

Abstract
Protein phosphatase types 1 α (PP1α/PPP1C) and 5 (PP5/PPP5C) are members of the PPP family of serine/threonine protein phosphatases. PP1 and PP5 share a common catalytic mechanism, and several natural compounds, including okadaic acid, microcystin, and cantharidin, act as strong inhibitors of both enzymes. However, to date there have been no reports of compounds that can selectively inhibit PP1 or PP5, and specific or highly selective inhibitors for either PP1 or PP5 are greatly desired by both the research and pharmaceutical communities. Here we describe the development and optimization of a sensitive and robust (representative PP5C assay data: Z′=0.93; representative PP1Cα assay data: Z′=0.90) fluorescent phosphatase assay that can be used to simultaneously screen chemical libraries and natural product extracts for the presence of catalytic inhibitors of PP1 and PP5.
Introduction
In higher eukaryotic organisms, the reversible phosphorylation of proteins represents an important and dynamic form of post-translational modification.1,2 Phosphorylation alters the biological functions of many proteins, notably by altering catalytic activities, targeting proteins for degradation, influencing the subcellular localization of proteins, and promoting or antagonizing protein–protein interactions. Because the phosphorylation state at any instant reflects the opposing activities of both protein kinases and protein phosphatases, the development of inhibitors targeting specific protein kinases or protein phosphatases should prove useful for both the study of disease processes and for the development of new agents for medical management of human ailments.
Indeed, a tremendous effort has already been devoted to the development of pharmacological agents that regulate the actions of “key” protein kinases, leading to the advent of an extensive arsenal of specific or selective inhibitors that can be employed to probe complex phosphorylation-regulated processes. In addition, specific inhibitors of certain kinases (e.g., STI-571/Gleevec)3,4 have proved useful for the medical management of human disease. In contrast, although protein phosphatases are likely to play an equally important role in human disease, little progress has been made in the development of specific phosphatase inhibitors.
PP1 (PPP1C) and PP5 (PPP5C) are members of the PPP family of ser/thr-specific protein phosphatases, which also includes PP2A (PPP2AC), calcineurin (PPP3C), PP4 (PPP4C), PP6 (PPP6C), and PP7 (PPP7C/PPEF). Currently, there are a number of natural compounds that act as potent inhibitors of these PPP-family phosphatases, including okadaic acid, microcystin, nodularin, tautomycetin, fostriecin, calyculin A, and cyclosporine A.5–7 Of these compounds, only cyclosporine A demonstrates class selectivity. Cyclosporine A acts to selectively inhibit calcineurin (PPP3C) activity, and selective calcineurin inhibitors have been developed into immunosuppressive drugs with a global market of ∼1 billion U.S. dollars per year. Fostriecin, a natural product produced by Streptomyces sp., is a highly selective inhibitor of PP2A/PP4 (IC50<2 nM) and a weak inhibitor of PP1 and PP5 (IC50>70 μM).8 Fostriecin demonstrated sufficient antitumor activity in animals to warrant Phase I human clinical trials.9,10 The other afore-mentioned natural products are strong inhibitors (IC50; low nM) of PP1/PP2A/PP4/PP5/PP6 that demonstrate modest or no selectivity. They have some utility as research reagents, but the combined inhibition of PP1–PP6 is toxic to most, if not all, eukaryotic cells.
In vivo, both PP1 and PP5 exist predominately in complexes with other proteins.11–13 Some PP1 binding partners (e.g., I-1, I-2, and NIPP1) function to inhibit PP1 catalytic activity by blocking substrate access to the catalytic site.14–16 Other partners control the intracellular location of PP1.17 In general PP1 regulatory proteins interact with PP1C via noncovalent interactions and are encoded by separate genes.
The catalytic domain of PP5 shares structural similarity with PP1C. However, PP5 has a unique N-terminal domain that both regulates catalytic activity and mediates the interaction with binding partners. Structural studies indicate that the N-terminal domain of PP5 is connected to the catalytic domain by a flexible 34-amino-acid “linker” that allows a tripartite tetratricopeptide-repeat (TPR) motif within the N-terminal domain to adopt a conformation that occludes the active site via the formation of stabilizing interactions with the catalytic domain and an adjacent C-terminal J-helix.18,19 When PP5 is displaced from its binding partners, this N-terminal/C-terminal interaction is inhibitory. The three TPR domains within the N-terminal domain also mediate the association of PP5 with other proteins.13,20,21 When in a complex with other proteins, a binding-induced conformational change opens the active site to substrates, thereby “activating” PP5.21–23 Removal of the autoinhibitory N-terminal domain by proteolysis results in a bare catalytic domain (PP5C) with increased activity.24 Since the physiological target of catalytic inhibitors of PP5 would be the “open” active form, the PP5 catalytic domain represents a better target for inhibitor screening than the autoinhibited full-length enzyme.
Aberrant PP1 activity has been linked to a number of human ailments,7,11,12,25–27 and many studies of PP5 function indicate pivotal roles in the regulation of cellular proliferation and stress-induced apoptosis that serve to validate it as an antitumor drug target.7,28–35 However, to date there are no reports of small-molecule inhibitors that can be used to distinguish the activity of PP1 versus PP5. Therefore, selective inhibitors of PP1 or PP5 are desired as probes to further characterize the biology associated with these enzymes, and specific or highly selective inhibitors may also serve as lead compounds for drug development.
To facilitate the identification of novel, specific inhibitors of PP1 or PP5, a homogeneous, fluorescence intensity (FLINT) biochemical assay was developed, which is amenable for miniaturization and ultra high-throughput screening (uHTS) of large compound libraries. To increase the chances of identifying specific inhibitors, we developed assays for both PP1C and PP5C using similar conditions and substrates. These assays measure the enzyme-catalyzed hydrolysis of a synthetic phosphomonoester substrate 3-O-methylfluorescein phosphate (OMFP). OMFP is a fluorogenic aryl phosphate originally developed ∼46 years ago to study alkaline phosphatase.36 Subsequently, OMFP has been utilized as a substrate for dual-specificity protein phosphatases and ATPases.37–40
Here we develop robust assay conditions in which OMFP is used as a substrate for ser/thr-specific protein phosphatases, whose natural substrates are alkyl-phosphate serine/threonine side-chains in target proteins. Although the assays described in the “Materials and Methods” section were primarily developed and optimized for PP1, only minor modifications were needed to optimize the assay for PP5. The methods utilized are quite general and should be readily adaptable for screening assays employing many other ser/thr-specific protein phosphatases from many different organisms. The optimized medium-throughput protocols are based on a 96-well format using manual pipettors for assembling reactions and serve as an example for phosphatase assay development. Both assays were easily scaled to the small volumes needed for uHTS in a 1,536-well format and successfully used to screen the Molecular Libraries Small Molecule Repository for inhibitors of PP1 and PP5.
Materials and Methods
Chemicals and Reagents
Dithiothreitol (43819), OMFP cyclohexylammonium salt (M2629), NaOH (S5881), KOH (P5958), dibasic potassium phosphate (P9666), cantharidin (C7632), Triton X-100 (T8787), sodium ascorbate (A4034), and manganese (II) chloride (MnCl2) (M3634) were purchased from Sigma-Aldrich Co. HEPES free acid (5320) was purchased from EMD. Bovine serum albumin (BSA) was purchased from CalBiochem (product number 12659). Dimethyl sulfoxide (DMSO; high-performance liquid chromatography grade) was purchased from Burdick & Jackson (product number 081-1). Sulfuric acid 97% (9681-33) was purchased from J.T. Baker. Tobacco Etch Virus (TEV) protease (12575-015) was purchased from Invitrogen. Enterokinase (P8070) was purchased from New England Biolabs. 3-O-Methylfluorescein (451770) was purchased from BD Biosciences.
Protein Expression and Purification
The expression and purification of the PP5 catalytic domain (PP5C) have been described previously.18 Briefly, PP5C (residues 169–499 of PP5) was expressed as an N-terminal fusion with maltose binding protein (MBP) and a linker sequence containing a hexahistidine tag and a TEV protease cleavage site. Biologically active MBP-PP5C was partially purified from isopropyl β-D-1-thiogalactopyranoside (IPTG)–induced bacterial cell lysates by ammonium sulfate fractionation and immobilized metal affinity chromatography on nickel-iminodiacetate media (GE Life Sciences). This was followed by cleavage of the linker with TEV protease digestion and purification of free PP5C by anion exchange chromatography. The final pooled active fractions were concentrated with centrifugal filters, aliquoted, and stored at −80°C.
PP1 catalytic subunits bind to heparin and the purification of PP1 isoforms by heparin affinity chromatography has been described many times.41–43 Briefly, for the present study, the coding sequence of PP1α was cloned into pMal-C2E (NEB) and expressed as an MBP fusion in a BL21 strain of Escherichia coli carrying the pRARE plasmid (Novagen). Active MBP-PP1α was partially purified from IPTG-induced bacterial cell lysates by ammonium sulfate fractionation and affinity chromatography on heparin sepharose high-performance media (GE Life Sciences). This was followed by proteolytic cleavage of the linker between MBP and PP1α by enterokinase digestion and purification of free PP1α via anion-exchange chromatography. The final pooled active fractions were aliquoted and stored at −80°C.
FLINT-Based Assay for PP1α and PP5C
A homogeneous FLINT-based assay for ser/thr protein phosphatases was developed using the artificial substrate OMFP and optimized for PP1α and PP5C in a 96-well format using black, flat-bottomed microtiter plates (Greiner; material No. 655209) with a final assay volume of 200 μL (for end point reads, kinetic reads performed without addition of stop solution use a volume of 150 μL). Enzyme and substrate concentrations, as well as appropriate buffer conditions, were optimized for both enzymes (see “Assay Development and Optimization” section).
