

I. Introduction
Universal and ubiquitous redox cofactors, nicotinamide adenine dinucleotide (NAD) and its phosphorylated analog (NADP), collectively contribute to ∼12% of all biochemical reactions included in the metabolic model of Escherichia coli K-12 (99). These reactions are driven by more than 80 distinct enzymes involved in all major metabolic pathways. In addition to interconversion between the oxidized (NAD(P)+) and reduced (NAD(P)H) forms, the pool of NAD cofactors undergoes a remarkably rapid turnover, with a half-life of ∼90 min (18). A homeostasis of the NAD pool faithfully maintained by the cells results from a dynamic balance in a network of NAD biosynthesis, utilization, decomposition, and recycling pathways that is subject to tight regulation at various levels.
In E. coli and Salmonella, the NAD molecule is assembled from the four major building blocks (Fig. 1), l-aspartic acid (l-Asp), dihydroxyacetone phosphate (DHAP), phosphoribosyl pyrophosphate (PRPP), and ATP supplied by central metabolic pathways, such as the citrate cycle, glycolysis, and the pentose phosphate pathway. Biosynthetic transformations comprising de novo biosynthesis of NAD and its phosphorylation to NADP are shared by many prokaryotes. They are well understood, and all respective genes and enzymes were characterized (see Section II, Table 1, and Fig.2). The main reactive center of NAD(P) is the C-3 atom of the pyridine ring involved in the reversible hydride transfer associated with all redox reactions. Several other types of reactions that involve phosphoryl, n-glycoside and amide bonds in NAD, lead to its breakdown to a series of products (Fig. 3) that can be utilized in NAD synthesis via recycling/salvage pathways. Some (but not all) of these pathways and respective genes were elucidated (see Section III, Table 1, and Fig. 2). Regulation of NAD metabolism appears to be quite variable in nature. The discovery of the multifunctional protein NadR in Salmonella provided the first insights into transcriptional regulation of NAD synthesis, a foundation that was later extended toward a broader range of Enterobacteriaceae (see Section IV and Fig. 4). A brief overview of NAD utilization processes is provided in Section IV, including some examples of nonredox utilization. This relatively unexplored area of NAD metabolism is gaining growing attention as it reveals new links between cellular metabolic and regulatory networks.
Fig. 1. Metabolic precursors (building blocks) involved in the de novo biosynthesis of NAD.
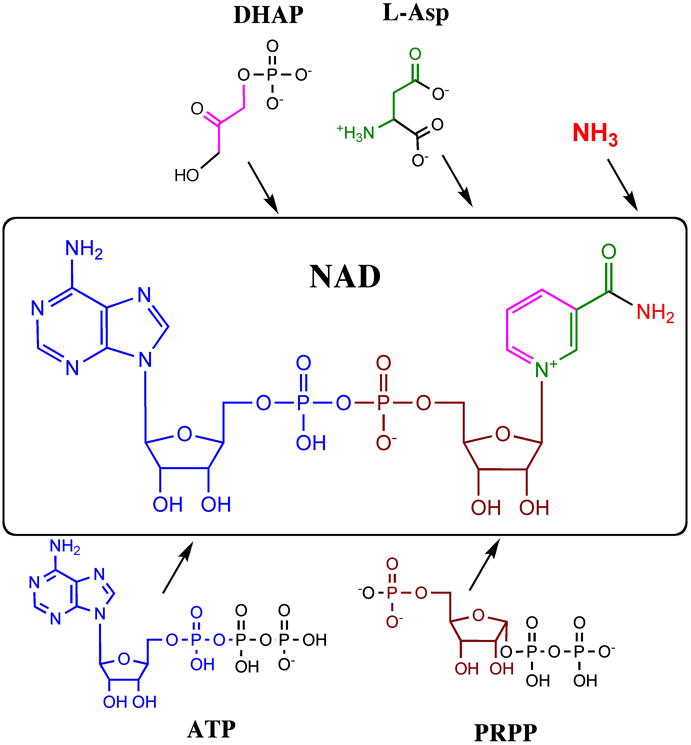
Table 1. Functional roles and corresponding genes involved in NAD(P) metabolism of E. coli K-12.
| Symbol | Functional Role | gene, ID | Ess* | PDB# |
|---|---|---|---|---|
| De novo synthesis: from Asp to NaMN | ||||
|
| ||||
| NadB | L-Aspartate oxidase (EC 1.4.3.16) | nadB, b2574 | N | 1KNP |
| NadA | Quinolinate synthetase (EC 4.1.99.-) | nadA, b0750 | N | 1WZU(1) |
| NadC | Quinolinate phosphoribosyltransferase (EC 2.4.2.19) | nadC, b0109 | N | 1QAP |
|
| ||||
| Common pathway: from NaMN to NAD(P) | ||||
|
| ||||
| NadD | Nicotinate-mononucleotide adenylyltransferase (EC 2.7.7.18) | nadD(ybeN),b0639 | E | 1K4K |
| NadE | NAD synthetase (EC 6.3.1.5) | nadE(efg), b1740 | E | 1WXE |
| NadK | NAD kinase (EC 2.7.1.23) | nadK(yfjB), b2615 | E | 2AN1 |
|
| ||||
| Salvage/recycling of Nm and Na | ||||
|
| ||||
| PncA | Nicotinamidase (EC 3.5.1.19) | pncA(ydjB), b1768 | N | 1IM5(2) |
| PncB | Nicotinate phosphoribosyltransferase (EC 2.4.2.11) | pncB, b0931 | N | 1VLP(3) |
|
| ||||
| Salvage of NMN and RNm | ||||
|
| ||||
| AphA | NMN phosphatase (EC 3.1.3.5) | aphA, b4055 | N | 2B8J |
| PnuC | Ribosyl nicotinamide transporter, PnuC family | pnuC, b0751 | N | - |
| NadR_K | Ribosylnicotinamide kinase (EC 2.7.1.22) | nadR, b4390 | N | 1LW7(4) |
| NadR_A | Nicotinamide-mononucleotide adenylyltransferase (EC 2.7.7.1) | |||
|
| ||||
| Regulation | ||||
|
| ||||
| NadR_R | NadR transcriptional regulator | nadR, b4390 | N | - |
|
| ||||
| Utilization | ||||
|
| ||||
| SthA | Soluble pyridine nucleotide transhydrogenase (EC 1.6.1.1) | sthA(udhA), b3962 | N | - |
| PntA, PntB | NAD(P) transhydrogenase, alpha and beta subunits (EC 1.6.1.2) | pntA, b1603 and pntB, b1602 | N,N | 1X13 |
| LigA, LigB | DNA ligase (EC 6.5.1.2), NAD-dependent | ligA, b2411 and ligB(yicF), b3647 | E,N | 2OWO |
| CobB | NAD-dependent protein deacetylase of SIR2 family | cobB, b1120 | N | 1S5P |
Gene essentiality assignments: essential (E) or nonessential (N) for the robust growth of E.coli K-12 in rich medium, as identified (in full agreement) by two independent studies (3, 33).
Protein Data Bank (PDB) accession numbers indicate the availability of 3D structural data for the representative proteins from E. coli or Salmonella, unless indicated otherwise:
- Pyrococcus horikoshii,
– Saccharomyces cerevisiae and
– Haemophilus influenzae.
Fig. 2. The key biosynthetic, salvage and recycling pathways of NAD(P) metabolism.

Committed metabolites (shaded circles) and functional roles (other shapes) are marked by abbreviations defined in the text and color coded to reflect the main pathways: de novo synthesis of NaMN from L-Asp (green); common pathway from NaMN to NAD(P) (red), pyridine salvage (blue), pyridine nucleoside salvage (magenta). Missing genes corresponding to functional roles implicated by substantial experimental data are shown by “?” and the corresponding transformations are sown by dashed arrows. Dashed arrows are also used to reflect undefined groups of transformations involved in NAD(P) utilization.
Fig. 3. Reactive centers in NAD molecule and primary products of NAD utilization reactions.

Metabolites undergoing recycling to NAD are indicated by arcs.
Fig.4. Regulation of NAD biosynthesis by NadR.

Domain structure of NadR protein and a consensus DNA motif are shown above the alignment of representative chromosomal loci containing genes regulated by NadR. In addition to that, two loci containing nonregulated paralogs of pnuC and nadR genes are shown. Orthologous genes (or, in case of NadR, matching domains) are shown by matching colors.
Although the first breakthroughs in NAD metabolic biochemistry—such as early discoveries by Warburg, Kornberg, and, later, by Preiss and Handler (57, 93)—came from mammalian and yeast systems, the identification of NAD biosynthetic genes was pioneered by studies in E. coli and Salmonella. The main results of these studies, which laid a foundation of our current understanding of NAD metabolism in all forms of cellular life, were reviewed in the earlier editions of this book (90, 125). In this chapter we will focus mostly on those aspects of NAD biogenesis and utilization in E. coli and Salmonella that emerged within the past 12 years since the chapter's previous edition. A revolutionary development of genomic sequencing and comparative analysis that coincided precisely with that period made a strong contribution to the progress in exploration of these and many other pathways. This development enabled identification, cloning, overexpression, and detailed characterization of all relevant genes in E. coli and Salmonella (see Table 1), including those that were missing at the time of the previous edition. It also allowed us to rapidly and accurately project the accumulated knowledge toward hundreds of diverse species with completely sequenced genomes. Genomic reconstruction of NAD biosynthesis in a broad range of bacterial pathogens implicated the most conserved and essential enzymes as potential targets for the development of novel antibiotics (34, 87). A comparative genomic approach also allowed us to utilize the knowledge of NAD metabolism acquired in other species to reveal previously uncharacterized features of E. coli.
Among other impacts of the recent technologic advancement is the comprehensive collection of 3D structures that were solved for all the NAD(P) biosynthetic enzymes from E. coli and/or some of their close orthologs from other species (Table 1). Significant progress in this direction together with accompanying mechanistic studies substantially advanced our understanding of the enzymology of NAD biosynthesis (for recent reviews, see (4, 5, 66, 101)). We refer readers to these reviews and original publications on individual enzymes listed in Table 1 for an in-depth discussion of these aspects. In this chapter we provide only a brief overview of pathways, reactions, and enzymes immediately involved in NAD biogenesis and associated with genes (proteins) presently identified and characterized in E. coli and/or Salmonella. A genomic projection of the NAD metabolic subsystem (88) toward ∼50 species from the group of Enterobacteriaceae with completely sequenced genomes.
