two molecule weights of Orai1 are likely to be the result of post-translational modification of Orai1 (glycosylation) (Gwack et al. 2007). Western blots with antibodies against Orai2 and Orai3 show weights of roughly 28 kDa (Orai2) and 33 kDa (Orai3), which are similar to those in other cell types (Motiani et al. 2010). Importantly, knockdown of STIM1, STIM2, Orai1, Orai2 and Orai3 with specific siRNAs dramatically decreases their mRNA and protein expression levels, further confirming the expression of the SOC family in dorsal horn neurons.
The calcium imaging data strongly support the finding that SOCs are functional in dorsal horn neurons. Like VGCCs, TG- or CPA-induced calcium entry was observed with various amplitudes in almost every single dorsal horn neuron. Neurons possess a variety of voltage-gated sodium and calcium channels which can be activated by depolarization. We eliminated interference of sodium channel activation by including TTX in the bath solution. VGCCs have been considered to be major routes of calcium entry. Low voltage-gated T-type calcium channels can open in response to smaller membrane voltage fluctuations. We therefore pretreated neurons with mibefradil, a specific T-type channel blocker, and found that it did not block TG-induced calcium entry, indicating T-type channels are not involved in SOCE. In addition, we tested the effects of calcium channel blockers for L-, N- and P/Q-types on TG-induced calcium entry. None of these blockers had any effect on SOCE, suggesting that SOCE is independent of VGCCs in dorsal horn neurons, consistent with previous studies in other cell types (McElroy et al. 2008; Gemes et al. 2011). TG- or CPA-induced calcium entry was dramatically blocked by SOC inhibitors 2-APB and GdCl3, suggesting that TG- or CPA-induced calcium response is SOCE. Because 2-APB and GdCl3 are not highly selective for SOCs, we also tested a more potent SOCE inhibitor, YM-58483. YM-58483 strongly blocks SOCE but not VGCC-mediated calcium entry, further indicating that SOCE as a voltage-independent calcium signal is functional in dorsal horn neurons.
Our electrophysiology results showed that depletion of calcium stores activates membrane currents with various amplitudes in dorsal horn neurons. Intracellular application of a high concentration of BAPTA slowly induces an inward current. This total inward current is reduced by replacing 10 mm Na+ with 10 mm Cs+. It has been reported that Cs+ is relatively impermeable to SOCs (Parekh & Putney, 2005), and blocks hyperpolarization/cyclic nucleotide-gated (HCN) cation channels (Oswald et al. 2009), which are carried by Na+ and K+. HCN channels are expressed in the spinal cord dorsal horn (Rivera-Arconada et al. 2013). These channels are open at the resting membrane potential (Day et al. 2005). Cs+-sensitive currents may be attributed to Ih currents. In the presence of Cs+, the currents are almost completely eliminated by removing extracellular Ca2+. BAPTA-induced currents are diminished by YM-58483 and GdCl3, suggesting that BAPTA-induced currents are mediated by SOCs in dorsal horn neurons. Since intracellular application of BAPTA alone only activated inward currents in some neurons, we bath applied TG to facilitate current activation. BAPTA/TG activated an inward current in most neurons, which was also blocked by 2-APB, YM-58483 and GdCl3. It is well known that VGCCs are highly selective for Ca2+ and Ba2+. In contrast to VGCCs, CRAC channels are highly selective for Ca2+, but are less permeable to Ba2+ in non-excitable cells (Hoth, 1995; Bakowski & Parekh, 2007). Our data from both patch-clamp and calcium imaging recordings show that calcium entry and currents induced by depletion of calcium stores are reduced by replacing Ca2+ with Ba2+, further confirming that CRAC channels mediate SOCE and SOC currents.
As cultured neurons may undergo phenotypic changes and neonatal neurons may go through developmental changes, the functions of the neonatal cultured neurons may differ from neurons to the intact adult tissue. We therefore confirmed functional expression of SOC currents in adult mice. SOC current recordings were performed in lamina I/II neurons from acutely prepared adult spinal cord slices. Similar to the data obtained from cultured dorsal horn neurons, SOC currents are present in adult mouse dorsal horn neurons, suggesting that SOCs do not undergo phenotypic or developmental changes. It is important to point out that SOCE is present in almost every neuron with calcium imaging recordings. SOC current was only observed in 86% of neurons with patch-clamp recordings. This discrepancy may be due to two different techniques. In general, the sensitivity of calcium imaging recording is higher than patch-clamp recording. Small calcium responses in some neurons may not be detected with the patch-clamp technique. Note that dorsal horn neurons represent a heterogeneous population and can be divided into projection neurons and interneurons (excitatory or inhibitory). Although every neuron has SOCE, the amplitudes vary. Also, amplitudes of SOC currents range from 5 to 125 pA. The functional expression of SOCs in different populations remains to be determined.