Stock solutions and storage
Stock solutions of 10× HEPES buffer (300 mM HEPES in milli-Q water, adjusted to pH 7.0 at room temperature with sodium hydroxide), 1 M MnCl2, and 1% Triton X-100 in milli-Q water were stored at room temperature. Aqueous stocks of DTT (100 mM), sodium ascorbate (1 M), and BSA (10 mg/mL) were aliquoted and stored at −80°C. Cantharidin stocks (10 mM or 100 mM) in DMSO were aliquoted and stored at −80°C.
Stop solution
Dibasic potassium phosphate (1 M in milli-Q water) was adjusted to pH 10 with potassium hydroxide and stored at room temperature. Potassium salts were used to avoid precipitation during storage.
Substrate
OMFP (100 mM) was dissolved in acidified DMSO. Acidified DMSO was made by dissolving 97%-grade sulfuric acid in DMSO to a final concentration of 100 mM H2SO4 immediately before use. The acid converts the rather insoluble monoanionic species of OMFP present in the commercially available compound to the free acid form, which is highly soluble in DMSO, and can then be aliquoted at high concentration and stored at −80°C. This is an important step for HTS development because it greatly aids stability and also reduces the amount of DMSO that must be introduced into the assay. The fluorescent product 3-O-methylfluorescein (10 mM) was dissolved in DMSO and stored at −80°C.
Assay buffers
The 10× HEPES buffer stock was diluted to a 1.5× concentration, along with the addition of the other components to a 1.5× concentration. That is, DTT to 1.5 mM, sodium ascorbate to 1.5 mM, and BSA to 0.45 mg/mL in the 1.5× buffer. The MnCl2 concentration varies with the enzyme (PP1α: 1.5 mM in the 1.5× buffer, PP5C: 0.15 mM or 0 mM in the 1.5× buffer). For substrate saturation studies, in which assays were started by the addition of enzyme in assay buffer to substrate solutions of various concentrations, a 3× assay buffer was made instead of 1.5×. The final optimized 1× assay buffer composition for PP5C is 30 mM HEPES, 1 mM DTT, 1 mM ascorbate, 0.1 mM MnCl2, and 0.3 mg/mL BSA (plus 1% DMSO and 0.01% Triton X-100 for the enzyme stability and assay variability studies). The final 1× assay buffer composition for PP1α is 30 mM HEPES, 1 mM DTT, 1 mM ascorbate, 1 mM MnCl2, and 0.3 mg/mL BSA (plus 1% DMSO and 0.01% Triton X-100 for the enzyme stability and assay variability studies).
Assay Development and Optimization
For the following, unless otherwise specified, all enzyme solutions were prepared from freshly thawed aliquots of enzyme stock solution that have been kept on ice for <3 h. Fluorescence measurements were performed with a Molecular Devices SpectraMax® M5 instrument with the following settings (unless otherwise noted): medium PMT sensitivity, excitation wavelength of 485 nm, emission wavelength of 525 nm, and emission cutoff filter of 515 nm.
Km Determination
For PP1α, a 3× enzyme solution (12.3 nM PP1α) was prepared in 3× assay buffer with BSA, DTT, ascorbate, and MnCl2 as described earlier (i.e., 90 mM HEPES, 3 mM DTT, 3 mM MnCl2, 3 mM ascorbate, and 0.9 mg/mL BSA). For PP5C, a 3× enzyme solution (4.5 nM PP5C) was prepared in 3× assay buffer with BSA, DTT, and ascorbate (i.e., 90 mM HEPES, 3 mM DTT, 3 mM ascorbate, and 0.9 mg/mL BSA). Blank solutions were made as described previously but omitting PP1α or PP5C, respectively. A series of dilutions of OMFP (at 100× the final concentrations of 1,000, 600, 300, 150, 100, 60, 30, 15, 10, 6, and 0 μM) in DMSO was prepared. The 100× DMSO stocks were then diluted 66.7-fold into milli-Q water to yield 1.5× substrate solutions. One hundred microliters of each 1.5× substrate solution was dispensed into the appropriate wells using a stepper pipette and a Plastibrand® positive-displacement syringe tip (PD-tip). For each substrate concentration, eight wells were used: two for blanks and six for enzyme-catalyzed reactions. Reactions were started by the addition of 50 μL of enzyme solution (or 50 μL of blank solution) to the appropriate wells with an 8-channel pipettor, followed as quickly as possible by mixing for 10 s via the plate shaker in the M5 and subsequent kinetic reads (at 30-s intervals) of each well for 5 min at room temperature (∼22°C) to obtain initial rate data. Reaction rates were obtained from linear least square fits to data and the rate versus substrate concentration was then fit to a hyperbola model with GraphPad Prism® to obtain Km.
Enzyme Titration
Twofold serial dilutions of PP5C were prepared in 1.5× assay buffer (with DTT, BSA, and ascorbate as described previously) from 4.8 to 0.2 nM (plus a 0 nM blank solution). Twofold serial dilutions of PP1α were similarly prepared in a 1.5× assay buffer from 18 to 1.125 nM (plus a 0 nM blank solution). One hundred microliters of each enzyme dilution was then aliquoted to the appropriate wells using PD tips with a stepper pipette (eight replicates for each enzyme concentration). Substrate solutions (3×; i.e., 150 μM OMFP for PP5C and 300 μM OMFP for PP1α) were prepared by diluting a 100 mM stock solution of OMFP into milli-Q water. Reactions were started by the addition of 50 μL of the appropriate substrate solution to the appropriate wells with an 8-channel pipettor, followed as quickly as possible by mixing for 10 s via the plate shaker in the M5 and subsequent kinetic reads (every 3 min) of each well for 30 min at room temperature. Data were analyzed with GraphPad Prism. An R-squared value of 0.995 or greater for a linear least square fit to the data was used as a criterion for linearity.
During the assay optimization process for PP5C, it became clear (see “Results” section) that the addition of MnCl2 to the PP5C assay buffer was necessary to ensure long-term room temperature stability of diluted PP5C solutions. Therefore, an additional time course experiment at the optimized PP5C concentration was performed with the addition of 0.1 mM MnCl2 to the buffer in order to determine whether linearity conditions were still met.
Effect of Triton X-100 upon OMF Fluorescence
Solutions of OMF (1 μM) were prepared in 1× assay buffer with or without the presence of 0.01% Triton X-100. The emission spectrum of each solution was measured in a quartz cuvette. The excitation wavelength was fixed at 485 nm and an emission scan was performed from 515 to 575 nm (515 nm cutoff filter). Data were plotted with GraphPad Prism.
Triton X-100 Tolerance
For PP1α, a 10 nM solution of PP1α was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), and MnCl2 (1.5 mM). This solution was divided into two equal volumes. One percent Triton X-100 (0.015 volume) was added to the first aliquot and 0.015 volume of milli-Q water was added to the second. A 3× substrate solution (300 μM OMFP) was prepared in milli-Q water. One hundred microliters of 3× substrate was added to 200 μL of PP1α solution in a quartz cuvette and mixed by inversion. Fluorescence was measured with the M5 every 3 s for 5 min.
For PP5C, a 1.5 nM solution of PP5C was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), and MnCl2 (0.15 mM). This solution was divided into two equal volumes. One percent Triton X-100 (0.015 volume) was added to the first aliquot and 0.015 volume of milli-Q water was added to the second. A 3× substrate solution (150 μM OMFP) was prepared in milli-Q water. One hundred microliters of 3× substrate was added to 200 μL of PP5C solution in a quartz cuvette and mixed by inversion. Fluorescence was measured every 3 s for 5 min. Data were plotted with GraphPad Prism and reaction progress curves were adjusted for starting fluorescence.
DMSO Tolerance
A 4.5 nM solution of PP1α was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (1.5 mM). One hundred microliter aliquots were transferred to 80 wells of a 96-well plate using a PD tip and stepper pipette. Finally, with a single-channel pipettor, 1.5 μL of milli-Q water was added to each of 40 wells and 1.5 μL of DMSO was added to each of the remaining 40 wells.
A 525 pM solution of PP5C was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (0.15 mM). One hundred microliter aliquots were transferred to 80 wells of a 96-well plate using a PD tip and stepper pipette. Finally, with a single-channel pipettor, 1.5 μL of milli-Q water was added to each of 40 wells and 1.5 μL of DMSO was added to each of the remaining 40 wells.
Substrate solutions (3.093×; i.e., 155 μM OMFP for PP5C and 309 μM OMFP for PP1α) were prepared by diluting a 100 mM stock solution of OMFP into milli-Q water. Reactions for each plate were started by the addition of 48.5 μL of the appropriate substrate solution to the appropriate wells with an 8-channel pipettor, followed as quickly as possible by mixing for 10 s via the shaker in the M5 and subsequent kinetic reads (every 30 s) of each well for 5 min at room temperature. Data were analyzed and reaction progress curves were plotted with GraphPad Prism.
Cantharidin-Dose Responses
A 6 nM solution of PP1α was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (1.5 mM). One hundred microliter aliquots were transferred to 66 wells of a 96-well plate using a PD tip and stepper pipette. Six of the remaining wells were filled with 100 μL of a blank solution prepared as earlier but omitting PP1α. A series of dilutions of cantharidin at 100× the final assay concentrations was prepared in DMSO from a 100 mM stock solution and, with a single-channel pipettor, 1.5 μL of the appropriate 100× cantharidin solution was added to the appropriate wells (six replicates for each concentration) and 1.5 μL of DMSO was added to each of the six blank wells and each of the six control wells. After adding cantharidin to the last well, ∼10 min was allowed to elapse before starting the reactions.
A 525 pM solution of PP5C was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (0.15 mM). One hundred microliter aliquots were transferred to 88 wells of a 96-well plate using a PD tip and stepper pipette. The remaining eight wells were filled with 100 μL of a blank solution prepared as earlier but omitting PP5C. Finally, with a single-channel pipettor, 1.5 μL of the appropriate 100× cantharidin solution was added to the appropriate wells (eight for each concentration) and 1.5 μL of DMSO was added to each of the eight blank wells and each of the eight control wells. After adding cantharidin to the last well, ∼10 min was allowed to elapse before starting the reactions.
