

The protein C anticoagulant system plays a critical role in the regulation of haemostasis and inflammation. Binding of protein C to endothelial cell protein C receptor (EPCR) and the subsequent activation of EPCR-bound protein C by thrombin/thrombomodulin are the key steps in the protein C anticoagulant system. Activated protein C (APC) -mediated cell signalling, supported by its binding to EPCR or independent of EPCR, activates anti-inflammatory and cytoprotective pathways [1]. Therefore, pathophysiologic conditions that impair the interaction of protein C with EPCR have the potential to decrease APC generation. Such reduced binding to EPCR may not only contribute to thrombotic disorders, but also result in the loss of endogenous APC-mediated cytoprotection..
EPCR shares sequence and three-dimensional homology with the major histocompatibility class 1/CD1 family of proteins, particularly CD1d [2,3]. Similar to the CD1 family proteins, EPCR has a tightly bound phospholipid in the antigen presenting groove [3]. This similarly led to an initial belief that EPCR may play a role in presenting protein C/APC or lipid antigen in inflammatory cells [2]. However, at present, there is no evidence that, similar to CD1d, EPCR plays a role in presenting lipid antigens to inflammatory cells. However, the presence of the lipid in EPCR seems to be essential for EPCR binding to protein C, as extraction of the lipid from EPCR abolishes protein C binding [3]. Molecular dynamic simulations of phosphatidylethanolamine-bound and -unbound forms of EPCR reveal that the lipid likely maintains the conformation of EPCR that is necessary for the interaction with its ligands [4]. Recently, Hermida and colleagues showed that phosphatidylcholine (PC) is the major phospholipid bound to human EPCR, and that lipid exchange can occur in EPCR, just as it can in CD1d [5]. They also showed that the exchange of PC in EPCR for lyso PC or platelet activating factor (PAF) impaired the ability of EPCR to bind protein C and FVII, indicating that EPCR function could be modulated by a change in the identity of the phospholipid in the hydrophobic groove of EPCR. Secretory group V phospholipase A2 (sPLA2-V), an enzyme that can be upregulated in a variety of inflammatory conditions and that metabolises PC into lyso PC, is capable of modulating both the binding of protein C to EPCR and the generation of APC on endothelial cells [5]. These data have raised the possibility that sPLA2-V may exert prothrombotic and proinflammatory effects through the modification of the bound lipid in EPCR.
In a study published in this issue of Journal of Thrombosis and Haemostasis, Tamayo et al. provide evidence that sPLA2-V, indeed, plays a thrombogenic role in vivo [6]. The data presented in the manuscript show that overexpression of sPLA2-V in mice by hydrodynamic gene delivery impairs the ability of mice to activate protein C. More importantly, sPLA2-V overexpression accelerates thrombus formation in a carotid artery laser thrombosis model. When EPCR was blocked with a blocking antibody, sPLA2-V overexpression no longer had a significant influence upon APC generation or thrombus formation. Furthermore, administration of manoalide, an inhibitor of sPLA2-V, significantly increased APC generation and moderately reduced thrombus formation in wild-type mice. From these data, the authors conclude that sPLA2-V downregulates protein C activation in vivo by encrypting EPCR, and thus promotes thrombus formation. The authors raise the possibility of targeting sPLA2-V activity as an antithrombotic strategy.
The present in vivo study builds on the authors’ earlier study using yeast expressed purified human soluble EPCR (sEPCR) and EPCR expressed on endothelial cells. To understand the true significance of the present study and its limitations, one should first know what the earlier study had shown, and more importantly, what it did not show. The earlier study showed the followings: (1) PC is the phospholipid located in the hydrophobic pocket of EPCR; (2) delipidated sEPCR had reduced ability to interact with its ligands; (3) lyso PC and PAF can locate into the hydrophobic pocket of sEPCR; (4) sEPCR containing lyso PC or PAF has impaired APC binding; (5) inhibition of sPLA2-V on endothelial cells, either by treating cells with sPLA2-V inhibitor or silencing sPLA2-V gene, increased the ligand binding to EPCR and enhanced APC generation on endothelial cells. The authors strongly imply from this data that sPLA2-V modulates the EPCR ability to generate APC by hydrolysing the PC in the EPCR to lyso PC. It is important to note here that there is no evidence presented in this study to show that the inhibition of the sPLA2-V activity actually changed the lipid in EPCR on endothelial cells.
