

Abstract
Introduction
Metastatic brain tumours remain an intractable clinical problem despite notable advances in the treatment of the primary cancers. It is estimated that 30–40% of breast and lung cancer patients will develop brain metastases. Typically, brain lesions are not diagnosed until patients exhibit neurological symptoms because there are currently no tests that can predict which patients will be afflicted. Brain metastases are resistant to current chemotherapies, and despite surgical resection and radiotherapy, the prognosis for these patients remains very poor with an average survival of only 6–9 months. Cancer is ultimately a genetic disease, involving patient genetics and aberrant tumour genomics; therefore the pursuit of an explanation for why or how brain metastases occur requires investigation of the associated somatic mutations.
In this article, we review the current literature surrounding the molecular and genome-based mechanistic evidence to indicate driver oncogenes that hold potential biomarkers for risk, or therapeutic targets for treatment of brain metastases.
Conclusion
Patients afflicted with metastatic brain tumours are in dire need of more effective therapies, and clinicians need predictive laboratory tests to identify patients at risk of developing metastatic brain tumours. The as yet unrealized comprehensive analysis of metastatic brain tumour genomics is necessary to meet these needs. Moreover, without improved understanding of the genomic aberrations that drive metastatic brain tumours, development of biomarkers and molecularly targeted therapies will remain stalled and patient outcomes will continue to be dismal.
Introduction
Incidence and current therapeutic options
There are approximately 200,000 newly diagnosed cases of brain metastases annually in the United States, 10-fold greater than primary brain cancer1,2. Current estimates indicate that up to 40% of patients with a systemic primary cancer will develop a metastatic brain tumour (MBT)3. A majority of brain metastases come from lung cancer (50–60%); secondarily from breast cancer (20–30%); third melanoma (5–10%) and various cancers, including gastrointestinal, oesophageal, prostate and ovarian that combined contribute 5–10%3–14 (summarized in Figure 1). Improved treatments for primary cancers are allowing patients to live longer, leading to increased incidence of MBTs4,15,16.
Figure 1.

Breakdown of brain metastatic tumors according to originating cancer.
Brain metastases are among the most feared complications for cancer patients because of diminished quality of life and a lack of effective chemotherapies. There are no predictive biomarkers to identify patients at risk to develop brain metastasis, and the healthcare system cannot bear the burden to screen all cancer patients for early detection of MBTs. Metastatic lesions are discovered when a patient presents with neurological symptoms; then, if left untreated, median survival is 1 month. The standard course of treatment for patients with controlled systemic disease is microsurgical resection plus radiation (whole-brain radiation therapy and/or stereotactic radio-surgery), extending median survival to 9–10 months17,18. Patients receiving radiation alone have a median survival of 4–6 months17,18. Chemotherapeutic agents are not effective, partly attributed to their limited ability to penetrate the blood–brain barrier (BBB)19,20. Although new therapies are in development to penetrate the BBB21,22, recent evidence also suggests that the BBB may not be intact in patients with brain tumours. Promising preliminary studies indicate that molecularly targeted therapies (small-molecule kinase inhibitors against activated oncogenes) can cross the BBB23,24 and correlate with modest improved outcomes25–28. Selected independent case studies report marked tumour regression and sustained response29–31.
The vast majority of MBTs develop from primary lung and breast cancers. The frequency at which lung and breast cancer patients develop brain metastases is approximately 25% and approximately 30%, respectively, but differs more specifically according to the histology or molecular subtypes of the systemic disease (described in detail in Figures 2 and 3)9,32–40. Stephen Paget first posited that the non random pattern of metastasis linked to specific primary cancers was not due to chance and that formation of metastatic tumours depends on interactions between the microenvironment of the metastatic site and the metastatic cancer cells41–43. Herein, we focus our review on this broadly accepted concept and what is known about the stages of metastasis as they relate to the understanding that cancer is ultimately a genetic disease with pathological phenotypes driven by oncogenic, somatic mutations.
Figure 2.

Frequency of occurrence for metastatic brain tumors according to breast cancer molecular subtype.
Figure 3.

Frequency of occurrence for metastatic brain tumors according to lung cancer subtype.
Discussion
Stages in the metastatic process
Metastatic biological processes, including the role of microenvironment and the ability of cancer cells to transverse the BBB, have been extensively reviewed in great detail elsewhere44–46. Briefly, the stages of metastasis comprise, in temporal order of occurrence, local invasion, survival in the circulation, intravasation, extravasation, micrometastasis formation and metastatic colonization. Invasion depends upon an epithelial to mesenchymal transition (EMT) where epithelial cell traits are suppressed and mesenchyme cell traits are activated, losing cell polarity and cell–cell adhesion. Underlying EMT is a gene expression program regulated by epigenetic changes across the genome and aberrant expression of key transcription factors and micro-RNAs45,47,48. New evidence describes further complexity, where a reversal of the EMT process is required for colonized metastatic cells to proliferate49,50. This potential phenotypic plasticity is consistent with epigenetic principles inherent to the hierarchically organized cancer stem cell model, where cancer stem cells undergo irreversible epigenetic changes to produce phenotypically diverse cancer cells51. Additional work does suggest that with the EMT process, cancer cells also acquire stem cell-like traits52. The resulting ‘cancer stem cells’ represent a minor subset of cells within a tumour. They maintain the capability for unlimited self-renewal, have the ability to seed new tumours and are a potential cell type from the primary tumour that initiates a metastatic lesion52.