Substrate solutions (3.093×; i.e., 155 μM OMFP for PP5C and 309 μM OMFP for PP1α) were prepared by diluting a 100 mM stock solution of OMFP into milli-Q water. Reactions for each plate were started by the addition of 48.5 μL of the appropriate substrate solution to the appropriate wells with an 8-channel pipettor, followed as quickly as possible by mixing for 10 s via the shaker in the M5 and subsequent kinetic reads (every 30 s) of each well for 5 min at room temperature to obtain initial rate data. GraphPad Prism was used to fit the data for each dose–response curve to a 4-parameter sigmoidal function to obtain IC50 estimates.
Maximum- and Minimum-Signal Stability
For PP1α, a 3.6 nM solution of enzyme was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (1.5 mM). One hundred microliter aliquots were transferred to the wells of two 96-well plates using a PD tip and stepper pipette. With a single-channel pipettor, 1.5 μL of DMSO was added to each well of one plate (maximum-signal plate) and 1.5 μL of 10 mM cantharidin (in DMSO) was added to each well of the remaining plate (minimum-signal plate). After adding cantharidin to the last well, ∼10 min was allowed to elapse before starting the reactions.
For PP5C, a 525 pM solution of enzyme was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (0.15 mM). One hundred microliter aliquots were transferred to each well of two 96-well plates using a PD tip and stepper pipette. With a single-channel pipettor, 1.5 μL of DMSO was added to each well of one plate (maximum-signal plate) and 1.5 μL of 10 mM cantharidin (in DMSO) was added to each well of the remaining plate (minimum-signal plate). After adding cantharidin to the last well, ∼10 min was allowed to elapse before starting the reactions.
Substrate solutions (3.093×; i.e., 155 μM OMFP for PP5C and 309 μM OMFP for PP1α) were prepared by diluting a 100 mM stock solution of OMFP into milli-Q water. Reactions for each plate were started by the addition of 48.5 μL of the appropriate substrate solution to the appropriate wells with an 8-channel pipettor. An eight-well column was started in sequence every 15 s with mixing by pipetting the solution in and out of the wells several times. Reactions were incubated at room temperature for 30 min and then stopped by the addition of 50 μL of 1 M potassium phosphate (pH 10). An eight-well column was stopped every 15 s in the same order in which the reactions were started. Thus, the reaction in each well was allowed to progress for 30 min.
After stopping all of the reactions, end point fluorescence measurements of wells were made as quickly as possible with the M5 after agitating with the plate shaker for 20 s (PMT sensitivity set to auto, 15 reads per measurement, excitation=485 nm, emission=525 nm, and emission filter=515 nm). This first set of fluorescence measurements was considered to be the t=0 read. Plates were then incubated in the dark at room temperature and fluorescence measurements were repeated for each plate at defined intervals over 6–8 h.
Enzyme Stability
A 4.5 nM solution of PP1α was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (1.5 mM). This solution was kept in the dark at room temperature. For the initial (t=0) activity assay, 100 μL aliquots were transferred to 32 wells of a 96-well plate using a PD tip and stepper pipette. With a single-channel pipettor, 1.5 μL of DMSO was added to each of the first 16 wells (maximum-signal wells) and 1.5 μL of 10 mM cantharidin (in DMSO) was added to each of the remaining 16 wells (minimum-signal wells). After adding cantharidin to the last well, ∼10 min was allowed to elapse before starting the reactions.
A 525 pM solution of PP5C was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (0.15 mM). This solution was kept in the dark at room temperature. For the initial (t=0) activity assay, 100 μL aliquots were transferred to 32 wells of a 96-well plate using a PD tip and stepper pipette. With a single-channel pipettor, 1.5 μL of DMSO was added to each of the first 16 wells (maximum-signal wells) and 1.5 μL of 10 mM cantharidin (in DMSO) was added to each of the remaining 16 wells (minimum-signal wells). After adding cantharidin to the last well, ∼10 min was allowed to elapse before starting the reactions.
Substrate solutions (3.093×; i.e., 155 μM OMFP for PP5C and 309 μM OMFP for PP1α) were prepared by diluting a 100 mM stock solution of OMFP into milli-Q water. Reactions for each plate were started by the addition of 48.5 μL of the appropriate substrate solution to the appropriate wells with an 8-channel pipettor. An eight-well column was started in sequence every 15 s with mixing by pipetting solution in and out of the wells several times. Reactions were incubated at room temperature for 30 min and then stopped by the addition of 50 μL of 1 M potassium phosphate (pH 10). An eight-well column was stopped every 15 s in the same order in which the reactions were started. Thus, the reaction in each well was allowed to progress for 30 min. End point fluorescence measurements were made as described previously. At four to five additional time points over a 24-h period, the enzyme solutions were assayed again according to the afore-mentioned procedure in order to determine the stability of the diluted enzyme at room temperature. OMFP substrate solutions were freshly prepared for each time point.
Assay Variability Studies
For each intraday variability study of the PP1α assay, a 4.5 nM solution of enzyme was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (1.5 mM). One hundred microliter aliquots were transferred to each well of four 96-well plates using a PD tip and stepper pipette. With a single-channel pipettor, 1.5 μL of DMSO was added to each well of two plates (maximum-signal plates) and 1.5 μL of 10 mM cantharidin (in DMSO) was added to each well of the remaining two plates (minimum-signal plates). After adding cantharidin to the last well, ∼10 min was allowed to elapse before starting the reactions.
For each intraday variability study of the PP5C assay, a 525 pM solution of enzyme was prepared in 1.5× assay buffer containing DTT (1.5 mM), BSA (0.45 mg/mL), ascorbate (1.5 mM), Triton X-100 (0.015%), and MnCl2 (0.15 mM). One hundred microliter aliquots were transferred to each well of four 96-well plates using a PD tip and stepper pipette. With a single-channel pipettor, 1.5 μL of DMSO was added to each well of two plates (maximum-signal plates) and 1.5 μL of 10 mM cantharidin (in DMSO) was added to each well of the remaining two plates (minimum-signal plates). After adding cantharidin to the last well, ∼10 min was allowed to elapse before starting the reactions.
Substrate solutions (3.093×; i.e., 155 μM OMFP for PP5C and 309 μM OMFP for PP1α) were prepared by diluting a 100 mM stock solution of OMFP into milli-Q water. Reactions for each plate were started by the addition of 48.5 μL of the appropriate substrate solution to the appropriate wells with an 8-channel pipettor. An eight-well column was started in sequence every 15 s with mixing by pipetting solution in and out of the wells several times. Reactions were incubated at room temperature for 30 min and then stopped by the addition of 50 μL of 1 M potassium phosphate (pH 10). An eight-well column was stopped every 15 s in the same order in which the reactions were started. Thus, the reaction in each well was allowed to progress for 30 min. End point fluorescence measurements were made as described previously.
The procedures specified before were repeated on different days for a total of 3 days of data for each enzyme assay. Means, standard deviations, and coefficients of variation were calculated for each plate; each day's maximum-signal plates; each day's minimum-signal plates; combined maximum-signal plates for days 1&2, 2&3, and 1&3; and combined minimum-signal plates for days 1&2, 2&3, and 1&3. Also, Z′ values and S:B were calculated from each day's data.
Results and Discussion
Enzyme Titration and Km Determination
Fusion of either PP1α or the PP5 catalytic domain (PP5C) with MBP allowed for the efficient production of large quantities of active enzymes in E. coli. As a fusion partner, MBP improves the soluble yield of many heterologously expressed proteins in E. coli.44 Indeed, our lab has had great success expressing ser/thr-specific protein phosphatases with good soluble yield in bacteria as MBP fusions. In our hands, the system works very well for expression of PP1α, PP2Cα, Wip1, and PP5C in an active form. It was not useful for production of active PP2A, PP4, or PP6, which are difficult to express in E. coli. In addition to MBP and accessory affinity tags (e.g., hexahistidine), the inclusion of a specific protease cleavage site (e.g., enterokinase) in the expression constructs allows for the straightforward removal of these non-native additions.
While we chose to develop assays using OMFP, other approaches based upon different artificial phosphatase substrates are also likely to be suitable for library screening in high-density formats. For example, a coupled-enzyme assay utilizing a fluorogenic rhodamine 110 bis-phosphopeptide substrate45 is commercially available from Promega (ProFluor®). Although providing a robust assay, this system is more expensive to implement than OMFP assays. The aminopeptidase coupled-enzyme protocol also requires a separate development step in which the dephosphorylated product is converted to unconjugated free rhodamine 110 dye and is thus limited to end point assays (unlike OMFP-based assays, which may also be utilized for kinetic reads). This step must fully inhibit the phosphatase by utilizing a specific inhibitor since simpler, cheaper methods of stopping the phosphatase reaction (e.g., with a pH jump) are likely to also inhibit aminopeptidase activity. An expensive inhibitor, okadaic acid, is suggested for use with PP1,45 though cheaper inhibitors, such as cantharidin, could probably be substituted. Another possible assay approach would be to utilize the fluorogenic substrate 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP), which is commercially available from Life Technologies (also available from the same company in a phosphatase assay kit; EnzChek® Phoshatase Assay). Although DiFMUP is an effective substrate for both PP1 and PP5,46,47 it is expensive.
The hydrolysis of the fluorogenic substrate OMFP by PP1α or PP5C, to yield O-methylfluorescein (OMF), can be detected in a spectrofluorometer or microtiter plate reader at 485 nm for the excitation wavelength and 525 nm for emission (using a 515 nm cutoff filter). OMFP was chosen as the substrate for the screening assays in part because of favorable kinetic parameters (low Km, robust rate of hydrolysis). The strong fluorescence exhibited by OMF over a wide range of pH affords the flexibility to perform the assay with either kinetic reads, in which rates are estimated from fluorescence measurements of a reaction taken at regular intervals over a period of time, or as an end point assay, in which only one measurement is made after a reaction is stopped (e.g., with a pH jump away from the optimum and/or the addition of an inhibitor). An initial drawback of OMFP was that the commercially available form is the cyclohexylammonium salt, which has very poor solubility in many organic solvents (including DMSO), requiring sonication to prepare stock solutions.48 However, we found that this can be easily overcome by making stocks with an acidified organic solvent, thereby forming the, quite soluble, OMFP free acid (see “Materials and Methods” section).
