

Abstract
MyD88 is the canonical adaptor for inflammatory signaling pathways downstream of members of the Toll-like receptor (TLR) and interleukin-1 (IL-1) receptor families. MyD88 links IL-1 receptor (IL-1R) or TLR family members to IL-1R-associated kinase (IRAK) family kinases via homotypic protein-protein interaction. Activation of IRAK family kinases leads to a variety of functional outputs, including the activation of nuclear factor-kappa B (NFκB), mitogen-activated protein kinases, and activator protein 1, making MyD88 a central node of inflammatory pathways. As more details of MyD88-dependent signaling have been elucidated, it has become clear that the functions of this critical signaling component can be influenced by multiple interaction partners in distinct subcellular compartments. In this review, we will focus on recent developments in the understanding of the assembly of MyD88 signaling complexes and the mechanisms leading to the diversification of MyD88-based signaling.
Identification of MyD88 as a proximal signaling adaptor
MyD88 was first described in 1990 as a gene upregulated during IL-6-induced myeloid differentiation [1], but its homology to the cytosolic domains of Drosophila Toll and mammalian IL-1Rs (whose homology had already been noted [2,3]) was not appreciated for another 4 years [4]. Eventually, MyD88 was implicated in signaling downstream of IL-1R and mammalian TLRs [5–7]. The C-terminal TIR (Toll IL-1R) domain mediates the interaction with other TIR domain-containing proteins (receptors or adaptors); the N-terminal death domain (DD) associates with the IRAK family members through homotypic DD interactions (Figure 1). Consistent with these roles, overexpression of the DD of MyD88 leads to spontaneous activation of NFκB and c-Jun N-terminal kinase (JNK), whereas the TIR domain can act as a dominant negative [5,7,8]. An intermediate (INT) domain of MyD88 links the TIR and DD. Although this domain does not appear to be involved in the direct interactions described above, it is necessary for IRAK4 activation. In fact, MyD88s, a splice variant of MyD88 lacking the INT domain, is induced upon activation and acts as a dominant negative form of MyD88 [9,10].
Figure 1. Organization of MyD88 domains.
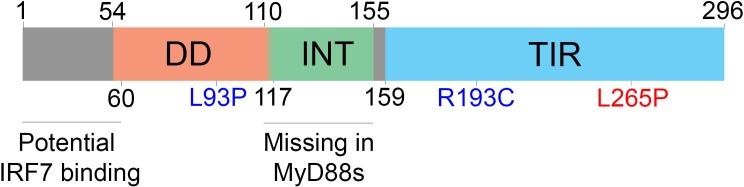
MyD88 is composed of three main domains: a death domain (DD) (54 to 109), intermediate domain (INT) (110 to 155), and Toll-interleukin-1 receptor domain (TIR) (159 to 296). Although the DD is annotated as 54 to 109, proper folding of the DD seems to require amino acids 110 to 117. Point mutations inducing a loss (blue) or gain (red) of function are indicated by amino acid number. The site implicated in interferon regulatory factor 7 (IRF-7) binding (1 to 59) and the domain lost in the splice variant MyD88s (110 to 155) are indicated in black.
Mice lacking MyD88 were reported in 1998, and the initial analysis of these mice confirmed the importance of this adaptor downstream of the IL-1R family [11]. In the following year, MyD88-deficient mice were shown to lack responsiveness to lipopolysaccharide (LPS), the ligand for TLR4 [12]. Although this muted response protected MyD88-deficient mice from endotoxic shock, MyD88-deficient cells retained some residual signaling when stimulated with LPS, suggesting MyD88-independent signaling mechanisms downstream of TLR4. Ultimately, this observation led to the discovery of TIR domain-containing adapter-inducing interferon-beta (TRIF) as an adaptor for TLR3 and 4 [13,14]. MyD88-deficient mice have been used extensively as a model for TLR deficiency and are susceptible to a large variety of bacterial pathogens or parasites [15,16]. However, these mice also lack the ability to signal through IL-1R family members. Comparisons between MyD88- and Caspase-1/11-deficient mice have been used to control for lack of IL-1R and IL-18R signaling in MyD88-deficient animals [17], but mice deficient for all TLR functions were recently described and may prove useful for evaluating the role of TLRs independently of receptors for IL-1 family cytokines [18].