Relatively large numbers of circulating tumour cells can be detected in the blood of carcinoma patients, including those who do not clinically develop distant metastases53. Of those numbers of circulating cells, approximately 80% undergo extravasation. However, once implanted at distant sites, fewer than 3% of cancer cells survive to form micrometastases and fewer than 0.1% persist to metastatic macro-colonization54,55. Macro-colonization is described as the rate-limiting step in metastatic tumour formation45. A related mechanistic hypothesis, consistent with the clinical observation of long latency periods for distant relapse following initial diagnosis56, posits that during the latent period, cancer cells, and likely also the surrounding microenvironment57, undergo gradual evolution to acquire molecular aberrations advantageous for metastatic colonization either because micrometastatic cell growth and death are equal or by discontinuous growth and periods of quiescence45,58. However, for brain metastasis from lung and breast cancers, the latency period is relatively short. The median latency from breast cancer diagnosis to MBTs is 34 months (with HER2 positive developing more quickly) and lung cancer-derived MBTs are detected within months of the initial diagnosis9,59–61. This suggests a different course for MBTs in which only a subset of cells (possibly stem cells) in the primary tumour has the molecular prerequisites for all metastatic stages. Alternative to a stem cell hypothesis, selection pressures within progressing primary tumours give rise to clonal evolution of tumour cells with acquired genetic variations and increased malignant or metastatic potential51,62. In line with this hypothesis, metastatic potential and inherent resistance against selective pressures are coincident with heightened genetic instability63.
The unique environment of the brain compared to other organs presents an optimum environment for metastatic progenitor cells arising from particular tissues, fitting with the nonrandom pattern of metastasis. While tropism and anatomical limitations are also thought to be pre-determinants, strong evidence suggests that the dominant influence is the nature of compatibility between the primary cell of origin and the site of metastatic colonization. et al. established this with a rodent xenograft model of human metastatic melanoma in which the same number of micrometastases was identified in the parenchyma of lung and kidney tissues embedded side by side; but metastatic tumours only developed in the lung fragment and not in the kidney fragment43.
Details of the biology of brain metastases are still emerging, but it appears that multiple widely expressed mechanisms in cancer cells are able to support intravasation, survival in circulation, extravasation and micrometastasis. However, a more select oncogenic mechanism drives colonization and growth. Putatative oncogenic drivers of brain metastasis and whether they exist in the primary tumour or are acquired in disseminated cells are further addressed below.
Oncogenes and tumour suppressors
Decades of research have contributed to the understanding that carcinogenesis is governed by the activation of oncogenes and inactivation of tumour suppressor genes. Oncogenes are activated by various mechanisms including gain-of-function sequence mutations, overexpression due to genomic amplification or deregulated epigenetics and genomic rearrangements64,65. Tumour suppressors, factors that otherwise slow normal cell growth processes or promote tissue differentiation, are inactivated by genomic lesions resulting in loss-of-function. Thus, within the tumour genome, the causes for initiation and progression of the disease can be found. Important clinical advances in primary cancer treatments are attributed to molecularly targeted treatments tailored to target activated oncogenes, e.g. HER2, EGFR, MET and BRAF64. Their aberrant presentation in the primary tumour genome presents a biomarker to indicate the efficacy of the targeted treatment. Moreover, oncogenic factors often possess kinase or ligand-binding function that present ‘druggable’ protein motifs66. The benefits of targeted treatments over traditional chemotherapy are clear, resulting in greater efficacy and less debilitating side effects67–69.
The success of driver oncogene-targeted therapies against specific primary cancers can also be attributed to the fact that oncogene-directed signalling promotes the many phenotypes that define the pathobiology of cancer cells including unrestricted growth, immune system evasion, metabolic transformation, invasive potential and cancer cell tumourigenicity (local and metastatic)70,71. This wide array of biological impacts suggests that somatic genome mutations effecting activated oncogenes and inactivated tumour suppressors are likely causally implicated in all stages of MBT development.
Genomic mutation landscape of metastatic brain tumours
Early studies indicate that molecularly targeted therapies in the form of small-molecule kinase inhibitors and monoclonal antibodies against activated oncogenes can elicit a response in the treatment of metastatic tumours and potentially reduce the rates of metastatic recurrence25–30,72,73. On the other hand, increased incidence of MBTs in patients with controlled primary cancer and peripheral metastasis has also been attributed to the inefficiency of targeted drug transfer across the BBB16. Although further investigation is required, each of these data indicates that an oncogene mechanistically linked to the primary cancer is also implicated in driving the incidence of MBTs. This scenario may apply to the mutated expression of BRAF oncogene in metastases from melanoma27, aberrant levels of HER2 oncogene in breast cancer brain metastases30 and mutated EGFR oncogene in lung cancer brain metastases28.
The potential importance of BRAF, HER2 and EGFR for brain metastasis was identified based on knowledge of their driver oncogene status and frequency of mutation in the related primary cancer. Their specific oncogenic role and frequency of mutation in MBTs remain to be ascertained. Additionally, it is unclear if for a given oncogene certain types of activating mutations are more often represented in brain metastases, as early evidence suggests74,75. Studying these unknowns will explain why brain metastases develop in cases where systemic disease is treated with targeted therapies and why treating MBTs has had only modest effects and no impact on therapy options. An important starting point is comprehensive genomic characterization of MBTs compared to their primary cancers, particularly the well-characterized molecular breast and lung cancers subtypes. Despite the large number of patients afflicted, the characterization of significant cancer-linked genomic copy number aberrations, rearrangements and sequence mutations in MBTs is only in its infancy.
