

Abstract
In addition to complications relating to the liver, patients with cirrhosis and portal hypertension develop extrahepatic functional disturbances of multiple organ systems. This can be considered a multiple organ failure that involves the heart, lungs, kidneys, the immune systems, and other organ systems. Progressive fibrosis of the liver and subsequent metabolic impairment leads to a systemic and splanchnic arteriolar vasodilatation. This affects both the haemodynamic and functional homeostasis of many organs and largely determines the course of the disease. With the progression of the disease, the circulation becomes hyperdynamic with cardiac, pulmonary as well as renal consequences for dysfunction and reduced survival. Infections and a changed cardiac function known as cirrhotic cardiomyopathy may be involved in further aggravation of other complications such as renal failure precipitating the hepatorenal syndrome. Patients with end-stage liver disease and related complications as for example the hepatopulmonary syndrome can only radically be treated by liver transplantation. As a bridge to this treatment, knowledge on the mechanisms of the pathophysiology of complications is essential for the choice of vasoactive drugs, antibiotics, drugs with specific effects on fibrogenesis and inflammation, and drugs that target specific receptors.
Keywords: Fibrogenesis, Splanchnic haemodynamics, Inflammation, Bacterial translocation, Infections, Systemic circulation, Cirrhotic cardiomyopathy, Hepatopulmonary syndrome, Portopulmonary hypertension, Hepatorenal syndrome, Ascites
Core tip: Patients with cirrhosis develop extrahepatic functional disturbances as a multiple organ failure that involves the heart, lungs, kidneys, the immune systems, and other organ systems. Fibrosis of the liver leads to a systemic vasodilatation that affects both the homeostasis of many organ systems. The circulation becomes hyperdynamic, which is often further aggravated by infections. Changes in organ function involve the heart as a cirrhotic cardiomyopathy, the kidneys such as the hepatorenal syndrome and the lungs with development of a hepatopulmonary syndrome. Liver transplantation is often the only radical treatment and as a bridge to this treatment, knowledge on the mechanisms of the pathophysiology of complications is essential.
INTRODUCTION
Patients with chronic liver failure typically present with symptoms relating to the diseased liver. In particular patients with cirrhosis show clinical, biochemical, and pathophysiological signs of structural and functional changes. Among these are alterations of the synthetic, excretory, and metabolic capacity, immunologic and regulatory function of hepatocytes, Kupffer cells, sinusoidal endothelial cells (SEC), biliary cells, and hepatic stellate cells (HSC). Impaired synthetic capacity of the hepatocytes leads to coagulopathy and to low circulating albumin level and often to jaundice related to compromised excretion of bilirubin and defects in conjugation[1]. When cirrhosis progresses the amount of fibrosis increases with development of regeneration nodules in the liver, which lead to portal hypertension[2]. Activation of the HSC by bioactive substances contributes to the elevated portal pressure[3]. One of the most important complications with relations to portal hypertension is bleeding from gastro-esophageal varices[4]. The combination of impaired hepatic degradation and porto-systemic shunting of vasodilators leads to a splanchnic and arterial vasodilatation with reduced splanchnic vascular resistance[5-7]. Over time a hyperdynamic, multi-organ failure syndrome develops with increased cardiac output and heart rate and decreased central blood volume[8-11]. Together with portal hypertension this leads to formation and perpetuation of ascites[12]. The hyperdynamic syndrome affects a variety of organ functions such as the lungs with development of the hepatopulmonary syndrome (HPS) and the heart with upcome of a cardiovascular dysfunction, including cirrhotic cardiomyopathy[13]. Due to the systemic and splanchnic vasodilatation, vasoactive systems like the sympatho-adrenergic, renin-angiotension-aldosterone, and the vasopressin system become activated[14]. This mediates vasoconstriction within the kidney with increased risk of development of hepatorenal syndrome (HRS)[15,16]. Translocation of bacteria from the gut to lymph nodes leads to complicating infections in relation to variceal bleeding and infected ascitic fluid as spontaneous bacterial peritonitis (SBP)[17,18]. Impaired phagocytic activity in cells belonging to the reticulo-endothelial system such as the Kuppfer cells may facilitate infections, which further aggravates the circulatory dysfunction[19,20].
During the last decade it has become clear that chronic liver failure is not only limited to a decreased liver function, but also involves impairment of most other organs in the body as part of a multi-organ syndrome particularly relating to haemodynamic and homoeostatic disturbances. This review imparts to highlight contemporary mechanisms of extrahepatic haemodynamic complications to chronic liver failure in patients with cirrhosis and portal hypertension.
PATHOPHYSIOLOGY OF THE LIVER
Structural and functional changes in cirrhosis
The microscopic human liver architecture is arranged in lobules with cords of liver cells radiated to a central vein. The portal tract consists of a triad containing a portal vein branch, a hepatic arteriole, and a bile duct[1]. Thus, the liver lobule is quantitatively formed by hepatocytes and cholangiocytes. In addition to these two primary epithelial cell populations the liver hosts SEC, Kupffer cells, and HSC, see Figure 1[21-23].
Figure 1.
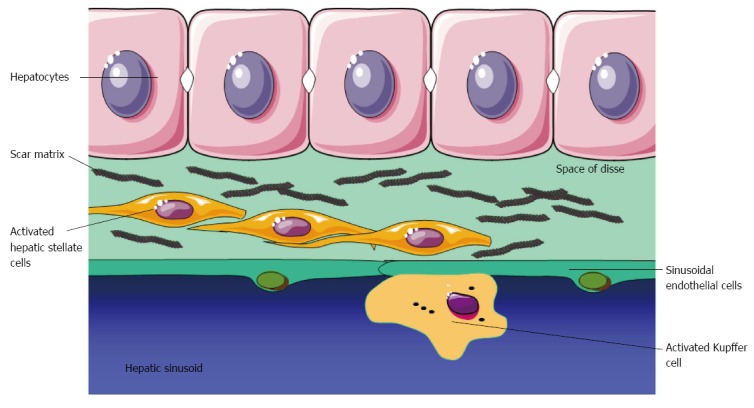
Fibrogenesis after liver injury. Hepatic stellate cells are activated into myofibroblasts that deposit scar matrix in the Space of Disse.
The hepatocytes accounts for approximately 80% of the total liver cell mass[24]. The hepatocytes have essential physiological functions, such as regulation of carbohydrate and lipid metabolism, clearance and inactivation of drugs, ethanol, toxins and various hormones and vasoactive substances. A very important function of the hepatocytes is the synthesis of plasma proteins, including albumin, C-reactive protein, fibrinogen, complement, and coagulation factors. In patients with acute and chronic liver injury hepatocyte apoptosis plays a major role for the impaired metabolic function[25]. When the liver is exposed for toxic substances such as drugs, alcohol, and hepatitis B or C virus, fibrogenesis increases and initiates the cirrhotic process with development of regeneration nodules with impairment of hepatocyte function[26,27]. The impaired metabolic function results in compromised degradation of various vasoactive substances, which is particularly important for the understanding of the systemic and splanchnic haemodynamic changes[28]. Atrial natriuretic peptide is cleared in the liver with extraction ratios of 22%-75%[29]. The hepatic extraction of glucagon has been determined by infusion studies and ranges 10%-20% and hepatic extraction ratios of adrenaline and noradrenaline are very high and ranges 63% to 67%[28]. Renin, angiotensin II, substance P, vasopressin, and aldosterone are other important other bioactive substances with haemodynamic implications that are degraded in the liver[28].
Kupffer cells are liver macrophages and constitute 80% of all tissue macrophages of the reticuloendothelial system on the body and about 15% of the liver cells[24]. The Kupffer cells are important for the immune function of the liver and act as antigen presenting cells of bacteria, endotoxins, liopolysaccharide (LPS) and other antigens together with natural killer (NK) cells[30,31]. In addition, the Kupffer cells play an important role in the expression of proinflammatory cytokines such as tumour necrosis factor-α (TNF-α), interleukin-1 (IL-1) and IL-6[32]. Furthermore, they play important roles in clearance of endotoxins, and in the defence of microbial infections and contribute to the production of the vasodilator nitric oxide[33].
HSC are located in the space of Disse and they are in close contact with hepatocytes and SEC, Figure 1. They are rich of vitamin A, which relates to their function as hepatic storage of retinyl esters[34]. The HSCs are furthermore actively involved in fibrogenesis and metabolism. They are involved in matrix degradation and tissue inhibitor of metalloproteinase-1 (TIMP-1) has been shown to be a survival factor for HSC, and inhibition of the actions of TIMP-1 may be a target for antifibrogenic treatment[35,36]. HSC has important contractile properties with relation to the development of portal hypertension[37]. Thus, vasoactive agents, such as endothelin-1 (ET-1), angiotensin-2, and trombin induces contraction[35,37]. From this point of view the contraction of HSC constitutes a reversible and dynamic component of the increase in the hepatic flow and regulation of the portal pressure[3]. This function is intimately associated with a counter regulation by nitric oxide and carbon monoxide, which induce relaxation of HSC and thereby ultimately reduces portal pressure[35,36]. The SECs possess highly specialised physiological functions as they serve as a source of several bioactive substances[38]. The cells are involved in regulation and production of proinflammatory cytokines and haemodynamically important vasoactive peptides and substances, including nitric oxide, ETs, prostanoids, and prostaglandins[39,40].
It can be concluded, that various cell types of the liver are deeply involved in the haemodynamic changes and extrahepatic complications. Thus, impaired hepatocyte degradation of vasoactive substances, production of vasodilators in the HSC and SEC, and impaired immune and clearance function of Kuppfer cells inducing production of proinflammatory cytokines and substances are of importance for the development of portal hypertension and directly involved in the pathophysiology of extrahepatic hemodynamic complications. Increased portal pressure leads to portosystemic shunts, and thereby increases the amount of vasodilators and other compounds that bypass the liver metabolism and degradation.
Fibrogenesis
The histological hallmark of chronic liver failure is development and perpetuation of fibrosis in the liver. Sustained fibrogenesis leads to cirrhosis, which is characterized by distortion of the liver parenchyma and reduction of vascular architecture. The fibrogenetic process is like a wound-healing response to injuries and a balance between formation and degradation of fibrotic tissue[35]. This process can be activated by injuries caused by for example viral hepatitis, alcohol intake, and autoimmune disorders. These stimuli primarily activate HSC into myofibroblasts-like cells with contractile, proliferative, and fibrogenic capacities and it is the primary cell type responsible for deposition of extracellular matrix in the liver[34]. The HSCs are located in the subendothelial space between the hepatocytes, Kupffer cells, and SECs so they mutually interact through numerous cellular processes extending across the space of Disse, see Figure 1[37]. Paracrine and autocrine activation of HSCs by tumour growth factor-β1, which is considered the most potent fibrogenic cytokine, initiates the fibrotic process in the liver[35]. In addition, the HSCs cover various physiological functions, including activation of the immune response, secretion of cytokines, and angiogenesis[34]. The activation of the HSC into myofibroblasts consists of an initiation phase and a perpetuation, followed by a resolution phase. Resolution of fibrosis refers to sequences of events such as apoptosis, senescence, or quiescence[27]. The inflammatory response plays an important role in fibrogenesis since inflammation always precedes fibrosis and immune activation induces fibrosis by bacterial LPSs[41]. Kupffer cells also activate HSC by increased NF-κB activity and secretion of pro-inflammatory cytokines, including TNF-α and monocyte chemoattractant protein[22,42]. Finally, NK cells have an anti-fibrotic effect by killing activated HSC. The contractile HSC contributes to the regulation of the sinusoidal blood flow and to angiogenesis in the cirrhotic liver[37]. Thereby, the HSCs contribute to regulate intrahepatic blood flow and portal pressure. Together with SECs that excert a paracrine effect through nitric oxide synthesis, the HSC represent an important dynamic component of the sinusoidal haemodynamic resistance in cirrhosis.
It can be concluded that the HSCs are the major fibrogenetic cells type in the injured liver and its numerous functions have disclosed important targets for effective antifibrotic therapies[36,43].
REGULATION OF SPLANCHNIC AND HEPATIC BLOOD FLOW
Homeostasis of the hepatic blood flow is essential since the liver plays a major role in the clearance of waste products, drugs, and hormones. The liver receives 25% of the total resting cardiac output through the the hepatic artery and portal vein, the latter being responsible for 75% of the total hepatic blood flow[44]. The hepatic blood flow equals the ratio of the hepatic venous pressure gradient and post-sinusoidal resistance. In case of portal hypertension, a substantial portal systemic collateral circulation develops together with an increased mesenterial inflow[45]. The hepatic blood flow can be measured by the indocyanine green clearance (ICG) method applying the Fick-principle by measurements of ICG-concentrations in the liver vein and in a peripheral artery[46].
To maintain metabolic homeostasis, the hepatic blood flow must be adjustable to full fill changing metabolic demands. For example increased regional oxygen consumption is followed by proportional increased blood flow to the splanchnic area[44]. However, the liver per se is not able to control the portal inflow and it is the splanchnic organs that drain into the portal vein that primarily determine changes in portal venous blood flow. Therefore, different regulatory mechanisms are essential to maintain haemodynamic and metabolic homeostasis.