II. De novo route of NAD(P) biosynthesis
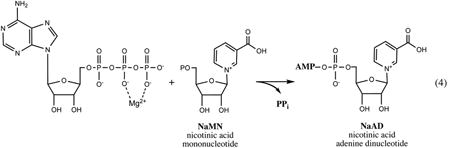
The de novo route to NAD in E. coli and Salmonella is composed of two biosynthetic blocks: (i) the upstream pathway from l-Asp to nicotinic acid mononucleotide (NaMN), often termed the l-Asp-DHAP pathway, and (ii) the downstream common pathway from NaMN to NAD(P). Separation of these two blocks is dictated by the topology of the entire NAD biosynthesis network (see Fig. 2). The three-step L-Asp-DHAP pathway is required only for the de novo synthesis of NAD, and therefore it is commonly referred to as the de novo pathway. All three genes (nadB, nadA, and nadC) of this pathway are dispensable when nicotinamide (Nm) or nicotinic acid (Na) are present in the growth medium. Mutations in each of these genes causing niacin auxotrophy could be readily obtained, which led to elucidation of the entire pathway in the early genetic studies (90). The downstream pathway converting NaMN to nicotinic acid adenine dinucleotide (NaAD) intermediate and then to NAD and NADP cofactors is common and required for both de novo and niacin salvage routes. The three genes of this pathway (nadD, nadE, and nadK) are indispensable in any growth conditions, and mapping of these genes presented a substantial challenge. Two of them (nadD and nadK) were ultimately identified and characterized only several years after the previous edition of this book (52, 72).
The decoupling of these two pathways is illustrated by their different phylogenomic distribution. The downstream pathway is largely conserved in both eukaryotes and prokaryotes, whereas the upstream pathway is a subject of substantial variations. Only the last step of this pathway, conversion of quinolinic acid (Qa) to NaMN, and the relevant enzyme, quinolinate phosphoribosyl transferase (NadC), are shared between the l-Asp-DHAP pathway and an otherwise unrelated five-step aerobic conversion of tryptophan to Qa. The latter pathway was elucidated prior to the l-Asp-DHAP pathway, and it was considered strictly eukaryotic until recently, when it was discovered in a small group of bacteria (58). Many groups of bacteria (most notably, Gram-positive pathogens) lack any de novo biosynthetic genes and require Nm or Na for growth but have an intact nadDEK gene set (87).
All six NAD biosynthetic genes are localized remotely from each other on the chromosome of E. coli and other species of the Enterobacteriaceae group. However, nadB and nadA genes are coregulated by the NAD-dependent transcriptional repressor NadR (Section IV). All three genes of the upstream pathway tend to form a conserved chromosomal cluster (operon) in many genomes, for example, in Bacillus subtilis and other Gram-positive bacteria, where the expression of the nadBCA operon is controlled by a recently identified niacin-responsive repressor NiaR (YrxA) (102, 104).
IIa. From L-Asp to NaMN
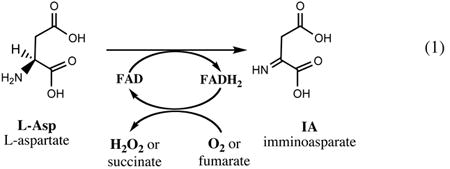
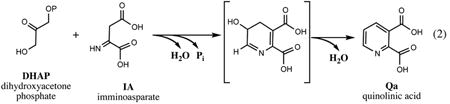
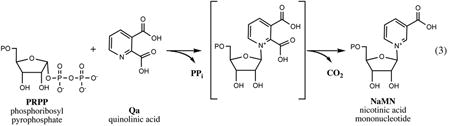
The conversion of l-Asp (derived from the TCA cycle intermediate, oxaloacetate), to NaMN is performed in three consecutive steps: (i) oxidation of l-Asp to an unstable intermediate, imminoaspartate (IA), catalyzed by l-aspartate oxidase (NadB), immediately followed by (ii) condensation of IA with DHAP, a central glycolytic intermediate, to form Qa catalyzed by quinolinate synthase (NadA), and (iii) phosphoribosylation of Qa by quinolinate phosphoribosyltransferase (NadC) using PRPP supplied by the pentose phosphate pathway as a second substrate. Each of these enzymes and respective reactions are briefly described below.
All three genes and their genomic context are conserved in nearly all representatives of Enterobacteriaceae, including E. coli, Salmonella, Erwinia, Photorabdus, Serratia, and Yersinia spp. Among remarkable exceptions is Proteus mirabilis, which lost the entire pathway while preserving all other genes related to NAD metabolism. Interestingly, the gene nadB and, in some cases, both nadB and nadA, appear to be disrupted or deleted in niacin auxotrophic virulent isolates of Shigella (94). This observation suggested that Qa synthesis may constitute one of the antivirulence factors that undergo selective elimination during the course of pathogen evolution from their commensal precursors (95). Although exogenously added Qa was shown to suppress invasion and cell-to-cell spread of Shigella flexneri, mechanistic details and physiological significance of these observations remain to be elucidated. Interestingly, an independent study revealed niacin auxotrophy due to nadA mutation in a series of isolates of a cattle pathogen, Salmonella enterica serovar Dublin (8).
l-Aspartate oxidase, a product of gene nadB, is an ∼60 kDa protein partially dimerizing in solution and containing a single molecule of FAD cofactor involved in the oxidation of l-Asp to IA. Importantly, NadB was shown to efficiently utilize both oxygen or fumarate as electron acceptors in vitro, which explains how the pathway may operate in anaerobic conditions (79, 122).
Genetic identification and mapping of the nadB locus followed by gene cloning, sequencing, and initial characterization of the purified protein from Salmonella and E. coli (24, 113) were previously reviewed (90). Among the important features of this enzyme is its stringent stereospecificity toward l-Asp and the observed inhibition by NAD, which appears to efficiently compete with FAD cofactor for the same binding site. This inhibition leading to ∼80-fold increase of the apparent Kd for FAD in the presence of physiological levels of NAD provides a rationale for the previously proposed mechanism of de novo biosynthesis feedback regulation (82, 121).
NadB shares substantial sequence similarity with FAD-containing subunits of fumarate reductase (Frd) and succinate dehydrogenase (Sdh) complexes, and all three enzymes seem to have a number of common mechanistic features. However, in contrast to multisubunit Frd and Sdh enzymes, NadB does not require a Fe-S cluster partner, as it appears to directly couple the reduction of fumarate to succinate to oxidation of l-Asp in vivo (74). The 3D structural analysis of the NadB apoenzyme (69) and, later, of the NadB R386L mutant in complex with FAD and succinate (13) substantially contributed to the mechanistic understanding of this enzyme. NadB protein consists of (i) an N-terminal FAD-binding domain, a characteristic dinucleotide fold composed of two segments with an insertion of a (ii) (α+β) capping domain, and (iii) a C-terminal helical domain. Binding of the FAD cofactor leads to a substantial conformational rearrangement leading to formation of the active site cavity at the interface between the capping and FAD-binding domains. This cavity contains a number of residues contributing to H-bonding of the substrate carboxylates and a conserved Glu-121 residue interacting with its α-amino group. Conservation of the overall topology and several FAD-binding residues between NadB and Frd enzymes further supports their likely mechanistic similarity. A mechanistic proposal for NadB based on the analogy with Frd includes a hydride transfer from the l-Asp Cα or Cβ atom to FAD in conjunction with proton abstraction mediated by the Arg-290 (13). This is in contrast with the classical d-amino acid oxidase mechanism, where the key electron transfer step involves a substrate α-amino group.
Quinolinate synthase was initially introduced as a common name for both NadB and NadA enzymes that were considered as subunits of the enzymatic complex generating Qa by condensation of DHAP with in situ–produced IA. This view is supported by the apparent instability of the IA intermediate, which in the absence of the downstream NadA activity gets readily hydrolyzed to oxaloacetate. On the other hand, there is no experimental evidence of a tight complex formation between NadA and NadB proteins or genomic evidence of the respective gene fusion. Moreover, several groups of bacterial species (e.g., some of the Corynebacteria and Helicobacter spp) that appear to have a functional l-Asp-DHAP pathway (and orthologs of nadA and nadC genes) lack the nadB gene in its recognizable form, suggesting that it was likely replaced by a yet-unknown gene (87). In the case of methanogenic archaea and the deep-branched bacterium Thermotoga maritima, the NadB function was shown to be performed by a nonhomologous NAD-dependent enzyme, aspartate dehydrogenase (132). In contrast to the observed NadB variability, the NadA enzyme appears to be conserved in all prokaryotes (as well as in plastids of plants (51)) harboring the l-Asp-DHAP pathway.
A ∼38-kDa product of gene nadA characterized by genetic and biochemical techniques in conjunction with the nadB gene (82) was initially reported to be functionally unstable due to the presence of the Fe-S cluster, a subject of oxidative damage in vitro and, possibly, in vivo, in the conditions of excessive aeration. In later studies the recombinant NadA was overexpressed, refolded from inclusion bodies, and purified, and its functional activity was reconstituted in vitro in a mixture with purified NadB protein, l-Asp, and DHAP (16). Formation of Qa occurred in the absence of Fe-S clusters that do not seem to have any obvious role in the mechanism of NadA, despite the apparent importance of Fe-S biogenesis and regeneration systems for its in vivo activity (63). Additional support for the latter observations was provided by a recent characterization of NadA enzyme from Arabidopsis thaliana whose additional SufE/CsdE-like N-terminal domain was shown to be responsible for regeneration of the highly oxygen-sensitive Fe-S cluster located in the C-terminal NadA enzymatic domain (84). The actual involvement of the Fe-S cluster in NadA catalysis remains a subject of active research. For example, substantial experimental evidence was recently presented to support a mandatory (albeit unspecified) participation of the Fe-S cluster in the enzymatic activity of E. coli NadA in vivo and in vitro (83).