Gene coding mutations
Most studies to date that have examined somatic DNA coding mutations in MBTs have been biased towards the activating mutations in oncogenes or tumour suppressors previously associated with the primary cancer. A limited study that included DNA from 10 melanoma metastases measured a panel of BRAF, NRAS, AKT, PIK3CA and KIT activating mutations using a highly sensitive primer extension, and mass spectrometry approach showed that BRAF activating mutations were the most frequent and secondarily NRAS, similar to primary melanoma76. A later study using Sanger sequencing identified BRAF and NRAS mutations in both primary melanoma and matched brain metastases from 44 patients at 80% consistency77. Another study measured mutations in 19 oncogenes, including BRAF, KRAS, NRAS and PIK3CA, by high-resolution DNA melting analysis of specimens from matched brain metastases and primary colorectal cancer. The study recapitulated that KRAS mutations were a more common event in colorectal cancer and that 9 of 10 matched brain metastases were 100% concordant with mutations observed in the primary cancer78. A breast cancer study that analysed 39 matched pairs of primary breast cancers and brain metastases using primer extension mass spectrometry investigated mutations in EGFR, HRAS, KRAS, NRAS and PIK3CA79. NRAS and PIK3CA mutations were identified independently in samples from two patients, in both the primary and metastatic specimens. An EGFR mutation was identified in breast cancer from a single patient, but not in the matched metastasis79.
From studies of Japanese populations, the frequencies of EGFR mutations in MBTs from non-small cell lung cancer (NSCLC) are similar to those reported for the primary tumour in the same population80,81. Results from studies investigating EGFR mutations in NSCLC-derived MBTs from Caucasian patients indicate that the mutation frequency in EGFR is ≤2%82–84, markedly less frequent than the estimated 10% in NSCLC primary tumours85. However, where a mutation was discovered in the metastasis, it was concordant with a mutation in the primary cancer. These results indicate that coding mutations in the investigated oncogenes are either equivalent or less represented in the MBT compared to the primary tumour, although the body of work is inadequate to draw any conclusions with certainty. However, evidence suggests that the incidence of mutations in tumour suppressor genes may increase in metastatic cancer86.
Whether or not an implicated driver oncogene is discovered in both the primary and metastatic cancer or solely in the brain metastasis is an important distinction that provides rationale for adapting targeted treatments, or developing predictive biomarkers for MBTs. Ding et al. directly investigated whether processes leading to macrometastases are driven by mutations that pre-exist in primary tumour cells or arise at the distant site using massively parallel DNA sequencing methods to analyse three related specimens: a primary triple negative basal-like breast cancer, the tumour derived from a mouse xenograft model and the matched MBT that developed within 8 months of primary diagnosis75. For coding mutations, 48 of 50 validated somatic gene mutations were detected in DNA isolated from all three tumour samples, supporting the notion that coding mutations are consistent for primary and metastatic cancer. The mutation allele frequency for 26 of those was significantly increased in the metastases or xenograft specimens, and the allelic frequency of two mutations was decreased. The observed levels of enrichment and loss of enrichment (but still detectable) for specific coding mutations in the metastasis and xenograft specimens may indicate that a subset of primary tumour cells carrying the array of mutations in the primary, not just a single clone, were contributing75,87. Unlike coding mutations, copy number aberrations were gained in the metastasis and xenograft specimens, such that 19% and 39%, respectively, were unique compared to the primary cancer75. Comparable work also using massively parallel sequencing to investigate an oestrogen receptor alpha-positive breast tumour arrived at a differing conclusion. Here, the metastasis was detected 9 years after the initial diagnosis88. Of the 32 non-synonymous coding mutations detected in the metastatic cancer, 19 were absent in the primary tumour, 5 were prevalent in the primary tumour and 6 were present at lower allelic frequency levels than the primary tumour88. The list of mutated genes identified in these studies of individuals has little translational value because a substantial number of specimens are required to elucidate specific, recurring causally implicated gene mutations. However, these two studies intimate highly valuable insights surrounding the potential for sequence and structural mutations and the potential for varied mechanisms driving metastatic cancer associated with short versus long latency.
Somatic copy number variations
The observation made by Ding et al. that the incidence of copy number alterations is increased in metastasis and xenograft specimens75 lends itself to the re-interpretation of other studies. For example, low-resolution arrayed, comparative genomic hybridization (aCGH) studies have highlighted recurring, broad copy number variation in primary cancers that develop brain metastases, positing that the specific loci represent risk markers of functional consequence89,90. Further validation is required as to whether the broad regions harbour true driver lesions, or if they represent greater genomic instability underlying other structural mutations with a role in metastasis. Nevertheless, studies do show significant frequencies of copy number aberrations in specific known cancer genes in brain metastases. Two studies of breast cancer brain metastasis suggest that gains in EGFR oncogene and loss of PTEN tumour suppressor are more frequent in metastases, one using aCGH methods and the other fluorescent in situ hybridization (FISH)91,92. Two additional studies reporting on FISH analysis of lung cancer brain metastases suggest a marginally higher frequency of EGFR copy number gain (by amplification or polysomy) in brain metastases compared to the matched primary cancer82,83.
Markedly high rates of brain metastases are observed for patients with HER2-amplified breast cancers93. Indications are that the frequency of HER2-positive status is increased in brain metastases over other peripheral metastases, although this may be confounded by the use of HER2-targeted treatments that may be more effective in the treatment of peripheral metastases and restricted in reaching brain tumours16,92. Additional work by Palmieri et al. that analysed HER2 expression in 124 specimens reinforces that the rate of HER2 overexpression largely associated with gene amplification is 36% in brain metastases, a ≥10% increase over rates of amplification and overexpression reported in primary breast cancers94,95. The functional consequence of HER2 amplification and overexpression was demonstrated by this same group using a human cancer cell line model of forced HER2 overexpression, which when introduced into mice yielded threefold greater numbers of brain macrometastases compared to the parent cell line xenografts95.