STIM1 and Orai1 are the key components of SOCs and STIM2 has a limited effect or inhibits STIM1-mediated SOCE in CD4+ T cells or overexpressing cell lines (Soboloff et al. 2006; Shaw & Feske, 2012). Given that the pharmacological properties of SOCE and SOC currents in dorsal horn neurons are similar to those in non-neuronal cells, we hypothesized that STIM1 and Orai1 play a key role in SOCE in dorsal horn neurons. Our knockdown results show that reduction of STIM1 or Orai1 markedly decreases SOCE, consistent with our hypothesis. However, reduction of STIM2 also significantly reduces SOCE and double knockdown of STIM1 and STIM2 further decreases SOCE in neurons, suggesting that STIM2 is an important contributor to SOCE in spinal cord neurons. These findings are in agreement with previous reports that STIM2 also functions as an ER Ca2+ sensor to activate SOCE (Kar et al. 2012; Thiel et al. 2013).
Under the resting state, dorsal horn neurons maintain the cytosolic calcium concentration at about 150 nm. It is well known that the basal calcium level is controlled by the activity of calcium pumps and Na+/Ca2+ exchangers. Little is known about contributions of the ER calcium stores and basal Ca2+ influx. Previous studies have indicated that STIM2 serves as a regulator of basal Ca2+ (Brandman et al. 2007; Berna-Erro et al. 2009). Indeed, we found that knockdown of STIM2 reduces the basal calcium level. However, knockdown of STIM1 and Orai1 also decreases the basal cytosolic calcium level. Our data suggest that STIM1, STIM2 and Orai1 play important roles in both SOCE and resting calcium homeostasis, indicating that SOCE may contribute to maintaining basal cytosolic calcium homeostasis. Our results support previous findings in myotubes and neurons that SOCs mediate SOCE and regulate resting calcium homeostasis (Berna-Erro et al. 2009; Li et al. 2010). The role of SOCs in maintaining basal calcium homeostasis appears to be complex and remains incompletely understood. Additional studies are needed to further establish the functional roles of SOCs in basal calcium homeostasis in neuronal cells.
We have previously demonstrated that inhibition of SOCs produces not only peripheral but also central analgesic effects (Gao et al. 2013), indicating that SOCs may be involved in central pain processing. Our current-clamp recordings revealed that activation of SOCs results in membrane depolarization and even induces action potentials in some dorsal horn neurons. This excitatory action fits well with our previous behavioural study that blockage of SOCs attenuated pain hypersensitivity (Gao et al. 2013). Note that the amount of depolarization induced by SOCE appears greater than expected. It would be surprising that activation of a small Ca2+ selective current could lead to such a robust membrane depolarization. Given dorsal horn neurons express numerous ion channels including calcium-activated non-specific cation channels (Morisset & Nagy, 1999), it is possible that the depolarization is partially attributed to SOCE-mediated secondary activation and modulation of non-selective or selective cation channels. It has been shown that SOCE is involved in neurotransmitter release and synaptic plasticity in hippocampal neurons (Bouron, 2000; Baba et al. 2003). SOCs can be activated by physiological agonists through G protein-coupled receptor signalling (Kar et al. 2012; Thiel et al. 2013). NK1 receptors are G-protein coupled receptors and are activated by the neurotransmitter SP, a well-known modulator of pain transmission. One would hypothesize that SOCs are involved in SP signalling. Our results are consistent with this hypothesis and suggest that SOCs can be activated by SP. In addition, chronic pain conditions are associated with an increase in resting cytoplasmic calcium concentration (Kawamata & Omote, 1996; Kruglikov et al. 2004). SOCs regulate the resting calcium homeostasis and may modulate the calcium-dependent pathological events related to chronic pain.