The details of the mechanism of NadA-catalyzed condensation of IA and DHAP, an intriguing combination of two condensation reactions, which includes the elimination of two water molecules and a phosphate moiety of DHAP, are yet to be elucidated. Two mechanistic proposals were formulated to account for the early labeling data (4, 82). The only structural data available for the NadA family were recently obtained for the enzyme from the hyperthermophilic archaeon Pyrococcus horikoshii crystallized in complex with malate, which is presumed to mimic the IA substrate (108). The core of the protein represents an unusual fold with a pseudo-three-fold symmetry composed of the three analogous α/β/α sandwiches. The observed sequence similarity within these domains reflects a possibility of two gene duplication events in the evolutionary history of this protein. So far this structure has provided only a limited mechanistic insight, mostly from the modeling of the substrate arrangement in the active site based on the position of bound malate. Although the authors tend to support one of the proposed mechanisms, which includes phosphate elimination during the first condensation at the C-3 position of DHAP, this conjecture is based mostly on the inability to accommodate the DHAP phosphate moiety in the structural model. However, this interpretation may not be sustained if DHAP binding is accompanied by substantial structural rearrangement, as hinted by some preliminary experiments (108).
Quinolinate phosphoribosyltransferase, an ∼36-kDa product of nadC gene initially identified and characterized in Salmonella (45, 48), catalyzes the last step in the de novo synthesis of NaMN. As mentioned above, this step is shared between prokaryotic and eukaryotic de novo pathways, and the NadC enzyme is conserved in the genomes of all species having one or the other pathway without a single detected case of nonorthologous displacement (as reflected in genomic reconstruction of NAD metabolism across >400 diverse species with completely sequenced genomes integrated in The SEED database (88)).
The enzyme exists as a homodimer in solution, which is of apparent functional importance, as shown by later structural analysis of NadC enzymes from various sources (23, 55, 112, 114). The crystallographic analysis confirmed that NadC is structurally unrelated to well-known phosphoribosyltransferases of type I involved in nucleotide synthesis. The 3D structure of NadC, the first characterized representative enzyme of type II phosphoribosyltransferases, revealed two domains, a mixed α/β sandwich N-terminal domain and a seven-stranded α/β barrel-like C-terminal domain (23). The active site identified from the structures of NadC complexed with substrate (Qa) or product (NaMN) is formed at the interface between the N-terminal and C-terminal domains supplied by different subunits. Both subunits contribute to each of the two shared active sites in a homodimer.
The mechanism of NadC catalysis was deduced from structural data and a detailed kinetic analysis of NadC from E. coli (9) and Salmonella (15). The results of the latter study support a sequential ordered kinetic mechanism, with Qa productive binding in the active site preceding interaction with PRPP. An apparent contradiction with the previously published model (9) appears to be mainly due to the existence of an alternative branch that involves a nonproductive binding of PRPP with an apoenzyme. The proposed catalytic mechanism (4) was to a large extent drawn from the analogy with two well-studied enzymes, orotate phosphoribosyltransferase and orotidine monophosphate decarboxylase, that catalyze two consecutive reactions with an overall outcome similar to that of the NadC enzyme. This mechanism includes activation of the pyrophosphate leaving the group of PRPP via coordinated Mg2+ ions and stabilization of the reactive oxocarbenium intermediate by the negative charge of the C-2 carboxylate in Qa substrate maintained by interactions with proximal Arg-139 and Arg-162 residues of the enzyme. Interaction of the oxocarbenium ion with the pyridine nitrogen of Qa apparently leads to the formation of an unstable quinolinate mononucleotide that undergoes rapid decarboxylation at C-2, assisted by the electrostatic destabilization effect of C-3 carboxylate and likely by a proton transfer from the proximal Lys-72 residue of the enzyme. A conservation of NadC overall structural organization and the functional importance of implicated conserved Arg and Lys residues is supported by site-directed mutagenesis and structural data recently reported for the human enzyme (61).
The ultimate product of the l-Asp-DHAP pathway, NaMN, is further converted to NAD(P) as described below. It is also utilized in the NaMN:5,6-dimethylbenzimidazole phosphoribosyltransferase reaction catalyzed by the CobT enzyme (64), which generates α-ribazole-phosphate, a precursor of the cobalamin lower ligand, releasing Na, a subject of pyridine recycling via the PncB enzyme (see Section III).
IIb. From NaMN to NAD(P)
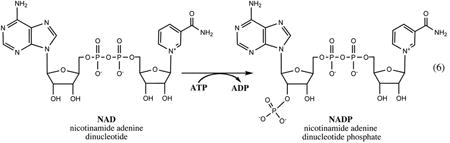
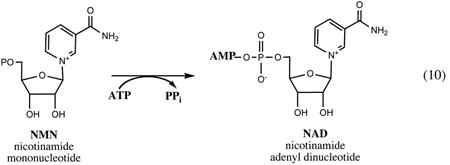
All enzymatic reactions that comprise this relatively conserved pathway utilize ATP as one of their substrates. All of the products and intermediates are phosphorylated, which effectively prevents their uptake from the growth medium, providing a rationale for the observed unconditional essentiality of the respective genes. A conversion of the universal NaMN intermediate to NAD is performed in two steps: (i) adenylation catalyzed by NaMN adenylyltransferase of the NadD family leads to a formation of NaAD, followed by (ii) amidation by the ATP-dependent NAD synthetase enzyme (NadE). The NAD is utilized as a cofactor in a variety of redox and undergoes a number of nonredox transformations (Fig.3 and Section V), including its phosphorylation by ATP-dependent kinase (NadK) to NADP, another indispensable redox cofactor utilized predominantly in anabolic pathways.
In the entire group of Enterobacteriaceae, the only species that do not have a full complement of all three nadDEK genes are Blochmannia spp. Of all genes related to NAD biogenesis, these intracellular endosymbionts with extremely truncated genomes have only nadK. The same gene pattern is observed in Rickettsia spp, suggesting the ability to uptake and salvage intact NAD from the host cells. Most Chlamydia spp also lack nadK, and they likely can salvage both, NAD and NADP) (34, 41, 87).
Nicotinic acid mononucleotide adenylyltransferase is a product of gene nadD that was approximately mapped on the Salmonella chromosome in the early studies of mutants resistant to 6-Amino-Nm, a toxic analog of Nm (46). However, the actual gene encoding this important enzyme was identified in the E. coli genome (former ybeN gene), cloned, and experimentally verified only ∼15 years later (72). Orthologs of E. coli NadD are present in a vast majority of diverse bacterial genomes, and many representatives of this family from various bacteria, including some pathogens, were cloned, expressed, and characterized, including 3D structural analysis (32, 34, 42, 85, 133, 136).
A reported crystal structure of the 24.5-kDa NadD protein from E. coli revealed a variation of a characteristic dinucleotide-binding (Rossman) fold, which in the case of NadD is composed of the central seven-stranded β-sheet surrounded by α-helices (136). The mechanism of NadD catalysis is likely similar to other nucleotidyltransferases of this class, including distantly related NaMN and NMN adenylyltransferases from archaea and eukaryotes reviewed in (65). It is thought to include a nucleophilic attack of the 5′-phosphoryl group of NaMN on the α-phosphate of ATP, followed by departure of pyrophosphate. This reaction is facilitated by Mg2+ and two conserved His residues of the ATP-binding sequence motif HYGH, characteristic of a large nucleotidyltransferase superfamily (2). Although the reaction is reversible in vitro, the equilibrium is likely shifted toward formation of NaAD in vivo due to the presence of inorganic pyrophosphatase and consumption of NaAD by downstream reactions.
A high-resolution structure of NadD in complex with its NaAD product provided a basis for the interpretation of its strict preference for NaMN substrate over its amidated analog, NMN (>1,000-fold based on comparison of kcat/Km values for each substrate). Substantial conformational changes observed upon ligand binding contribute to a network of interactions with NaAD, and some of these interactions, such as H-bonding of a nicotinosyl carboxy-group with several backbone amides, appear to favor the deamidated form of the ligand. The preference of NaMN over NMN displayed by all characterized members of the NadD family is consistent with the housekeeping role of this enzyme in NAD synthesis of E. coli and most other bacteria, where NaMN is a key intermediate in both major routes to NAD, de novo synthesis from l-Asp and salvage of Nm or Na (Fig. 2). Remarkably, an NMN adenylyltransferase domain of the multifunctional NadR protein, a distantly related member of the same family, has a strong preference for NMN over NaMN substrates (35, 59, 98), consistent with its role in salvage/recycling of amidated precursors (as discussed in Section III). Other known families of adenylyltransferases involved in NAD metabolism have either a dual substrate specificity (such as in eukaryotes and archaea (65)) or a strong preference for NMN (as in some characterized bacterial representatives of the NadM family(44, 97)).
NAD synthetase, an ∼30-kDa product of gene nadE forming a homodimer in solution, catalyzes the last amidation step in the synthesis of NAD. This gene was originally localized on the chromosome of Salmonella using sophisticated genetic techniques (47). It was later shown to be identical to an essential E. coli gene, efg, previously known to complement the ammonia-dependent growth mutants (adgA) of Rhodobacter capsulatus (130). Overexpression, crystallization, and preliminary structural data for the E. coli NadE enzyme were reported in 1999 (89), and a high-resolution structure of the apoenzyme and several complexes with its substrates and products recently became available (49). The latter study together with an insightful structural analysis of NadE from B. subtilis (100, 120) contributed to a detailed mechanistic understanding of this enzyme.
NadE belongs to a large family of ATP-dependent amidotransferases (134), and its structural core represents a typical Rossman fold characteristic of this superfamily. In addition to topological similarity with NadD, a mechanistic analogy between these two enzyme families lies in the fact that despite different outcomes (adenylation versus amidation), they share the key steps, ATP hydrolysis and transfer of the adenylyl moiety. In NadE this process is utilized for the activation of pyridine carboxylate, which is followed by a nucleophilic attack by ammonia and displacement of AMP.
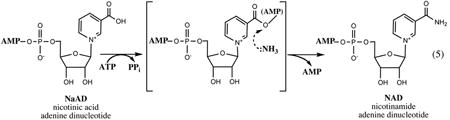
Both subunits of the NadE homodimer appear to contribute to the formation of the two NaAD/NAD binding sites located at the dimer interface, whereas each subunit contains its own ATP-binding site. Despite some differences between E. coli and B. subtilis enzymes, especially in the composition of their NAD-binding sites, the key aspects of catalytic mechanism are shared by these enzymes. Those likely include the important role of coordinated Mg2+ ions in (i) activation of the ATP α-phosphate for the nucleophilic attack by the carboxylate of NaAD, (ii) stabilization of the pyrophosphate leaving group, and, likely, (iii) interaction with the AMP leaving the group to facilitate the collapse of tetrahedral intermediate formed after the addition of ammonia. An additional feature reported only for E. coli NadE is a substantial structural rearrangement between NaAD and NAD-bound states, which may reflect a reaction progression from substrate binding to product release.