Whole-genome expression studies
The variety of possible structural and coding mutations and epigenetic deregulation establish the foundation for aberrant gene expression that is the biomolecular link from the aberrant genome to transformed cancer cell function. Whole-genome and transcriptome studies of primary cancers are used to identify genes or transcripts displaying significant cancer-specific expression and are especially useful to elucidate cancer cell-specific systems and pathwaylevel dysfunction. Additionally, diagnostics based on gene expression signatures have proven effective at predicting lymph node metastases and improving patient outcomes96. Limited recent gene expression data also suggest mechanisms underlying brain metastasis. A study from Da Silva et al. compared 15 matched primary triple negative, basal breast cancers and their matched brain metastatic cancers using microarrays that simultaneously analysed 512 cancer-linked genes. Twenty-seven genes differed significantly, notably HIF1A and HER3, which were markedly upregulated79. This suggests a role for the epidermal growth factor receptor family in even the triple negative breast cancer subtype. Also, the HIF1A transcription factor activates genes encoding glucose transporters and glycolytic pathway genes, and an important role for HIF1A is supported by independent work showing more frequent overexpression in patient brain metastases compared to primary breast cancer tissues and enhanced aerobic glycolysis in cells migrating to or implanted in the brain versus parent cells in mouse models of human cancer cell line xenografts97,98. Multiple lines of whole-genome investigation in cell lines and patient specimens by Bos et al. identified upregulated expression of an EGFR ligand HBEGF and cyclooxygenase PTGS2 and the ectopic expression of the brain-specific gene α2,6-sialyltransferase ST6GALNAC5 in breast cancer cells as factors contributing to their ability to cross the BBB99. Increased PTGS2 expression is also identified as an important factor in brain metastasizing melanoma100.
While the described studies highlight what is unique to the metastatic cancer and do not address when or how the features arise, multiple studies present evidence for many, potentially coexisting possibilities. Studies combining analysis of methylation and gene expression to elucidate recurring features in breast cancer specimens associated with the incidence of brain metastasis do suggest pre-existing features supportive of metastasis, particularly related to genes implicated in EMT101–103. Analysis of substantial numbers of primary and metastatic breast tumours using whole-genome expression assays was successful in identifying gene expression signatures preserved in both primary and metastatic cancers104,105, suggesting that the bulk of tumour cells in the primary lesion and the metastasis are similar. Work by Park et al. following a xenograft model of metastases presents specific evidence that metastatic breast cancer cells acquire expression patterns found in neuronal cells, thus indicating that an evolutionary process underlies MBT development106.
Conclusion
Outlook for developing novel therapeutic strategies
The current lack of insight into brain metastases spawns many questions (depicted graphically in Figure 4), including whether or not the primary cancer drivers are also the drivers of the related MBT. Conversely, early data suggests that unique drivers may be responsible for MBT development, raising additional key questions. Does a new driver/s emerge with the gain of mutations at the metastatic site? Or alternatively, do cells derived from a minor subclone in the original tumour harbour lesions with the capacity to evade primary adjuvant therapy and seed the metastatic occurrence. Much research is still needed to fill the void and answer these critical questions.
Figure 4.
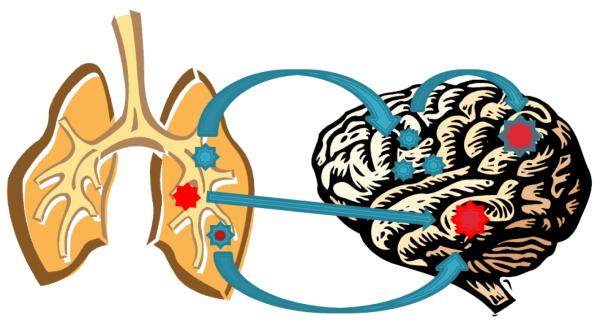
Potential pathways from cancer cell somatic mutations to metastatic colonization and metastatic tumor growth. Upper arrows in sequence depict how disseminated cancer cells may acquire further metastatic growth promoting mutations. Middle and lower arrows depict how cells from a major or minor subclone fraction in the original tumor may harbor mutations thatspecifically support metastatic tumor growth.
Patients afflicted with MBTs are in dire need of more effective therapies, and clinicians need predictive laboratory tests to identify patients at risk of developing MBTs. The as yet unrealized comprehensive analysis of MBT genomics is necessary to meet these needs. Moreover, without improved understanding of the genomic aberrations that drive MBTs, development of biomarkers and molecularly targeted therapies will remain stalled and patient outcomes will continue to be dismal.