In summary, we demonstrate that the SOC family is functionally expressed in dorsal horn neurons. We identified STIM1, STIM2 and Orai1 as major players mediating SOCE and maintaining resting calcium homeostasis. Our results indicate that SOCs are involved in the function of NK1 receptors and that activation of SOCs produces an excitatory action in dorsal horn neurons. These findings provide a potential mechanism for the YM-58483-induced central analgesic effect reported previously (Gao et al. 2013), and reveal a new calcium signal in dorsal horn neurons. Our study opens an exciting new area in pain research and may suggest new avenues for the treatment of chronic pain.
Acknowledgments
We thank Dr J. E. Barrett for critical reading and valuable comments.
Glossary
- ACSF
artificial cerebrospinal fluid
- 2-APB
2-aminoethyl diphenyl borate
- ANOVA
analysis of variance
- [Ca2+]i
intracellular free calcium concentration
- CPA
cyclopiazonic acid
- CTX
ω-conotoxin MVIIC
- CRAC channels
Ca2+ release-activated Ca2+ channels
- DMSO
dimethyl sulfoxide
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- HBSS
Hanks balance salt solution
- HCN
hyperpolarization/cyclic nucleotide-gated
- NK1
neurokinin 1
- NMDG
N-methyl-d-glucamine
- RIPA
radio immunoprecipitation assay
- RP 67580
(3aR,7aR)-octahydro-2-[1-imino-2-(2-methoxyphenyl)ethyl]-7,7-diphenyl-4H-isoindol
- siRNA
small inhibitory RNA
- SOCE
store-operated Ca2+ entry
- SOCs
store-operated calcium channels
- SP
substance P
- STIM
stromal cell-interaction molecule
- TG
thapsigargin
- TTX
tetrodotoxin
- VGCCs
voltage-gated calcium channels
- YM-58483
N-[4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl]-4-methyl-1,2,3-thiadiazole-5-carboxamide
Key points
A previous study indicates that store-operated calcium channels (SOCs) play a role in pain hypersensitivity. Here we report for the first time that SOCs are expressed and functional in spinal cord dorsal horn neurons.
Using the small inhibitory RNA knockdown approach, we have demonstrated that Orai1 is necessary, and both STIM1 and STIM2 are important for SOC entry and SOC current in dorsal horn neurons.
Our findings demonstrate that STIM1, STIM2 and Orai1 play an important role in resting Ca2+ homeostasis.
Our results also indicate that SOCs are involved in the function of neurokinin 1 receptors and activation of SOCs produces an excitatory action in dorsal horn neurons.
The present study reveals that a novel calcium signal mediated by SOCs is present in dorsal horn neurons and provides a potential mechanism for SOC inhibition-induced central analgesia.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
H.H. and J.X. conceived the project and designed experiments. J.X., R.P. and X.G. performed all experiments, and analysed the data. H.H., J.X., X.G. and O.M. prepared the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by NIH/NINDS grant R21 NS077330-01A1.
References
- Baba A, Yasui T, Fujisawa S, Yamada RX, Yamada MK, Nishiyama N, Matsuki N. Ikegaya Y. Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J Neurosci. 2003;23:7737–7741. doi: 10.1523/JNEUROSCI.23-21-07737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowski D. Parekh AB. Voltage-dependent Ba2+ permeation through store-operated CRAC channels: implications for channel selectivity. Cell Calcium. 2007;42:333–339. doi: 10.1016/j.ceca.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Berna-Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G. Nieswandt B. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal. 2009;2:ra67. doi: 10.1126/scisignal.2000522. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I. Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Bird GS, DeHaven WI, Smyth JT. Putney JW., Jr Methods for studying store-operated calcium entry. Methods. 2008;46:204–212. doi: 10.1016/j.ymeth.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouron A. Activation of a capacitative Ca2+ entry pathway by store depletion in cultured hippocampal neurones. FEBS Lett. 2000;470:269–272. doi: 10.1016/s0014-5793(00)01340-5. [DOI] [PubMed] [Google Scholar]
- Brandman O, Liou J, Park WS. Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T. Surmeier DJ. Dendritic excitability of mouse frontal cortex pyramidal neurons is shaped by the interaction among HCN, Kir2, and Kleak channels. J Neurosci. 2005;25:8776–8787. doi: 10.1523/JNEUROSCI.2650-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Wu J, Lin Q. Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22:4196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Lirk P, Stucky C, Abram SE. Hogan QH. Painful nerve injury decreases resting cytosolic calcium concentrations in sensory neurons of rats. Anesthesiology. 2005;102:1217–1225. doi: 10.1097/00000542-200506000-00023. [DOI] [PubMed] [Google Scholar]