Extrinsic regulatory mechanisms
The splanchnic blood flow increases normally after a meal[47]. The mechanisms are largely unknown but splanchnic blood vessels are richly innervated by sympathetic nerves from the prevertebral sympathetic ganglia and an increase in sympathetic nervous outflow is partly responsible for the initial increase in cardiac output together with mediators such as glucose and long-chain fatty acids[48]. The extrinsic neural control of intestinal blood flow is predominantly through sympathetic vasoconstriction mediated by alfa adrenoceptors. Sympathetic activity reduces intestinal blood flow by increasing the vascular resistance of the arterioles and veins, and this is among the major effects on beta-blocking agents on hepatic blood flow and portal pressure[4,14,49].
In normal conditions, the vascular compliance of the liver is sufficient to maintain pressure homeostasis, but in cirrhotic patients with increased systemic vascular compliance and reduced hepatic and portal compliance, the portal pressure and degree of porto-systemic shunting increases and thereby the risk of bleeding from oesophageal varices[4,50,51]. This risk is further increased following a meal that augment the hepatic inflow[52]. Inversely, changes in splanchnic haemodynamics may affect function of other organs and the existence of a hepatorenal reflex has been debated for years. Experimental and clinical studies have provided support for a direct link between the liver and the kidneys[53,54]. In human cirrhosis, reduced renal blood flow following an increase in portal pressure and a concurrent increase in renal venous ET-1 suport this assumption[55,56]. There are now both clinical and experimental evidence of a hepatic blood flow-dependent hepatorenal reflex and this is a primary pathophysiological mechanism for renal dysfunction in liver disease[57]. This reflex is activated by adenosine in the space of Mall and is regulated by hepatic blood flow[54] (Figure 2).
Figure 2.
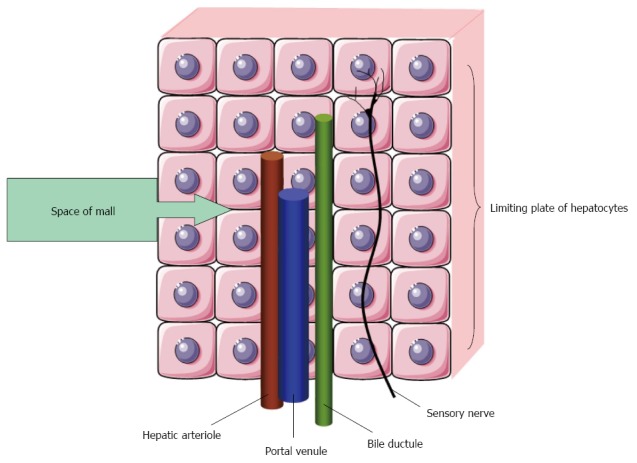
Space of Mall is a small space of fluid surrounding a hepatic arteriole, a portal venule and a bile ductule. Adenosine is secreted into the space of Mall. A reduction in portal blood flow increases adenosine levels and leads to hepatic arteriolar vasodilataion and activation of sensory nerves after[54].
The liver receives blood from both the hepatic artery and the portal vein. From a homeostatic point of view this is unique, since it secures a constant blood flow to the liver through a hepatic arterial buffer system[58]. According to the hepatic arterial buffer hypothesis a reduction in portal blood flow will cause a local accumulation of adenosine not washed away from the space of Mall surrounding the hepatic arterial resistance vessels and this will lead to local vasodilatation[54,59]. The metabolic homeostatic mechanisms maintain constant oxygen delivery, whereas the myogenic homeostatic mechanisms maintain a constant intravascular pressure[60].
Local regulatory mechanisms
A number of hormones with vasoactive effects such as gastrin, vasoactive intestinal polypeptide, cholecystokinin, secretin, and glucagon are implicated in the fine-tuning of the splanchnic haemodynamics of the liver[11,61]. As previously mentioned, SECs and HSCs are intimately involved in the regulation of the sinusoidal blood flow and potent vasodilators such as nitric oxide, ET-1, and tromboxan-A2 play a role in the vasodilation/vasoconstriction balance and the dynamic component of the increase in portal pressure in cirrhosis[40,62-65].
In conclusion, several homeostatic mechanism are involved in maintaining blood flow and metabolism in the normal liver. During development of cirrhosis and portal hypertension these mechanisms may not be adequate to meet the requirements of the body and liver failure, metabolic insufficiency, and portal hypertension may develop.
ROLE OF INFLAMMATION, BACTERIAL TRANSLOCATION, AND INFECTION IN CIRRHOSIS
Patients with cirrhosis are more susceptible to bacterial infections and in particular SBP, urinary tract and pulmonary infections, and Clostridium difficile infections[66,67]. The reduced ability to effective clearance of bacteria primarily relates to impaired cellular immune function of macrophages and monocytes, depressed neutrophil phagocytic and intracellular killing and deficiencies in the complement system[68-70]. Together with increased intestinal permeability and gut bacterial overgrowth this immune dysfunction facilitates transition of bacteria from the gastrointestinal tract to mesenterial lymph nodes, see Figure 3[18,70]. Bacterial translocation of especially Escherichia coli from the gut plays a significant role for the development of spontaneous infections, in particular SBP[71]. Presence of bacteria or bacterial products in splanchnic lymph nodes, ascitic fluid, or in the circulation significantly challenges the homeostatic haemodynamic balance and the hyperdynamic circulatory state. Bacterial translocation activates monocytes and lymphocytes and increase the circulating levels of pro-inflammatory cytokines such as TNF-α and IL-6 as a “cytokine storm” and subsequent activation of nitric oxide[18,72]. This inflammatory response further augments the circulatory dysfunction and aggravates the vasodilatory state[73]. Several surrogate markers of bacterial translocation has been proposed. Lipopolysaccharide binding protein is synthesised in the liver in response to bacteria and increased levels in cirrhotic patients correlate to the activation of different vasoactive systems, pro-inflammatory cytokines and nitric oxide[74]. Findings of bacterial DNA may reflect presence of bacterial components in body fluids and plasma, and preliminary studies have been promising[75], but the assessment is associated with significant technical challenges[76]. In patients with cirrhosis, infection of the ascitic fluid as in SBP is frequent, and it is an important risk factor for the development of circulatory and renal dysfunction[71,77,78]. Some patients may develop a systemic inflammatory response syndrome with fever, increased heart rate, respiratory failure and activated immune system[79] and sepsis in case of a confirmed bacterial etiology[73]. Severe bacterial infections and in particular SBP are main causes for the development of HRS as about 33% of patients with SBP develop HRS[80,81].
Figure 3.

Illustration of bacterial translocation from the gastrointestinal lumen through the epithelial layers and capillaries to the lymphalic vessels.
Bacterial translocation may be affected by measures that ameliorate bacterial overgrowth, intestinal permeability, and immunity, Figure 3. Thus, anti-, pre,- and probiotics and drugs that reduce the gastro-intestinal transit time may be of benefit[82,83]. Beta-blockers may also reduce the gastrointestinal transit time and reduce bacterial overgrowth, and results of a recent metaanalysis suggest that beta-blockers may even prevent SBP[84].
In conclusion, bacterial infections are important complications in cirrhosis. The mechanisms are complex and bacterial translocation lead through activation of pro-inflammatory mediators to a homeostatic imbalance that may precipitate renal and circulatory failure and lead to a multiorgan failure syndrome.
SYSTEMIC CIRCULATION
The circulatory homeostasis is largely intact in patients with early cirrhosis and portal hypertension. But with the progression of the disease over the portal hypertensive, preascitic to the decompensated, portal hypertensive, ascitic stage, there is an overall direct relation between the severity of cirrhosis e.g., reflected by the Child or MELD scores and the degree of hyperdynamic circulation[85-89].
The pathophysiological origin for a variety of extrahepatic complications is a splanchnic and systemic arteriolar vasodilatation that precedes renal sodium and water retention and plasma volume expansion[90]. According to the “peripheral arterial vasodilation hypothesis”[91], primary splanchnic arteriolar vasodilation leads to reduction of the overall systemic vascular resistance and in advanced disease, to avid arterial underfilling with low arterial blood pressure. A modification of this, “the forward theory of ascites formation” combines arterial underfilling with a forward increase in hepatosplanchnic capillary pressure and filtration with increased lymph formation[90]. A reduced effective blood volume, which is that part of the blood volume where volume and baroreceptors are located, leads to activation of vasoconstrictor systems and secondary sodium-water retention[90-93]. A number of potent intrinsic vasodilators are implicated in this and summarised in Table 1. Particular focus has been given to nitric oxide, calcitonin gene-related peptide, and adrenomedullin. Other substances with vasodilating properties which have been implicated are natriuretic peptides, TNF-α, IL-6, substance P, vascular endothelial growth factor, and cannabinoids[65,94-102]. The delicate homeostatic balance between primary vasodilatating and counterregulatory vasoconstriction is significantly deviated towards a sustained systemic vasodilatation, in spite of highly activation of all vasoconstrictor systems. This is most likely related to a combination of changes in receptor affinity, down regulation of receptors and several post-receptor defects but future research should further disclose the pathophysiology[13,103].
Table 1.
Vasodilating and vasoconstricting forces involved in disturbed haemodynamics in cirrhosis (Alphabetic order)
| Vasodilator systems |
| Adenosine |
| Adrenomedullin |
| Atrial natriuretic peptide |
| Bradykinin |
| Brain natriuretic peptide |
| Calcitonin gene-related peptide |
| Carbon monoxide |
| Endocannabinoids |
| Endothelin-3 |
| Endotoxin |
| Enkephalins |
| Glucagon |
| Histamine |
| Hydrogen sulphide |
| Interleukins |
| Natriuretic peptide of type C |
| Nitric oxide |
| Prostacyclin (PGI2) |
| Substance P |
| Tumour necrosis factor-α |
| Vasoactive intestinal polypeptide |
| Vasoconstrictor systems |
| Angiotensin II |
| Adrenaline and noradrenaline |
| Endothelin-1 |
| Neuropeptide Y |
| Renin-angiotensin-aldosterone system |
| Sympathetic nervous system |
| Vasopressin |
In general, an increase in cardiac output can be attributed to an increase in venous return, heart rate, and myocardial contractility, all of which are controlled by the autonomic nervous system. Arteriolar dilatation, the presence of arteriovenous communications, expanded blood volume, and enhanced sympathetic nervous activity may further raise the cardiac output; most of these pathophysiological mechanisms may operate in advanced cirrhosis[6,11,14]. In early cirrhosis, the presence of a hyperdynamic circulation is often not apparent. But with the progression of the liver disease, there is an overall association to the degree of hyperdynamic circulation. Studies on circulatory changes with posture suggest that the patients are mostly hyperdynamic in the supine position[104-107]. Blood and plasma volumes are expanded in advanced cirrhosis but the distribution between central and non-central vascular areas is imbalanced[108-111]. Thus, by different techniques it has been established that the central and arterial blood volume is most often decreased, whereas the non-central blood volume, in particular the splanchnic blood volume is increased in animals and patients with cirrhosis[6,109,112,113]. The effective arterial blood volume and the central circulation time (i.e., central blood volume relative to cardiac output) are substantially reduced and bear a significant relation to poorer survival in advanced cirrhosis[114]. The haemodynamic changes pertaining to specific vascular beds are shown in Table 2.
Table 2.
Circulatory changes in specific vascular beds in cirrhosis
| Systemic circulation |
| Plasma volume ↑ |
| Total blood volume ↑ |
| Non-central blood volume ↑ |
| Central and arterial blood volume ↓ (→) |
| Cardiac output ↑ |
| Arterial blood pressure ↓ (→) |
| Heart rate ↑ |
| Systemic vascular resistance ↓ |
| Arterial and total vascular compliance ↑ |
| Heart |
| Left atrial volume ↑ |
| Left ventricular volume → (↑) |
| Right atrial volume → ↑ ↓ |
| Right ventricular volume → ↑ ↓ |
| Right atrial pressure → ↑ |
| Right ventricular end diastolic pressure → |
| Pulmonary artery pressure → ↑ |
| Left ventricular end diastolic pressure → |
| Hepatic and splanchnic circulation |
| Hepatic blood flow ↓ → (↑) |
| Hepatic venous pressure gradient ↑ |
| Postsinusoidal resistance ↑ |
| Renal circulation |
| Renal blood flow ↓ |
| Glomerular filtration rate ↓ → |
| Pulmonary circulation |
| Pulmonary blood flow ↑ |
| Pulmonary vascular resistance ↓ (↑)1 |
| Cutaneous and skeletal muscle circulation |
| Skeletal muscular blood flow ↑ → ↓ |
| Cutaneous blood flow ↑ → ↓ |
Portopulmonary syndrome. ↑ → ↓ denote: Increased, unchanged, or decreased, respectively. Parenthesis denotes less frequent changes.
In the decompensated state, plasma volume expansion is a prevailing feature and should be considered secondary to the activation of neurohumoral mechanisms consequent on mainly splanchnic vasodilatation, low arterial blood pressure, and reduced central and arterial blood volume.
Evaluation of systemic haemodynamic changes in clinical practice is complex. Even patients with early cirrhosis may exhibit a hyperdynamic circulatory state and a few patients with decompensated cirrhosis with considerable fluid retention may present a relatively normal circulation[86]. Recently, it has become apparent that patients with advanced disease and refractory ascites may have a suppressed cardiac output[115,116]. Moreover, pharmacological treatment e.g., with β-blockers may attenuate a hyperdynamic circulatory state[86,117,118]. However, on a whole, there is a direct relationship between the state of vasodilatation, the systemic circulatory derangement and the progression of the liver disease, development of complications and prognosis.