The in vivo source of ammonia required for NaAD amidation was among the most intriguing puzzles of NadE enzymology. By analogy with other characterized ATP-dependent amidotransferases, NadE was expected to utilize glutamine as a natural source of ammonia. Indeed, all eukaryotic members of the NadE family possess an additional N-terminal nitrilase-like domain (Nit) that was shown to hydrolyze glutamine and channel ammonia directly to the amidotransferase domain (11). A similar Nit-domain is present in many bacterial NadE enzymes, and for some of them (e.g., from M. tuberculosis and Synechocystis sp.) the ability to utilize glutamine in vitro was demonstrated (7, 32). On the other hand, NadE enzymes from many other bacteria, including E. coli and B. subtilis (as well as all archaea), do not contain Nit-domain and can utilize only ammonia (but not glutamine) for NAD synthesis in vitro. One possible interpretation of this observation could be the existence of a yet-unknown glutaminase subunit that would combine with NadE to form an enzymatic complex. Indeed, some amidotransferases are composed of two subunits, and several cases of such an arrangement were established within the NadE family (e.g., from Thermus thermophilus, K. Shatalin and A. Osterman, unpublished data). However, in all these cases both subunits are nearly always located next to each other on the chromosome. No such chromosomal clustering that would reveal candidate genes for NadE-specific glutaminase was observed in a comparative analysis of hundreds of genomes that contain NadE without Nit-domain. This genomic consideration is consistent with a conclusion that free ammonia (and not glutamine) is a natural substrate of NadE in Salmonella, which was deduced from the analysis of mutants defective in nitrogen assimilation (111).
Such a conclusion makes NadE of Salmonella and E. coli a second example (after glutamine synthase) of an amido-ligase that uses ammonia in vivo. Moreover, it likely applies to many other bacterial species containing single-domain NadE enzymes. A mosaic phylogenetic distribution of long (glutamine-utilizing) and short (ammonia-utilizing) NadE enzymes in bacteria suggests a convoluted evolutionary history with numerous gene acquisitions and losses. In the group of Enterobacteriaceae, Yersinia and several related species of Serratia, Photorabdus, and Proteus genus contain a long form of nadE with a seemingly functional Nit-domain, instead of a short nadE gene. A global sequence similarity analysis suggests that the long NadE enzyme is actually an ancestral form for the entire Enterobacteriaceae group. Close homologs of the Yersinia-type NadE are found in other Gammaproteobacteria (e.g., in Alteromonadales), whereas all the closest homologs of a short NadE characteristic of E. coli and Salmonella are from Gram-positive bacteria (Listeria spp and other Bacillales), pointing to a likely horizontal gene transfer (HGT) event. Such a gene-acquisition event in a common ancestor of E. coli and Salmonella could be followed by loss of a functionally redundant long nadE gene (e.g., YP2544 in Y. pestis). The hypothesized gene-loss event can be traced to a precise location between genes yfhA (YP2543) and glnB (YP2545) within a locus otherwise highly conserved between all the compared species. Functional significance of such a gene replacement in the group of Enterobacteriaceae is unclear.
NAD kinase, an essential enzyme catalyzing the last step in the synthesis of NADP, was also the one eluding gene identification for the longest time. A number of important features of this enzyme, such as allosteric inhibition by the reduced cofactors NADH and NADPH, were revealed by genetic (19) and biochemical studies (135), but the cloning and experimental characterization of E. coli gene nadK (previously yfjB) were reported only in 2001 (52). However, the first representative of this family was identified in M. tuberculosis and named ppnK for its ability to utilize polyphosphates (poly(P)) as phosphoryl donors in conversion of NAD to NADP (53). Whereas the same dual specificity was later confirmed in other NadK enzymes from Gram-positive bacteria and archaea (29, 62, 92, 107), the enzymes from E. coli and S. enterica (37), as well as their distant orthologs from eukaryotes (67), could utilize only ATP and, less efficiently, other nucleoside triphosphates.
Although the 3D structure of an ∼30-kDa NadK protein from E. coli recently became available in the PDB (see Table 1), most of the structure–function studies that contributed to the detailed mechanistic understanding of the NadK family were performed with enzymes from M. tuberculosis and Listeria monocytgenes (78, 92, 96). The 3D structural analysis confirmed an earlier prediction about NadK sharing the overall fold and some details of the active site topology with other members of the phosphofructokinase (Pfk) superfamily (60). Oligomerization observed in solution and in crystals may be of importance for catalysis and/or allosteric regulation, as some of the conserved active site components are supplied by different subunits. The N-terminal domain of NadK, composed of five β-strands and three α-helices, is particularly similar to the corresponding domain of Pfk, and it contains the GGDGT sequence motif highly conserved in the entire Pfk superfamily. However, the mechanistic role proposed for the universally conserved Asp residue in the activation of 2′-hydroxyl of the NAD substrate is drastically different from its role in Pfk, where this residue contributes to the activation of ATP (92). This provides a rationale for the 2′-OH specificity of NAD phosphorylation by NadK. Among other interesting features of the NadK mechanism proposed in this study is a substrate-assisted catalysis via participation of the NAD diphosphate moiety in coordination of catalytic Mg2+ ions. Despite this remarkable progress, some important details of NadK catalysis, such as the aforementioned difference in phosphoryl donor specificity between different members of the NadK family, are yet to be elucidated.
Allosteric regulation of NadK appears to be of crucial physiological importance due to its role in modulating and maintaining a delicate balance between the pools of NAD and NADP cofactors. These two cofactors play remarkably different roles in the cellular metabolism, as manifested by the groups of their cognate enzymes and their respective pathways. A recent study in S. enterica provided the evidence that an allosteric inhibition of NadK by the reduced cofactors and possibly mediated by a shift in the equilibrium between dimeric and tetrameric forms of the enzyme is a major factor controlling the NAD(P)/NAD(P)H pools in response to variations in growth conditions (e.g., aerobic versus anaerobic) and to oxidative stress (37). The proposed model, based on comparison of NadK kinetic parameters with in vivo pool sizes, suggests that the enzyme activity is suppressed mainly by NADPH during the normal aerobic growth or by NADH during the anaerobic growth. A drop in the concentration of either reduced cofactor activates NadK and ultimately leads to the increase in the NADP(H) pool, which allows cells to sustain the stress and maintain the needed level of reductive biosynthesis.
III. Salvage and recycling
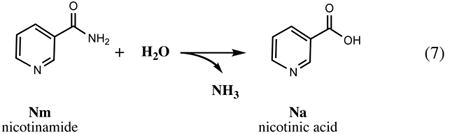
A network of biochemical transformation that allows the cell to utilize the pyridine ring and other byproducts of NAD consumption (Fig.3 and Section V) within the cell (recycling) or available from the medium (salvage) was traditionally referred to as a pyridine nucleotide cycle (PNC) (90)). The first PNC originally identified in mammalian systems and termed the Preiss-Handler pathway includes a single-step conversion of niacin (Na) to NaMN by nicotinic acid phosphoribosyltransferase (PncB) in a reaction very similar to the reaction of NadC in the de novo route (Fig.2). In E. coli and many other prokaryotes, this enzyme, together with nicotinamide deamidase (PncA), compose the major pathway for utilization of the pyridine ring in the form of amidated (Nm) or deamidated (Na) precursors. In many (mostly Gram-positive) bacterial pathogens, Nm salvage via the PncA-PncB route is the single and essential pathway supplying NaMN precursor for NAD synthesis (87).
The most economical salvage pathway, one which includes uptake of exogenous ribosyl nicotinamide (RNm), followed by a two-step conversion to NAD via phosphorylation and adenylation, was originally elucidated in H. influenzae, where it is the only route of NAD biogenesis (59, 73). Orthologs of both genes composing this pathway (pnuC and nadR) are conserved in E. coli and most other Enterobacteriaceae, although their role in the context of a much more complicated NAD metabolic machinery of these species appears to be somewhat different (see below). The importance of the RNm salvage pathway, albeit driven by a nonhomologous gene, was recently recognized in eukaryotic systems (6, 10, 123).
Among other PNC enzymes, NMN deamidase and NMN glycohydrolase involved in utilization of endogenous and exogenous NMN, respectively (see Fig. 2), were inferred from physiological and biochemical studies in Salmonella, but their respective genes have not been identified (18, 36). In contrast to NAD biosynthetic machinery, salvage and recycling pathways appear to be subject to substantial variations, even between closely related species. With the exception of the two pathways described in this section, our knowledge of these aspects of NAD metabolism is limited.
IIIa. Salvage of the pyridine ring
Unlike other important vitamins, such as riboflavin (vitamin B1, a committed precursor of flavin cofactors) or pantothenate (vitamin B5, a precursor of Coenzyme A), niacin (vitamin B3) is not equivalent to any intermediate in the NAD de novo synthesis. Therefore, its salvage is not limited by the mere vitamin uptake (as in the cases of B1 and B5), but requires additional biochemical transformations. In E. coli and Salmonella, these transformations are performed by the PncA-PncB pathway merging with the main NAD biosynthetic route at the formation of NaMN (Fig. 2). These bacteria have access to a substantial pool of niacin in their habitats, and the Nm/Na salvage pathway appears to play a substantial role in their lifestyle, taking precedence over the l-Asp-DHAP pathway whenever these precursors are available in the medium. Moreover, despite the existence of a nonhydrolytic (and seemingly more economical) PnuC-NadR salvage pathway, the default route for the utilization of exogenous NMN includes its cleavage by a yet-unidentified NMN glycohydrolase followed by the uptake of released Nm and its conversion to NaMN via the PncA-PncB pathway. A puzzling feature of the microbial Nm salvage is that it begins with deamidation of pyridine carboxylate, which ultimately has to undergo an energetically expensive amidation in the last step of NAD synthesis. Although an alternative nondeamidating salvage of Nm via nicotinamide phosphoribosyltransferase (NadV) was discovered in H. ducreyi (68) and then identified in a number of species, including Homo sapiens (32, 103), it is not present in Enterobacteriaceae.