An examination of the reaction progress curves for an enzyme and substrate provides a basis for choosing appropriate conditions for the biochemical screening assay: concentrations of substrate and enzyme, and length of reaction. To determine the proper substrate concentration for the assay, we first performed substrate saturation plots to determine the kinetic parameters of the substrate with the two enzymes (Fig. 1). Kinetic reads were used for this step in order to ensure that only data in the linear range were used. The Km of OMFP was 113±2.85 μM for PP1α and 71.3±2.31 μM for PP5C. Typically, a substrate concentration at or below the Km should be chosen for inhibitor screening assays since substrate concentrations high relative to the Km can significantly reduce the apparent affinity (i.e., increase the IC50) of a competitive inhibitor by a factor of 1+[S]/Km (rarely occurring uncompetitive inhibition is affected in the opposite fashion).49 Very high, saturating, substrate concentration can thus mask the effects of bona fide inhibitors and drastically reduce the hit rate in a screening project. Very high substrate levels are also likely to decrease signal to background since reaction velocity will asymptotically approach a finite maximum with increasing substrate concentration, while background fluorescence from substrate (and/or contaminating fluorescent product in the substrate preparation) will tend to increase linearly. Bearing these considerations in mind, the OMFP concentrations chosen for the screening assays were 100 μM for PP1α and 50 μM for PP5C.
Fig. 1.

Determination of the Km of OMFP for PP5C and PP1α. Recombinant phosphatase was incubated with the indicated concentrations of OMFP in 150 μL reactions as described in “Materials and Methods” section. Fluorescence intensity (485-nm excitation/525-nm emission) was measured every 30 s over 5 min with an M5 plate reader. Initial rates were determined from linear regression analyses of the raw fluorescence data with SoftMax Pro® and the mean change in fluorescence over time (milli-RFU/min)±SD of 6 replicates is plotted versus OMFP concentration and fit to the Michaelis-Menten equation with GrahPad Prism® to obtain Km estimates. (A) Substrate saturation plot for PP5C (Km=71.3±2.31 μM). (B) Substrate saturation plot for PP1α (Km=113±2.85 μM).
For a robust, high-quality screening assay, the chosen enzyme concentration should yield a reasonable signal-to-background ratio and produce a linear response over the duration of the assay. For the PP1α and PP5C enzyme titration plots, reactions set up over a range of enzyme concentrations were performed with the substrate concentrations identified previously (Fig. 2). The reaction progress curves were subjected to linear least-square regression analysis and an R-squared value of 0.995 or greater was used as a criterion for judging linearity. Based on the data shown in Figure 2, 3 nM was chosen for the PP1α assay and 350 pM was chosen for the PP5C assay.
Fig. 2.

Enzyme titration studies. The indicated concentrations of recombinant phosphatase were incubated with either 50 μM (for PP5C) or 100 μM (for PP1α) OMFP substrate in 150 μL reactions at room temperature as described in “Materials and Methods” section. Fluorescence intensity (485-nm excitation/525-nm emission) was measured every 3 min over 30 min with an M5 plate reader. The mean fluorescence intensity±SD of 8 replicates is plotted with GraphPad Prism and linear regression analyses were performed to assess linearity of the progress curves at each concentration. (A) PP5C-dependent progress curves of OMFP hydrolysis at the indicated enzyme concentrations (0 nM [●], 0.2 nM [■], 0.4 nM [▲], 0.8 nM [▼], 1.6 nM [◆], and 3.2 nM [◯]). (B) PP1α-dependent progress curves of OMFP hydrolysis at the indicated enzyme concentrations (0 nM [●], 0.75 nM [■], 1.5 nM [▲], 3 nM [▼], 6 nM [◆], and 12 nM [◯]). (C) PP5C-dependent progress curve at 350 pM PP5C with the addition of 0.1 mM MnCl2 to the reaction buffer. PP5C (350 pM) was incubated at room temperature with 50 μM OMFP in 150 μL reactions. Fluorescence intensity (485-nm excitation/525-nm emission) was measured every 3 min over 30 min with an M5 plate reader. The mean fluorescence intensity±SD of 12 replicates is plotted with GraphPad Prism and linear regression analysis was performed to assess linearity of the progress curve.
In the course of later optimization steps it was found that PP5C, while quite stable over the 30-min duration of the assay, was slightly unstable under the buffer conditions used in Figure 2A, with activity slowly decreasing over a period of many hours such that a substantial loss would occur after 16–24 h at room temperature (data not shown). It was found that addition of 0.1 mM MnCl2 (final assay concentration) to the buffer significantly improved stability to at least 24 h (see “Reagent Stability”). When such changes in assay conditions have to be made during optimization, it is prudent to reassess the reaction progress curve to ensure that S:B and linearity criteria are still met (Fig. 2C).
Selecting Assay Controls and pH
Identifying appropriate assay controls is crucial to the development of reproducible and robust assays. High-signal controls, for the OMFP-based assay, represent product formation from uninhibited phosphatase and define the upper limit of the assay signal window. High-signal controls for this case include enzyme, OMFP substrate, 0.01% Triton X-100, reaction buffer components, and the solvent (DMSO) in which compound libraries are typically dissolved. Low-signal controls form the lower limit of the signal window, representing the product formation from fully inhibited phosphatase, and are dominated by spontaneous OMFP hydrolysis. Previous research indicates that cantharidin—a readily available, cheap, nonspecific inhibitor of several PPP phosphatases—would make an excellent inhibitor for the low-signal controls for both PP1α and PP5C.5 Dose–response assays performed with OMFP at the chosen assay pH confirmed this, indicating submicromolar IC50s for both phosphatases (Fig. 3). Essentially complete inhibition was observed at 100 μM cantharidin, which was subsequently chosen for low-signal control reactions.
Fig. 3.

Cantharidin-dose responses. Reactions containing recombinant phosphatase in the presence of the indicated concentrations of cantharidin were conducted as described in “Materials and Methods” section. Fluorescence intensity (485-nm excitation/525-nm emission) was measured every 30 s over 5 min with an M5 plate reader. Initial rates were determined from linear regression analyses of the raw fluorescence data with SoftMax Pro and the mean change in fluorescence over time (milli-RFU/min)±SD of six replicates was normalized to uninhibited controls and plotted as % inhibition versus cantharidin concentration. Inhibition curves were fit to a 4-parameter sigmoidal function with Prism® to estimate IC50s. (A) Inhibition curve of PP5C in the presence of cantharidin (IC50=328 nM; 95% CI: 271–396 nM). (B) Inhibition curve of PP1α in the presence of cantharidin (IC50=139 nM; 95% CI: 114–170 nM).
The assay controls should, if possible, allow for a signal window wide enough for changes in FLINT signal caused by inhibitory library compounds to be reliably distinguished from noise (i.e., inherent variability in measurements). Typically, this criterion of assay robustness is assessed by the Z′ parameter,50 which takes into account both the assay signal's dynamic range and measurement variability.
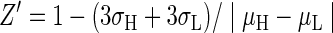 |
Where σH and σL are the standard deviations and μH and μL are the means of the high- and low-signal controls, respectively. Typical intraday Z′ values (Fig. 4) for the final PP1α and PP5C assays, optimized for end point reads, in a 96-well format are 0.90 and 0.93, respectively. This is well above the value of 0.5, which is generally considered to indicate a high-quality assay for screening purposes.
Fig. 4.

Representative scatter plots of 3-day assay variability and Z′ factor determinations. To assess the assay signal windows and variabilities, we performed, for each enzyme, two full 96-well plates of maximum-signal controls (1% DMSO) and two full plates of minimum-signal controls (100 μM cantharidin and 1% DMSO) on 3 consecutive days. Representative assay data for 1-day run were analyzed with Origin® and are presented as a scatter plot of fluorescence intensity versus well number (two 96-well plates numbered consecutively from 1 to 192 for each set of controls); closed circles are maximum-signal controls and closed squares are minimum-signal controls. The lines going through and bracketing the maximum signal controls indicate the mean (solid line) and ±3 SD (dashed lines) of the maximum control fluorescence intensity. Similarly, the lines going through and bracketing the minimum-signal controls indicate the mean (solid line) and±3 SD (dashed lines) of the maximum control fluorescence intensity, although these are, to a great extent, obscured by the symbols in the plot. (A) Representative PP5C assay data for 1-day run: Z′=0.93. (B) Representative PP1α assay data for 1-day run: Z′=0.90.
The pH at which an assay is performed can have a great impact upon its sensitivity and robustness. pH can affect not only enzyme activity and stability, but also the quantum yield and absorption/emission maxima of fluorescent reaction products, and binding affinities of any inhibitors used for control reactions. A pH that allows for maximum signal window size (i.e., the difference between 100% enzyme activity and fully inhibited background signal) represents a trade-off between the pH optimum for the enzyme's catalytic activity, the pH range that maximizes FLINT signal from the fluorescent product (for kinetic reads, end point reads would allow for a pH jump as part of the stopping conditions), and the pH range that allows for complete inhibition of the enzyme by the chosen control inhibitor. However, it is important to realize that, for a particular enzyme/substrate/control inhibitor, the pH that maximizes the signal window may be far from physiologically relevant conditions (e.g., cytosolic pH of ∼7–7.451) and pH can affect ionization states of groups on the target protein (as well as on any putative inhibitors) that may alter binding affinity. Thus, screening compound libraries for inhibitors using a pH far from that likely in the target's native environment may lead to identifying hits that fail to work in vivo or to rejecting compounds that might have worked in vivo but do not under the nonphysiological assay conditions. These considerations dictate a compromise between maximizing the signal window and choosing a pH relevant to conditions in which newly identified inhibitors and their derivatives are expected to operate.