- Fukushima M, Tomita T, Janoshazi A. Putney JW. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J Cell Sci. 2012;125:4354–4361. doi: 10.1242/jcs.104919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Gao X, Xia J, Tian Y, Barrett JE, Dai Y. Hu H. Potent analgesic effects of a store-operated calcium channel inhibitor. Pain. 2013;154:2034–2044. doi: 10.1016/j.pain.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garret C, Carruette A, Fardin V, Moussaoui S, Peyronel JF, Blanchard JC. Laduron PM. Pharmacological properties of a potent and selective nonpeptide substance P antagonist. Proc Natl Acad Sci U S A. 1991;88:10208–10212. doi: 10.1073/pnas.88.22.10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM. Hogan QH. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci. 2011;31:3536–3549. doi: 10.1523/JNEUROSCI.5053-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M. Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG. Rao A. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- Heinke B, Balzer E. Sandkuhler J. Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. Eur J Neurosci. 2004;19:103–111. doi: 10.1046/j.1460-9568.2003.03083.x. [DOI] [PubMed] [Google Scholar]
- Hoth M. Calcium and barium permeation through calcium release-activated calcium (CRAC) channels. Pflugers Arch. 1995;430:315–322. doi: 10.1007/BF00373905. [DOI] [PubMed] [Google Scholar]
- Hu HJ. Gereau RW. ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J Neurophysiol. 2003;90:1680–1688. doi: 10.1152/jn.00341.2003. [DOI] [PubMed] [Google Scholar]
- Ishikawa J, Ohga K, Yoshino T, Takezawa R, Ichikawa A, Kubota H. Yamada T. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J Immunol. 2003;170:4441–4449. doi: 10.4049/jimmunol.170.9.4441. [DOI] [PubMed] [Google Scholar]
- Kar P, Bakowski D, Di Capite J, Nelson C. Parekh AB. Different agonists recruit different stromal interaction molecule proteins to support cytoplasmic Ca2+ oscillations and gene expression. Proc Natl Acad Sci U S A. 2012;109:6969–6974. doi: 10.1073/pnas.1201204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata M. Omote K. Involvement of increased excitatory amino acids and intracellular Ca2+ concentration in the spinal dorsal horn in an animal model of neuropathic pain. Pain. 1996;68:85–96. doi: 10.1016/S0304-3959(96)03222-8. [DOI] [PubMed] [Google Scholar]
- Koss DJ, Riedel G, Bence K. Platt B. Store-operated Ca2+ entry in hippocampal neurons: regulation by protein tyrosine phosphatase PTP1B. Cell Calcium. 2013;53:125–138. doi: 10.1016/j.ceca.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Kruglikov I, Gryshchenko O, Shutov L, Kostyuk E, Kostyuk P. Voitenko N. Diabetes-induced abnormalities in ER calcium mobilization in primary and secondary nociceptive neurons. Pflugers Arch. 2004;448:395–401. doi: 10.1007/s00424-004-1263-8. [DOI] [PubMed] [Google Scholar]
- Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- Li H, Ding X, Lopez JR, Takeshima H, Ma J, Allen PD. Eltit JM. Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J Biol Chem. 2010;285:39171–39179. doi: 10.1074/jbc.M110.149690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ. Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–DDCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma J, McCarl CA, Khalil S, Luthy K. Feske S. T-cell-specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur J Immunol. 2010;40:3028–3042. doi: 10.1002/eji.201040614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RL, Lee JH, Cribbs LL, Perez-Reyes E. Hanck DA. Mibefradil block of cloned T-type calcium channels. J Pharmacol Exp Ther. 2000;295:302–308. [PubMed] [Google Scholar]
- McDonough SI, Boland LM, Mintz IM. Bean BP. Interactions among toxins that inhibit N-type and P-type calcium channels. J Gen Physiol. 2002;119:313–328. doi: 10.1085/jgp.20028560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy SP, Gurney AM. Drummond RM. Pharmacological profile of store-operated Ca2+ entry in intrapulmonary artery smooth muscle cells. Eur J Pharmacol. 2008;584:10–20. doi: 10.1016/j.ejphar.2008.01.018. [DOI] [PubMed] [Google Scholar]