The mechanism of Nm and Na uptake remains unclear. Although the evidence of an active energy-dependent transport was presented (105), the actual gene(s) has (have) not been identified in any studies. Therefore, a popular point of view is that niacin uptake in E. coli and Salmonella occurs via a passive diffusion (90). However, an alternative interpretation of the failure of genetic studies to map transport machinery may be related to its possible genetic redundancy. For example, the inactivation of the niacin transporter gene niaP recently identified in some Gram-positive bacteria only partially suppressed the sensitivity of B. subtilis to toxic niacin analogs, suggesting the existence of an additional, yet-unknown, uptake mechanism (which may as well be a passive diffusion) (102). An endogenous Nm for recycling may be supplied by NAD-consuming enzymes such as ADP ribosyltransferases and/or NAD-dependent deacetylase (CobB). In some serovars of S. enterica subsp enterica and S. bongori, an additional flux of Nm may be provided by the reaction of RNm phosphorylase (RnmP, a paralog of uridine phosphorylase Udp). A candidate rnmP gene forms a conserved chromosomal cluster with a second copy of pnuC transporter, supporting its proposed role in the alternative (kinase-independent) RNm salvage pathway (see the next subsection, Fig. 4, such as recently described in yeast (6).
Nicotinamide deamidase encoded by E. coli gene pncA (former ydjB), is an ∼20-kDa protein that belongs to a large superfamily of thiol-dependent amidases that are often present as multiple paralogs in many organisms. N-Carbamoylsarcosine amidase (CSHse) and isochorismatase catalyze the hydrolysis of similar substrates, and they are structurally related to PncA. Among pncA paralogs in E. coli are isochorismatase (gene entB) involved in siderophore biosynthesis, as well as two putative hydrolases of unknown specificity, yecD and ycaC. A 3D structure of the protein product of the latter gene, which is the closest pncA homolog, was solved, revealing a putative active site organization similar to CSHse (20). It is tempting to consider ycaC a candidate for a presently unknown gene of NMN deamidase (J. Roth, personal communication). Although the pncA mutations affecting Nm salvage were described in early studies (25), the identification of the actual gene and characterization of its protein product were reported only after publication of the previous edition of this book (26). This enzyme is often referred to as pyrazinamidase (PZAse) due to its role in hydrolytic activation of an antituberculosis prodrug pyrazinamide (PZA, a close analog of Nm) into its active form, pyrazinoic acid, which appears to suppress the synthesis of long-chain fatty acids characteristic of M. tuberculosis. Therefore, PncA studies were largely driven by clinical implications for tuberculosis, including PZA resistance, which in many cases is caused by pncA mutations (116). The name PZAse is obviously a misnomer in context of other species (such as E. coli), as PZA is neither a natural metabolite nor a relevant antibiotic for most bacteria.
No 3D structure data are presently available for PncA from E. coli or any other bacteria. However, the mechanistic interpretation of PncA based on the analogy with CSHse is supported by recent structural studies of the PncA ortholog from Pyrococcus horikoshii (22). The 3D structure of this protein (37% identity with E. coli PncA) revealed an overall α/β fold composed of a six-stranded parallel β-sheet with helices packed on both sides and a conserved active site C156-K111-D10 triad (numeration as in E. coli PncA), similar to those in CSHse from Arthrobacter sp (PDB code 1NBA). Unexpectedly, the 3D structure revealed the presence of Zn2+ in the active site, which appears to interact with D10, facilitating its proposed role in proton abstraction from C-156 to form a reactive thiolate for nucleophilic attack on the amide bond.
Nicotinic acid phosphoribosyltransferase (PncB) catalyzes conversion of Na to NaMN, which is a common step in salvage of both, Nm and Na. Orthologs of the pncB gene are present in all bacteria containing pncA genes but also in many species that do not have pncA, including mammals, where PncB participates in the Preiss-Handler pathway of Na salvage. Among Enterobacteriaceae, such a gene pattern (pncB without pncA) is found in some Buchnera spp. Whereas in many prokaryotes pncA and pncB genes form an operon, they are located remotely in E. coli and other Enterobacteriaceae. Moreover, only the pncB gene is regulated by the NadR repressor (see Section IV) so that at high concentration of NAD excessive Na would accumulate and, likely, get excreted from the cells (90). The pncB gene was mapped in Salmonella Nm salvage mutants (25), and its sequence encoding an ∼45-kDa protein was reported (128, 131).
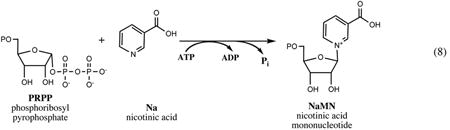
The PncB enzyme from Salmonella was a subject of the most detailed kinetic and mechanistic studies that revealed a new paradigm of energy coupling in catalysis (38-40, 127). Although the chemical reaction catalyzed by PncB is very similar to the reaction of NadC (except for the decarboxylation step involved in the de novo synthesis of NaMN from Qa), a unique feature of PncB is its ability to couple the energy of ATP-hydrolysis to >10-fold acceleration of phosphoribosyl transfer, which is relatively slow in the absence of ATP. Of no less importance, in the presence of ATP, this reversible reaction is almost entirely shifted toward formation of NaMN product from the nearly equimolar substrate/product ratio at thermodynamic equilibrium. An elegant interpetation of this interesting phenomenon and the proposed kinetic model includes autophosphorylation of the H219 residue of PncB by ATP (38), which leads to a substantially increased affinity of the phosphorylated enzyme toward its PRPP substrate, as well as a more efficient product release concomitant with the enzyme dephosphorylation (39).
Although no structural data are yet available for the E. coli or Salmonella PncB enzymes, a recently solved 3D structure of an ortholog from yeast (∼36% identity) provided an additional support for this model (17). Despite a substantial amino acid sequence divergence, PncB shares the overall architecture characteristic of the type II phosphoribosyltransferases, with NadC and NadV (54), supporting the proposed common evolutionary origin of all three phosphoribosyltransferases that contribute to various aspects of NAD synthesis (14). However`, substantial differences between PncB and NadC are observed at the level of quaternary structure and domain arrangement. An additional C-terminal domain of the apparently monomeric PncB comprises a likely ATP binding site involved in autophosphorylation of the conserved H219 (H232 in the yeast enzyme). In Salmonella, NadC enzyme`, the corresponding position is occupied by E214 whose side-chain carboxylate possibly contributes to the stabilization of the positively charged oxocarbenium intermediate together with C-2 carboxylate of Qa substrate. In the mechanism of PncB`, the absence of either carboxylate is thought to be compensated by the phosphohistidine providing a rationale for its ATP-dependent activation. Such an activation does not seem to be a part of NadV mechanism`, and it may not be even universally conserved within the PncB family`, as suggested by the lack of the foregoing His residue in a recently reported 3D structure of PncB from the archaeon Themoplasma acidophilum (115). Several 3D structures of PncB orthologs from diverse bacteria are presently available in the PDB (e.g., 2IM5, 1YBE, 2F7F), and their analysis may shed more light on various mechanistic aspects of this interesting enzyme family.
IIIb. Salvage of the pyridine nucleoside
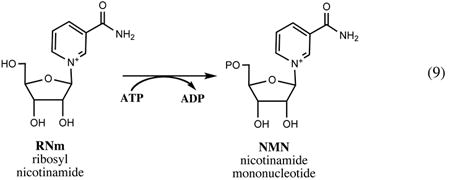
The existence of a salvage pathway for the uptake and utilization of the exogenous RNm without hydrolysis of the N-glycoside bond was recognized as a part of the PNC machinery from the analysis of Salmonella mutants with deficiencies in de novo and Nm salvage pathways (90). This analysis implicated both genes (pnuC and nadR) that are presently known to implement one of such pathways. However, their actual functional roles had not been fully understood until later genomics-driven studies (36, 59). The detailed comparative sequence analysis of NadR protein showed that, in addition to the putative DNA-binding N-terminal domain (NadR_R), its central domain is distantly related to archaeal-like NMN adenylyltransferases (80). A predicted NMN adenylyltransferase activity of NadR (NadR_A) was soon experimentally confirmed in an independent study (98). A complete PnuC-NadR pathway, uptake of NmR followed by its phosphorylation to NMN and adenylation to NAD (see Fig. 2), was initially elucidated in H. influenzae. This organism was known to rely on the so-called V-factors (NAD, NMN, or RNm) for growth and survival. A comparative genome analysis revealed that H. influenzae and several related Pasteurellaceae lack most of the common NAD biosynthetic machinery except for the orthologs of genes pnuC and nadR. Although the role of the PnuC-like transporter in RNm uptake as well as the role of NadR in NMN adenylation could be inferred by homology, no genes encoding a required RNm kinase acitivity were previously identified. Remarkably, this enzymatic activity was identified in the C-terminal domain of the NadR protein (NadR_K) (59). The physiological roles of the PnuC trasporter and both enzymatic activities of the NadR protein in RNm salvage by H. influenzae were further confirmed (73). Interestingly, the originally established NadR function in transcriptional regulation of NAD synthesis appears to be relevant only for Enterobacteriaceae (see Section IV), as NadR homologs that occur in a limited number of other bacterial species lack the N-terminal NadR_R domain.
Although both enzymatic activities were confirmed for NadR proteins from E. coli and Salmonella (35, 59), arguments were presented that only one of them, NadR_K, is physiologically relevant for RNm salvage (35). Whereas RNm phosphorylation is essential for trapping it within the cell upon the PnuC-mediated uptake, further utilization of the NMN intermediate is believed to proceed via a different route, deamidation to NaMN by a yet-unidentified NMN deamidase (see Fig. 2). Interestingly, three strains of E. coli contain an additional chromosomal cluster of pnuC'-nadR' genes (see Fig. 4 and that may constitute a fully functional nondeamidating RNm salvage pathway as in H. influenzae. The NadR′ protein encoded in this cluster does not contain a NadR_R domain, and, thus, it cannot participate in transcriptional regulation. The comparative sequence and genome context analysis of this pnuC'-nadR' cluster suggests a likely HGT event rather than internal duplication, as the closest (and similarly clustered) homologs of both genes occur in Stenotrophomonas maltophilia, Hahella chejuensis, and Pseudoalteromonas atlantica genomes.