In the case of several PPP family phosphatases—lambda phosphatase,52 PP1,53 calcineurin,54 and PP5 (unpublished observations)—much research on the reaction mechanisms and kinetics of these enzymes has established the importance of two active-site ionizable groups for enzyme activity, producing inverted-U-shaped pH rate profiles with broad optima between ∼pH 6 and 7. The mammalian PPP family shows high sequence homology among its members, with the catalytically important residues being absolutely conserved throughout, suggesting similar pH optima for the entire family. Further, our chosen control inhibitor, cantharidin, has been previously reported to bind with high affinity at near-neutral pH to several members of the PPP family.5 This is confirmed in Figure 3 for PP1α and PP5C. Therefore, for our particular screening assays, a pH of 7 (HEPES buffer) was chosen.
Triton X-100 Tolerance
One serious persistent problem of HTS efforts is the nonspecific inhibition by promiscuous molecules that plague screening libraries and appear on hit lists from screening projects.55 At least some of these nonspecific compounds are thought to inhibit by forming submicrometer aggregates that adsorb and sequester the enzyme(s) being assayed. False-positive hits due to nonspecific adsorption can, to some extent, be suppressed by the presence of small concentrations of detergents, such as Triton X-100.55,56 Since detergents might affect enzyme activity/stability and/or product fluorescence, it is important to determine the effects of the chosen detergent concentration (in this case, 0.01% v/v Triton X-100) upon the assay. Addition of surfactants to the assay buffer may affect surface tension enough to alter menisci shape and effective path lengths. Path length changes from meniscus effects can lead to differences in FLINT signal that are not caused by any surfactant-induced changes in enzyme activity or in the spectral properties of the fluorescent reaction products. Such was the case for the addition of 0.01% Triton X-100 in our assay system (data not shown), making it difficult to directly test for differences in enzyme activity in the presence or absence of Triton X-100 using a 96-well-plate format. However, by using a cuvette to fix the path length, we confirmed that Triton X-100 has no apparent effect upon OMF fluorescence or upon the activities of PP1α and PP5C (Fig. 5).
Fig. 5.

Triton X-100 tolerance. (A) Triton X-100 (0.01% v/v) has no discernible effect upon fluorescence intensity measurements of 3-O-methylfluorescein (OMF) when path length is fixed by using a cuvette. Fluorescence emission scans (485-nm excitation) were performed in quartz cuvettes on solutions of 1 μM OMF prepared in reaction buffer with (▼) and without (▲) the presence of 0.01% Triton X-100. Emission spectra for each solution are plotted together on the same graph and are almost perfectly superimposable. (B) Triton X-100 (0.01% v/v) has no significant effect upon fluorescence intensity measurements for PP5C reaction progress curves when path length is fixed. PP5C (1 nM) was incubated as described in “Materials and Methods” section with 50 μM OMFP in assay buffer with (▼) and without (▲) the presence of 0.01% Triton X-100. Fluorescence intensity measurements of the reaction mixture in a quartz cuvette were conducted every 3 s for 5 min (485-nm excitation/525-nm emission) with the M5. Reaction progress curves were plotted together (adjusting starting fluorescence intensity to zero) with GraphPad Prism and are almost perfectly superimposable. (C) Triton X-100 (0.01% v/v) has no significant effect upon fluorescence intensity measurements for PP1α reaction progress curves when path length is fixed. PP1α (6.67 nM) was incubated as described in “Materials and Methods” section with 100 μM OMFP in assay buffer with (▼) and without (▲) the presence of 0.01% Triton X-100. Fluorescence intensity measurements of the reaction mixture in a quartz cuvette were conducted every 3 s for 5 min (485-nm excitation/525-nm emission) with the M5. Reaction progress curves are plotted together (adjusting starting fluorescence intensity to zero) with GraphPad Prism and are almost perfectly superimposable.
DMSO Tolerance
As compound libraries are typically dissolved in DMSO, it is inevitable that enzymes and other assay components will be exposed to DMSO. It is therefore important to ascertain whether or not DMSO, in concentrations likely to be encountered, causes any significant interference in the assay (e.g., by affecting enzyme activity or product fluorescence). In most cases, it should be sufficient to determine the effects of a final concentration of 1% DMSO upon the assay results by a head-to-head comparison of assays (high- and low-signal controls with a large number of replications) with and without the presence of DMSO. The results of assays performed in the presence and absence of DMSO revealed no apparent effect upon FLINT signal for PP1α or PP5C (Fig. 6).
Fig. 6.

DMSO tolerance. (A) DMSO (1% v/v) has no apparent effect upon fluorescence intensity measurements for PP5C reaction progress curves. PP5C (350 pM) was incubated as described in “Materials and Methods” section with 50 μM OMFP in buffer with (▼) and without (▲) the presence of 1% DMSO. Fluorescence intensity measurements were conducted on reactions in each well (40 replicates for each condition) every 30 s over 5 min (485-nm excitation/525-nm emission) with the M5. Reaction progress curves are plotted together with GraphPad Prism and are almost perfectly superimposable. (B) DMSO (1% v/v) has no apparent effect upon fluorescence intensity measurements for PP1α reaction progress curves. PP1α (3 nM) was incubated as described in “Materials and Methods” section with 100 μM OMFP in buffer with (▼) and without (▲) the presence of 1% DMSO. Fluorescence intensity measurements were conducted on reactions in each well (40 replicates for each condition) every 30 s over 5 min (485-nm excitation/525-nm emission) with the M5. Reaction progress curves are plotted together with GraphPad Prism and are almost perfectly superimposable.
Maximum- and Minimum-Signal Stability
Optimizing assays for end point reads requires the addition of a stopping reagent to each well after the reaction has been allowed to proceed for a certain specified time (30 min for the assays under consideration). The purpose of the stopping reagent is to dramatically suppress (i.e., essentially stop) enzymatic activity upon the substrate by one or more mechanisms. For the PP1α and PP5C assays, we chose to suppress activity by two separate mechanisms: (1) shifting away from the enzymes' pH optima to a very basic pH, and (2) adding a high concentration of the nonspecific inhibitor orthophosphate. This is accomplished by the addition, at the appropriate times, of a stop solution consisting of 1 M potassium phosphate at pH 10. If the fluorescence signal stability of the stopped reactions is less than the time required to read multiple plates with the appropriate detector, batch processing of plates may be necessary and throughput will be reduced.
High-signal controls consist of vehicle (i.e., DMSO)–treated enzyme under the optimized reaction conditions and low-signal controls contain 100 μM cantharidin. Figure 7 shows FLINT measurements of high- and low-signal controls of PP1α (Fig. 7A) and PP5C (Fig. 7B) subsequent to addition of stop solution (after 30-min reactions) and continuing at additional time points over the course of several hours. These results show that the FLINT signal from stopped reactions is reasonably stable over a period of an hour or so, but that there is still a significant amount of substrate hydrolysis occurring. As low OMFP hydrolysis occurs even in the absence of enzyme (data not shown), the continued increase in FLINT signal of the stopped reactions is likely due primarily to the spontaneous hydrolysis of OMFP.
Fig. 7.

Stability of end point fluorescence intensity for controls. Maximum (1% DMSO) and minimum (100 μM cantharidin and 1% DMSO) controls (one 96-well plate per condition) for each phosphatase were prepared as described in “Material and Methods” section. After incubating for 30 min at room temperature, reactions were stopped by the addition of 50 μL of 1 M potassium phosphate (pH 10). Fluorescence intensity (485-nm excitation/525-nm emission) of each replicate was acquired at the indicated times with the M5 plate reader. The first set of measurements (t=0 h) were acquired essentially immediately after stopping the reactions. Plates were subsequently stored in the dark at room temperature except for the brief periods required to acquire FLINT measurements at the indicated times. Due to limitations upon the number of subcolumns in Prism, only data for half of each plate (i.e., 48 replicates for each condition) are displayed here. For both enzymes, after stopping reactions, fluorescence intensities of both maximum- and minimum-signal controls increase steadily at a low rate due to spontaneous hydrolysis of OMFP. (A) Stability of PP5C maximum (■) and minimum (●) controls. (B) Stability of PP1α maximum (■) and minimum (●) controls.
Reagent Stability
If screening large compound libraries in an HTS setting, then enzyme/buffer solutions may sit for many hours in the liquid-handling system before being completely dispensed into assay plates. Stability of reagents over the course of typical daily operations must be assessed and any special requirements are addressed with appropriate procedures. The stabilities of diluted PP1α and PP5C enzyme/buffer solutions were tested at room temperature over a 24-h period using the final optimized assay conditions with freshly prepared OMFP substrate solutions. Although BSA has a known tendency to nonspecifically bind a wide variety of small molecules,57 we included it in our buffers because it is a standard component in the literature for stabilizing dilute solutions of ser/thr protein phosphatases.58,59 Stability of both enzyme activity (DMSO-treated, uninhibited high-signal controls) and inhibitor sensitivity (cantharidin inhibited low-signal controls) was assessed (Fig. 8). As shown in the first several hours of the PP1α time course, unstable room temperature can also lead to fluctuations in activity.
Fig. 8.

Enzyme stability. Solutions of PP5C and PP1α at 1.5× concentration were prepared in the final optimized 1.5× assay buffers as described in “Materials and Methods” section and stored in the dark at room temperature except as noted below. At each of the indicated time points, aliquots of the enzyme solutions were transferred to black 96-well plates. Maximum- (■) and minimum- (●) signal controls were prepared as described (16 replicates of each) at each time point by addition of DMSO and cantharidin, respectively. Reactions were started by addition of OMFP substrate and subsequently stopped by addition of 1 M potassium phosphate after 30-min incubation at room temperature. Fluorescence intensity (485-nm excitation/525-nm emission) measurements of the stopped reactions were then immediately acquired on a SpectraMax M5. (A) PP5C stability. Over the 24-h time frame of this experiment, no deterioration was observed for either the PP5C enzyme activity with OMFP or the sensitivity of PP5C to the control inhibitor cantharidin. (B) PP1α stability. Over the 24-h time frame of this experiment, no significant deterioration was observed for either the PP1α enzyme activity with OMFP or the sensitivity of PP1α to the control inhibitor cantharidin. PP1α activity was observed to be slightly lower during the initial few hours of the experiment. This is thought to be due to poor climate control in the lab that led to relatively lower room temperature during the first part of the experiment.