CHANGES IN CARDIAC FUNCTION
Subclinical impairment of the function of the cirrhotic heart has been known for 60 years[119]. However, it is only recently that the reduced function of the heart has been associated with the development of various complications of cirrhosis. Results of experimental and clinical studies have shown impaired myocardial contractility as well as electrophysiological abnormalities in cirrhosis, which have crystalized the clinical entity cirrhotic cardiomyopathy[13,120,121]. This term denotes a chronic cardiac dysfunction, characterised by blunted contractile responsiveness to stress and altered diastolic relaxation with electrophysiological abnormalities, such as prolongation of the Q-T interval, all occurring in the absence of any other cardiac disease, Table 3[122,123]. This cardiac dysfunction may affect the prognosis of the patients and aggravate the course during invasive procedures such as surgery, insertion of a transjugular intrahepatic portosystemic shunt (TIPS), and liver transplantation[124-126]. The pathophysiological mechanisms include changes in the cardiomyocyte plasma membrane, attenuated function of the beta-adrenergic pathway, and greater activity of inhibitory systems[123,127]. Other studies have focused on negative inotropic effects of nitric oxide, nitration of cardiac proteins, carbon monoxide, endogenous cannabinoids, bile acids, and endotoxins[128,129]. The mechanisms of cirrhotic cardiomyopathy are summarised in Figure 4.
Table 3.
Characterization of cirrhotic cardiomyopathy
| Definition |
| A cardiac dysfunction in patients with cirrhosis characterised by impaired contractile responsiveness to stress and/or altered diastolic relaxation with electrophysiological abnormalities in the absence of other known cardiac disease |
| Diagnostic criteria |
| Systolic dysfunction |
| Blunted increase in cardiac output with exercise, volume challenge or pharmacological stimuli |
| Resting EF < 55% |
| Diastolic dysfunction |
| E/A ratio < 1.0 (age-corrected) |
| Prolonged deceleration time (> 200 ms) |
| Prolonged isovolumetric relaxation time (> 80 ms) |
| Supportive criteria |
| Electrophysiological abnormalities |
| Abnormal chronotropic response |
| Electromechanical uncoupling/dyssynchrony |
| Prolonged Q-T interval |
| Enlarged left atrium |
| Increased myocardial mass |
| Increased BNP and pro-BNP |
| Increased troponin I |
BNP: Brain natriuretic peptide; E/A: Early diastolic/atrial filling ratio; EF: Left ventricular ejection fraction.
Figure 4.

Mechanisms of cirrhotic cardiomyopathy. The figure reviews the most important mechanisms involved in cirrhotic cardiomyopathy: Desensitisation and downregulation of β-adrenergic receptors with decreased content of G-protein (Gαi: inhibitory G protein; Gαs: stimulatory G protein) and following impaired intracellular signalling; alterations in particular in M2 muscarinic receptors; upregulation of cannabinoid 1-receptor stimulation; altered plasma membrane cholesterol/phospholipid ratio; increased inhibitory effects of haemooxygenase (HO), carbon monoxide (CO), nitric oxide (NO), and tumour necrosis factor-α (TNF-α); reduced density of potassium channels; changed function and fluxes through L-type calcium channels; altered ratio and function of collagens and titins. Many post-receptor effects are mediated by adenylcyclase (AC) inhibition or stimulation. PKA: Protein kinase A.
Systolic dysfunction
In patients with cirrhotic cardiomyopathy, cardiac failure may become manifest only under conditions of haemodynamic stress. Thus, the left ventricular end-diastolic pressure increases after exercise, but the expected increases in cardiac stroke index and left ventricular ejection fraction (LVEF) are absent or subnormal, which indicates an inadequate response of the ventricular reserve to a rise in filling pressure[130]. A vasoconstrictor-induced increase of 30% in the left ventricular afterload results in a doubling in pulmonary capillary wedged-pressure, with no change in cardiac output[131]. This response may be useful in diagnosing cirrhotic cardiomyopathy. A similar pattern is seen after insertion of TIPS, but the raised cardiac pressures tend to normalise with time[132,133]. Some of these patients (12%) may develop manifest cardiac failure in association with the TIPS insertion[134]. A failure to increase cardiac output, despite increased ventricular filling pressure, indicates that normalisation of the afterload impairs cardiac performance and unmasks left ventricular dysfunction[131].
LVEF reflects systolic function, even though it is very much influenced by preload and afterload. It has been reported to be normal at rest in some studies and reduced in one study of a subgroup of patients with ascites[130,131,135]. After exercise, LVEF increases less in cirrhotic patients than in controls[130,136,137]. The reduced functional capacity may be attributed to a combination of blunted heart rate response to exercise, reduced myocardial reserve, and profound skeletal muscle wasting with impaired oxygen extraction[138,139]. By modern techniques like tissue-Doppler and speckle tracking echocardiography, it is possible also to detect systolic and diastolic dysfunction at rest[140,141] .
Diastolic dysfunction
The clinical significance of diastolic dysfunction and its importance in cirrhotic cardiomyopathy has been questioned, as overt cardiac failure is not a prominent feature of cirrhosis. However, there are several reports of unexpected death from heart failure following liver transplantation, surgical portocaval shunts, and TIPS[134,142]. These procedures involve a rapid increase in cardiac preload. In a less compliant heart, the diastolic dysfunction could be enough to cause pulmonary oedema and heart failure. This is consistent with the findings of an increase in pulmonary artery pressure, pre-load, and diastolic dysfunction after TIPS[132]. Diastolic dysfunction affecting left ventricular filling may progress to systolic dysfunction[123,143]. The pathological basis of the increased stiffness of the left ventricle seems to be cardiac hypertrophy, patchy fibrosis, and subendothelial oedema[131,136,144]. Decreased E/A ratio and delayed early diastolic transmitral filling with prolonged deceleration and isovolumetric relaxation times indicate diastolic dysfunction on the Doppler echocardiogram and corresponding characteristics on the tissue-Doppler and speckle tracking echocardiography[131,140,141,145].
Q-T interval prolongation
In cirrhotic patients, the Q-T interval is prolonged and significantly related to the severity of the liver disease, portal hypertension, portosystemic shunts, elevated brain type natriuretic peptide (BNP) and pro-BNP, elevated plasma noradrenaline, decreased heart rate variability, and reduced survival[122,124,146-149]. The prolongation of the Q-T interval is partly reversible after liver transplantation and beta-blocker treatment[146,150]. Recently, it has been documented that gastrointestinal bleeding further prolongs the Q-T interval in cirrhosis and independently predicts bleeding-induced mortality[151]. The prolonged Q-T interval in cirrhosis should be considered an element in the cirrhotic cardiomyopathy and may be of potential use in the stratification and identification of patients at risk[149,152].
Taken together, cirrhotic cardiomyopathy encompasses impaired contractility and diastolic relaxation, and electrophysiological abnormalities in particular prolonged Q-T interval. Independent of aetiology of cirrhosis, systolic dysfunction can be diagnosed at rest by for example tissue-Doppler imaging or demasked by physical or pharmacological stress. Diastolic dysfunction can be detected by echocardiography or tissue-Doppler imaging. Cirrhotic cardiomyopathy may aggravate complications such as gastro-intestinal bleeding and development of the HRS. Liver transplantation may revert the cardiac dysfunction but surgery and TIPS insertion may also aggravate the condition.
CHANGES IN PULMONARY FUNCTION
Pulmonary dysfunction involves diffusing abnormalities with development of the HPS and portopulmonary hypertension (PoPH) in some patients with cirrhosis. The circulatory and neuroendocrine derangements seem to play important roles in the hepatopulmonary dysfunction and these aspects should be taken into account in the management of diffusing and oxygenation-related complications of cirrhosis. Below are the two main entities HPS and PoPH shortly considered and the pathophysiological differences summarised in Table 4.
Table 4.
Diagnostic criteria for the hepatopulmonary syndrome and portopulmonary hypertension
| HPS | PoPH |
| Presence of liver disease | Presence of liver disease and portal hypertension |
| PA-aO2 > 15 mmHg (> 2 kPa) | Mean pulmonary arterial pressure > 25 mmHg |
| Positive contrast enhanced echocardiography1 | Pulmonary vascular resistance > 240 dyn·s·cm-5 left atrial pressure < 15 mmHg |
Visualisation of microbubbles in the left heart chambers within three or more cardiac cycles implies definite intrapulmonary vascular dilatation. PA-aO2: Alveolar-arterial oxygen gradient; PoPH: Portopulmonary hypertension; HPS: Hepatopulmonary syndrome.
HPS
A condition with reduced diffusing capacity, abnormal ventilation/perfusion ratio and intrapulmonary vascular dilatations, and low arterial oxygen saturation in association with liver disease and absence of cardiopulmonary disease is termed HPS[153-156]. Arterial deoxygenation is reflected by a widened alveolar-arterial oxygen gradient (PA-aO2)[88,157]. The frequency of HPS in patients with cirrhosis is variably reported from 5%-50% according to the type of population with respect to aetiology, geography, etc [154,156,158-160]. The pathophysiological hallmark of HPS, is dilatations of capillaries near the alveolar areas, see Figure 5. Fallon et al[153] have recently reported increased pulmonary vascular endothelial nitric oxide synthase (NOS) and increased production of cholangiocyte ET-1 and increased expression of ET-B receptors[161,162]. Recently, pulmonary angiogenesis have been implemented in the development of this complicated pathophysiology[163].
Figure 5.
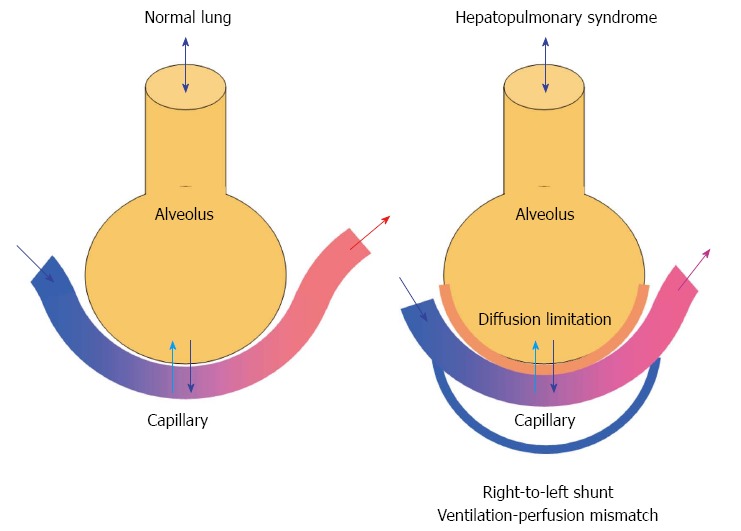
Gas exchange in the normal lung (left) and mechanism of hepatopulmonary syndrome (right). The hepatopulmonary syndrome comprises an increased alveolar-arterial oxygen gradient owing to diffusion limitations and development of intrapulmonary right-to-left shunts leading to arterial hypoxaemia.
From a practical point of view HPS is defined by arterial hypoxaemia [PaO2 < 70 mmHg (or 9.3 kPa)], an age-adjusted increase in PA-aO2 > 15 mmHg (or 2.0 kPa) and presence of intrapulmonary vasodilatations[154,162]. A large proportion of patients with HPS present with insidious onset of dyspnea, plathypnea (upon standing), orthodeoxia, clubbing, and cyanosis[164]. The diagnosis requires arterial blood gas measurements and calculation of PA-aO2, Contrast-enhanced echocardiography (CEE) is today considered the most sensitive test and method of choice in the diagnosis HPS[156], but also injection of macroaggregated albumin with estimated the extra-pulmonary shunt fraction > 6% indicates presence of HPS[165]. Currently, the following criteria for HPS are: (1) Presence of liver disease; (2) PA-aO2 ≥ 15 mmHg (2.0 kPa); and (3) a positive CEE (Table 4).
At present there is no effective medical therapy for HPS. Insertion of TIPS has been reported to be successful, but it may result in increased pulmonary pressures and is therefore not recommended[166,167]. Since HPS is reversible after liver transplantation, it has become an indication for urgent liver transplantation and the long-term outcome after LT in HPS is increasingly favorable[168,169].
Portopulmonary hypertension
PoPH is defined as a mean pulmonary artery pressure > 25 mmHg and pulmonary vascular resistance >240 dyn•s•cm-5, and normal left atrial pressure (< 15 mmHg), see Table 4[154,170,171]. The histological appearance of pulmonary vessels is similar to that seen in primary pulmonary artery hypertension, and includes smooth muscle proliferation, hypertrophy, and fibrosis[154]. Various pathophysiological aspects seem to be involved in the development of portopulmonary hypertension, including vasoproliferation, genetics, and inflammation with increased pulmonary phagocytosis[154]. Of particular interest is the activation of potent local vasoconstrictor systems, like serotonin and the ET system. ET-1 is produced in the pulmonary endothelium and binding to ETA and ETB receptors on the pulmonary smooth muscle cells leads to vasoconstriction[172-175]. Symptoms are typically progressive and include fatigue, chest discomfort, exertional dyspnoea, oedema, and syncope[175,176]. The median survival in patients with PoPH is considered low, about six months[177] and lower than in patients with idiopathic pulmonary hypertension[171]. The diagnosis can be based on radiological, echocardiographic, lung functional, and haemodynamic findings. Pulmonary function tests often show reduced lung volumes, diffusing capacity, and forced vital capacities and widened PA-aO2[178]. For purposes of screening, Doppler-echocardiography can be used to estimate pulmonary pressures and right ventricular systolic pressure. Right ventricular systolic pressure thresholds from > 30 to > 50 mmHg as cutt-off limits are still discussed[156,179].