Abbreviations list
- aCGH
arrayed, comparative genomic hybridization
- BBB
blood–brain barrier
- EMT
epithelial to mesenchymal transition
- FISH
fluorescent in situ hybridization
- MBT
metastatic brain tumour
- NSCLC
non-small cell lung cancer
References
- 1.Gavrilovic IT, Posner JB. Brain metastases: epidemiology and pathophysiology. J Neurooncol. 2005 Oct;75(1):5–14. doi: 10.1007/s11060-004-8093-6. [DOI] [PubMed] [Google Scholar]
- 2.Patchell RA. The management of brain metastases. Cancer Treat Rev. 2003 Dec;29(6):533–40. doi: 10.1016/s0305-7372(03)00105-1. [DOI] [PubMed] [Google Scholar]
- 3.Nathoo N, Chahlavi A, Barnett GH, Toms SA. Pathobiology of brain metastases. J Clin Pathol. 2005 Mar;58(3):237–42. doi: 10.1136/jcp.2003.013623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Norden AD, Wen PY, Kesari S. Brain metastases. Current Opin Neurol. 2005 Dec;18(6):654–61. doi: 10.1097/01.wco.0000191514.37498.2b. [DOI] [PubMed] [Google Scholar]
- 5.Wen PY, Loeffler JS. Brain metastases. Current Treat Options Oncol. 2000 Dec;1(5):447–58. doi: 10.1007/s11864-000-0072-3. [DOI] [PubMed] [Google Scholar]
- 6.Shi AA, Digumarthy SR, Temel JS, Halpern EF, Kuester LB, Aquino SL. Does initial staging or tumor histology better identify asymptomatic brain metastases in patients with non-small cell lung cancer? J Thorac Oncol. 2006 Mar;1(3):205–10. doi: 10.1016/s1556-0864(15)31569-0. [DOI] [PubMed] [Google Scholar]
- 7.Caffo O, Veccia A, Fellin G, Mussari S, Russo L, Tomio L, et al. Frequency of brain metastases from prostate cancer: an 18-year single-institution experience. J Neurooncol. 2013 Jan;111(2):163–7. doi: 10.1007/s11060-012-0994-1. [DOI] [PubMed] [Google Scholar]
- 8.Niwinska A, Murawska M, Pogoda K. Breast cancer subtypes and response to systemic treatment after whole-brain radiotherapy in patients with brain metastases. Cancer. 2010 Sep;116(18):4238–47. doi: 10.1002/cncr.25391. [DOI] [PubMed] [Google Scholar]
- 9.Chang EL, Lo S. Diagnosis and management of central nervous system metastases from breast cancer. Oncologist. 2003;8(5):398–410. doi: 10.1634/theoncologist.8-5-398. [DOI] [PubMed] [Google Scholar]
- 10.Weinberg JS, Suki D, Hanbali F, Cohen ZR, Lenzi R, Sawaya R. Metastasis of esophageal carcinoma to the brain. Cancer. 2003;98(9):1925–33. doi: 10.1002/cncr.11737. [DOI] [PubMed] [Google Scholar]
- 11.York JE, Stringer J, Ajani JA, Wildrick DM, Gokaslan ZL. Gastric cancer and metastasis to the brain. Ann Surg Oncol. 1999 Dec;6(8):771–6. doi: 10.1007/s10434-999-0771-3. [DOI] [PubMed] [Google Scholar]
- 12.Go PH, Klaassen Z, Meadows MC, Chamberlain RS. Gastrointestinal cancer and brain metastasis: a rare and ominous sign. Cancer. 2011 Aug;117(16):3630–40. doi: 10.1002/cncr.25940. [DOI] [PubMed] [Google Scholar]
- 13.Tremont-Lukats IW, Bobustuc G, Lagos GK, Lolas K, Kyritsis AP, Puduvalli VK. Brain metastasis from prostate carcinoma: The M. D. Anderson Cancer Center experience. Cancer. 2003 Jul;98(2):363–8. doi: 10.1002/cncr.11522. [DOI] [PubMed] [Google Scholar]
- 14.Cohen ZR, Suki D, Weinberg JS, Marmor E, Lang FF, Gershenson DM, et al. Brain metastases in patients with ovarian carcinoma: prognostic factors and outcome. J Neurooncol. 2004 Feb;66(3):313–25. doi: 10.1023/b:neon.0000014516.04943.38. [DOI] [PubMed] [Google Scholar]
- 15.Caffo O, Veccia A, Russo L, Galligioni E. Brain metastases from prostate cancer: an emerging clinical problem with implications for the future therapeutic scenario. Future Oncol. 2012 Dec;8:1585–95. doi: 10.2217/fon.12.156. [DOI] [PubMed] [Google Scholar]
- 16.Burstein HJ, Lieberman G, Slamon DJ, Winer EP, Klein P. Isolated central nervous system metastases in patients with HER2-overexpressing advanced breast cancer treated with first-line trastuzumab-based therapy. Ann Oncol. 2005 Nov;16(11):1772–7. doi: 10.1093/annonc/mdi371. [DOI] [PubMed] [Google Scholar]
- 17.Patchell RA, Tibbs PA, Walsh JW, Dempsey RJ, Maruyama Y, Kryscio RJ, et al. A randomized trial of surgery in the treatment of single metastases to the brain. N Engl J Med. 1990 Feb;322(8):494–500. doi: 10.1056/NEJM199002223220802. [DOI] [PubMed] [Google Scholar]
- 18.Vecht CJ, Haaxma-Reiche H, Noordijk EM, Padberg GW, Voormolen JH, Hoekstra FH, et al. Treatment of single brain metastasis: radiotherapy alone or combined with neurosurgery? Ann Neurol. 1993 Jun;33:583–90. doi: 10.1002/ana.410330605. [DOI] [PubMed] [Google Scholar]
- 19.Greig NH, Soncrant TT, Shetty HU, Momma S, Smith QR, Rapoport SI. Brain uptake and anticancer activities of vincristine and vinblastine are restricted by their low cerebrovascular permeability and binding to plasma constituents in rat. Cancer Chemother Pharmacol. 1990;26(4):263–8. doi: 10.1007/BF02897227. [DOI] [PubMed] [Google Scholar]
- 20.Genka S, Deutsch J, Stahle PL, Shetty UH, John V, Robinson C, et al. Brain and plasma pharmacokinetics and anticancer activities of cyclophosphamide and phosphoramide mustard in the rat. Cancer Chemother Pharmacol. 1990;27(1):1–7. doi: 10.1007/BF00689268. [DOI] [PubMed] [Google Scholar]