While enzymes and assay buffer components appear to be stable out to ∼24 h, the OMFP substrate is, unfortunately, not very stable in aqueous solution over the same time frame. The high rate of spontaneous hydrolysis steadily produces fluorescent OMF, introducing a steadily increasing background signal to the assays. To overcome this problem in an HTS setting, it would be necessary to either periodically introduce freshly prepared substrate solution (i.e., DMSO stock freshly diluted with water) into the liquid-handling system, or find a way of suppressing the background hydrolysis that does not interfere with performance of the assays.
The rapid hydrolysis of OMFP at pH dominated by the dianionic species is likely due to its very good leaving group (pKa=4.6). Kirby and Varvoglis60 have shown, for hydrolysis of a series of aryl phosphates, that the dependence of the rate upon the leaving group pKa is much greater for dianions than for monoanions.60 Thus, for very low leaving group pKa, the monoanionic species is expected to be significantly more stable than the dianion (just the opposite of what is typically the case for phosphomonoesters).61 These considerations suggest that aqueous solutions of OMFP can be stabilized by lowering the pH to a level where the phosphoryl group is predominately monoprotonated (i.e., pH=2–3). If this is done by preparing substrate solution in dilute HCl (e.g., 1 mM), then the amount of acid added to the final assembled phosphatase reaction can be kept low enough (∼0.33 mM; or less if substrate volume ratio is reduced) to avoid lowering the assay buffer pH significantly. Indeed, lowering the pH of the substrate solution was found to reduce background OMFP hydrolysis by about an order of magnitude (data not shown). Further reductions in background hydrolysis can be achieved by cooling the substrate solution reservoir below room temperature. Through these methods it is possible to dramatically increase the substrate stability and slow down degradation of assay S:B over time.
Assay Variability
In HTS and uHTS settings, assay variability can have many sources, from limits on the precision and accuracy of equipment to intra- and interlot differences in reagents and supplies. After optimizing all relevant parameters, the suitability of an assay for screening must be assessed. The assay must be robust (giving consistent and reproducible results) and it must be sufficiently sensitive to identify active compounds with reasonable confidence. Having a large signal window, and low variability, is vital for detecting bona fide hits. High-signal control and low-signal control plates for PP1α and PP5C, respectively, were assayed according to optimized procedures and assessed for reproducibility and variability. Variability measures (e.g., coefficient of variation [CV]) were determined both within each assay plate (intraplate), as well as between different plates of the same type (interplate). Additionally, assay control plates done on different days were compared to determine day-to-day variability. Figure 4 and Tables 1 and 2 show the variabilities and statistical analyses of the PP1α and PP5C assays. For each enzyme, interplate, intraplate, and day-to-day comparisons yielded low CV values and high Z′ values (>0.8 for PP1α and >0.9 for PP5C). An overview of the basic assay protocol steps for the final optimized conditions and procedures are given in Tables 3 and 4 for PP5C and PP1α, respectively.
Table 1.
PP5C Assay 3-Day Variability and Performance Statistics
| Class | Day | Plate | Maximum/Minimum | Mean | SD | CV | Z′a | S:Bb |
|---|---|---|---|---|---|---|---|---|
| Intraplate | 1 | 1 | Maximum | 4,710 | 104 | 2.20 | ||
| 2 | Maximum | 4,730 | 65.5 | 1.39 | ||||
| 3 | Minimum | 177 | 3.40 | 1.92 | ||||
| 4 | Minimum | 174 | 6.54 | 3.76 | ||||
| All | 0.94 | 27.0 | ||||||
| 2 | 1 | Maximum | 5,180 | 69.5 | 1.34 | |||
| 2 | Maximum | 5,130 | 82.4 | 1.61 | ||||
| 3 | Minimum | 184 | 4.03 | 2.19 | ||||
| 4 | Minimum | 180 | 4.22 | 2.35 | ||||
| All | 0.95 | 28.3 | ||||||
| 3 | 1 | Maximum | 5,550 | 104 | 1.88 | |||
| 2 | Maximum | 5,480 | 133 | 2.42 | ||||
| 3 | Minimum | 178 | 4.00 | 2.26 | ||||
| 4 | Minimum | 176 | 4.26 | 2.42 | ||||
| All | 0.93 | 31.2 | ||||||
| Interplate | 1 | 1 & 2 | Maximum | 4,720 | 86.6 | 1.84 | ||
| 3 & 4 | Minimum | 175 | 5.42 | 3.09 | ||||
| 2 | 1 & 2 | Maximum | 5,150 | 81.1 | 1.57 | |||
| 3 & 4 | Minimum | 182 | 4.62 | 2.54 | ||||
| 3 | 1 & 2 | Maximum | 5,520 | 125 | 2.27 | |||
| 3 & 4 | Minimum | 177 | 4.17 | 2.36 | ||||
| Day to day | 1&2 | All | Maximum | 5,120 | 413 | 8.07 | ||
| Minimum | 176 | 4.88 | 2.77 | |||||
| 2&3 | All | Maximum | 5,330 | 210 | 3.93 | |||
| Minimum | 180 | 5.15 | 2.87 | |||||
| 1&3 | All | Maximum | 5,120 | 413 | 8.07 | |||
| Minimum | 176 | 4.88 | 2.77 |
aZ′ parameter (see “Materials and Methods” section).
bSignal-to-background ratio: S:B=(mean of maximum controls)/(mean of minimum controls).
CV, coefficient of variation; SD, standard deviation.
Table 2.
PP1α Assay 3-Day Variability and Performance Statistics
| Class | Day | Plate | Maximum/Minimum | Mean | SD | CV | Z′ | S:B |
|---|---|---|---|---|---|---|---|---|
| Intraplate | 1 | 1 | Maximum | 3,250 | 109 | 3.35 | ||
| 2 | Maximum | 3,250 | 178 | 5.48 | ||||
| 3 | Minimum | 388 | 16.8 | 4.32 | ||||
| 4 | Minimum | 382 | 11.6 | 3.05 | ||||
| All | 0.83 | 8.44 | ||||||
| 2 | 1 | Maximum | 3,460 | 83.2 | 2.40 | |||
| 2 | Maximum | 3,470 | 64.7 | 1.86 | ||||
| 3 | Minimum | 317 | 5.21 | 1.64 | ||||
| 4 | Minimum | 307 | 11.7 | 3.80 | ||||
| All | 0.92 | 11.1 | ||||||
| 3 | 1 | Maximum | 3,090 | 68.4 | 2.21 | |||
| 2 | Maximum | 3,040 | 84.5 | 2.78 | ||||
| 3 | Minimum | 336 | 5.67 | 1.69 | ||||
| 4 | Minimum | 313 | 6.71 | 2.15 | ||||
| All | 0.90 | 9.48 | ||||||
| Interplate | 1 | 1 & 2 | Maximum | 3,250 | 147 | 4.53 | ||
| 3 & 4 | Minimum | 385 | 14.7 | 3.82 | ||||
| 2 | 1 & 2 | Maximum | 3,470 | 74.6 | 2.15 | |||
| 3 & 4 | Minimum | 312 | 10.3 | 3.30 | ||||
| 3 | 1 & 2 | Maximum | 3,070 | 80.3 | 2.62 | |||
| 3 & 4 | Minimum | 324 | 13.0 | 4.00 | ||||
| Day to day | 1&2 | All | Maximum | 3,360 | 160 | 4.77 | ||
| Minimum | 348 | 38.7 | 11.1 | |||||
| 2&3 | All | Maximum | 3,270 | 216 | 6.60 | |||
| Minimum | 318 | 13.3 | 4.18 | |||||
| 1&3 | All | Maximum | 3,160 | 150 | 4.74 | |||
| Minimum | 355 | 33.3 | 9.39 |
Table 3.
PP5C Assay Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Enzyme mix | 100 μL | PP5C in 1.5× assay buffer |
| 2 | Test compounds, control inhibitor, or vehicle control | 1.5 μL | Dissolved in DMSO at 100× final concentration |
| 3 | Substrate mix | 48.5 μL | 155 μM OMFP in water |
| 4 | Incubation time | 30 min | Room temperature |
| 5 | Stop solution | 50 μL | 1 M potassium phosphate (pH 10) |
| 6 | Assay readout | Ex: 485 nm, Em: 525 nm | Fluorescence of OMF product |
Step Notes
1. Dispensed with stepper pipettor and positive-displacement tip.
2. Dispensed with single-channel pipettor. About 1.5 μL of 10 mM cantharidin was added to low-signal control wells. About 1.5 μL of DMSO was added to high-signal control wells.
3. Dispensed with multichannel pipettor.
4. Final assay mix in 150 μL total volume (30 mM HEPES [pH 7], 0.01% Triton X-100, 0.3 mg/mL BSA, 1 mM sodium ascorbate, 0.1 mM MnCl2, 1 mM DTT, 50 μM OMFP, and 350 pM PP5C).
5. Dispensed with multichannel pipettor.
6. SpectraMax® M5: 515-nm emission filter.
Table 4.
PP1α Assay Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Enzyme mix | 100 μL | PP1α in 1.5× assay buffer |
| 2 | Test compounds, control inhibitor, or vehicle control | 1.5 μL | Dissolved in DMSO at 100× final concentration |
| 3 | Substrate mix | 48.5 μL | 309 μM OMFP in water |
| 4 | Incubation time | 30 min | Room temperature |
| 5 | Stop solution | 50 μL | 1 M potassium phosphate (pH 10) |
| 6 | Assay readout | Ex: 485 nm, Em: 525 nm | Fluorescence of OMF product |
Step Notes
1. Dispensed with stepper pipettor and positive-displacement tip.
2. Dispensed with single-channel pipettor. About 1.5 μL of 10 mM cantharidin was added to low-signal control wells. About 1.5 μL of DMSO was added to high-signal control wells.