Different medical treatments that modify the circulation have been applied. These include prostacyclin analogs such as epoprosterol[180], ET receptor antagonists such as bosentan[180,181], and phosphodiesterase-5 inhibitors such as sildenafil[182]. The effects of these drugs are however modest. TIPS and liver transplantation are not treatment options of choice in these patients sice a mean pulmonary artery pressure > 35 mmHg is associated with increased mortality following liver transplantation[168,183,184].
In conclusion, a considerable number of cirrhotic patients present with changes in pulmonary vascular resistance, impaired ventilation, and hypoxaemia as part of HPS or PoPH. Although, the prevalence varies, the circulatory and neuroendocrine derangements seem to play important roles in the clinical aggravation, hepatopulmonary dysfunction, and circulatory reactivity of these entities. Therefore these pathophysiological aspects should be taken into account in the clinical management of the patient with cirrhosis and pulmonary dysfunction.
CHANGES IN RENAL FUNCTION
Acute kidney injury (AKI) is frequent in patients with cirrhosis and determines the prognosis in advanced cirrhosis. HRS denotes a functional and partly reversible impairment of renal function. HRS is precipitated by factors that aggravate the effective hypovolaemia in decompensated cirrhosis, by lowering arterial pressure and cardiac output and enhance sympathetic nervous activity.
Acute kidney injury in cirrhosis
Acute renal failure is estimated to occur in approximately 20% of hospitalized patients with cirrhosis[185]. Several attempts have been made to achieve agreement on a more precise definition of AKI. AKI has been defined as an increase in serum creatinine ≥ 133 μmol/L (≥ 1.5 mg/dL) or an increase > 50%, but initiatives from the Acute Kidney Injury Network and others have defined AKI as an absolute increase in serum creatinine > 26.4 μmol/L (≥ 0.3 mg/dL) (or a 50% increase over 48 h)[186]. Differential diagnosis of AKI in cirrhosis is difficult as it ranges from pre-renal AKI (45%), intra-renal AKI including acute tubular necrosis and glomerulonephritis (32%), HRS (23%), and seldom post-renal AKI (< 1%).
HRS
Approximately 20% of the cirrhotic patients with ascites who are resistant to diuretics, progress to HRS[187]. HRS denotes a functional prerenal failure that is unresponsive to volume expansion in patients with chronic liver disease and ascites without significant morphological changes in renal histology, and with a largely normal tubular function (Table 5)[78,188]. The prognosis of patients with a full-blown HRS is poor ranging from days to weeks and liver transplantation is the only radical treatment for the HRS[187,189]. Two types of HRS have been defined depending on the rapidness and the extent of the renal failure[78,190,191]. Type-1 HRS is an acute form with a rapid decrease in renal function and renal failure as an independent predictive factor; type-2 HRS is a chronic form with a more stable renal dysfunction[191,192].
Table 5.
New diagnostic criteria for the hepatorenal syndrome from the International Ascites Club (2013)[192]
| Cirrhosis with ascites |
| Serum creatinine > 133 μmol/L (1.5 mg/dL) |
| No improvement of serum creatinine (decrease to a level of < 133 μmol/L) after at least 2 d with diuretic withdrawal and volume expansion with albumin. 1 g/kg of body weight per day up to a maximum of 100 g/d |
| Absence of shock |
| No current treatment with nephrotoxic drugs |
| Absence of parenchymal kidney disease as indicated by proteinuria > 500 mg/d, or microhaematuria, (> 50 red blood cells per high power field) and /or a normal renal ultrasonography |
Pathophysiology of HRS
The major elements in the development of HRS are the diseased liver, the circulatory dysfunction with vasodilatation and lowering of the arterial blood pressure, the abnormal systemic neuro-humoral regulation with activation of the sympathetic nervous system which alters renal autoregulation, a cardiac dysfunction due to cirrhotic cardiomyopathy with a pre-terminal decline in cardiac output[40,193,194]. These aspects are summarised in Figure 6.
Figure 6.
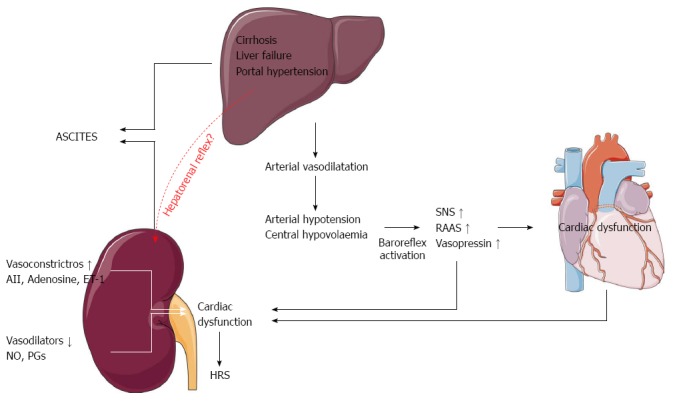
Pathophysiological mechanisms in the development of ascites and the hepatorenal syndrome. SNS: Sympathetic nervous system; RAAS: The renin-angiotensin-aldosterone system; AII: Angiotensin II; ET-1: Endothelin-1; NO: Nitric oxide; PGs: Prostaglandins.
Low systemic vascular resistance, central hypovolaemia, reduced baroreflex sensitivity, and abnormal renal autoregulation play a pivotal role in the circulatory dysfunction[111,193,195]. In patients with increased sympathetic nervous activity, the autoregulation curve may be shifted towards the right side[193]. Because of this, even minor reductions in arterial blood pressure may be harmful to renal perfusion. Thus, the renal blood flow decreases with the advancement of the clinical stage of liver dysfunction[196] and in these patients low arterial pressure relates to survival[11,85,197]. Cirrhotic cardiomyopathy has been described as a condition with impaired contractile responsiveness to stress and altered diastolic relaxation[123]. With the progression of the disease, the reduction in the systemic vascular resistance becomes so severe that the hyperdynamic cirrhotic heart is unable further to increase the high cardiac output, which leads to an underfilling of the central vascular bed and effective central hypovolaemia[111,188,198]. There is now evidence from several studies of a relation between the terminal decline in cardiac output and the progression of the disease, development of HRS, and survival[115,116]. We have therefore recently hypothesized a cardiorenal interaction in patients with advanced cirrhosis and renal dysfunction that refers to a condition where cardiac dysfunction in cirrhosis is a major determinant of the course of patients who develop HRS[199] (Figure 7).
Figure 7.
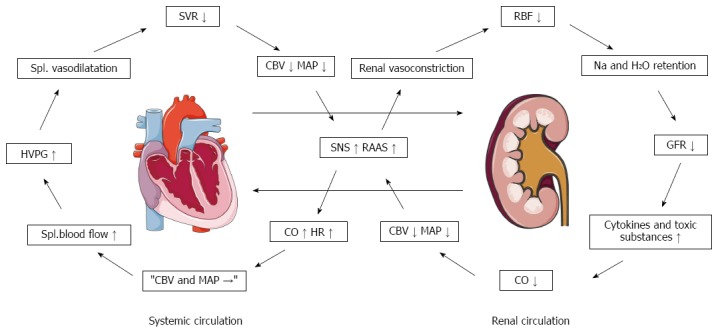
Pathophysiological proposal for the background of a cardiorenal syndrome in cirrhosis[194]. CBV: Central blood volume; CO: Cardiac output; GFR: Glomerular filtration rate; HR: Heart rate; HVPG: Hepatic venous pressure gradient; MAP: Mean arterial pressure; RAAS: Renin-angiotensin-aldosterone system; RBF: Renal blood flow; SNS: Sympathetic nervous system; SVR: Systemic vascular resistance.
Bacterial translocation from the gut plays a significant role for spontaneous infections and the circulatory dysfunction characterised by aggravation of vasodilatation elicited by an inflammatory response with the production of proinflammatory cytokines such as TNF-α and IL-6 as a “cytokine storm”[72]. SBP is frequent in patients with cirrhosis and is an important risk factor for the development of circulatory dysfunction and HRS (Figure 3)[71,77,78].
The future in terms of therapy will probably attack different aspects in the pathophysiological process. A multi-target strategy should seek efficiently to counteract the arterial vasodilatation, central hypovolaemia, and arterial hypotension by administration of potent vasoconstrictors such as terlipressin combined with human serum albumin. Development of orally long-acting systemic vasoconstrictors should be encouraged. All patients with HRS including those who respond to terlipressin and albumin should be properly prioritized on the waiting list for liver transplantation.
CHANGES IN ADRENAL FUNCTION
Increased cortisol levels due to activation of the hypothalamus-pituitary-adrenal axis represent an important adaptive mechanism in critical illness, regulating inflammation and cardiovascular response to the sympathetic nervous system. Circulating cytokines, such as TNF-α and IL-6, might impair pituitary responsiveness leading to inadequate cortisol secretion[200,201]. This has been termed relative adrenal insufficiency (RAI), which is associated with more pronounced hemodynamic instability, vasopressor dependency and increased mortality in critically ill patients[202,203].
In addition, RAI has been reported in cirrhosis as part of a hepato-adrenal syndrome with inhibition of ACTH and CRH due to high levels of pro-inflammatory cytokines[203,204]. Since patients with adrenal insufficiency may exhibit similar characteristics in terms of cardiac dysfunction, we recently hypothesized that adrenal insufficiency may contribute to cirrhotic cardiomyopathy and to precipitate HRS[200]. The relation between cardiac dysfunction and development of HRS should therefore be focus for treatment strategies that seek to improve cardiac function[116].
CONCLUDING SUMMARY
The recent years have considerably improved our knowledge on the mechanisms of disease processes in chronic liver disease. The cellular and humoral responses related to the fibrogenesis, inflammation, and bacterial translocation have been shown to be highly involved in the development of portal, splanchnic, and systemic haemodynamic complications to chronic liver disease. These extra-hepatic complications comprise changes in numerous organ systems as a multi-organ failure syndrome. The arterial vasodilatation and the general increased vascular compliance are directly linked to the cardiac and circulatory changes and to pulmonary haemodynamics and function. In conditions such as HRS and PoPH a preferential reactive and counter-regulatory vasoconstriction is prevailing. The function of the heart in cirrhosis is disturbed, with a hyperdynamic circulation with increased cardiac output and heart rate. Cardiac performance and the systolic and diastolic functions are clearly impaired and may contribute to other complications such as HRS as part of a cardio-renal syndrome.
Knowledge on the mechanisms of development of complications is crucial with respect to choice of vasoactive drugs, drugs with specific effects on fibrogenesis and inflammation, and drugs that target specific receptors. Although major unsolved questions remain, the circulatory and humoral derangements play important roles in the clinical aggravation of renal complications, cardio-pulmonary dysfunction, and circulatory reactivity. This aspect is important to take into account in futures research programmes and in the clinical handling of the cirrhotic patient.