- 21.Park EJ, Zhang YZ, Vykhodtseva N, McDannold N. Ultrasound-mediated blood-brain/blood-tumor barrier disruption improves outcomes with trastuzumab in a breast cancer brain metastasis model. J Control Release. 2012 Nov;163(3):277–84. doi: 10.1016/j.jconrel.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cote J, Bovenzi V, Savard M, Dubuc C, Fortier A, Neugebauer W, et al. Induction of selective blood-tumor barrier permeability and macromolecular transport by a biostable kinin B1 receptor agonist in a glioma rat model. PLoS One. 2012;7(5):e37485. doi: 10.1371/journal.pone.0037485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heimberger AB, Learn CA, Archer GE, McLendon RE, Chewning TA, Tuck FL, et al. Brain tumors in mice are susceptible to blockade of epidermal growth factor receptor (EGFR) with the oral, specific, EGFR-tyrosine kinase inhibitor ZD1839 (iressa) Clin Cancer Res. 2002 Nov;8(11):3496–502. [PubMed] [Google Scholar]
- 24.Gluck S, Castrellon A. Lapatinib plus capecitabine resolved human epidermal growth factor receptor 2-positive brain metastases. Am J Ther. 2009 Nov-Dec;16:585–90. doi: 10.1097/MJT.0b013e31818bee2b. [DOI] [PubMed] [Google Scholar]
- 25.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006 Dec;355(26):2733–43. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 26.Lin NU, Dieras V, Paul D, Lossignol D, Christodoulou C, Stemmler HJ, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res. 2009 Feb;15(4):1452–9. doi: 10.1158/1078-0432.CCR-08-1080. [DOI] [PubMed] [Google Scholar]
- 27.Narayana A, Mathew M, Tam M, Kannan R, Madden KM, Golfinos JG, et al. Vemurafenib and radiation therapy in melanoma brain metastases. J Neurooncol. 2013 Jul;113(3):411–6. doi: 10.1007/s11060-013-1127-1. [DOI] [PubMed] [Google Scholar]
- 28.Mok T, Yang JJ, Lam KC. Treating patients with EGFR-sensitizing mutations: first line or second line--is there a difference? J Clin Oncol. 2013 Mar;31(8):1081–8. doi: 10.1200/JCO.2012.43.0652. [DOI] [PubMed] [Google Scholar]
- 29.Abboud M, Saghir NS, Salame J, Geara FB. Complete response of brain metastases from breast cancer overexpressing Her-2/neu to radiation and concurrent Lapatinib and Capecitabine. Breast J. 2010 Dec;16(6):644–6. doi: 10.1111/j.1524-4741.2010.00980.x. [DOI] [PubMed] [Google Scholar]
- 30.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001 Mar;344(11):783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 31.Gounant V, Wislez M, Poulot V, Khalil A, Lavole A, Cadranel J, et al. Subsequent brain metastasis responses to epidermal growth factor receptor tyrosine kinase inhibitors in a patient with non-small-cell lung cancer. Lung Cancer. 2007 Dec;58(3):425–8. doi: 10.1016/j.lungcan.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Langer CJ, Mehta MP. Current management of brain metastases, with a focus on systemic options. Clin Oncol. 2005 Sep;23(25):6207–19. doi: 10.1200/JCO.2005.03.145. [DOI] [PubMed] [Google Scholar]
- 33.Cox JD, Scott CB, Byhardt RW, Emami B, Russell AH, et al. Addition of chemotherapy to radiation therapy alters failure patterns by cell type within non-small cell carcinoma of lung (NSCCL): analysis of radiation therapy oncology group (RTOG) trials. Int J Radiat Oncol Biol Phys. 1999 Feb;43(3):505–9. doi: 10.1016/s0360-3016(98)00429-5. [DOI] [PubMed] [Google Scholar]
- 34.Howlander NNA, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, et al. SEER Cancer Statistics Review, 1975–2008. National Cancer Institute; Bethesda, MD: 2011. [Google Scholar]
- 35.Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia Sk, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008 Mar 1;14(5):1368–7. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 36.Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010 Jul;28(20):3271–7. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 37.Pauletti G, Godolphin W, Press MF, Slamon DJ. Detection and quantitation of HER-2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene. 1996 Jul;13(1):63–72. [PubMed] [Google Scholar]
- 38.Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Brown PO, et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999 Sep;23(1):41–6. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- 39.Press MF, Sauter G, Bernstein L, Villalobos IE, Mirlacher M, Zhou JY, et al. Diagnostic evaluation of HER-2 as a molecular target: an assessment of accuracy and reproducibility of laboratory testing in large, prospective, randomized clinical trials. Clin Cancer Res. 2005 Sep;11(18):6598–607. doi: 10.1158/1078-0432.CCR-05-0636. [DOI] [PubMed] [Google Scholar]
- 40.Eroles P, Bosch A, Perez-Fidalgo JA, Lluch A. Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev. 2012 Oct;38(6):698–707. doi: 10.1016/j.ctrv.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 41.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989 Aug;8(2):98–101. [PubMed] [Google Scholar]
- 42.Ramakrishna R, Rostomily R. Seed, soil, and beyond: The basic biology of brain metastasis. Surg Neurol Int. 2013 May;4(Suppl 4):S256–64. doi: 10.4103/2152-7806.111303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res. 1980 Jul;40(7):2281–7. [PubMed] [Google Scholar]
- 44.Fidler IJ. The role of the organ microenvironment in brain metastasis. Semin Cancer Biol. 2011 Apr;21(2):107–12. doi: 10.1016/j.semcancer.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 45.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011 Oct;147(2):275–92. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang Y, Pantel K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell. 2013 May;23(5):573–81. doi: 10.1016/j.ccr.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011 Jul;18(8):867–74. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008 Apr;22(7):894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012 Dec;22:709–24. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 50.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012 Dec;22:725–36. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009 Sep;138(5):822–9. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 52.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008 May;133(4):704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007 Dec;450:1235–9. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998 Sep;153:865–73. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, et al. Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000 May;60(9):2541–6. [PubMed] [Google Scholar]