3. Dispensed with multichannel pipettor.
4. Final assay mix in 150 μL total volume (30 mM HEPES [pH 7], 0.01% Triton X-100, 0.3 mg/mL BSA, 1 mM sodium ascorbate, 1 mM MnCl2, 1 mM DTT, 50 μM OMFP, and 3 nM PP1α).
5. Dispensed with multichannel pipettor.
6. SpectraMax® M5: 515-nm emission filter.
Assay Miniaturization and uHTS
The assays described previously are easily scalable, and, with minor modifications, were miniaturized for compatibility with a high-density, 1,536-well format. For primary screening, compounds (>315,000) from the Molecular Libraries Small Molecule Repository were tested in singlicate at a final concentration of 6.7 μM according to the following protocols.
PP5C assay
Three microliters of assay buffer (40 mM HEPES, pH 7.0; 0.13 mg/mL BSA; 0.013% Triton-X100; 1.3 mM DTT; 1.3 mM sodium ascorbate; and 0.13 mM MnCl2) containing 310 pM PP5c was dispensed into each well of a 1,536-well microtiter plate. Next, 27 nL of test compound in DMSO, PP5C inhibitor (cantharidin: 100 μM final concentration) in DMSO, or DMSO alone (0.7% final concentration) was added to the appropriate wells. The plates were incubated for 10 min at room temperature, and the assay was started by dispensing 1 μL of OMFP (200 μM in 3 mM HCl) to each well. After 30 min of incubation at room temperature, 2 μL of 300 mM potassium phosphate at pH 10.0 was added to each well to stop the assay. Well fluorescence was measured on a PerkinElmer Viewlux® using fluorescein filters: excitation wavelength of 480 nm (with 20-nm bandwidth) and emission wavelength of 540 nm (with 20-nm bandwidth).
PP1α assay
Three microliters of assay buffer (40 mM HEPES, pH 7.0; 0.13 mg/mL BSA; 0.013% Triton X-100; 1.3 mM DTT; 1.3 mM sodium ascorbate; and 1.3 mM MnCl2) containing 670 pM PP1α was dispensed into each well of a 1,536-well microtiter plate. Next, 27 nL of test compound in DMSO, PP5C inhibitor (cantharidin: 100 μM final concentration) in DMSO, or DMSO alone (0.7% final concentration) was added to the appropriate wells. The plates were incubated for 10 min at room temperature, and the assay was started by dispensing 1 μL of OMFP (400 μM in 3 mM HCl) to each well. After 30 min of incubation at room temperature, 2 μL of 300 mM potassium phosphate at pH 10.0 was added to each well to stop the assay. Well fluorescence was measured on a PerkinElmer Viewlux using fluorescein filters: excitation wavelength of 480 nm (with 20-nm bandwidth) and emission wavelength of 540 nm (with 20-nm bandwidth).
The % inhibition for each compound was then calculated as follows:
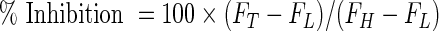 |
where FT is the measured FLINT signal of the well containing test compound. FH is the median FLINT of the high-inhibition control wells containing 100 μM cantharidin. FL is the median FLINT of the low-inhibition control wells containing the DMSO vehicle (0.7%).
A mathematical algorithm was used to determine nominally inhibiting compounds in the primary screens. Two values were calculated: (1) the average percent inhibition of all compounds tested, and (2) three times their standard deviation. The sum of these two values was used as a cutoff parameter; that is, any compound that exhibited greater % inhibition than the cutoff parameter was declared hits. The PP1α and PP5C assays proved to be robust and sensitive. PP5C run statistics: n=257 plates; 315,000 compounds tested; average Z′=0.95±0.01; average Z=0.93±0.05; average S:B=12.54±0.80; and hit cutoff=9.61% inhibition, hit rate=0.18% (564 hits). The data from this uHTS have been deposited in PubChem (AID: 1987). PP1α run statistics: n=257 plates; 315,000 compounds tested; average Z′=0.93±0.03; average Z=0.86±0.04; average S:B=6.04±0.16; and hit cutoff=15.17% inhibition, hit rate=0.90% (2,841 hits). The data from this uHTS have been deposited in PubChem (AID: 2235).
Those hits from the primary screens deemed chemically suitable for further probe development and subsequently validated by confirmatory OMFP-based screens (assays in triplicate at 6.7 μM as well as dose–response studies) were then purchased or synthesized for low-throughput in-house confirmatory screening. Purchased or synthesized compounds were first retested with OMFP assays and active compounds were subsequently validated with 32P or 33P-labeled phosphoprotein substrates. The primary and confirmatory screens as well as further probe development efforts will be described in much more detail in future publications.
Conclusions
Here we describe the development and optimization of homogeneous FLINT-based biochemical screening assays for PP1α and PP5C in a low-density format utilizing the artificial substrate OMFP. The optimized assay conditions yield a large signal window and low variability for both enzymes, with typical Z′ values >0.8 for PP1α and >0.9 for PP5C. The assay is quite tolerant of additives, such as DMSO (the typical vehicle for small-molecule libraries) and Triton X-100 (an additive commonly used to suppress nonspecific inhibition). Additionally, diluted enzyme/buffer solutions were shown to be quite stable (to at least 24 h at room temperature) and, therefore, are compatible with the long shifts typical of daily operations within an HTS campaign. As discussed previously, these assays are thus suitable for HTS and have been successfully miniaturized to higher density formats and used to screen the Molecular Libraries Small Molecule Repository for inhibitors of PP5 and PP1α (see PubChem AID 1987 and 2235). Conversely, these assays may also be employed as robust, low-cost counterscreens in ligand discovery campaigns conducted upon other phosphatase targets.
Abbreviations Used
- BSA
bovine serum albumin
- DiFMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- DMSO
dimethyl sulfoxide
- FLINT
fluorescence intensity
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- MBP
maltose binding protein
- OMF
O-methylfluorescein
- OMFP
3-O-methylfluorescein phosphate
- TEV
tobacco etch virus
- TPR
tetratricopeptide repeat
- uHTS
ultra high-throughput screening
Disclosure Statement
No competing financial interests exist.
References
- 1.Cohen PT: Novel protein serine/threonine phosphatases: variety is the spice of life. Trends Biochem Sci 1997;22:245–251 [DOI] [PubMed] [Google Scholar]
- 2.Sefton BM: Overview of protein phosphorylation. Curr Protoc Cell Biol 2001;14.1:14.1.1–14.1.3 [DOI] [PubMed] [Google Scholar]
- 3.Ha HT, Lee JS, Urba S, et al. : A phase II study of imatinib in patients with advanced anaplastic thyroid cancer. Thyroid Off J Am Thyroid Assoc 2010;20:975–980 [DOI] [PubMed] [Google Scholar]
- 4.Au A, Baba AA, Azlan H, Norsa'adah B, Ankathil R: Clinical impact of ABCC1 and ABCC2 genotypes and haplotypes in mediating imatinib resistance among chronic myeloid leukaemia patients. J Clin Pharm Ther 2014;39:685–690 [DOI] [PubMed] [Google Scholar]
- 5.Swingle M, Ni L, Honkanen RE: Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol Clifton NJ 2007;365:23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azzi JR, Sayegh MH, Mallat SG: Calcineurin inhibitors: 40 years later, can't live without. J Immunol Baltim Md 1950 2013;191:5785–5791 [DOI] [PubMed] [Google Scholar]
- 7.Honkanen RE, Golden T: Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem 2002;9:2055–2075 [DOI] [PubMed] [Google Scholar]
- 8.Swingle MR, Amable L, Lawhorn BG, et al. : Structure-activity relationship studies of fostriecin, cytostatin, and key analogs, with PP1, PP2A, PP5, and(beta12-beta13)-chimeras (PP1/PP2A and PP5/PP2A), provide further insight into the inhibitory actions of fostriecin family inhibitors. J Pharmacol Exp Ther 2009;331:45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lê LH, Erlichman C, Pillon L, et al. : Phase I and pharmacokinetic study of fostriecin given as an intravenous bolus daily for five consecutive days. Invest New Drugs 2004;22:159–167 [DOI] [PubMed] [Google Scholar]
- 10.De Jong RS, Mulder NH, Uges DR, et al. : Phase I and pharmacokinetic study of the topoisomerase II catalytic inhibitor fostriecin. Br J Cancer 1999;79:882–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen PTW: Protein phosphatase 1- Targeted in many directions. J Cell Sci 2002;115:241–256 [DOI] [PubMed] [Google Scholar]
- 12.Ceulemans H, Bollen M: Functional Diversity of Protein Phosphatase-1, a Cellular Economizer and Reset Button. Physiol Rev 2004;84:1–39 [DOI] [PubMed] [Google Scholar]
- 13.Skarra DV, Goudreault M, Choi H, et al. : Label-free quantitative proteomics and SAINT analysis enable interactome mapping for the human Ser/Thr protein phosphatase 5. Proteomics 2011;11:1508–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Endo S, Zhou X, Connor J, Wang B, Shenolikar S: Multiple structural elements define the specificity of recombinant human inhibitor-1 as a protein phosphatase-1 inhibitor. Biochemistry (Mosc.) 1996;35:5220–5228 [DOI] [PubMed] [Google Scholar]
- 15.Alessi DR, Street AJ, Cohen P, Cohen PT: Inhibitor-2 functions like a chaperone to fold three expressed isoforms of mammalian protein phosphatase-1 into a conformation with the specificity and regulatory properties of the native enzyme. Eur J Biochem FEBS 1993;213:1055–1066 [DOI] [PubMed] [Google Scholar]
- 16.Beullens M, Vulsteke V, Van Eynde A, Jagiello I, Stalmans W, Bollen M: The C-terminus of NIPP1 (nuclear inhibitor of protein phosphatase-1) contains a novel binding site for protein phosphatase-1 that is controlled by tyrosine phosphorylation and RNA binding. Biochem J 2000;352 Pt 3:651–658 [PMC free article] [PubMed] [Google Scholar]