Footnotes
Supported by Novo Nordisk Foundation and the University of Copenhagen
P- Reviewer: Gupta S, Procopet B S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.11th ed. Oxford: Wiley-Blackwell; 2002. Sherlock’s Diseases of the Liver and Biliary system. [Google Scholar]
- 2.Sethasine S, Jain D, Groszmann RJ, Garcia-Tsao G. Quantitative histological-hemodynamic correlations in cirrhosis. Hepatology. 2012;55:1146–1153. doi: 10.1002/hep.24805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.García-Pagán JC, Gracia-Sancho J, Bosch J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J Hepatol. 2012;57:458–461. doi: 10.1016/j.jhep.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Bendtsen F, Krag A, Møller S. Treatment of acute variceal bleeding. Dig Liver Dis. 2008;40:328–336. doi: 10.1016/j.dld.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Bosch J, Abraldes JG, Fernández M, García-Pagán JC. Hepatic endothelial dysfunction and abnormal angiogenesis: new targets in the treatment of portal hypertension. J Hepatol. 2010;53:558–567. doi: 10.1016/j.jhep.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 6.Iwakiri Y, Groszmann RJ. The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule. Hepatology. 2006;43:S121–S131. doi: 10.1002/hep.20993. [DOI] [PubMed] [Google Scholar]
- 7.Henriksen JH, Fuglsang S, Bendtsen F, Christensen E, Møller S. Arterial compliance in patients with cirrhosis: stroke volume-pulse pressure ratio as simplified index. Am J Physiol Gastrointest Liver Physiol. 2001;280:G584–G594. doi: 10.1152/ajpgi.2001.280.4.G584. [DOI] [PubMed] [Google Scholar]
- 8.Henriksen JH, Bendtsen F, Sørensen TI, Stadeager C, Ring-Larsen H. Reduced central blood volume in cirrhosis. Gastroenterology. 1989;97:1506–1513. doi: 10.1016/0016-5085(89)90396-x. [DOI] [PubMed] [Google Scholar]
- 9.Ginès P, Fernández J, Durand F, Saliba F. Management of critically-ill cirrhotic patients. J Hepatol. 2012;56 Suppl 1:S13–S24. doi: 10.1016/S0168-8278(12)60003-8. [DOI] [PubMed] [Google Scholar]
- 10.Gatta A, Bolognesi M, Merkel C. Vasoactive factors and hemodynamic mechanisms in the pathophysiology of portal hypertension in cirrhosis. Mol Aspects Med. 2008;29:119–129. doi: 10.1016/j.mam.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Møller S, Henriksen JH. The systemic circulation in cirrhosis. In: Gines P, Arroyo V, Rodes J, Schrier RW, editors. Ascites and renal dysfunction in liver disease. 2nd ed. Malden: Blackwell; 2005. pp. 139–155. [Google Scholar]
- 12.Ginès P, Cárdenas A. The management of ascites and hyponatremia in cirrhosis. Semin Liver Dis. 2008;28:43–58. doi: 10.1055/s-2008-1040320. [DOI] [PubMed] [Google Scholar]
- 13.Møller S, Henriksen JH. Cardiovascular complications of cirrhosis. Gut. 2008;57:268–278. doi: 10.1136/gut.2006.112177. [DOI] [PubMed] [Google Scholar]
- 14.Tage-Jensen U, Henriksen JH, Christensen E, Widding A, Ring-Larsen H, Christensen NJ. Plasma catecholamine level and portal venous pressure as guides to prognosis in patients with cirrhosis. J Hepatol. 1988;6:350–358. doi: 10.1016/s0168-8278(88)80053-9. [DOI] [PubMed] [Google Scholar]
- 15.Lebrec D, Moreau R. Pathogenesis of portal hypertension. Eur J Gastroenterol Hepatol. 2001;13:309–311. doi: 10.1097/00042737-200104000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Angeli P, Gines P. Hepatorenal syndrome, MELD score and liver transplantation: an evolving issue with relevant implications for clinical practice. J Hepatol. 2012;57:1135–1140. doi: 10.1016/j.jhep.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 17.Bernardi M. Spontaneous bacterial peritonitis: from pathophysiology to prevention. Intern Emerg Med. 2010;5 Suppl 1:S37–S44. doi: 10.1007/s11739-010-0446-x. [DOI] [PubMed] [Google Scholar]
- 18.Wiest R, Cadelina G, Milstien S, McCuskey RS, Garcia-Tsao G, Groszmann RJ. Bacterial translocation up-regulates GTP-cyclohydrolase I in mesenteric vasculature of cirrhotic rats. Hepatology. 2003;38:1508–1515. doi: 10.1016/j.hep.2003.09.039. [DOI] [PubMed] [Google Scholar]
- 19.Bellot P, Francés R, Such J. Pathological bacterial translocation in cirrhosis: pathophysiology, diagnosis and clinical implications. Liver Int. 2013;33:31–39. doi: 10.1111/liv.12021. [DOI] [PubMed] [Google Scholar]
- 20.Neugebauer H, Hartmann P, Krenn S, Glück T, Schölmerich J, Straub R, Wiest R. Bacterial translocation increases phagocytic activity of polymorphonuclear leucocytes in portal hypertension: priming independent of liver cirrhosis. Liver Int. 2008;28:1149–1157. doi: 10.1111/j.1478-3231.2008.01829.x. [DOI] [PubMed] [Google Scholar]
- 21.Yokomori H, Oda M, Yoshimura K, Watanabe S, Hibi T. Aberrant expressions of aquaporin-1 in association with capillarized sinusoidal endothelial cells in cirrhotic rat liver. Med Mol Morphol. 2010;43:6–12. doi: 10.1007/s00795-009-0475-6. [DOI] [PubMed] [Google Scholar]
- 22.Steib CJ. Kupffer cell activation and portal hypertension. Gut. 2011;60:1307–1308. doi: 10.1136/gut.2011.242560. [DOI] [PubMed] [Google Scholar]
- 23.Reynaert H, Thompson MG, Thomas T, Geerts A. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut. 2002;50:571–581. doi: 10.1136/gut.50.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marra F, Parola M. Cells in the Liver - Functions in Health and Disease. In: Gines P, Kamath PS, Arroyo V, editors. Chronic Liver Failure. Mechanisms and Management. 1 ed. New York: Springer; 2011. pp. 3–32. [Google Scholar]
- 25.Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151:1247–1256. doi: 10.1083/jcb.151.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rockey DC. The cell and molecular biology of hepatic fibrogenesis. Clinical and therapeutic implications. Clin Liver Dis. 2000;4:319–355. doi: 10.1016/s1089-3261(05)70113-6. [DOI] [PubMed] [Google Scholar]
- 27.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henriksen JH. Degradation of Bioactive Substances: Physiology and Pathophysiology. Boca Raton: CRC Press; 1991. [Google Scholar]
- 29.Gerbes AL, Witthaut R, Gülberg V, Thibault G, Bilzer M, Jüngst D. Role of the liver in splanchnic extraction of atrial natriuretic factor in the rat. Hepatology. 1992;16:790–793. doi: 10.1002/hep.1840160327. [DOI] [PubMed] [Google Scholar]
- 30.Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol. 2009;51:212–223. doi: 10.1016/j.jhep.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nolan JP. The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology. 2010;52:1829–1835. doi: 10.1002/hep.23917. [DOI] [PubMed] [Google Scholar]
- 32.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–1186. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 33.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol. 2010;16:1321–1329. doi: 10.3748/wjg.v16.i11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167sr1. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 37.Rockey DC. Hepatic fibrosis, stellate cells, and portal hypertension. Clin Liver Dis. 2006;10:459–79, vii-viii. doi: 10.1016/j.cld.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 38.DeLeve LD. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest. 2013;123:1861–1866. doi: 10.1172/JCI66025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1:1. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwakiri Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int. 2012;32:199–213. doi: 10.1111/j.1478-3231.2011.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao B, Seki E, Brenner DA, Friedman S, Cohen JI, Nagy L, Szabo G, Zakhari S. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G516–G525. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mortensen C, Andersen O, Krag A, Bendtsen F, Møller S. High-sensitivity C-reactive protein levels predict survival and are related to haemodynamics in alcoholic cirrhosis. Eur J Gastroenterol Hepatol. 2012;24:619–626. doi: 10.1097/MEG.0b013e328351db6e. [DOI] [PubMed] [Google Scholar]
- 43.Rockey DC. Antifibrotic therapy in chronic liver disease. Clin Gastroenterol Hepatol. 2005;3:95–107. doi: 10.1016/s1542-3565(04)00445-8. [DOI] [PubMed] [Google Scholar]
- 44.Henriksen JH, Møller S. Hemodynamics, distribution of blood volume, and kinetics of vasoactive substances in cirrhosis. In: Epstein M, editor. The kidney in liver disease. 4th ed. Philadelphia: Hanley and Belfus; 1996. pp. 241–58. [Google Scholar]
- 45.Bosch J, García-Pagán JC. Complications of cirrhosis. I. Portal hypertension. J Hepatol. 2000;32:141–156. doi: 10.1016/s0168-8278(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 46.Henriksen JH, Winkler K. Hepatic blood flow determination. A comparison of 99mTc-diethyl-IDA and indocyanine green as hepatic blood flow indicators in man. J Hepatol. 1987;4:66–70. doi: 10.1016/s0168-8278(87)80011-9. [DOI] [PubMed] [Google Scholar]
- 47.Madsen JL, Søndergaard SB, Møller S. Meal-induced changes in splanchnic blood flow and oxygen uptake in middle-aged healthy humans. Scand J Gastroenterol. 2006;41:87–92. doi: 10.1080/00365520510023882. [DOI] [PubMed] [Google Scholar]
- 48.Benoit JN, Granger DN. Splanchnic hemodynamics in chronic portal hypertension. Semin Liver Dis. 1986;6:287–298. doi: 10.1055/s-2008-1040611. [DOI] [PubMed] [Google Scholar]
- 49.Hobolth L, Møller S, Grønbæk H, Roelsgaard K, Bendtsen F, Feldager Hansen E. Carvedilol or propranolol in portal hypertension? A randomized comparison. Scand J Gastroenterol. 2012;47:467–474. doi: 10.3109/00365521.2012.666673. [DOI] [PubMed] [Google Scholar]
- 50.Albillos A, Bañares R, González M, Catalina MV, Pastor O, Gonzalez R, Ripoll C, Bosch J. The extent of the collateral circulation influences the postprandial increase in portal pressure in patients with cirrhosis. Gut. 2007;56:259–264. doi: 10.1136/gut.2006.095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berzigotti A, De Gottardi A, Vukotic R, Siramolpiwat S, Abraldes JG, García-Pagan JC, Bosch J. Effect of meal ingestion on liver stiffness in patients with cirrhosis and portal hypertension. PLoS One. 2013;8:e58742. doi: 10.1371/journal.pone.0058742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bendtsen F, Simonsen L, Henriksen JH. Effect on hemodynamics of a liquid meal alone and in combination with propranolol in cirrhosis. Gastroenterology. 1992;102:1017–1023. doi: 10.1016/0016-5085(92)90191-z. [DOI] [PubMed] [Google Scholar]
- 53.Morita H, Abe C. Negative feedforward control of body fluid homeostasis by hepatorenal reflex. Hypertens Res. 2011;34:895–905. doi: 10.1038/hr.2011.88. [DOI] [PubMed] [Google Scholar]
- 54.Lautt WW. Regulatory processes interacting to maintain hepatic blood flow constancy: Vascular compliance, hepatic arterial buffer response, hepatorenal reflex, liver regeneration, escape from vasoconstriction. Hepatol Res. 2007;37:891–903. doi: 10.1111/j.1872-034X.2007.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jalan R, Forrest EH, Redhead DN, Dillon JF, Hayes PC. Reduction in renal blood flow following acute increase in the portal pressure: evidence for the existence of a hepatorenal reflex in man? Gut. 1997;40:664–670. doi: 10.1136/gut.40.5.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kapoor D, Redhead DN, Hayes PC, Webb DJ, Jalan R. Systemic and regional changes in plasma endothelin following transient increase in portal pressure. Liver Transpl. 2003;9:32–39. doi: 10.1053/jlts.2003.50007. [DOI] [PubMed] [Google Scholar]
- 57.Jiménez-Sáenz M, Soria IC, Bernardez JR, Gutierrez JM. Renal sodium retention in portal hypertension and hepatorenal reflex: from practice to science. Hepatology. 2003;37:1494; author reply 1494–1495. doi: 10.1053/jhep.2003.50226. [DOI] [PubMed] [Google Scholar]
- 58.Gülberg V, Haag K, Rössle M, Gerbes AL. Hepatic arterial buffer response in patients with advanced cirrhosis. Hepatology. 2002;35:630–634. doi: 10.1053/jhep.2002.31722. [DOI] [PubMed] [Google Scholar]
- 59.Zipprich A, Steudel N, Behrmann C, Meiss F, Sziegoleit U, Fleig WE, Kleber G. Functional significance of hepatic arterial flow reserve in patients with cirrhosis. Hepatology. 2003;37:385–392. doi: 10.1053/jhep.2003.50065. [DOI] [PubMed] [Google Scholar]