Several inactivating mutations in MyD88 have also been identified in humans with recurrent infections with pyogenic bacteria [19]. These mutations, as well as some rare missense polymorphisms [20], are associated with reduced IRAK4 activation, leading to impaired responses through TLRs and IL-1 family members. MyD88-deficient humans do not appear abnormally susceptible to many viral, fungal, or parasitic infections. Whether MyD88 is somehow less central to human immunity is complicated by the vaccinations and antibiotic regimens received by these patients. As our understanding of innate immunity has grown, it has become clear that other MyD88-independent signals can mediate innate and adaptive immune responses to many pathogens, especially viruses. It is possible that these pathways compensate for MyD88 deficiency [21].
Initiation of a MyD88 signaling complex: from receptors to IRAK kinases
The first structure of a TIR domain was elucidated shortly after the first descriptions of the function of MyD88 as a signaling adaptor [22]. This first study solved the structure of the human TLR1 TIR domain and demonstrated that TIR domains can oligomerize, but with an affinity in the millimolar range. Of note, additional studies showed potentially significant differences in the structures of other TIR domains, in particular the BB loop of the TIR domain of MyD88 [23,24]. This structural difference results in the inability of the TIR domain of MyD88 to dimerize with itself, which may be critical to prevent ligand-independent activation.
Because of the relative weakness of TIR-TIR interactions, it has been postulated that conformational changes upon TLR ligand recognition bring TIR domains in close proximity. This change of avidity would allow for the initiation of MyD88 signaling. In the case of TLR9, it has indeed been shown that the receptor occurs as a homodimer and that ligand binding brings the cytoplasmic tails containing the TIR domains in close proximity [25]. The structure of the extracellular domain of TLR3 has been determined in the absence [26,27] and presence [28] of ligand. Although the TIR domain was not part of the crystallized protein, modeling shows that ligand binding by a TLR3 homodimer brings the TIR domains in close proximity. Similarly, the structure of the extracellular domains of a TLR1/2 heterodimer formed in the presence of ligand suggests a close association between the TIR domains of each TLR [29]. The recently solved structure of the extracellular domain of TLR8 [30] also supports this idea. Indeed, binding of a TLR8 homodimer to its ligand induces a conformational change in the extracellular domains, bringing the membrane-proximal regions together. This conformational change most likely promotes the association of the transmembrane domains and cytoplasmic tails that contain the TIR domain, although those were not included in the structure. Together, all of the structures of TLR and their ligands solved so far support the model outlined above.
MyD88 can associate directly with receptor TIR domains, so it was unexpected when the TIR-domain-containing adaptor protein (TIRAP [also named Mal]), which contains its own TIR domain and a lipid-binding motif, was shown to facilitate the association of MyD88 and TLR4 [31,32]. Initial studies showed that TIRAP was required for TLR4 and TLR2 signaling but was dispensable for TLR3, 7, or 9 signaling [33,34]. Of note, the requirement for TIRAP does not appear absolute and can depend on ligand concentration as well as cell type [33,35]. However, recent evidence suggests that TIRAP can also be involved in TLR9 signaling [36]. Because the requirement for TIRAP during TLR9 signaling was evident only with particular ligands (viruses but not the more stable and commonly used phosphorothioate-linked CpG DNA) and cell types (immortalized macrophages but not the more phagocytic bone marrow-derived macrophages), it is possible that TIRAP is necessary only when few TLRs are engaged, suggesting that a certain threshold of TIR domains must be achieved to trigger downstream MyD88 signaling. It is also possible that different types of ligands induce different degrees of conformational changes and TIR clustering in TLR9 homodimers. In the case of TLR4, where the requirement for TIRAP is most evident, several non-exclusive hypotheses have been proposed to explain the role of TIRAP. TIRAP appears to be necessary because of its membrane localization and can be bypassed by targeting MyD88 directly to the membrane [37]. On the other hand, there is evidence for a preferential interaction between TIRAP and MyD88 TIRs but not TLR4 and MyD88 TIRs [24].