- 56.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007 Nov;7(11):834–46. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013 Jul;15(7):807–17. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demicheli R, Terenziani M, Bonadonna G. Estimate of tumor growth time for breast cancer local recurrences: rapid growth after wake-up? Breast Cancer Res Treat. 1998 Sep;51(12):133–7. doi: 10.1023/a:1005887422022. [DOI] [PubMed] [Google Scholar]
- 59.Feld R, Rubinstein LV, Weisenberger TH. Sites of recurrence in resected stage I non-small-cell lung cancer: a guide for future studies. J Clin Oncol. 1984 Dec;2(12):1352–8. doi: 10.1200/JCO.1984.2.12.1352. [DOI] [PubMed] [Google Scholar]
- 60.Hoffman PC, Mauer AM, Vokes EE. Lung cancer. Lancet. 2000 Feb;355(9202):479–85. doi: 10.1016/S0140-6736(00)82038-3. [DOI] [PubMed] [Google Scholar]
- 61.Bendell JC, Domchek SM, Burstein HJ, Harris L, Younger J, Kuter I, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer. 2003 Jun;97(12):2972–7. doi: 10.1002/cncr.11436. [DOI] [PubMed] [Google Scholar]
- 62.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976 Oct;194(4260):23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 63.Cifone MA, Fidler IJ. Increasing metastatic potential is associated with increasing genetic instability of clones isolated from murine neoplasms. Proc Natl Acad Sci U S A. 1981 Nov;78(11):6949–52. doi: 10.1073/pnas.78.11.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007 Dec;21:3214–31. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- 65.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006 Jan;439(7074):353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 66.Russ AP, Lampel S. The druggable genome: an update. Drug Discov Today. 2005 Dec;10(23–24):1607–10. doi: 10.1016/S1359-6446(05)03666-4. [DOI] [PubMed] [Google Scholar]
- 67.Blackwell KL, Burstein HJ, Storniolo AM, Rugo HS, Sledge G, Aktan G, et al. Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 Study. J Clin Oncol. 2012 Jul;30(21):2585–92. doi: 10.1200/JCO.2011.35.6725. [DOI] [PubMed] [Google Scholar]
- 68.Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol. 2011 May;8(7):426–33. doi: 10.1038/nrclinonc.2011.69. [DOI] [PubMed] [Google Scholar]
- 69.Jordan VC. Tamoxifen: catalyst for the change to targeted therapy. Eur J Cancer. 2008 Jan;44(1):30–8. doi: 10.1016/j.ejca.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 71.Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: implications for efficacy of adjuvant trastuzumab. Cancer Res. 2013 Mar;73(5):1635–46. doi: 10.1158/0008-5472.CAN-12-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gluck S, Castrellon A. Lapatinib plus capecitabine resolved human epidermal growth factor receptor 2-positive brain metastases. Am J Ther. 2009 Nov-Dec;16(6):585–90. doi: 10.1097/MJT.0b013e31818bee2b. [DOI] [PubMed] [Google Scholar]
- 73.Lin NU, Carey LA, Liu MC, Younger J, Come SE, Ewend M, et al. Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2008 Apr;26(12):1993–9. doi: 10.1200/JCO.2007.12.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.El-Osta H, Falchook G, Tsimberidou A, Hong D, Naing A, Kim K, et al. BRAF mutations in advanced cancers: clinical characteristics and outcomes. PLoS One. 2011;6(10):e25806. doi: 10.1371/journal.pone.0025806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010 Apr;464(7291):999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davies MA, Stemke-Hale K, Lin E, Tellez C, Deng W, Gopal YN, et al. Integrated molecular and clinical analysis of AKT activation in metastatic melanoma. Clin Cancer Res. 2009 Dec;15:7538–46. doi: 10.1158/1078-0432.CCR-09-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. 2012 Jul;30(20):2522–9. doi: 10.1200/JCO.2011.41.2452. [DOI] [PubMed] [Google Scholar]
- 78.Tie J, Lipton L, Desai J, Gibbs P, Jorissen RN, Christie M, et al. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin Cancer Res. 2011 Mar;17(5):1122–30. doi: 10.1158/1078-0432.CCR-10-1720. [DOI] [PubMed] [Google Scholar]
- 79.Da Silva L, Simpson PT, Smart CE, Cocciardi S, Waddell N, Lane A, et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. 2010;12(4):R46. doi: 10.1186/bcr2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matsumoto S, Takahashi K, Iwakawa R, Matsuno Y, Nakanishi Y, Kohno T, et al. Frequent EGFR mutations in brain metastases of lung adenocarcinoma. Int J Cancer. 2006 Sep;119:1491–4. doi: 10.1002/ijc.21940. [DOI] [PubMed] [Google Scholar]
- 81.Gow CH, Chang YL, Hsu YC, Tsai MF, Wu CT, Yu CJ, et al. Comparison of epidermal growth factor receptor mutations between primary and corresponding metastatic tumors in tyrosine kinase inhibitor-naive non-small-cell lung cancer. Ann Oncol. 2009 Apr;20(4):696–702. doi: 10.1093/annonc/mdn679. [DOI] [PubMed] [Google Scholar]