The data presented in the current report [6] clearly demonstrates that overexpression of sPLA2-V inhibits APC generation, whereas inhibition of endogenous sPLA2-V increases APC generation in vivo. sPLA2-V overexpression did not alter the expression levels of EPCR or TM, and appears to have no significant effect on overall haemostatic balance as measured in whole blood thromboelastometry. However, this study does not provide evidence that sPLA2-V actually modifies the lipid bound to EPCR, or that such lipid modification is responsible for the observed decrease in the APC generation in mice overexpressing sPLA2-V. With the current existing methodologies, it may not be feasible to show in vivo that sPLA2-V alters the lipid in the EPCR on the endothelium. However, lack of direct evidence for such a mechanism even in the in vitro cell system makes it difficult to conclude definitively that sPLA2-V-mediated lipid modification in EPCR is responsible for the reduced APC generation thus results in accelerated thrombus formation in mice overexpressing sPLA2-V. There are a number of other caveats and limitations in the present study. Some of them are probably unavoidable, but others are avoidable. Lack of a suitable method to effectively measure the plasma levels of sPLA2-V makes it difficult to determine any correlation exists between sPLA2-V and APC levels in the above group of mice. There is no information in the present study whether sPLA2-V overexpression actually impairs protein C binding to EPCR on the endothelium. This could have been easily examined by measuring protein C levels in circulation in wild-type and sPLA2-V overexpressing mice. This study measures APC levels in mice following PC/phosphatidylserine/factor Xa challenge and not in control unchallenged mice. Since the reduction of APC levels in sPLA2-V overexpressing mice is proposed to be responsible for the accelerated thrombus formation following the laser injury in mice that are not challenged with PC/phosphatidylserine/factor Xa prior to the laser injury, it is more appropriate to measure APC levels in unchallenged mice. To substantiate that EPCR is needed for sPLA2-V to exert its prothrombotic effect, sPLA2-V overexpressing mice were administered with anti-EPCR mAb that blocks protein C/APC binding to EPCR. This experiment showed that sPLA2-V overexpression was unable to accelerate the thrombus formation when EPCR was blocked with the antibody. But the EPCR blocking antibody treatment exhibits a stronger prothrombotic effect than sPLA2-V overexpression (i.e., greater reduction in APC generation and increased thrombus formation). Thus, blocking protein C binding to EPCR is not an appropriate experiment to assess the role of EPCR in sPLA2-V-mediated prothrombotic effect.
Despite the above caveats and limitations, the present study provides valuable and novel information, i.e. sPLA2-V plays a thrombogenic role in vivo. sPLA2-V is constitutively expressed by endothelial cells, and its expression is increased by inflammatory stimuli, such as tumour necrosis factor-α and vascular endothelial growth factor [7,8]. sPLA2-V is expressed in all stages of atherosclerosis development and thought to play an atherogenic role through degradation of phospholipids [9,10]. Therefore, it is entirely possible that upregulation of sPLA2-V in inflammation and other diseases may contribute, at least partly, to thrombotic disorders associated with these diseases. If so, inhibition of sPLA2-V activity in such patients may represent an effective antithrombotic therapeutic strategy. To further investigate this possibility, it is important to determine first whether sPLA2-V levels correlate with thrombotic risk in specific patient groups. This requires development of a suitable assay to measure sPLA2-V levels in plasma.
APC plays an influential role in many disease processes, including thrombosis, sepsis, inflammation, diabetes and cancer [1,11–13]. Protein C binding to EPCR is critical for the generation of APC. APC-mediated anti-inflammatory and cytoprotective effects most frequently require APC binding to EPCR. In addition to protein C and APC, many other ligands bind EPCR. They include factor VIIa, proteinase-3, β2 integrin Mac-1, Plasmodium falciparum erythrocyte membrane protein 1, and γδ T cell antigen receptor [1]. Recent studies elucidating the importance of interactions between these novel ligands and EPCR indicate new roles for EPCR in haemostasis, the pathogenesis of severe malaria, innate immunity and cancer [1]. Interestingly, most of the ligands that bind EPCR do so at sites that overlap with the binding site for protein C. It will therefore be very interesting to ascertain whether EPCR lipid modification also influences the binding of other ligands to EPCR. If the proposed mechanism of sPLA2-V-mediated impairment of protein C activation is proven to be correct, i.e. sPLA2-V-mediated lipid modification in EPCR results in the loss of the ligand binding to EPCR, it opens an exciting new area of research investigation related to regulation of EPCR function through lipid modifications within EPCR. If the present findings were confirmed and extended, use of sPLA2-V inhibitors to treat various thrombotic, inflammatory and vascular diseases may become a viable option in the future. For this, we have to build a stronger basic foundation by convincingly demonstrating that sPLA2-V modifies the lipid in EPCR in vivo, and this lipid modification is responsible for EPCR encryption.
Fig. 1.