Clustering of TIR domains is thought to provide a stable platform for MyD88 binding and oligomerization. This oligomerization of MyD88 was apparent in early overexpression studies [8], but the complex formed by MyD88 DDs and IRAK DD was crystallized only recently [38]. This structure, termed the Myddosome [39], is composed of a ring of six MyD88 DDs that serve as a seed for assembly of a ring of four IRAK4 DDs and ultimately four IRAK2 DDs in a helix. This helical assembly was observed in other DD-based oligomeric complexes, such as the PIDDosome [40] and the death-inducing signaling complex (DISC) [41]. All assemblies rely on three distinct types of interactions between DDs, allowing sequential assembly of a large complex. Interestingly, the Myddosome structure was obtained in the absence of most of the INT domain (residues 1 to 117, the annotated INT domain starts at residue 110), but a similar stoichiometry was observed in the presence of the full INT domain (residues 1 to 157) [39]. The structure of the Myddosome provides important insight into the initiation of MyD88 signaling. However, because the structure was crystallized in the absence of TIR domains, several questions remain unanswered. For example, it is unclear whether all MyD88 molecules in the Myddosome are bound to a TLR or IL-1R through their TIR domain or whether the clustering of two TIR domains is enough to allow stable DD association that seeds binding of additional MyD88 DDs and assembly of a full Myddosome. It will also be interesting to investigate whether Myddosomes can connect multiple dimerized TLRs, as suggested by recent work on tumor necrosis factor (TNF)-induced DD complexes.
In addition to showing MyD88 functions induced by TIR-containing receptors, a few studies have shown a role for MyD88 downstream of receptors that do not contain TIR domains, like interferon gamma receptor (IFNγR) [42] and TNF receptor superfamily member 13b (also named TACI) [43]. In those cases, it is unclear whether the same molecular mechanisms are involved in MyD88 signaling, as TACI but not IFNγR appears to signal through the usual partners used during TLR/IL-1R signaling.
On the other hand, although most of the MyD88-dependent signaling is mediated by DD-dependent IRAK recruitment, MyD88 can also directly bind to interferon regulatory factor 7 (IRF-7) to promote type I IFN production [44]. This association is mediated by the N-terminal portion of MyD88 but is not dependent on a DD fold (as a MyD88 Δ60-296 can still interact with IRF-7).
It is interesting to note that a gain-of-function variant of MyD88 (L256P) has been described in some classes of B-cell lymphomas [45]. This mutant can spontaneously assemble and lead to persistent NFκB and signal transducer and activator of transcription 3 (STAT3) activation. The mutation lies within the TIR domain of MyD88, suggesting that Myddosome assembly and signal initiation may be affected. At least two models could explain the oncogenic potential of this mutation: either the mutation promotes the direct binding of MyD88 TIR homodimers, allowing efficient self-assembly, or the mutation enhances the affinity of MyD88 for TLR or IL-1R TIR domains. Importantly, this mutation was observed in the absence of well-characterized oncogenic events (for example, Myc translocation), suggesting that this mutant of MyD88 can act as an oncogenic driver [46]. Of note, it is also possible that the overexpression of wildtype MyD88 is sufficient to promote tumor growth in other cell types [47]. In light of the oncogenic potential of MyD88, it may be interesting to revisit its role during tumorigenesis [48] (for example, by comparing MyD88-deficient mice to TLR-deficient mice).
Location and outcomes of signaling
MyD88 acts as a central hub in inflammatory responses [49] and can induce signaling from several receptors, located either at the plasma membrane or in endosomes. Additionally, MyD88 signaling can lead to the production of pro- or anti-inflammatory cytokines as well as type I IFNs [50]. Two TNF receptor-associated factors (TRAFs)—TRAF3 and TRAF6—can be activated by IRAK family members and lead to the production of type I IFN or pro-inflammatory cytokines, respectively [51,52]. Although the MyD88-IRAK4-IRAK2 axis of the pathway can theoretically interact with both TRAF3 and TRAF6 complexes, it is likely that only some receptors, adaptors, or signaling molecules are available at given subcellular locations or in specific cell types. Here, we will discuss some established examples as well as new research avenues in the interplay between the initiation and localization of MyD88 signaling.
TLR4 is unique among TLRs because it can signal both through MyD88 to induce pro-inflammatory cytokines via NFκB and through TRIF to induce type I IFN through IRF3 [13]. Interestingly, it was found that those signaling events are spatially separated, with TIRAP/MyD88 signaling occurring from the surface whereas signaling from TRIF-related adaptor molecule/TRIF (TRAM/TRIF) occurs from endosomes [53]. In that specific case, receptor internalization induces a switch from MyD88 to TRIF signaling because of a change of adaptors, from TIRAP to the other TIR-containing adaptor TRAM [54]. Because of the distinct lipid composition of the plasma membrane and endosomes, and of the affinity of TIRAP for the plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PI[4,5]P2) [37], internalization limits TIRAP affinity and MyD88 activation. Given the promiscuity of TIRAP binding to lipids [36], it is unclear whether the difference in affinities of TIRAP for PI(4,5)P2 and phosphatidylinositol (3,4,5)-trisphosphate (PI[3,4,5]P3) is sufficient to mediate the switch from TIRAP to TRAM or whether other mechanisms can act simultaneously to promote TRAM association.