- 82.Daniele L, Cassoni P, Bacillo E, Cappia S, Righi L, Volante M, et al. Epidermal growth factor receptor gene in primary tumor and metastatic sites from nonsmall cell lung cancer. J Thorac Oncol. 2009 Jun;4(6):684–8. doi: 10.1097/JTO.0b013e3181a52359. [DOI] [PubMed] [Google Scholar]
- 83.Sun M, Behrens C, Feng L, Ozburn N, Tang X, Yin G, et al. HER family receptor abnormalities in lung cancer brain metastases and corresponding primary tumors. Clin Cancer Res. 2009 Aug;15(15):4829–37. doi: 10.1158/1078-0432.CCR-08-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cortot AB, Italiano A, Burel-Vandenbos F, Martel-Planche G, Hainaut P. KRAS mutation status in primary nonsmall cell lung cancer and matched metastases. Cancer. 2010 Jun;116(11):2682–7. doi: 10.1002/cncr.25014. [DOI] [PubMed] [Google Scholar]
- 85.Marchetti A, Martella C, Felicioni L, Barassi F, Salvatore S, Chella A, et al. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol. 2005 Feb;23(4):857–65. doi: 10.1200/JCO.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 86.Lo Nigro C, Vivenza D, Monteverde M, Lattanzio L, Gojis O, Garrone O, et al. High frequency of complex TP53 mutations in CNS metastases from breast cancer. Br J Cancer. 2012 Jan;106(2):397–404. doi: 10.1038/bjc.2011.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gray J. Cancer: Genomics of metastasis. Nature. 2010 Apr;464(7291):989–90. doi: 10.1038/464989a. [DOI] [PubMed] [Google Scholar]
- 88.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009 Oct;461(7265):809–13. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 89.Wrage M, Ruosaari S, Eijk PP, Kaifi JT, Hollmen J, Yekebas EF, et al. Genomic profiles associated with early micrometastasis in lung cancer: relevance of 4q deletion. Clin Cancer Res. 2009 Mar;15(5):1566–74. doi: 10.1158/1078-0432.CCR-08-2188. [DOI] [PubMed] [Google Scholar]
- 90.Pecina-Slaus N, Nikuseva Martic T, Zeljko M, Bulat S. Brain metastases exhibit gross deletions of the APC gene. Brain Tumor Pathol. 2011 Jul;28(3):223–8. doi: 10.1007/s10014-011-0030-8. [DOI] [PubMed] [Google Scholar]
- 91.Wikman H, Lamszus K, Detels N, Uslar L, Wrage M, Benner C, et al. Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast Cancer Res. 2012 Mar;14(2):R49. doi: 10.1186/bcr3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hohensee I, Lamszus K, Riethdorf S, Meyer-Staeckling S, Glatzel M, Matschke J, et al. Frequent genetic alterations in EGFR- and HER2-driven pathways in breast cancer brain metastases. Am J Pathol. 2013 Jul;183(1):83–95. doi: 10.1016/j.ajpath.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 93.Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010 Jul;28(20):3271–7. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 94.Press MF, Sauter G, Bernstein L, Villalobos IE, Mirlacher M, Zhou JY, et al. Diagnostic evaluation of HER-2 as a molecular target: an assessment of accuracy and reproducibility of laboratory testing in large, prospective, randomized clinical trials. Clin Cancer Res. 2005 Sep;11(18):6598–607. doi: 10.1158/1078-0432.CCR-05-0636. [DOI] [PubMed] [Google Scholar]
- 95.Palmieri D, Bronder JL, Herring JM, Yoneda T, Weil RJ, Stark AM, et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007 May;67(9):4190–8. doi: 10.1158/0008-5472.CAN-06-3316. [DOI] [PubMed] [Google Scholar]
- 96.Rouzier R, Pronzato P, Chereau E, Carlson J, Hunt B, Valentine WJ. Multigene assays and molecular markers in breast cancer: systematic review of health economic analyses. Breast Cancer Res Treat. 2013 Jun;139(3):621–37. doi: 10.1007/s10549-013-2559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen EI, Hewel J, Krueger JS, Tiraby C, Weber MR, Kralli A, et al. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007 Feb;67(4):1472–86. doi: 10.1158/0008-5472.CAN-06-3137. [DOI] [PubMed] [Google Scholar]
- 98.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999 Nov;59(22):5830–5. [PubMed] [Google Scholar]
- 99.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009 Jun;459(7249):1005–9. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Izraely S, Sagi-Assif O, Klein A, Meshel T, Tsarfaty G, Pasmanik-Chor M, et al. The metastatic microenvironment: brain-residing melanoma metastasis and dormant micrometastasis. Int J Cancer. 2012 Sep;131(5):1071–82. doi: 10.1002/ijc.27324. [DOI] [PubMed] [Google Scholar]
- 101.Rodenhiser DI, Andrews J, Kennette W, Sadikovic B, Mendlowitz A, Tuck AB, et al. Epigenetic mapping and functional analysis in a breast cancer metastasis model using whole-genome promoter tiling microarrays. Breast Cancer Res. 2008;10(4):R62. doi: 10.1186/bcr2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Andrews J, Kennette W, Pilon J, Hodgson A, Tuck AB, Chambers AF, et al. Multi-platform whole-genome microarray analyses refine the epigenetic signature of breast cancer metastasis with gene expression and copy number. PLoS One. 2010 Jan;5(1):e8665. doi: 10.1371/journal.pone.0008665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011 Mar;3(75):75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003 Jan;33(1):49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 105.Weigelt B, Hu Z, He X, Livasy C, Carey LA, Ewend MG, et al. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005 Oct;65(20):9155–8. doi: 10.1158/0008-5472.CAN-05-2553. [DOI] [PubMed] [Google Scholar]
- 106.Park ES, Kim SJ, Kim SW, Yoon SL, Leem SH, Kim SB, et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc Natl Acad Sci U S A. 2011 Oct;108(42):17456–61. doi: 10.1073/pnas.1114210108. [DOI] [PMC free article] [PubMed] [Google Scholar]