- 60.Lautt WW, Greenway CV, Legare DJ. Effect of hepatic nerves, norepinephrine, angiotensin, and elevated central venous pressure on postsinusoidal resistance sites and intrahepatic pressures in cats. Microvasc Res. 1987;33:50–61. doi: 10.1016/0026-2862(87)90006-9. [DOI] [PubMed] [Google Scholar]
- 61.Møller S, Bendtsen F, Henriksen JH. Vasoactive substances in the circulatory dysfunction of cirrhosis. Scand J Clin Lab Invest. 2001;61:421–429. doi: 10.1080/00365510152567059. [DOI] [PubMed] [Google Scholar]
- 62.Iwakiri Y. The molecules: mechanisms of arterial vasodilatation observed in the splanchnic and systemic circulation in portal hypertension. J Clin Gastroenterol. 2007;41 Suppl 3:S288–S294. doi: 10.1097/MCG.0b013e3181468b4c. [DOI] [PubMed] [Google Scholar]
- 63.Alam I, Bass NM, Bacchetti P, Gee L, Rockey DC. Hepatic tissue endothelin-1 levels in chronic liver disease correlate with disease severity and ascites. Am J Gastroenterol. 2000;95:199–203. doi: 10.1111/j.1572-0241.2000.01684.x. [DOI] [PubMed] [Google Scholar]
- 64.Zipprich A, Mehal WZ, Ripoll C, Groszmann RJ. A distinct nitric oxide and adenosine A1 receptor dependent hepatic artery vasodilatatory response in the CCl-cirrhotic liver. Liver Int. 2010;30:988–994. doi: 10.1111/j.1478-3231.2010.02278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiest R, Groszmann RJ. The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatology. 2002;35:478–491. doi: 10.1053/jhep.2002.31432. [DOI] [PubMed] [Google Scholar]
- 66.Fasolato S, Angeli P, Dallagnese L, Maresio G, Zola E, Mazza E, Salinas F, Donà S, Fagiuoli S, Sticca A, et al. Renal failure and bacterial infections in patients with cirrhosis: epidemiology and clinical features. Hepatology. 2007;45:223–229. doi: 10.1002/hep.21443. [DOI] [PubMed] [Google Scholar]
- 67.Bajaj JS, O’Leary JG, Wong F, Reddy KR, Kamath PS. Bacterial infections in end-stage liver disease: current challenges and future directions. Gut. 2012;61:1219–1225. doi: 10.1136/gutjnl-2012-302339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mookerjee RP, Stadlbauer V, Lidder S, Wright GA, Hodges SJ, Davies NA, Jalan R. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology. 2007;46:831–840. doi: 10.1002/hep.21737. [DOI] [PubMed] [Google Scholar]
- 69.Grønbaek H, Sandahl TD, Mortensen C, Vilstrup H, Møller HJ, Møller S. Soluble CD163, a marker of Kupffer cell activation, is related to portal hypertension in patients with liver cirrhosis. Aliment Pharmacol Ther. 2012;36:173–180. doi: 10.1111/j.1365-2036.2012.05134.x. [DOI] [PubMed] [Google Scholar]
- 70.Fernández J, Gustot T. Management of bacterial infections in cirrhosis. J Hepatol. 2012;56 Suppl 1:S1–12. doi: 10.1016/S0168-8278(12)60002-6. [DOI] [PubMed] [Google Scholar]
- 71.Wiest R, Krag A, Gerbes A. Spontaneous bacterial peritonitis: recent guidelines and beyond. Gut. 2012;61:297–310. doi: 10.1136/gutjnl-2011-300779. [DOI] [PubMed] [Google Scholar]
- 72.Gustot T, Durand F, Lebrec D, Vincent JL, Moreau R. Severe sepsis in cirrhosis. Hepatology. 2009;50:2022–2033. doi: 10.1002/hep.23264. [DOI] [PubMed] [Google Scholar]
- 73.Tandon P, Garcia-Tsao G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin Liver Dis. 2008;28:26–42. doi: 10.1055/s-2008-1040319. [DOI] [PubMed] [Google Scholar]
- 74.Albillos A, de la Hera A, González M, Moya JL, Calleja JL, Monserrat J, Ruiz-del-Arbol L, Alvarez-Mon M. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology. 2003;37:208–217. doi: 10.1053/jhep.2003.50038. [DOI] [PubMed] [Google Scholar]
- 75.Bellot P, García-Pagán JC, Francés R, Abraldes JG, Navasa M, Pérez-Mateo M, Such J, Bosch J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology. 2010;52:2044–2052. doi: 10.1002/hep.23918. [DOI] [PubMed] [Google Scholar]
- 76.Mortensen C, Karlsen S, Grønbæk H, Nielsen DT, Frevert S, Clemmesen JO, Møller S, Jensen JS, Bendtsen F. No difference in portal and hepatic venous bacterial DNA in patients with cirrhosis undergoing transjugular intrahepatic portosystemic shunt insertion. Liver Int. 2013;33:1309–1315. doi: 10.1111/liv.12205. [DOI] [PubMed] [Google Scholar]
- 77.Ruiz-del-Arbol L, Urman J, Fernández J, González M, Navasa M, Monescillo A, Albillos A, Jiménez W, Arroyo V. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 2003;38:1210–1218. doi: 10.1053/jhep.2003.50447. [DOI] [PubMed] [Google Scholar]
- 78.European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53:397–417. doi: 10.1016/j.jhep.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 79.Leithead JA, Ferguson JW, Bates CM, Davidson JS, Lee A, Bathgate AJ, Hayes PC, Simpson KJ. The systemic inflammatory response syndrome is predictive of renal dysfunction in patients with non-paracetamol-induced acute liver failure. Gut. 2009;58:443–449. doi: 10.1136/gut.2008.154120. [DOI] [PubMed] [Google Scholar]
- 80.Follo A, Llovet JM, Navasa M, Planas R, Forns X, Francitorra A, Rimola A, Gassull MA, Arroyo V, Rodés J. Renal impairment after spontaneous bacterial peritonitis in cirrhosis: incidence, clinical course, predictive factors and prognosis. Hepatology. 1994;20:1495–1501. doi: 10.1002/hep.1840200619. [DOI] [PubMed] [Google Scholar]
- 81.Fagundes C, Ginès P. Hepatorenal syndrome: a severe, but treatable, cause of kidney failure in cirrhosis. Am J Kidney Dis. 2012;59:874–885. doi: 10.1053/j.ajkd.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 82.Pardo A, Bartolí R, Lorenzo-Zúñiga V, Planas R, Viñado B, Riba J, Cabré E, Santos J, Luque T, Ausina V, et al. Effect of cisapride on intestinal bacterial overgrowth and bacterial translocation in cirrhosis. Hepatology. 2000;31:858–863. doi: 10.1053/he.2000.5746. [DOI] [PubMed] [Google Scholar]
- 83.Tazi KA, Moreau R, Hervé P, Dauvergne A, Cazals-Hatem D, Bert F, Poirel O, Rabiller A, Lebrec D. Norfloxacin reduces aortic NO synthases and proinflammatory cytokine up-regulation in cirrhotic rats: role of Akt signaling. Gastroenterology. 2005;129:303–314. doi: 10.1053/j.gastro.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 84.Senzolo M, Cholongitas E, Burra P, Leandro G, Thalheimer U, Patch D, Burroughs AK. beta-Blockers protect against spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. Liver Int. 2009;29:1189–1193. doi: 10.1111/j.1478-3231.2009.02038.x. [DOI] [PubMed] [Google Scholar]
- 85.Llach J, Ginès P, Arroyo V, Rimola A, Titó L, Badalamenti S, Jiménez W, Gaya J, Rivera F, Rodés J. Prognostic value of arterial pressure, endogenous vasoactive systems, and renal function in cirrhotic patients admitted to the hospital for the treatment of ascites. Gastroenterology. 1988;94:482–487. doi: 10.1016/0016-5085(88)90441-6. [DOI] [PubMed] [Google Scholar]
- 86.Braillon A, Cales P, Valla D, Gaudy D, Geoffroy P, Lebrec D. Influence of the degree of liver failure on systemic and splanchnic haemodynamics and on response to propranolol in patients with cirrhosis. Gut. 1986;27:1204–1209. doi: 10.1136/gut.27.10.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Møller S. Systemic haemodynamics in cirrhosis and portal hypertension with focus on vasoactive substances and prognosis. Dan Med Bull. 1998;45:1–14. [PubMed] [Google Scholar]
- 88.Møller S, Hillingsø J, Christensen E, Henriksen JH. Arterial hypoxaemia in cirrhosis: fact or fiction? Gut. 1998;42:868–874. doi: 10.1136/gut.42.6.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Møller S, Hobolth L, Winkler C, Bendtsen F, Christensen E. Determinants of the hyperdynamic circulation and central hypovolaemia in cirrhosis. Gut. 2011;60:1254–1259. doi: 10.1136/gut.2010.235473. [DOI] [PubMed] [Google Scholar]
- 90.Arroyo V, Colmenero J. Ascites and hepatorenal syndrome in cirrhosis: pathophysiological basis of therapy and current management. J Hepatol. 2003;38 Suppl 1:S69–S89. doi: 10.1016/s0168-8278(03)00007-2. [DOI] [PubMed] [Google Scholar]
- 91.Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodés J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151–1157. doi: 10.1002/hep.1840080532. [DOI] [PubMed] [Google Scholar]
- 92.Møller S, Henriksen JH. Circulatory abnormalities in cirrhosis with focus on neurohumoral aspects. Semin Nephrol. 1997;17:505–519. [PubMed] [Google Scholar]
- 93.Schrier RW, Ecder T. Gibbs memorial lecture. Unifying hypothesis of body fluid volume regulation: implications for cardiac failure and cirrhosis. Mt Sinai J Med. 2001;68:350–361. [PubMed] [Google Scholar]
- 94.Martin PY, Ginès P, Schrier RW. Nitric oxide as a mediator of hemodynamic abnormalities and sodium and water retention in cirrhosis. N Engl J Med. 1998;339:533–541. doi: 10.1056/NEJM199808203390807. [DOI] [PubMed] [Google Scholar]
- 95.Møller S, Bendtsen F, Schifter S, Henriksen JH. Relation of calcitonin gene-related peptide to systemic vasodilatation and central hypovolaemia in cirrhosis. Scand J Gastroenterol. 1996;31:928–933. doi: 10.3109/00365529609052004. [DOI] [PubMed] [Google Scholar]
- 96.Hori N, Okanoue T, Sawa Y, Kashima K. Role of calcitonin gene-related peptide in the vascular system on the development of the hyperdynamic circulation in conscious cirrhotic rats. J Hepatol. 1997;26:1111–1119. doi: 10.1016/s0168-8278(97)80120-1. [DOI] [PubMed] [Google Scholar]
- 97.Guevara M, Ginès P, Jiménez W, Sort P, Fernández-Esparrach G, Escorsell A, Bataller R, Bosch J, Arroyo V, Rivera F, et al. Increased adrenomedullin levels in cirrhosis: relationship with hemodynamic abnormalities and vasoconstrictor systems. Gastroenterology. 1998;114:336–343. doi: 10.1016/s0016-5085(98)70486-x. [DOI] [PubMed] [Google Scholar]
- 98.Bátkai S, Járai Z, Wagner JA, Goparaju SK, Varga K, Liu J, Wang L, Mirshahi F, Khanolkar AD, Makriyannis A, et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat Med. 2001;7:827–832. doi: 10.1038/89953. [DOI] [PubMed] [Google Scholar]
- 99.Moezi L, Gaskari SA, Lee SS. Endocannabinoids and liver disease. V. endocannabinoids as mediators of vascular and cardiac abnormalities in cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2008;295:G649–G653. doi: 10.1152/ajpgi.90352.2008. [DOI] [PubMed] [Google Scholar]
- 100.Ros J, Clària J, To-Figueras J, Planagumà A, Cejudo-Martín P, Fernández-Varo G, Martín-Ruiz R, Arroyo V, Rivera F, Rodés J, et al. Endogenous cannabinoids: a new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat. Gastroenterology. 2002;122:85–93. doi: 10.1053/gast.2002.30305. [DOI] [PubMed] [Google Scholar]
- 101.Fernandez M, Mejias M, Angermayr B, Garcia-Pagan JC, Rodés J, Bosch J. Inhibition of VEGF receptor-2 decreases the development of hyperdynamic splanchnic circulation and portal-systemic collateral vessels in portal hypertensive rats. J Hepatol. 2005;43:98–103. doi: 10.1016/j.jhep.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 102.Huang HC, Haq O, Utsumi T, Sethasine S, Abraldes JG, Groszmann RJ, Iwakiri Y. Intestinal and plasma VEGF levels in cirrhosis: the role of portal pressure. J Cell Mol Med. 2012;16:1125–1133. doi: 10.1111/j.1582-4934.2011.01399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iwakiri Y, Grisham M, Shah V. Vascular biology and pathobiology of the liver: Report of a single-topic symposium. Hepatology. 2008;47:1754–1763. doi: 10.1002/hep.22203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bernardi M, Fornalè L, Di Marco C, Trevisani F, Baraldini M, Gasbarrini A, De Collibus C, Zacà F, Ligabue A, Colantoni A. Hyperdynamic circulation of advanced cirrhosis: a re-appraisal based on posture-induced changes in hemodynamics. J Hepatol. 1995;22:309–318. doi: 10.1016/0168-8278(95)80284-3. [DOI] [PubMed] [Google Scholar]
- 105.Laffi G, Barletta G, La Villa G, Del Bene R, Riccardi D, Ticali P, Melani L, Fantini F, Gentilini P. Altered cardiovascular responsiveness to active tilting in nonalcoholic cirrhosis. Gastroenterology. 1997;113:891–898. doi: 10.1016/s0016-5085(97)70184-7. [DOI] [PubMed] [Google Scholar]
- 106.Gentilini P, Romanelli RG, Laffi G, Barletta G, Del Bene R, Messeri G, La Villa G. Cardiovascular and renal function in normotensive and hypertensive patients with compensated cirrhosis: effects of posture. J Hepatol. 1999;30:632–638. doi: 10.1016/s0168-8278(99)80193-7. [DOI] [PubMed] [Google Scholar]