Exogenous sources of pyridine nucleoside may include NAD and NMN that cannot be directly taken up by E. coli or Salmonella and need to be converted to RNm or Nm prior to uptake and salvage. It was recently shown that a conversion of the exogenous NMN to RNm in Salmonella is performed by acid phosphatase AphA (36). Orthologs of the aphA gene are present in all genomes of the E. coli/Salmonella branch and in Proteus mirabilis but not in Yersinia and related spp, where this role may be performed by a nonhomologous gene. An extracellular hydrolase converting exogenous NAD to NMN was identified in H. influenzae, but its counterpart in E. coli or Salmonella remains unknown. DNA ligase is the only well-characterized source of endogenous NMN, which may undergo recycling via NMN deamidase or, in some cases, NadR_A-mediated routes. It was hypothesized that cells may respond to oxidative stress by partially degrading NAD and excreting RNm via a PnuC facilitator (36).
As of today, the only two well-described enzymatic activities involved in the salvage/recycling of pyridine nucleoside are NadR_A and NadR_K. The 3D structural data contributing to their mechanistic understanding are presently available only for the NadR protein from H. influenzae (117).
RNm kinase domain (NadR_K) corresponding to aa 235 - 417 of E. coli NadR adopts a three-layered α/β/α-fold with a different arrangement of β-sheets in the central layer, reminiscent of several typical P-loop kinases, such as yeast thymidylate kinase (TMK). An analogy with the latter enzyme was used to map catalytically important components of the putative active site of NadR_K, which did not contain any bound ligand. Although the detailed mechanism of NadR_K catalysis is yet to be elucidated, it is likely quite similar to the well-studied mechanism of TMK. A recently reported structure–function analysis of a nonhomologous eukaryotic RNm kinase (human Nrk1) revealed a substantial difference from its bacterial counterpart, not only in the overall structure, but also in substrate specificity (123). Whereas NadR_K has a very stringent substrate preference for RNm, the human Nrk1 can phosphorylate both RNm and its deamidated analog ribosyl nicotinic acid (RNa) with nearly equal efficiency. The latter observation prompted the authors to propose the existence of yet another salvage pathway in eukaryotes that feeds a hypothesized RNa intermediary metabolite directly to the Preiss-Handler pathway via its phosphorylation by the Nrk1 enzyme.
Despite the fact that the two enzymatic domains of the H. influenzae NadR protein are involved in the consecutive reactions, there is no indication of any mechanistic coupling or substrate channeling between them, as evidenced by their spatial separation (117) and mutagenesis data (59). On the other hand, the detailed kinetic studies of NadR from S. enterica revealed an appreciable inhibition of NadR_K activity by NAD (35). Moreover, the functional analysis of a series of missense mutations in the nadR gene allowed the authors to suggest the allosteric model where NAD binding in the NadR_A domain leads to both inhibition of NadR_K activity and superrepression of NAD biosynthetic genes controlled by NadR_R. Both these regulatory effects would suppress NAD production when its level exceeds the requirements of the cell.
NMN adenylyltransferase domain (NadR_A) that corresponds to aa 64 - 234 of the E. coli NadR protein shares a characteristic Rossman-like fold and several aspects of the active site organization with other enzymes of this family involved in NAD synthesis(65). Despite a substantial sequence divergence, its three-layered α/β/α-fold with the central five-stranded parallel β-sheet closely resembles archaeal enzymes of the NadM family. The analysis of interactions with the NAD molecule bound in the NadR_A active site revealed the presence of the key contacts with the nicotinamide moiety, providing a structural basis for the observed strict preference of NMN over its deamidated analog NaMN, characteristic of the NadR family (35, 59, 98).
The enzymatic function of the NadR_A domain in RNm salvage is of undisputable physiological importance in H. influenzae and other species that contain a bi-functional version of the NadR protein without a regulatory NadR_R domain. However, in E. coli, Salmonella, and other Enterobateriaceae that contain a tri-functional version of NadR, the NadR_A domain could have adopted a function of an effector domain modulating the DNA-binding activity of the NadR_R domain in response to changes in the NAD pool. This conjecture is supported by several lines of evidence, including a relatively low specific activity of NadR_A from S. enterica, as well as genetic and physiological data showing that this enzymatic activity is neither required nor sufficient for supporting the growth of the organism when RNm salvage operates as the only route of NAD synthesis (35). At the same time, an enzymatic function of the S. enterica NadR_K domain remains essential for RNm salvage as it contributes to capturing the NMN intermediate within the cell for its further utilization, presumably via the NMN deamidase-dependent pathway.
IV. Regulation
The existence of various regulatory mechanisms and checkpoints that control the NAD biosynthetic machinery reflects the importance of maintaining NAD homeostasis in a variety of growth conditions. Among the most important regulatory mechanisms at the level of individual enzymes are a classic feedback inhibition of NadB, the first enzyme of NAD de novo biosynthesis, by NAD and a metabolic regulation of NadK by reduced cofactors. The latter mechanism appears to be responsible for substantial changes in the distribution of NAD(P)/NAD(P)H components within the NAD pool between aerobic and anaerobic growth conditions. For example, the ratio of the phosphorylated over nonphosphorylated cofactor levels increased by more than eightfold, while the fraction of reduced cofactors within the overall NAD pool decreased by more than sixfold upon transition of S. enterica from anaerobic to aerobic growth conditions (37). An additional regulatory fine-tuning is conceivable for other enzymes and respective pathways involved in NAD biogenesis. For example, a flux through the Nm/Na salvage pathway may be suppressed in the extreme ATP-limiting conditions due to the already-described role of ATP in modulation of PncB activity. Likewise, the RNm salvage pathway may be suppressed by excessive NAD due to allosteric inhibition of NadR_K.
A transcriptional regulation of NAD biogenesis in E. coli and Salmonella is controlled by the tri-functional NadR protein containing the N-terminal helix-turn-helix DNA-binding domain (NadR_R). The regulatory function of nadR was established in early genetic studies (see (90) for the historic perspective and detailed overview), long before its two enzymatic activities, NadR_A and Nad_K, were discovered. It was shown that NadR operates as a transcriptional repressor for the three genomic loci containing genes nadA-pnuC, nadB, and pncB, which allows it to suppress all three major routes of NAD biogenesis. In vitro DNA-binding studies confirmed that recombinant purified ∼45-kDa NadR protein from both Salmonella and E. coli, can specifically bind to DNA fragments that correspond to the upstream regions of the regulated genes via recognition of a palindromic DNA motif, a so-called NAD-box (see Fig.4 for a consensus sequence) (35, 91, 98). These studies also confirmed earlier conjecture that NAD is a physiologically relevant corepressor, thus providing a clear regulatory mechanism responsive to the overall level of NAD in the cell. Remarkably, NadR-DNA binding was shown to be NAD-dependent only in the presence of ATP. In the absence of NAD, ATP operates as an antirepressor, and the observed NAD dependency may be a result of competition between NAD and ATP for binding at the same (or overlapping) site(s) in the NadR protein (35). This consideration together with an unusually low Km of NadR_A for ATP (∼1 microM, substantially lower than a typical level of ATP in the cell) and other observations mentioned in the previous section support the proposed mechanistic model of NAD-dependent transcriptional regulation. According to this model, NadR_A operates as an effector domain that stimulates or suppresses the DNA-binding affinity of the NadR_R domain (via conformational changes and/or oligomerization), depending on the nature of the bound ligand, NAD or ATP, respectively. Structural analysis of a tri-functional NadR protein (such as from E. coli and Salmonella) and its complex with DNA, NAD, and ATP ligands would be instrumental in direct testing of this model.
Conservation of a tri-functional NadR protein and cognate DNA signals (NAD-boxes) in the entire group of Enterobacteriaceae, except intracellular endosymbionts , implies great importance of this regulatory mechanism. A comparative genome analysis approach was applied for computational identification of NAD-boxes and reconstruction of NadR regulon in all available genomes from this group (31). This analysis revealed a remarkable difference in the NadR regulon content between the E. coli/Salmonella and Yersinia branches illustrated in Fig. 4. Despite the conservation of chromosomal neighborhoods in all three loci listed above, two of them, nadB and pncB, do not contain recognizable NadR-binding DNA motifs in Yersinia and other available genomes outside of the E. coli/Salmonella branch. At the same time, all these genomes contain an additional NAD-box upstream of the nadR gene, pointing to an autoregulatory loop characteristic of many bacterial transcription factors. Evolutionary and physiological implications of these variations are yet to be elucidated. The only invariant member of the NadR regulon in all Enterobacteriaceae is the nadA-PnuC operon. Not surprisingly, paralogous pnuC′ and nadR′ genes found in several recently analyzed genomes (Fig.4) do not belong to the NadR regulon (D. Rodionov, personal communication).
A combination of various data from comparative genomics, physiological, regulatory, and enzymatic studies suggest a plausible evolutionary scenario for the tri-functional NadR, a versatile and, until recently, mysterious protein, which includes two gene-fusion events. The first event, a fusion of the ancestral NMN adenylyltransferase (NadR_A domain) of the archaeal NadM-type with the NadR_K domain distantly related to other P-loop kinases, could have occurred within the Gammaproteobacteria lineage, prior to separation of Enterobacteraceae and Pasteurellaceae. The emerged bi-functional enzyme would provide an efficient mechanism of nonhydrolytic salvage of pyridine nucleoside, which is of vital importance for organisms that went through a radical truncation of NAD biosynthetic machinery, such as H. influenzae. The second and more recent event, fusion with a typical DNA-binding domain, could have occurred in a common ancestor of extant Enterobacteriaceae, giving birth to the new function of NadR in transcriptional regulation of NAD biosynthesis. The importance of NadR_K activity in assisting the PnuC-mediated RNm uptake could have exerted sufficient evolutionary pressure to maintain this domain in its enzymatically active form even in the context of redundant NAD biosynthetic machinery of Enterobacteriaceae. On the other hand, the existence of an alternative and, apparently, very efficient NMN deamidase-dependent processing of NMN would make a NadR_A enzymatic function less important, and the role of this domain could have shifted from purely enzymatic (as in Pasteurellaceae) to regulatory (as in Enterobacteriaceae).