- Morisset V. Nagy F. Ionic basis for plateau potentials in deep dorsal horn neurons of the rat spinal cord. J Neurosci. 1999;19:7309–7316. doi: 10.1523/JNEUROSCI.19-17-07309.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motiani RK, Abdullaev IF. Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285:19173–19183. doi: 10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Dougherty KJ. Johnston D. Calcium store depletion induces persistent perisomatic increases in the functional density of h channels in hippocampal pyramidal neurons. Neuron. 2010;68:921–935. doi: 10.1016/j.neuron.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai J, Burbassi S, Rubin J. Meucci O. CXCL12 inhibits expression of the NMDA receptor's NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Dis. 2010;1:e33. doi: 10.1038/cddis.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald MJ, Oorschot DE, Schulz JM, Lipski J. Reynolds JN. Ih current generates the afterhyperpolarisation following activation of subthreshold cortical synaptic inputs to striatal cholinergic interneurons. J Physiol. 2009;587:5879–5897. doi: 10.1113/jphysiol.2009.177600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Putney JW. Pharmacology of store-operated calcium channels. Mol Interv. 2010;10:209–218. doi: 10.1124/mi.10.4.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza M, Deshpande LS, Blair RE, Carter DS, Sombati S. DeLorenzo RJ. Aging is associated with elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons. Neurosci Lett. 2007;418:77–81. doi: 10.1016/j.neulet.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Arconada I, Roza C. Lopez-Garcia JA. Characterization of hyperpolarization-activated currents in deep dorsal horn neurons of neonate mouse spinal cord in vitro. Neuropharmacology. 2013;70:148–155. doi: 10.1016/j.neuropharm.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Selvaraj S, Watt JA. Singh BB. TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP+ Cell Calcium. 2009;46:209–218. doi: 10.1016/j.ceca.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ. Feske S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J Physiol. 2012;590:4157–4167. doi: 10.1113/jphysiol.2012.233221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siau C. Bennett GJ. Dysregulation of cellular calcium homeostasis in chemotherapy-evoked painful peripheral neuropathy. Anesth Analg. 2006;102:1485–1490. doi: 10.1213/01.ane.0000204318.35194.ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinska-Kijek A, Wisniewska MB, Gruszczynska-Biegala J, Methner A. Kuznicki J. Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol Exp (Wars) 2009;69:413–428. doi: 10.55782/ane-2009-1753. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA. Gill DL. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr Biol. 2006;16:1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- Spinelli AM, Gonzalez-Cobos JC, Zhang X, Motiani RK, Rowan S, Zhang W, Garrett J, Vincent PA, Matrougui K, Singer HA. Trebak M. Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflugers Arch. 2012;464:481–492. doi: 10.1007/s00424-012-1160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targos B, Baranska J. Pomorski P. Store-operated calcium entry in physiology and pathology of mammalian cells. Acta Biochim Pol. 2005;52:397–409. doi: 10.18388/abp.2005_3452. [DOI] [PubMed] [Google Scholar]
- Thiel M, Lis A. Penner R. STIM2 drives Ca2+ oscillations through store-operated Ca2+ entry caused by mild store depletion. J Physiol. 2013;591:1433–1445. doi: 10.1113/jphysiol.2012.245399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH. Zhuo M. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. J Neurosci. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ. Dziadek MA. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357:673–685. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissenbach U, Philipp SE, Gross SA, Cavalie A. Flockerzi V. Primary structure, chromosomal localization and expression in immune cells of the murine ORAI and STIM genes. Cell Calcium. 2007;42:439–446. doi: 10.1016/j.ceca.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Wojda U. Kuznicki J. Alzheimer's disease modeling: ups, downs, and perspectives for human induced pluripotent stem cells. J Alzheimers Dis. 2013;34:563–588. doi: 10.3233/JAD-121984. [DOI] [PubMed] [Google Scholar]
- Wojda U, Salinska E. Kuznicki J. Calcium ions in neuronal degeneration. IUBMB Life. 2008;60:575–590. doi: 10.1002/iub.91. [DOI] [PubMed] [Google Scholar]