- 107.Møller S, Nørgaard A, Henriksen JH, Frandsen E, Bendtsen F. Effects of tilting on central hemodynamics and homeostatic mechanisms in cirrhosis. Hepatology. 2004;40:811–819. doi: 10.1002/hep.20416. [DOI] [PubMed] [Google Scholar]
- 108.Henriksen JH, Bendtsen F, Gerbes AL, Christensen NJ, Ring-Larsen H, Sørensen TI. Estimated central blood volume in cirrhosis: relationship to sympathetic nervous activity, beta-adrenergic blockade and atrial natriuretic factor. Hepatology. 1992;16:1163–1170. [PubMed] [Google Scholar]
- 109.Brinch K, Møller S, Bendtsen F, Becker U, Henriksen JH. Plasma volume expansion by albumin in cirrhosis. Relation to blood volume distribution, arterial compliance and severity of disease. J Hepatol. 2003;39:24–31. doi: 10.1016/s0168-8278(03)00160-0. [DOI] [PubMed] [Google Scholar]
- 110.Schrier RW. Water and sodium retention in edematous disorders: role of vasopressin and aldosterone. Am J Med. 2006;119:S47–S53. doi: 10.1016/j.amjmed.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 111.Møller S, Henriksen JH, Bendtsen F. Central and noncentral blood volumes in cirrhosis: relationship to anthropometrics and gender. Am J Physiol Gastrointest Liver Physiol. 2003;284:G970–G979. doi: 10.1152/ajpgi.00521.2002. [DOI] [PubMed] [Google Scholar]
- 112.Møller S, Bendtsen F, Henriksen JH. Effect of volume expansion on systemic hemodynamics and central and arterial blood volume in cirrhosis. Gastroenterology. 1995;109:1917–1925. doi: 10.1016/0016-5085(95)90759-9. [DOI] [PubMed] [Google Scholar]
- 113.Kiszka-Kanowitz M, Henriksen JH, Møller S, Bendtsen F. Blood volume distribution in patients with cirrhosis: aspects of the dual-head gamma-camera technique. J Hepatol. 2001;35:605–612. doi: 10.1016/s0168-8278(01)00175-1. [DOI] [PubMed] [Google Scholar]
- 114.Møller S, Bendtsen F, Christensen E, Henriksen JH. Prognostic variables in patients with cirrhosis and oesophageal varices without prior bleeding. J Hepatol. 1994;21:940–946. doi: 10.1016/s0168-8278(05)80599-9. [DOI] [PubMed] [Google Scholar]
- 115.Ruiz-del-Arbol L, Monescillo A, Arocena C, Valer P, Ginès P, Moreira V, Milicua JM, Jiménez W, Arroyo V. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005;42:439–447. doi: 10.1002/hep.20766. [DOI] [PubMed] [Google Scholar]
- 116.Krag A, Bendtsen F, Henriksen JH, Møller S. Low cardiac output predicts development of hepatorenal syndrome and survival in patients with cirrhosis and ascites. Gut. 2010;59:105–110. doi: 10.1136/gut.2009.180570. [DOI] [PubMed] [Google Scholar]
- 117.Bendtsen F, Henriksen JH, Sørensen TI. Long-term effects of oral propranolol on splanchnic and systemic haemodynamics in patients with cirrhosis and oesophageal varices. Scand J Gastroenterol. 1991;26:933–939. doi: 10.3109/00365529108996245. [DOI] [PubMed] [Google Scholar]
- 118.Krag A, Møller S, Burroughs AK, Bendtsen F. Betablockers induce cardiac chronotropic incompetence. J Hepatol. 2012;56:298–299. doi: 10.1016/j.jhep.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 119.Kowalski HJ, Abelmann WH. The cardiac output at rest in Laennec’s cirrhosis. J Clin Invest. 1953;32:1025–1033. doi: 10.1172/JCI102813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alqahtani SA, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Semin Liver Dis. 2008;28:59–69. doi: 10.1055/s-2008-1040321. [DOI] [PubMed] [Google Scholar]
- 121.Zaky A, Lang JD, Treggiari M. Towards a better characterization of cirrhosis-associated cardiomyopathy? J Hepatol. 2013;59:192–193. doi: 10.1016/j.jhep.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 122.Zambruni A, Trevisani F, Caraceni P, Bernardi M. Cardiac electrophysiological abnormalities in patients with cirrhosis. J Hepatol. 2006;44:994–1002. doi: 10.1016/j.jhep.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 123.Møller S, Henriksen JH. Cirrhotic cardiomyopathy. J Hepatol. 2010;53:179–190. doi: 10.1016/j.jhep.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 124.Bernardi M, Calandra S, Colantoni A, Trevisani F, Raimondo ML, Sica G, Schepis F, Mandini M, Simoni P, Contin M, et al. Q-T interval prolongation in cirrhosis: prevalence, relationship with severity, and etiology of the disease and possible pathogenetic factors. Hepatology. 1998;27:28–34. doi: 10.1002/hep.510270106. [DOI] [PubMed] [Google Scholar]
- 125.Rabie RN, Cazzaniga M, Salerno F, Wong F. The use of E/A ratio as a predictor of outcome in cirrhotic patients treated with transjugular intrahepatic portosystemic shunt. Am J Gastroenterol. 2009;104:2458–2466. doi: 10.1038/ajg.2009.321. [DOI] [PubMed] [Google Scholar]
- 126.Saner FH, Neumann T, Canbay A, Treckmann JW, Hartmann M, Goerlinger K, Bertram S, Beckebaum S, Cicinnati V, Paul A. High brain-natriuretic peptide level predicts cirrhotic cardiomyopathy in liver transplant patients. Transpl Int. 2011;24:425–432. doi: 10.1111/j.1432-2277.2011.01219.x. [DOI] [PubMed] [Google Scholar]
- 127.Liu H, Gaskari SA, Lee SS. Cardiac and vascular changes in cirrhosis: pathogenic mechanisms. World J Gastroenterol. 2006;12:837–842. doi: 10.3748/wjg.v12.i6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mani AR, Ippolito S, Ollosson R, Moore KP. Nitration of cardiac proteins is associated with abnormal cardiac chronotropic responses in rats with biliary cirrhosis. Hepatology. 2006;43:847–856. doi: 10.1002/hep.21115. [DOI] [PubMed] [Google Scholar]
- 129.Møller S, Hove JD, Dixen U, Bendtsen F. New insights into cirrhotic cardiomyopathy. Int J Cardiol. 2013;167:1101–1108. doi: 10.1016/j.ijcard.2012.09.089. [DOI] [PubMed] [Google Scholar]
- 130.Wong F, Girgrah N, Graba J, Allidina Y, Liu P, Blendis L. The cardiac response to exercise in cirrhosis. Gut. 2001;49:268–275. doi: 10.1136/gut.49.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Møller S, Henriksen JH. Cardiovascular dysfunction in cirrhosis. Pathophysiological evidence of a cirrhotic cardiomyopathy. Scand J Gastroenterol. 2001;36:785–794. doi: 10.1080/003655201750313289. [DOI] [PubMed] [Google Scholar]
- 132.Huonker M, Schumacher YO, Ochs A, Sorichter S, Keul J, Rössle M. Cardiac function and haemodynamics in alcoholic cirrhosis and effects of the transjugular intrahepatic portosystemic stent shunt. Gut. 1999;44:743–748. doi: 10.1136/gut.44.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Merli M, Valeriano V, Funaro S, Attili AF, Masini A, Efrati C, De CS, Riggio O. Modifications of cardiac function in cirrhotic patients treated with transjugular intrahepatic portosystemic shunt (TIPS) Am J Gastroenterol. 2002;97:142–148. doi: 10.1111/j.1572-0241.2002.05438.x. [DOI] [PubMed] [Google Scholar]
- 134.Ginès P, Uriz J, Calahorra B, Garcia-Tsao G, Kamath PS, Del Arbol LR, Planas R, Bosch J, Arroyo V, Rodés J. Transjugular intrahepatic portosystemic shunting versus paracentesis plus albumin for refractory ascites in cirrhosis. Gastroenterology. 2002;123:1839–1847. doi: 10.1053/gast.2002.37073. [DOI] [PubMed] [Google Scholar]
- 135.Pozzi M, Carugo S, Boari G, Pecci V, de Ceglia S, Maggiolini S, Bolla GB, Roffi L, Failla M, Grassi G, et al. Evidence of functional and structural cardiac abnormalities in cirrhotic patients with and without ascites. Hepatology. 1997;26:1131–1137. doi: 10.1002/hep.510260507. [DOI] [PubMed] [Google Scholar]
- 136.Torregrosa M, Aguadé S, Dos L, Segura R, Gónzalez A, Evangelista A, Castell J, Margarit C, Esteban R, Guardia J, et al. Cardiac alterations in cirrhosis: reversibility after liver transplantation. J Hepatol. 2005;42:68–74. doi: 10.1016/j.jhep.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 137.Møller S, Henriksen JH. Cirrhotic cardiomyopathy: a pathophysiological review of circulatory dysfunction in liver disease. Heart. 2002;87:9–15. doi: 10.1136/heart.87.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Grose RD, Nolan J, Dillon JF, Errington M, Hannan WJ, Bouchier IA, Hayes PC. Exercise-induced left ventricular dysfunction in alcoholic and non-alcoholic cirrhosis. J Hepatol. 1995;22:326–332. doi: 10.1016/0168-8278(95)80286-x. [DOI] [PubMed] [Google Scholar]
- 139.Epstein SK, Ciubotaru RL, Zilberberg MD, Kaplan LM, Jacoby C, Freeman R, Kaplan MM. Analysis of impaired exercise capacity in patients with cirrhosis. Dig Dis Sci. 1998;43:1701–1707. doi: 10.1023/a:1018867232562. [DOI] [PubMed] [Google Scholar]
- 140.Kazankov K, Holland-Fischer P, Andersen NH, Torp P, Sloth E, Aagaard NK, Vilstrup H. Resting myocardial dysfunction in cirrhosis quantified by tissue Doppler imaging. Liver Int. 2011;31:534–540. doi: 10.1111/j.1478-3231.2011.02468.x. [DOI] [PubMed] [Google Scholar]
- 141.Sampaio F, Pimenta J, Bettencourt N, Fontes-Carvalho R, Silva AP, Valente J, Bettencourt P, Fraga J, Gama V. Systolic dysfunction and diastolic dysfunction do not influence medium-term prognosis in patients with cirrhosis. Eur J Intern Med. 2014;25:241–246. doi: 10.1016/j.ejim.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 142.Myers RP, Lee SS. Cirrhotic cardiomyopathy and liver transplantation. Liver Transpl. 2000;6:S44–S52. doi: 10.1002/lt.500060510. [DOI] [PubMed] [Google Scholar]
- 143.Pozzi M, Redaelli E, Ratti L, Poli G, Guidi C, Milanese M, Calchera I, Mancia G. Time-course of diastolic dysfunction in different stages of chronic HCV related liver diseases. Minerva Gastroenterol Dietol. 2005;51:179–186. [PubMed] [Google Scholar]
- 144.Gaskari SA, Honar H, Lee SS. Therapy insight: Cirrhotic cardiomyopathy. Nat Clin Pract Gastroenterol Hepatol. 2006;3:329–337. doi: 10.1038/ncpgasthep0498. [DOI] [PubMed] [Google Scholar]
- 145.Finucci G, Desideri A, Sacerdoti D, Bolognesi M, Merkel C, Angeli P, Gatta A. Left ventricular diastolic function in liver cirrhosis. Scand J Gastroenterol. 1996;31:279–284. doi: 10.3109/00365529609004879. [DOI] [PubMed] [Google Scholar]
- 146.Henriksen JH, Bendtsen F, Hansen EF, Møller S. Acute non-selective beta-adrenergic blockade reduces prolonged frequency-adjusted Q-T interval (QTc) in patients with cirrhosis. J Hepatol. 2004;40:239–246. doi: 10.1016/j.jhep.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 147.Henriksen JH, Gøtze JP, Fuglsang S, Christensen E, Bendtsen F, Møller S. Increased circulating pro-brain natriuretic peptide (proBNP) and brain natriuretic peptide (BNP) in patients with cirrhosis: relation to cardiovascular dysfunction and severity of disease. Gut. 2003;52:1511–1517. doi: 10.1136/gut.52.10.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Genovesi S, Prata Pizzala DM, Pozzi M, Ratti L, Milanese M, Pieruzzi F, Vincenti A, Stella A, Mancia G, Stramba-Badiale M. QT interval prolongation and decreased heart rate variability in cirrhotic patients: relevance of hepatic venous pressure gradient and serum calcium. Clin Sci (Lond) 2009;116:851–859. doi: 10.1042/CS20080325. [DOI] [PubMed] [Google Scholar]
- 149.Bernardi M, Maggioli C, Dibra V, Zaccherini G. QT interval prolongation in liver cirrhosis: innocent bystander or serious threat? Expert Rev Gastroenterol Hepatol. 2012;6:57–66. doi: 10.1586/egh.11.86. [DOI] [PubMed] [Google Scholar]
- 150.Mohamed R, Forsey PR, Davies MK, Neuberger JM. Effect of liver transplantation on QT interval prolongation and autonomic dysfunction in end-stage liver disease. Hepatology. 1996;23:1128–1134. doi: 10.1002/hep.510230529. [DOI] [PubMed] [Google Scholar]
- 151.Trevisani F, Di Micoli A, Zambruni A, Biselli M, Santi V, Erroi V, Lenzi B, Caraceni P, Domenicali M, Cavazza M, et al. QT interval prolongation by acute gastrointestinal bleeding in patients with cirrhosis. Liver Int. 2012;32:1510–1515. doi: 10.1111/j.1478-3231.2012.02847.x. [DOI] [PubMed] [Google Scholar]
- 152.Møller S, Bernardi M. Interactions of the heart and the liver. Eur Heart J. 2013;34:2804–2811. doi: 10.1093/eurheartj/eht246. [DOI] [PubMed] [Google Scholar]
- 153.Fallon MB, Abrams GA. Pulmonary dysfunction in chronic liver disease. Hepatology. 2000;32:859–865. doi: 10.1053/jhep.2000.7519. [DOI] [PubMed] [Google Scholar]