V. Utilization
Once synthesized, the NAD molecule can participate in a number of biochemical transformations that involve several reactive centers (Fig. 3). Phosphorylation at 2′-OH of the adenylyl moiety generating NADP cofactor was discussed above in the context of de novo synthesis. The reverse reaction was described, and the existence of NADP phosphatase was inferred, but a committed gene was not identified in any species. Both cofactors undergo conversion between their oxidized (NAD+ and NADP+) and reduced (NADH and NADPH) forms in a variety of redox reactions that compose a core of the cellular metabolic machinery. Despite obvious chemical and mechanistic similarities of these two cofactors and respective catalyzed reactions, they play fundamentally distinct physiological roles in the cell. A brief survey of the published metabolic reconstruction of E. coli K-12 (99) confirms a commonly recognized bias in distribution of NAD and NADP-dependent enzymes between catabolic and anabolic pathways. Indeed, whereas the fractions of enzymes preferentially utilizing NAD or NADP are comparable (42 and 31 from the analyzed set of 82 enzymes, with 9 enzymes using both cofactors), nearly 88% of NAD-dependent enzymes are involved in the catabolic pathway. Nearly half of them are associated with carbohydrate utilization, with another half contributing to oxidative consumption of other substrates such as carboxylic acids, amino acids, and fatty acids. At the same time, more than 80% of NADP-dependent enzymes utilize the reductive power of NADPH in anabolic processes, mostly in the biosynthesis of amino acids, as well as fatty acids, cofactors, and nucleotides. Off-note, at least 25% of genes encoding NADP-dependent enzymes are essential for E. coli survival and growth as defined in single-gene deletion studies (3), whereas the fraction of essential genes among NAD-dependent enzymes is much lower (∼5%). A global separation of metabolic functions implemented by NAD and NADP cofactors provides a foundation for decoupling of the catabolic and anabolic aspects of cellular networks. A delicate balance between phosphorylated and nonphosphorylated as well as reduced and oxidized forms within the NAD(P) cofactor pool is a product of multiple reactions within the cell. Among them are reactions directly interconverting the main components of the NAD(P) pool catalyzed by NadK and NAD transhydrogenase enzymes (see below). These enzymes contribute to maintaining the balance within the pool as well as modulating this balance in response to changes in growth conditions (e.g., aerobic versus anaerobic) (37, 109).
Another dynamic aspect of NAD homeostasis is associated with nonredox degradative reactions where NAD is used as a substrate rather than a cofactor. Reactions of that type affect mostly the pyrophosphate and the N-glycoside bond of the pyridine ring, generating intermediates (see Figure 3) that may undergo further hydrolysis (including deamidation and dephosphorylation), recycling, or, in certain conditions, excretion. In contrast to NAD(P) biogenesis and redox interconversions, relatively little is known about degradative aspects of the NAD metabolism. Nevertheless, an observed rapid turnover of the NAD(P) pool suggests its intensive nonredox consumption emphasizing the importance of NAD regeneration machinery described in the previous sections. Among a few known NAD-consuming enzymes, NAD-dependent DNA ligase (LigA and LigB) and protein deacetylase (CobB) are conserved in nearly all Eneterobacteriaceae, whereas ADP-ribosyl transferases were identified in only a limited number of strains and in the bacteriophage T4.
Va. Redox reactions
The key mechanistic aspects of redox catalysis are shared by many NAD(P)-dependent enzymes despite the diversity of involved metabolites. The main common step of NAD(P) catalyzed oxidation is the hydride transfer from the reduced form of a substrate (such as alcohol or aldehyde) to the C-3 atom in the pyridine ring of the oxidized cofactor (NAD(P)+) as a stereospecific conjugate nucleophilic addition (71). The process is accomplished by the enzyme-assisted transfer of a second H-atom from a substrate to the cofactor in the form of a proton. The formal net result of this process, often referred to as dehydrogenase reaction, is the transfer of two protons and two electrons, which produces a reduced cofactor (NAD(P)H+) and, in case of alcohol dehydrogenase, a carbonyl product. A reduction of substrates containing carbonyl groups or carbon-carbon double bonds (as in the reaction of enoyl-ACP reductase in fatty acid synthesis) proceeds as a reversal of an oxidation reaction. Not surprisingly, many divergent oxidoreductases of this class share structurally related NAD(P)-binding domains. For example, within the same set of 82 NAD(P)-dependent enzymes in the E. coli metabolic network, more than 90% contain domains that belong to a common α/β class (according to SCOP classification (1)). Nearly two-thirds of those domains share a so-called NAD(P)-binding Rossman fold. These common topological and mechanistic features notwithstanding, many details of stereochemistry, substrate selectivity, and other aspects characteristic of individual reactions are the subject of substantial variations.
Pyridine nucleotide (or NAD(P)) transhydrogenase is an interesting special case of oxidoreductase that catalyzes the interconversion between the reduced and oxidized forms of both NAD and NADP cofactors without involving any additional substrates. The membrane-associated enzyme of E. coli and Salmonella is a complex composed of two subunits (α- and β-) that are encoded by an operon, pntA-pntB, conserved in all Enterobacteriaceae (except endosymbionts). This enzymatic complex performs the reduction of NADP to NADPH+ coupled with oxidation of NADH+ to NAD driven by an electrochemical gradient, with a 1:1 ratio of hydride transfer and proton translocation equivalents.
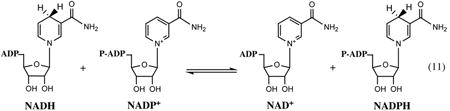
This enzyme is present in a broad range of bacteria and in eukaryotic mitochondria playing a crucial role in maintaining the balance between catabolic NADH production and anabolic NADPH consumption. An overall topology of integral membrane (proton translocating) and cytoplasmic (NAD(P)-binding) domains is conserved, whereas their connectivity at the level of sequence of individual subunits varies between the species. In E. coli, the active form of the enzyme complex is an α2 β2 -heterotetramer, in which the N-terminal segment of each α- and β-subunit form a composite membrane domain (dII), and their C-terminal segments form NAD-(dI)- and NADP(dIII)-binding domains, respectively. Detailed structure–function studies of this enzyme from different species, including an extensive mapping of the membrane dII domain(75) and determination of the 3D structure of the dI domain (50) of the E. coli enzyme, provided a rather detailed mechanistic model. A presently accepted, so-called binding change mechanism assumes the existence of two alternating states for each monomer. The closed state of one monomer promotes hydride transfer between NADH+ and NADP, whereas the open state of another monomer allows nucleotide exchange. The transition between these states is triggered by proton translocation coupled to the redox reaction (50).
Unlike most other bacteria, Enterobacteriaceae contain a second, soluble NAD(P) transhydrogenase encoded by a single gene, sthA (formerly udhA) (12). Enzymes of this family originally identified in Pseudomonadales catalyze a similar hydride transfer reaction, but they are structurally unrelated and mechanistically distinct from PntAB-type enzymes. The closest structural analogs of SthA enzymes are flavoprotein-disulfide oxidoreductases such as dihydrolipoamide dehydrogenase. Although the 3D structure has not been reported for any member of the SthA family, the existing data indicate the presence of three domains: an N-terminal FAD binding domain, a central NAD(P) binding domain, and a C-terminal domain involved in functionally important dimerization. A recent study of metabolic fluxes in E. coli pntAB and sthA mutants provided a rationale for the existence of two NAD(P) transhydrogenases (109). The membrane-associated enzymatic complex PntAB is involved in energy-dependent generation of NADPH. During aerobic growth on glucose, PntAB contributed up to 45% to the total NADPH pool, with the rest of it supplied by isocitrate dehydrogenase and the pentose phosphate pathway. On the other hand, the main function of the SthA enzyme appears to be an energy-independent reoxidation of NADPH, which is particularly important during growth on acetate and in other conditions leading to excessive formation of NADPH. Therefore, the existence of the two distinct and differentially regulated NAD(P) transhydrogenases allows E. coli to efficiently adjust cellular metabolism in response to varying environmental conditions.
Vb. Nonredox consumption
Recent progress in understanding enzymatic reactions consuming NAD as a substrate has led to recognition of the important role of the NAD(P) pool in linking metabolic and regulatory networks (6, 119). Some of these reactions, such as formation of poly(ADP)-ribose associated with DNA damage response and apoptosis, or cyclic ADP-ribose involved in Ca2+ mobilization and signaling, are characteristic of eukaryotic systems. NAD-dependent mono(ADP)-ribosylation of proteins is also quite common for eukaryotes, whereas in bacteria this activity is usually associated with virulence factors modifying target proteins of the host cells. Among a few examples described in the group of Enterobacteriaceae is the SpvB protein secreted by S. enterica, which was shown to ADP-ribosylate certain Arg residues in actin (43). As in the case of the well-studied clostridial C2 toxin, this modification likely leads to depolymerization of actin filaments and, ultimately, to decomposition of the entire cytoskeleton. SpvB orthologs can be detected in only a few strains, including S. dublin and S. typhimurium LT2. Such a limited phylogenetic distribution appears to be typical for these types of protein toxins (106).
Although no enzymes of this type were identified in E. coli K-12, three ADP-ribosyltransferases (ModA, ModB, and Alt) expressed by the bacteriophage T4 collectively modify a range of E. coli proteins involved in transcription, translation, and metabolism (21). The best known among them is ModA-catalyzed ADP-ribosylation of the host RNA polymerase, which reroutes it toward preferential transcription of the bacteriophage genes. The importance of NAD consumption and regeneration for the bacteriophage life cycle is emphasized by the presence of NAD recycling genes in a number of recently sequenced genomes of T4-like bacteriophages (76), most frequently nadV and nadM genes. Orthologs of these genes form a conserved operon in a number of bacterial genomes, encoding a two-step conversion: Nm→NMN→NAD (32). These observations point to a likely role of bacteriophages in evolution and HGT of this efficient (albeit absent in Enterobacteriaceae) NAD recycling pathway (S. Gerdes, personal communication). Although a rapid turnover of the NAD pool (even in the absence of bacteriophage infection) suggests the existence of an active NAD degradation machinery, only two NAD-consuming enzymes, DNA ligase and protein-lysine deacetylase, have been so far identified and characterized in E. coli.