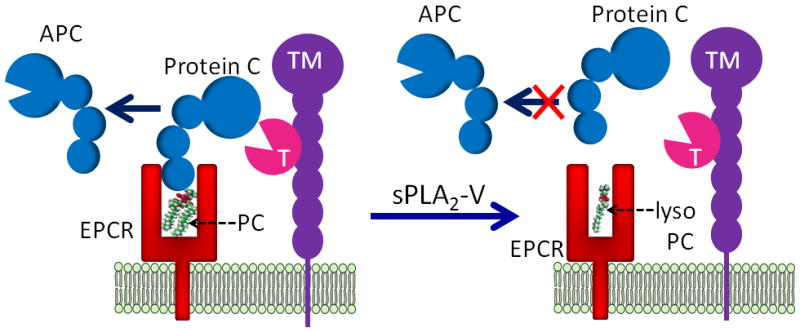
A hypothetical model of sPLA2-V impairment of protein C activation. Functional EPCR contains a phosphatidylcholine (PC) in its hydrophobic groove. In this conformation, EPCR can bind protein C. Thrombin (T):thrombomodulin (TM) complex activates the protein C bound to EPCR to generate activated protein C (APC). Secreted phospholipase A2, group V (sPLA2-V) hydrolyses the PC in the EPCR to lyso PC. The lipid modification in the hydrophobic groove of EPCR leads to a structural rearrangement in the helical region of EPCR, which probably results in narrowing of the pocket of the ligand-binding groove. The structurally rearranged EPCR is unable to bind protein C. The loss of protein C binding to EPCR translates into impaired protein C activation by thrombin:thrombomodulin complex.
Footnotes
Disclosure of Conflicts of Interest
This work was supported by National Institutes of Health grant HL1074830 (L.V.M.R.). The author states that he has no conflict of interest. For the record, the author wants to disclose that he has ongoing collaboration with Esmon, who was one of the coauthors of the article that was commented on here, on projects that are not related to the present study.
References
- 1.Rao LV, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multi-liganded and multi-functional receptor. Blood. 2014 [Google Scholar]
- 2.Fukudome K, Esmon CT. Identification, cloning, and regulation of a novel endothelial cell protein c/activated protein c receptor. J Biol Chem. 1994;269:26486–91. [PubMed] [Google Scholar]
- 3.Oganesyan V, Oganesyan N, Terzyan S, Qu D, Dauter Z, Esmon NL, Esmon CT. The crystal structure of the endothelial protein C receptor and a bound phospholipid. JBC. 2002;277:24851–4. doi: 10.1074/jbc.C200163200. [DOI] [PubMed] [Google Scholar]
- 4.Chiappori F, Merelli I, Milanesi L, Rovida E. Exploring the role of the phospholipid ligand in endothelial protein C receptor: a molecular dynamics study. Proteins. 2010;78:2679–90. doi: 10.1002/prot.22782. [DOI] [PubMed] [Google Scholar]
- 5.Lopez-Sagaseta J, Puy C, Tamayo I, Allende M, Cervero J, Velasco SE, Esmon CT, Montes R, Hermida J. sPLA2-V inhibits EPCR anticoagulant and antiapoptotic properties by accommodating lysophosphatidylcholine or PAF in the hydrophobic groove. Blood. 2012;119:2914–21. doi: 10.1182/blood-2011-05-353409. [DOI] [PubMed] [Google Scholar]
- 6.Tamayo I, Velasco SE, Puy C, Esmon CT, Dichiara MG, Montes R, Hermida J. sPLA2-V impairs EPCR-dependent protein C activation and accelerates thrombosis in vivo. J Thromb Haemost. 2014 doi: 10.1111/jth.12676. [DOI] [PubMed] [Google Scholar]
- 7.Sawada H, Murakami M, Enomoto A, Shimbara S, Kudo I. Regulation of type V phospholipase A2 expression and function by proinflammatory stimuli. Eur J Biochem. 1999;263:826–35. doi: 10.1046/j.1432-1327.1999.00565.x. [DOI] [PubMed] [Google Scholar]
- 8.Sonoki K, Iwase M, Ohdo S, Ieiri I, Matsuyama N, Takata Y, Kitazono T. Telmisartan and N-acetylcysteine suppress group V secretory phospholipase A2 expression in TNFalpha-stimulated human endothelial cells and reduce associated atherogenicity. J Cardiovasc Pharmacol. 2012;60:367–74. doi: 10.1097/FJC.0b013e3182646ccc. [DOI] [PubMed] [Google Scholar]
- 9.Kimura-Matsumoto M, Ishikawa Y, Komiyama K, Tsuruta T, Murakami M, Masuda S, Akasaka Y, Ito K, Ishiguro S, Morita H, Sato S, Ishii T. Expression of secretory phospholipase A2s in human atherosclerosis development. Atherosclerosis. 2008;196:81–91. doi: 10.1016/j.atherosclerosis.2006.08.062. [DOI] [PubMed] [Google Scholar]
- 10.Murakami M, Kudo I. New phospholipase A(2) isozymes with a potential role in atherosclerosis. Curr Opin Lipidol. 2003;14:431–6. doi: 10.1097/00041433-200310000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–72. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 12.Rezaie AR. Regulation of the protein C anticoagulant and antiinflammatory pathways. Curr Med Chem. 2010;17:2059–69. doi: 10.2174/092986710791233706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esmon CT. Protein C anticoagulant system--anti-inflammatory effects. Semin Immunopathol. 2012;34:127–32. doi: 10.1007/s00281-011-0284-6. [DOI] [PMC free article] [PubMed] [Google Scholar]