- 154.Rodríguez-Roisin R, Krowka MJ, Hervé P, Fallon MB. Pulmonary-Hepatic vascular Disorders (PHD) Eur Respir J. 2004;24:861–880. doi: 10.1183/09031936.04.00010904. [DOI] [PubMed] [Google Scholar]
- 155.Grace JA, Angus PW. Hepatopulmonary syndrome: update on recent advances in pathophysiology, investigation, and treatment. J Gastroenterol Hepatol. 2013;28:213–219. doi: 10.1111/jgh.12061. [DOI] [PubMed] [Google Scholar]
- 156.Machicao VI, Balakrishnan M, Fallon MB. Pulmonary complications in chronic liver disease. Hepatology. 2014;59:1627–1637. doi: 10.1002/hep.26745. [DOI] [PubMed] [Google Scholar]
- 157.Scarlata S, Conte ME, Cesari M, Gentilucci UV, Miglioresi L, Pedone C, Picardi A, Ricci GL, Incalzi RA. Gas exchanges and pulmonary vascular abnormalities at different stages of chronic liver disease. Liver Int. 2011;31:525–533. doi: 10.1111/j.1478-3231.2011.02467.x. [DOI] [PubMed] [Google Scholar]
- 158.Gaines DI, Fallon MB. Hepatopulmonary syndrome. Liver Int. 2004;24:397–401. doi: 10.1111/j.1478-3231.2004.0944.x. [DOI] [PubMed] [Google Scholar]
- 159.Deibert P, Allgaier HP, Loesch S, Müller C, Olschewski M, Hamm H, Maier KP, Blum HE. Hepatopulmonary syndrome in patients with chronic liver disease: role of pulse oximetry. BMC Gastroenterol. 2006;6:15. doi: 10.1186/1471-230X-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Møller S, Krag A, Madsen JL, Henriksen JH, Bendtsen F. Pulmonary dysfunction and hepatopulmonary syndrome in cirrhosis and portal hypertension. Liver Int. 2009;29:1528–1537. doi: 10.1111/j.1478-3231.2009.02103.x. [DOI] [PubMed] [Google Scholar]
- 161.Luo B, Tang L, Wang Z, Zhang J, Ling Y, Feng W, Sun JZ, Stockard CR, Frost AR, Chen YF, et al. Cholangiocyte endothelin 1 and transforming growth factor beta1 production in rat experimental hepatopulmonary syndrome. Gastroenterology. 2005;129:682–695. doi: 10.1016/j.gastro.2005.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Machicao VI, Fallon MB. Hepatopulmonary syndrome. Semin Respir Crit Care Med. 2012;33:11–16. doi: 10.1055/s-0032-1301730. [DOI] [PubMed] [Google Scholar]
- 163.Zhang J, Fallon MB. Hepatopulmonary syndrome: update on pathogenesis and clinical features. Nat Rev Gastroenterol Hepatol. 2012;9:539–549. doi: 10.1038/nrgastro.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Grace JA, Burrell LM, Patel SK. Angiotensin-converting enzyme 2 polymorphisms and cardiovascular risk. Intern Med J. 2012;42:1167. doi: 10.1111/j.1445-5994.2012.02909.x. [DOI] [PubMed] [Google Scholar]
- 165.Krishnamurthy GT, Krishnamurthy S. Nuclear Hepatology. a Textbook of Hepatobiliary Diseases. Berlin: Springer; 2000. [Google Scholar]
- 166.Schwartz JM, Beymer C, Althaus SJ, Larson AM, Zaman A, Glickerman DJ, Kowdley KV. Cardiopulmonary consequences of transjugular intrahepatic portosystemic shunts: role of increased pulmonary artery pressure. J Clin Gastroenterol. 2004;38:590–594. doi: 10.1097/00004836-200408000-00010. [DOI] [PubMed] [Google Scholar]
- 167.Boyer TD, Haskal ZJ. The Role of Transjugular Intrahepatic Portosystemic Shunt (TIPS) in the Management of Portal Hypertension: update 2009. Hepatology. 2010;51:306. doi: 10.1002/hep.23383. [DOI] [PubMed] [Google Scholar]
- 168.Murray KF, Carithers RL. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology. 2005;41:1407–1432. doi: 10.1002/hep.20704. [DOI] [PubMed] [Google Scholar]
- 169.Iyer VN, Swanson KL, Cartin-Ceba R, Dierkhising RA, Rosen CB, Heimbach JK, Wiesner RH, Krowka MJ. Hepatopulmonary syndrome: favorable outcomes in the MELD exception era. Hepatology. 2013;57:2427–2435. doi: 10.1002/hep.26070. [DOI] [PubMed] [Google Scholar]
- 170.Katsuta Y, Zhang XJ, Kato Y, Shimizu S, Komeichi K, Ohsuga M, Higashi H, Satomura K, Takano T. Hemodynamic features and impaired arterial oxygenation in patients with portopulmonary hypertension. Hepatol Res. 2005;32:79–88. doi: 10.1016/j.hepres.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 171.Kawut SM, Taichman DB, Ahya VN, Kaplan S, Archer-Chicko CL, Kimmel SE, Palevsky HI. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl. 2005;11:1107–1111. doi: 10.1002/lt.20459. [DOI] [PubMed] [Google Scholar]
- 172.Battistini B, Dussault P. Biosynthesis, distribution and metabolism of endothelins in the pulmonary system. Pulm Pharmacol Ther. 1998;11:79–88. doi: 10.1006/pupt.1998.0148. [DOI] [PubMed] [Google Scholar]
- 173.Lüscher TF. Endothelin: systemic arterial and pulmonary effects of a new peptide with potent biologic properties. Am Rev Respir Dis. 1992;146:S56–S60. doi: 10.1164/ajrccm/146.5_Pt_2.S56. [DOI] [PubMed] [Google Scholar]
- 174.Luo B, Liu L, Tang L, Zhang J, Stockard CR, Grizzle WE, Fallon MB. Increased pulmonary vascular endothelin B receptor expression and responsiveness to endothelin-1 in cirrhotic and portal hypertensive rats: a potential mechanism in experimental hepatopulmonary syndrome. J Hepatol. 2003;38:556–563. doi: 10.1016/s0168-8278(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 175.Giusca S, Jinga M, Jurcut C, Jurcut R, Serban M, Ginghina C. Portopulmonary hypertension: from diagnosis to treatment. Eur J Intern Med. 2011;22:441–447. doi: 10.1016/j.ejim.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 176.Krowka MJ, Cartin-Ceba R. Portopulmonary hypertension: formidable dual threat versus hopeful dual therapy. Liver Transpl. 2014;20:635–636. doi: 10.1002/lt.23885. [DOI] [PubMed] [Google Scholar]
- 177.Krowka MJ. Portopulmonary hypertension and the issue of survival. Liver Transpl. 2005;11:1026–1027. doi: 10.1002/lt.20494. [DOI] [PubMed] [Google Scholar]
- 178.Kuo PC, Plotkin JS, Johnson LB, Howell CD, Laurin JM, Bartlett ST, Rubin LJ. Distinctive clinical features of portopulmonary hypertension. Chest. 1997;112:980–986. doi: 10.1378/chest.112.4.980. [DOI] [PubMed] [Google Scholar]
- 179.Swanson KL, Krowka MJ. Screen for portopulmonary hypertension, especially in liver transplant candidates. Cleve Clin J Med. 2008;75:121–12, 125-30, 133 passim. doi: 10.3949/ccjm.75.2.121. [DOI] [PubMed] [Google Scholar]
- 180.Hoeper MM, Seyfarth HJ, Hoeffken G, Wirtz H, Spiekerkoetter E, Pletz MW, Welte T, Halank M. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J. 2007;30:1096–1102. doi: 10.1183/09031936.00032407. [DOI] [PubMed] [Google Scholar]
- 181.Grander W, Eller P, Fuschelberger R, Tilg H. Bosentan treatment of portopulmonary hypertension related to liver cirrhosis owing to hepatitis C. Eur J Clin Invest. 2006;36 Suppl 3:67–70. doi: 10.1111/j.1365-2362.2006.01687.x. [DOI] [PubMed] [Google Scholar]
- 182.Hollatz TJ, Musat A, Westphal S, Decker C, D’Alessandro AM, Keevil J, Zhanhai L, Runo JR. Treatment with sildenafil and treprostinil allows successful liver transplantation of patients with moderate to severe portopulmonary hypertension. Liver Transpl. 2012;18:686–695. doi: 10.1002/lt.23407. [DOI] [PubMed] [Google Scholar]
- 183.Rodríquez-Roisin R, Krowka MJ, Hervé P, Fallon MB. Highlights of the ERS Task Force on pulmonary-hepatic vascular disorders (PHD) J Hepatol. 2005;42:924–927. doi: 10.1016/j.jhep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 184.Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: implications for liver transplantation. Clin Chest Med. 2005;26:587–97, vi. doi: 10.1016/j.ccm.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 185.Garcia-Tsao G, Parikh CR, Viola A. Acute kidney injury in cirrhosis. Hepatology. 2008;48:2064–2077. doi: 10.1002/hep.22605. [DOI] [PubMed] [Google Scholar]
- 186.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fede G, D’Amico G, Arvaniti V, Tsochatzis E, Germani G, Georgiadis D, Morabito A, Burroughs AK. Renal failure and cirrhosis: a systematic review of mortality and prognosis. J Hepatol. 2012;56:810–818. doi: 10.1016/j.jhep.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 188.Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009;361:1279–1290. doi: 10.1056/NEJMra0809139. [DOI] [PubMed] [Google Scholar]
- 189.Stremitzer S, Tamandl D, Kaczirek K, Maresch J, Abbasov B, Payer BA, Ferlitsch A, Gruenberger T. Value of hepatic venous pressure gradient measurement before liver resection for hepatocellular carcinoma. Br J Surg. 2011;98:1752–1758. doi: 10.1002/bjs.7672. [DOI] [PubMed] [Google Scholar]
- 190.Salerno F, Gerbes A, Ginès P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Postgrad Med J. 2008;84:662–670. doi: 10.1136/gut.2006.107789. [DOI] [PubMed] [Google Scholar]
- 191.Wong F, Nadim MK, Kellum JA, Salerno F, Bellomo R, Gerbes A, Angeli P, Moreau R, Davenport A, Jalan R, et al. Working Party proposal for a revised classification system of renal dysfunction in patients with cirrhosis. Gut. 2011;60:702–709. doi: 10.1136/gut.2010.236133. [DOI] [PubMed] [Google Scholar]
- 192.Angeli P, Sanyal A, Moller S, Alessandria C, Gadano A, Kim R, Sarin SK, Bernardi M. Current limits and future challenges in the management of renal dysfunction in patients with cirrhosis: report from the International Club of Ascites. Liver Int. 2013;33:16–23. doi: 10.1111/j.1478-3231.2012.02807.x. [DOI] [PubMed] [Google Scholar]
- 193.Stadlbauer V, Wright GA, Banaji M, Mukhopadhya A, Mookerjee RP, Moore K, Jalan R. Relationship between activation of the sympathetic nervous system and renal blood flow autoregulation in cirrhosis. Gastroenterology. 2008;134:111–119. doi: 10.1053/j.gastro.2007.10.055. [DOI] [PubMed] [Google Scholar]
- 194.Møller S, Krag A. Cardiorenal syndrome - A new entity? In: Gerbes A, editor. Hyponatremia and hepatorenal syndrome: Progress in treatment. Basel: Karger; 2011. pp. 102–111. [Google Scholar]
- 195.Møller S, Iversen JS, Henriksen JH, Bendtsen F. Reduced baroreflex sensitivity in alcoholic cirrhosis: relations to hemodynamics and humoral systems. Am J Physiol Heart Circ Physiol. 2007;292:H2966–H2972. doi: 10.1152/ajpheart.01227.2006. [DOI] [PubMed] [Google Scholar]
- 196.Schmidt LE, Ring-Larsen H. Vasoconstrictor therapy for hepatorenal syndrome in liver cirrhosis. Curr Pharm Des. 2006;12:4637–4647. doi: 10.2174/138161206779010413. [DOI] [PubMed] [Google Scholar]
- 197.Henriksen JH, Fuglsang S, Bendtsen F. Fourier analysis of arterial pulse in patients with advanced cirrhosis indicates reduced wave reflections that may protect against manifest cardiac dysfunction. Gut. 2012;61:1239–1240. doi: 10.1136/gutjnl-2011-301598. [DOI] [PubMed] [Google Scholar]
- 198.Schrier RW. Decreased effective blood volume in edematous disorders: what does this mean? J Am Soc Nephrol. 2007;18:2028–2031. doi: 10.1681/ASN.2006111302. [DOI] [PubMed] [Google Scholar]
- 199.Krag A, Bendtsen F, Burroughs AK, Møller S. The cardiorenal link in advanced cirrhosis. Med Hypotheses. 2012;79:53–55. doi: 10.1016/j.mehy.2012.03.032. [DOI] [PubMed] [Google Scholar]
- 200.Theocharidou E, Krag A, Bendtsen F, Møller S, Burroughs AK. Cardiac dysfunction in cirrhosis - does adrenal function play a role? A hypothesis. Liver Int. 2012;32:1327–1332. doi: 10.1111/j.1478-3231.2011.02751.x. [DOI] [PubMed] [Google Scholar]
- 201.Galbois A, Rudler M, Massard J, Fulla Y, Bennani A, Bonnefont-Rousselot D, Thibault V, Reignier S, Bourrier A, Poynard T, et al. Assessment of adrenal function in cirrhotic patients: salivary cortisol should be preferred. J Hepatol. 2010;52:839–845. doi: 10.1016/j.jhep.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 202.Trifan A, Chiriac S, Stanciu C. Update on adrenal insufficiency in patients with liver cirrhosis. World J Gastroenterol. 2013;19:445–456. doi: 10.3748/wjg.v19.i4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Acevedo J, Fernández J, Prado V, Silva A, Castro M, Pavesi M, Roca D, Jimenez W, Ginès P, Arroyo V. Relative adrenal insufficiency in decompensated cirrhosis: Relationship to short-term risk of severe sepsis, hepatorenal syndrome, and death. Hepatology. 2013;58:1757–1765. doi: 10.1002/hep.26535. [DOI] [PubMed] [Google Scholar]
- 204.Jalan R, Gines P, Olson JC, Mookerjee RP, Moreau R, Garcia-Tsao G, Arroyo V, Kamath PS. Acute-on chronic liver failure. J Hepatol. 2012;57:1336–1348. doi: 10.1016/j.jhep.2012.06.026. [DOI] [PubMed] [Google Scholar]