- 17.Heroes E, Lesage B, Görnemann J, Beullens M, Van Meervelt L, Bollen M: The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J 2013;280:584–595 [DOI] [PubMed] [Google Scholar]
- 18.Swingle MR, Honkanen RE, Ciszak EM: Structural basis for the catalytic activity of human serine/threonine protein phosphatase-5. J Biol Chem 2004;279:33992–33999 [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Roe SM, Cliff MJ, et al. : Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. EMBO J 2005;24:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee A, Periyasamy S, Wolf IM, et al. : Control of glucocorticoid and progesterone receptor subcellular localization by the ligand-binding domain is mediated by distinct interactions with tetratricopeptide repeat proteins. Biochemistry (Mosc.) 2008;47:10471–10480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Connarn JN, Assimon VA, Reed RA, et al. : The molecular chaperone Hsp70 activates protein phosphatase 5 (PP5) by binding the tetratricopeptide repeat (TPR) domain. J Biol Chem 2014;289:2908–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramsey AJ, Chinkers M: Identification of potential physiological activators of protein phosphatase 5. Biochemistry (Mosc.) 2002;41:5625–5632 [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi Y, Katoh H, Mori K, Negishi M: Galpha(12) and Galpha(13) interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr Biol CB 2002;12:1353–1358 [DOI] [PubMed] [Google Scholar]
- 24.Chen MX, Cohen PT: Activation of protein phosphatase 5 by limited proteolysis or the binding of polyunsaturated fatty acids to the TPR domain. FEBS Lett 1997;400:136–140 [DOI] [PubMed] [Google Scholar]
- 25.Gregório LK, Esteves SLC, Fardilha M: Protein phosphatase 1 catalytic isoforms: specificity toward interacting proteins. Transl Res J Lab Clin Med 2014. [Epub ahead of print]; DOI: 10.1016/j.trsl.2014.07.001 [DOI] [PubMed] [Google Scholar]
- 26.LaPointe NE, Morfini G, Pigino G, et al. : The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res 2009;87:440–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fardilha M, Esteves SLC, Korrodi-Gregório L, Da Cruz E.Silva OAB, Da Cruz E.Silva EF: The physiological relevance of protein phosphatase 1 and its interacting proteins to health and disease. Curr Med Chem 2010;17:3996–4017 [DOI] [PubMed] [Google Scholar]
- 28.Golden T, Aragon IV, Rutland B, et al. : Elevated levels of Ser/Thr protein phosphatase 5 (PP5) in human breast cancer. Biochim Biophys Acta 2008;1782:259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuo Z, Urban G, Scammell JG, et al. : Ser/Thr protein phosphatase type 5 (PP5) is a negative regulator of glucocorticoid receptor-mediated growth arrest. Biochemistry (Mosc.) 1999;38:8849–8857 [DOI] [PubMed] [Google Scholar]
- 30.Golden T, Swingle M, Honkanen RE: The role of serine/threonine protein phosphatase type 5 (PP5) in the regulation of stress-induced signaling networks and cancer. Cancer Metastasis Rev 2008;27:169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaughan CK, Mollapour M, Smith JR, et al. : Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol Cell 2008;31:886–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyata Y, Nakamoto H, Neckers L: The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des 2013;19:347–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou G, Golden T, Aragon IV, Honkanen RE: Ser/Thr protein phosphatase 5 inactivates hypoxia-induced activation of an apoptosis signal-regulating kinase 1/MKK-4/JNK signaling cascade. J Biol Chem 2004;279:46595–46605 [DOI] [PubMed] [Google Scholar]
- 34.Wechsler T, Chen BPC, Harper R, et al. : DNA-PKcs function regulated specifically by protein phosphatase 5. Proc Natl Acad Sci U S A 2004;101:1247–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urban G, Golden T, Aragon IV, et al. : Identification of a functional link for the p53 tumor suppressor protein in dexamethasone-induced growth suppression. J Biol Chem 2003;278:9747–9753 [DOI] [PubMed] [Google Scholar]
- 36.Hill HD, Summer GK, Waters MD: An automated fluorometric assay for alkaline phosphatase using 3-O-methylfluorescein phosphate. Anal Biochem 1968;24:9–17 [DOI] [PubMed] [Google Scholar]
- 37.Johnston PA, Foster CA, Tierno MB, et al. : Cdc25B dual-specificity phosphatase inhibitors identified in a high-throughput screen of the NIH compound library. Assay Drug Dev Technol 2009;7:250–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnston PA, Foster CA, Shun TY, et al. : Development and implementation of a 384-well homogeneous fluorescence intensity high-throughput screening assay to identify mitogen-activated protein kinase phosphatase-1 dual-specificity protein phosphatase inhibitors. Assay Drug Dev Technol 2007;5:319–332 [DOI] [PubMed] [Google Scholar]
- 39.Gottlin EB, Xu X, Epstein DM, et al. : Kinetic analysis of the catalytic domain of human cdc25B. J Biol Chem 1996;271:27445–27449 [DOI] [PubMed] [Google Scholar]
- 40.Davis RL, Robinson JD: Characteristics of 3-O-methylfluorescein phosphate hydrolysis by the (Na+ +K+)-ATPase. J Bioenerg Biomembr 1988;20:571–584 [DOI] [PubMed] [Google Scholar]
- 41.Gergely P, Erdödi F, Bot G: Heparin inhibits the activity of protein phosphatase-1. FEBS Lett 1984;169:45–48 [DOI] [PubMed] [Google Scholar]
- 42.Zhang AJ, Bai G, Deans-Zirattu S, Browner MF, Lee EY: Expression of the catalytic subunit of phosphorylase phosphatase (protein phosphatase-1) in Escherichia coli. J Biol Chem 1992;267:1484–1490 [PubMed] [Google Scholar]
- 43.Szöór B, Dombrádi V, Gergely P, Fehér Z: Purification and characterization of the catalytic subunit of protein phosphatase 1 from Neurospora crassa. Acta Biol Hung 1997;48:289–302 [PubMed] [Google Scholar]
- 44.Raran-Kurussi S, Waugh DS: The ability to enhance the solubility of its fusion partners is an intrinsic property of maltose-binding protein but their folding is either spontaneous or chaperone-mediated. PloS One 2012;7:e49589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kupcho K, Hsiao K, Bulleit B, Goueli SA: A homogeneous, nonradioactive high-throughput fluorogenic protein phosphatase assay. J Biomol Screen 2004;9:223–231 [DOI] [PubMed] [Google Scholar]
- 46.Gee KR, Sun WC, Bhalgat MK, et al. : Fluorogenic substrates based on fluorinated umbelliferones for continuous assays of phosphatases and beta-galactosidases. Anal Biochem 1999;273:41–48 [DOI] [PubMed] [Google Scholar]
- 47.Ni L, Swingle M, Bourgeois AC, Honkanen RE: High yield expression of serine/threonine protein phosphatase type 5, and a fluorescent assay suitable for use in the detection of catalytic inhibitors. Assay Drug Dev Technol 2007;5:645–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tierno MB, Johnston PA, Foster C, et al. : Development and optimization of high-throughput in vitro protein phosphatase screening assays. Nat Protoc 2007;2:1134–1144 [DOI] [PubMed] [Google Scholar]
- 49.Wu G, Yuan Y, Hodge CN: Determining appropriate substrate conversion for enzymatic assays in high-throughput screening. J Biomol Screen 2003;8:694–700 [DOI] [PubMed] [Google Scholar]
- 50.Zhang JH, Chung TD, Oldenburg KR: A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4:67–73 [DOI] [PubMed] [Google Scholar]
- 51.Bright GR, Fisher GW, Rogowska J, Taylor DL: Fluorescence ratio imaging microscopy: temporal and spatial measurements of cytoplasmic pH. J Cell Biol 1987;104:1019–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoff RH, Mertz P, Rusnak F, Hengge AC: The transition state of the phosphoryl-transfer reaction catalyzed by the lambda Ser/Thr protein phosphatase. J Am Chem Soc 1999;121:6382–6390 [Google Scholar]
- 53.McWhirter C, Lund EA, Tanifum EA, et al. : Mechanistic study of protein phosphatase-1 (PP1), a catalytically promiscuous enzyme. J Am Chem Soc 2008;130:13673–13682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hengge AC, Martin BL: Isotope effect studies on the calcineurin phosphoryl-transfer reaction: transition state structure and effect of calmodulin and Mn2+. Biochemistry (Mosc.) 1997;36:10185–10191 [DOI] [PubMed] [Google Scholar]
- 55.McGovern SL, Helfand BT, Feng B, Shoichet BK: A specific mechanism of nonspecific inhibition. J Med Chem 2003;46:4265–4272 [DOI] [PubMed] [Google Scholar]
- 56.Pohjala L, Tammela P: Aggregating behavior of phenolic compounds—a source of false bioassay results? Mol Basel Switz 2012;17:10774–10790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peña I, Domínguez JM: Thermally denatured BSA, a surrogate additive to replace BSA in buffers for high-throughput screening. J Biomol Screen 2010;15:1281–1286 [DOI] [PubMed] [Google Scholar]
- 58.Watanabe T, Huang HB, Horiuchi A, et al. : Protein phosphatase 1 regulation by inhibitors and targeting subunits. Proc Natl Acad Sci U S A 2001;98:3080–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McAvoy T, Nairn AC: Serine/threonine protein phosphatase assays. Curr Protoc Mol Biol Ed Frederick M Ausubel Al 2010;Chapter 18:Unit1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kirby AJ, Varvoglis AG: The Reactivity of phosphate esters. Monoester hydrolysis. J Am Chem Soc 1967;89:415–423 [Google Scholar]
- 61.Lad C, Williams NH, Wolfenden R: The rate of hydrolysis of phosphomonoester dianions and the exceptional catalytic proficiencies of protein and inositol phosphatases. Proc Natl Acad Sci U S A 2003;100:5607–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]