Aside from the case of TLR4, there is evidence that the site of ligand recognition influences the signaling outcome even for receptors that rely solely on MyD88. The existence of distinct signaling pathways under the control of a single receptor is evident in the case of TLR9, which recognizes CpG motifs and can trigger the production of pro-inflammatory cytokines or type I IFN. It has been appreciated for quite some time that different classes of CpG motifs (A or B) favor type I IFN or pro-inflammatory cytokine production, respectively [55]. Analysis of plasmacytoid dendritic cells from adaptor protein 3 (AP-3)-deficient mice revealed that this adaptor complex is critical for type I IFN production but dispensable or detrimental for pro-inflammatory cytokine production [56]. Similarly, a localization motif in the cytoplasmic tail of TLR9 is required for pro-inflammatory cytokine but not type I IFN production [57]. Again, lipid composition of the signaling compartment appears to be critical for specificity, as fusion of TRAF3 (a critical signaling node downstream of IRAK kinases) to a pleckstrin homology (PH) domain that allows localization to phosphatidylinositol 3,5-bisphosphate (PI[3,5]P2)-containing vesicles can bypass the requirement for AP-3 [56]. Although these bifurcation pathways have mostly been identified in the case of TLR9, they may occur during signaling through other receptors and may be underlying the ability of TLR2 to induce type I IFN during viral infection in inflammatory monocytes [58].
In addition to the spatial regulation of adaptors (such as TIRAP) and signaling molecules (such as TRAF3), it is clear that TLRs themselves are spatially regulated [59]. The chaperone Unc93b is responsible for the trafficking of all endosomal TLRs [60,61], but some TLRs can also use their own localization motifs for trafficking (such as TLR7 and AP-4 [61]), providing a basis for differential trafficking of distinct receptors. Ligands themselves can also be trafficked through different cellular compartments depending on their nature, as is the case with DNA in the form of immune complexes, for example [62,63]. A key challenge for future years is to understand the interplay between the trafficking of receptors, adaptors, and ligands and how localization impacts signaling initiation and the interaction between MyD88 and other specialized signaling components.
Concluding remarks
Overall, since its discovery about 20 years ago, MyD88 has emerged as a central and well-conserved node of innate immune responses through its role as a bridge between TIR-containing receptors and DD-containing kinases. The characterization of the Myddosome suggests a clear molecular mechanism for signal transduction upon the local clustering of TIR domain-containing receptors. MyD88 signaling can lead to distinct outputs depending on the context, usually leading to pro-inflammatory cytokine or type I IFN production. Distinct pathways downstream of IRAK family members regulate these outputs, and the outcome of signaling can be influenced by the cell type and location of signal initiation. In the years ahead, a more dynamic view of MyD88 interaction networks, from ligands to receptors to downstream signaling components, may provide us with additional insight into the regulation of diverse MyD88 functions.
Acknowledgments
Research in the Barton Lab is supported by the National Institutes of Health, the Lupus Research Institute, and the Burroughs Wellcome Fund. Jacques Deguine is supported by a Long-Term Fellowship from the Human Frontier Science Program (LT-000081/2013-L).
Abbreviations
- AP-3
adaptor protein 3
- DD
death domain
- IFNγR
interferon gamma receptor
- IRF
interferon regulatory factor
- IL
interleukin
- INT
intermediate
- IRAK
interleukin-1 receptor-associated kinase
- LPS
lipopolysaccharide
- NFκB
nuclear factor-kappa B
- TIR
Toll interleukin-1 receptor
- TIRAP
TIR-domain-containing adaptor protein
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- TRAF
tumor necrosis factor receptor-associated factor
- TRAM
TRIF-related adaptor molecule
- TRIF
TIR-domain-containing adapter-inducing interferon-beta
Disclosures
The authors declare that they have no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/b/6/97
Contributor Information
Jacques Deguine, Email: jdeguine@berkeley.edu.
Gregory M. Barton, Email: barton@berkeley.edu.
References
- 1.Lord KA, Hoffman-Liebermann B, Liebermann DA. Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene. 1990;5:1095–7. [PubMed] [Google Scholar]
- 2.Gay NJ, Keith FJ. Drosophila Toll and IL-1 receptor. Nature. 1991;351:355–6. doi: 10.1038/351355b0. [DOI] [PubMed] [Google Scholar]
- 3.Schneider DS, Hudson KL, Lin TY, Anderson KV. Dominant and recessive mutations define functional domains of Toll, a transmembrane protein required for dorsal-ventral polarity in the Drosophila embryo. Genes Dev. 1991;5:797–807. doi: 10.1101/gad.5.5.797. [DOI] [PubMed] [Google Scholar]
- 4.Hultmark D. Macrophage differentiation marker MyD88 is a member of the Toll/IL-1 receptor family. Biochem Biophys Res Commun. 1994;199:144–6. doi: 10.1006/bbrc.1994.1206. [DOI] [PubMed] [Google Scholar]
- 5.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–5. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 6.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. Pillars article: MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997. 7: 837-847. J Immunol. 2013;190:5–15. [PubMed] [Google Scholar]
- 7.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–8. doi: 10.1016/S1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 8.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem. 1998;273:12203–9. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 9.Janssens S, Burns K, Vercammen E, Tschopp J, Beyaert R. MyD88S, a splice variant of MyD88, differentially modulates NF-kappaB- and AP-1-dependent gene expression. FEBS Lett. 2003;548:103–7. doi: 10.1016/S0014-5793(03)00747-6. [DOI] [PubMed] [Google Scholar]
- 10.Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–8. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–50. doi: 10.1016/S1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–22. doi: 10.1016/S1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1014540
- 14.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. 2003;4:161–7. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1011894
- 15.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–6. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 16.Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, Medzhitov R, Sher A. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1007396
- 17.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–50. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1001479
- 18.Sivick KE, Arpaia N, Reiner GL, Lee BL, Russell BR, Barton GM. Toll-like receptor-deficient mice reveal how innate immune signaling influences Salmonella virulence strategies. Cell Host Microbe. 2014;15:203–13. doi: 10.1016/j.chom.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku C, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, Vasconcelos J, Sirvent N, Guedes M, Vitor AB, Herrero-Mata MJ, Aróstegui JI, Rodrigo C, Alsina L, Ruiz-Ortiz E, Juan M, Fortuny C, Yagüe J, Antón J, Pascal M, Chang H, Janniere L, Rose Y, Garty B, Chapel H, Issekutzy A, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–6. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1119814
- 20.George J, Motshwene PG, Wang H, Kubarenko AV, Rautanen A, Mills TC, Hill Adrian VS, Gay NJ, Weber Alexander NR. Two human MYD88 variants, S34Y and R98C, interfere with MyD88-IRAK4-myddosome assembly. J Biol Chem. 2011;286:1341–53. doi: 10.1074/jbc.M110.159996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Picard C, Casanova J, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin Microbiol Rev. 2011;24:490–7. doi: 10.1128/CMR.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, Tong L. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408:111–5. doi: 10.1038/35047056. [DOI] [PubMed] [Google Scholar]
- 23.Khan JA, Brint EK, O'Neill Luke AJ, Tong L. Crystal structure of the Toll/interleukin-1 receptor domain of human IL-1RAPL. J Biol Chem. 2004;279:31664–70. doi: 10.1074/jbc.M403434200. [DOI] [PubMed] [Google Scholar]
- 24.Ohnishi H, Tochio H, Kato Z, Orii KE, Li A, Kimura T, Hiroaki H, Kondo N, Shirakawa M. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc Natl Acad Sci USA. 2009;106:10260–5. doi: 10.1073/pnas.0812956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein Dionne CG, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, Espevik T, Golenbock DT. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–9. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1088202
- 26.Bell JK, Botos I, Hall PR, Askins J, Shiloach J, Segal DM, Davies DR. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci USA. 2005;102:10976–80. doi: 10.1073/pnas.0505077102. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1028586
- 27.Choe J, Kelker MS, Wilson IA. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science. 2005;309:581–5. doi: 10.1126/science.1115253. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1027224
- 28.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–81. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1112028
- 29.Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik S, Lee H, Lee J. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–82. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1091080
- 30.Tanji H, Ohto U, Shibata T, Miyake K, Shimizu T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science. 2013;339:1426–9. doi: 10.1126/science.1229159. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/717991295
- 31.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, McMurray D, Smith DE, Sims JE, Bird TA, O'Neill LA. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1000958
- 32.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–41. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1001154
- 33.Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–336. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1011324
- 34.Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–9. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1011323
- 35.Kenny EF, Talbot S, Gong M, Golenbock DT, Bryant CE, O'Neill Luke AJ. MyD88 adaptor-like is not essential for TLR2 signaling and inhibits signaling by TLR3. J Immunol. 2009;183:3642–51. doi: 10.4049/jimmunol.0901140. [DOI] [PubMed] [Google Scholar]
- 36.Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, Iwasaki A, Knipe DM, Kagan JC. A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell. 2014;156:705–16. doi: 10.1016/j.cell.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718278271
- 37.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–55. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 38.Lin S, Lo Y, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–90. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718046819
- 39.Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, Robinson CV, Latz E, Gay NJ. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem. 2009;284:25404–11. doi: 10.1074/jbc.M109.022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007;128:533–46. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1068788
- 41.Wang L, Yang JK, Kabaleeswaran V, Rice AJ, Cruz AC, Park AY, Yin Q, Damko E, Jang SB, Raunser S, Robinson CV, Siegel RM, Walz T, Wu H. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol. 2010;17:1324–9. doi: 10.1038/nsmb.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/6295958
- 42.Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–81. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1031788
- 43.He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, Shan M, Xiong H, Bussel JB, Chiu A, Puel A, Reichenbach J, Marodi L, Döffinger R, Vasconcelos J, Issekutz A, Krause J, Davies G, Li X, Grimbacher B, Plebani A, Meffre E, Picard C, Cunningham-Rundles C, Casanova J, Cerutti A. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010;11:836–45. doi: 10.1038/ni.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/5233961
- 44.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh W, Taniguchi T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci USA. 2004;101:15416–21. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–9. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/8044956
- 46.Kraan W, Horlings HM, van Keimpema M, Schilder-Tol EJM, Oud MECM, Scheepstra C, Kluin PM, Kersten MJ, Spaargaren M, Pals ST. High prevalence of oncogenic MYD88 and CD79B mutations in diffuse large B-cell lymphomas presenting at immune-privileged sites. Blood Cancer J. 2013;3:e139. doi: 10.1038/bcj.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dimicoli S, Wei Y, Bueso-Ramos C, Yang H, Dinardo C, Jia Y, Zheng H, Fang Z, Nguyen M, Pierce S, Chen R, Wang H, Wu C, Garcia-Manero G. Overexpression of the toll-like receptor (TLR) signaling adaptor MYD88, but lack of genetic mutation, in myelodysplastic syndromes. PLoS ONE. 2013;8:e71120. doi: 10.1371/journal.pone.0071120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swann JB, Vesely MD, Silva A, Sharkey J, Akira S, Schreiber RD, Smyth MJ. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci USA. 2008;105:652–6. doi: 10.1073/pnas.0708594105. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1097742
- 49.Oda K, Kitano H. A comprehensive map of the toll-like receptor signaling network. Mol Syst Biol. 2006;2:2006–0015. doi: 10.1038/msb4100057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105–8. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- 51.Häcker H, Redecke V, Blagoev B, Kratchmarova I, Hsu L, Wang GG, Kamps MP, Raz E, Wagner H, Häcker G, Mann M, Karin M. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–7. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1029831
- 52.Häcker H, Tseng P, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11:457–68. doi: 10.1038/nri2998. [DOI] [PubMed] [Google Scholar]
- 53.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–8. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1103067
- 54.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–50. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1016387
- 55.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 56.Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science. 2010;329:1530–4. doi: 10.1126/science.1187029. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/5297956
- 57.Chockalingam A, Rose WA, Hasan M, Ju C, Leifer CA. Cutting edge: a TLR9 cytoplasmic tyrosine motif is selectively required for proinflammatory cytokine production. J Immunol. 2012;188:527–30. doi: 10.4049/jimmunol.1102713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10:1200–7. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1165537
- 59.Lee BL, Barton GM. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 2014;24:360–9. doi: 10.1016/j.tcb.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, Beutler B. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–64. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1030536
- 61.Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, Barton GM. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife. 2013;2:e00291. doi: 10.7554/eLife.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717986717
- 62.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–17. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, Coyle AJ, Kolbeck R, Green DR, Sanjuan MA. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37:986–97. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717970385