NAD-dependent DNA ligase is a NAD-consuming enzyme characteristic of most bacteria. It is not present in eukaryotes, where the same function is performed by an ATP-dependent enzyme. Many aspects of DNA ligase catalytic mechanism are shared by both NAD- and ATP-dependent enzyme families (124, 129). The main housekeeping DNA ligase involved in most aspects of DNA repair in E. coli and Salmonella is an ∼74-kDa protein composed of several domains and encoded by an essential gene, ligA. The N-terminal NAD-binding domain is the unique feature of LigA distinguishing it from ATP-dependent ligases. LigA cleaves the pyrophosphate bond of NAD in a reaction reminiscent of many other nucleotidyl pyrophosphatases and transferases. The AMP moiety is transferred to the conserved Lys residue in the active site of the central core nucleotidyltransferase domain, which is shared between NAD- and ATP-dependent DNA ligases. This activation step occurs in the absence of DNA substrate and, in the case of LigA, leads to the release of the NMN byproduct.
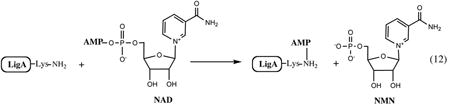
The following steps include another adenylyl transfer from Lys residue to the 5′-phosphate of the nicked DNA substrate, followed by the attack of the DNA 3′-OH end, regeneration of the phosphodiester bond, and expulsion of AMP. A recently reported 3D structure of LigA complex with adenylated nicked DNA substrate revealed a massive movement and rearrangement of LigA domains associated with reaction progression, contributing to better mechanistic understanding of this important enzyme (81). Interestingly, E. coli and many (but not all) other Enterobacteriaceae contain an additional ligA homolog, a nonessential gene termed ligB (formerly ycjF). The LigB protein lacks the C-terminal domain and some of the conserved residues of LigA. The NAD-dependent enzymatic activity of the recombinant LigB is at least 100-fold lower compared to LigA, and its actual physiological role is unclear. Inhibition of LigA by excessive NMN is thought to be one of the major reasons why this intermediate is toxic for the cells and requires rapid clearing via degradation or recycling. At the same time, neither LigA nor LigB seems to be a major source of intracellular NMN, suggesting the existence of another (yet-unidentified) NAD pyrophosphatase activity in the cell.
NAD-dependent protein-lysine deacetylase, an ∼26-kDa protein encoded by the cobB gene, was initially identified and characterized in S. typhimurium LT2 as a phosphoribosyltransferase partially compensating for the deficiency of CobT enzyme in the synthesis of a cobalamin precursor (hence, the cobB gene name), alpha-ribazole-5′-phosphate, from 5,6-dimethylbenzimidazole and NaMN (126). This observation, albeit falling short of establishing the actual function of this interesting protein, provided the first hint of the connection with NAD metabolism for the entire sirtuin (or SIR2) family broadly conserved in eukaryotes, archaea, and many bacteria. An exciting development of the CobB/SIR2 saga at the turn of the millennium provided us with the most remarkable illustration of the power of a comparative genomic approach applied across all three domains of cellular life. An integration of evidence from biochemical and genetic studies in bacteria with the established role of SIR2 in chromatin silencing in yeast and other eukaryotic systems and with 3D structural data for the archaeal SIR2 homolog ultimately led to functional and mechanistic understanding of this enzyme family (119). Despite the substantial divergence of sequences and actual physiological functions, the members of the CobB/SIR2 family share the newly discovered enzymatic activity, an NAD-dependent deacetylation of N-acetyl-Lys residues in various proteins. This discovery, among other impacts, led to recognition of Lys N-acetylation as an important and ubiquitous protein posttranslational modification of significant regulatory importance (especially in eukaryotes), beyond its previously known role in modulation of histone–DNA binding. Numerous publications implicate acetylation/NAD-dependent deacetylation in regulation of activity and stability of a growing number of regulatory and signaling mammalian proteins (including global regulators such as p53) (119). These studies revealed links connecting SIR2 activity with regulatory cascades that control longevity and programmed cell death, sensing the overall metabolic status of the cell via changes in the NAD pool. Such a rediscovery of NAD triggered a new wave of studies that filled in many gaps in the knowledge of NAD synthesis and recycling in eukaryotic cells (5).
A mechanistic model derived from detailed studies with eukaryotic, bacterial, and archaeal members of the SIR2 family (110) revealed its fundamental difference from the previously described ADP-ribosyl transferases. In contrast to these enzymes (also referred to as NAD glycohydrolases) that generate β1'-substituted ADPR via a nucleophile attack on the ADP-ribooxacarbenium intermediate, the actual product of CobB/SIR2 enzymes is a 2′-O-Ac-ADPR. This product is generated via an attack of the ribosyl 2′-OH group on the α1'-OAc-ADPR intermediate formed upon interaction of NAD with NAc-Lys and cleavage of the nicotinamide bond. A recently reported 3D structure of CobB from E. coli confirmed the similarity of its enzymatic core to other members of the SIR2 family (137). This core is composed of two domains, a larger catalytic domain with a prototypic Rossman fold and a smaller and less-conserved Zn2+-binding domain, likely contributing to recognition of protein substrates. The latter aspect of SIR2 in vivo functional activity remains a subject of active research, which is of particular importance for mammalian cells that contain several members of the SIR2 family and multiple acetylated protein substrates.
In contrast to mammalian systems, E. coli and Salmonella contain a single copy of cobB and only two unambiguously established deacetylation protein substrates. One of these substrates is the enzyme Acetyl-Coenzyme A synthetase (Acs), whose activity is regulated by N-acetylation and NAD-dependent deacetylation of the active-site Lys residue (119). Remarkably, Acetyl-Coenzyme A (Ac-CoA), the product of Acs, is the substrate of a protein acetyl transferase (Pat) enzyme that was shown to be responsible for N-acetylation of the active-site Lys in Acs, leading to its inactivation (118). Moreover, Nm, a byproduct of CobB-mediated deacetylation and reactivation of Acs, is a potent inhibitor of CobB. A combination of these feedback loops creates a metabolic system with an exquisitely sophisticated autoregulation. Remarkably, the same CobB-mediated regulatory mechanism was demonstrated for the yeast ortholog of Acs (119), and it is conceivable that a similar mechanism exists in mammalian cells. A propionyl-CoA synthetase (PrpE), an enzyme involved in utilization of propionate in S. enterica, was recently proven to be a subject of regulation by propionylation and NAD-dependent depropionylation also mediated by the CobB enzyme (30).
The reaction products of a NAD-dependent deacetylase, 2′-OAc-ADPR, and its spontaneously formed 3′-regioisomer are novel metabolites whose physiological role and cellular fate are yet to be elucidated. It is conceivable that these metabolites, as well as the products of various ADP-ribosyltransferase reactions, are ultimately hydrolyzed to a more stable ADPR. Further hydrolysis of ADPR by Nudix-like ADPR pyrophosphatase yields AMP and ribose-5-phosphate (Fig. 3) that may be reused in NAD biogenesis after conversion to ATP and PRPP. The existence of a family of bacterial NadM-like NMN adenylyltransferases that are fused with a Nudix-like ADPR pyrophosphatase domain provides evidence of its functional coupling with NAD metabolism (44, 97). In E. coli, the ADPR pyrophosphatase activity was reported for another member of the Nudix hydrolase superfamily encoded by gene nudF (formerly yqiE) (27, 28). However, a recent study provided the evidence that the major physiological substrate of NudF is another (albeit similar) metabolite, ADP-glucose, linked to glycogen biosynthesis (77). This is not surprising as many members of the Nudix hydrolase superfamily seem to display a broad substrate specificity (70).
VI. Conclusions and future directions
A new wave of studies over the last decade facilitated by the advent of genomic technologies and protein crystallography brought about a substantially enriched and refined picture of NAD biogenesis and utilization in E. coli and Salmonella. A number of key biosynthetic genes that were unknown or only approximately mapped at the time of the previous edition of this book were identified, and all the respective proteins were overexpressed, purified, and characterized in detail, including structural and mechanistic analysis. Among them are the three enzymes—NadD, NadE, and NadK—of the common pathway from NaMN to NAD(P) conserved in the majority of known bacterial species and, with minor variations, in archaea and eukaryotes. A comparative genome analysis allowed us to efficiently apply the wealth of knowledge accumulated in E. coli and Salmonella for accurate reconstruction of NAD metabolism in hundreds of diverse species. This analysis revealed remarkable patterns of conservation and variation, leading to better understanding of physiological, biochemical, and evolutionary aspects of NAD biosynthetic machinery in all forms of cellular life. Moreover, it facilitated identification of novel components of NAD metabolism in E. coli and Salmonella. For example, the ultimate elucidation of the mysterious tri-functional NadR protein was facilitated by the studies in H. Influenzae, whereas the unforeseen function of CobB in NAD-dependent protein deacetylation emerged largely from the work in yeast. An appreciable advancement of mechanistic enzymology of NAD biosynthesis may be easily recognized by a brief comparison with the comprehensive survey published in the beginning of the century (4). Nearly all open structural and mechanistic problems formulated therein for several key enzymes—NadC, NadD, NadK, and NadR—have been successfully addressed as briefly outlined in this chapter. Among a few remaining problems are incomplete understanding of the catalytic mechanism and the actual role of Fe-S clusters for the activity of the NadA enzyme.
Although our current knowledge of NAD biosynthetic and recycling pathways in E. coli is quite comprehensive, several challenging aspects require further analysis. At least one well-documented enzymatic activity immediately involved in pyridine recycling, NMN deamidase, remains in the category of missing genes. Genes encoding additional components of the intracellular NAD degradative machinery, such as NAD(P) pyrophosphatase, NADP phosphatase, and enzymes generating ADP-ribose and its derivatives, are yet to be identified. Likewise, extracytoplasmic enzymes processing exogenous pyridine nucleotides, such as NMN glycohydrolase and NAD(P) pyrophosphatase, are presently unknown. The mechanism of vitamin B3 uptake (in the form of Nm or Na) and the very existence of respective transporter(s) are still unclear. The availability of powerful comparative and functional genomic tools, including genome-scale collections of knockout mutants and overexpression strains (3, 56), will contribute to accelerated resolution of many remaining problems. The emerging data connecting NAD metabolism with regulatory and signaling networks point to a likely existence of an even larger variety of these connections. Some of them may involve NAD and its derivatives in yet-unknown nonredox reactions, such as posttranslational modifications of proteins, interactions with nucleic acids, etc. We expect comparative genomics and genome context analysis techniques that infer functional coupling of genes from their chromosomal clustering, fusion events, and co-regulation (86) to contribute to future developments in this direction. It is tempting to speculate that some of the presently uncharacterized genes that form chromosomal clusters with NAD biosynthetic genes conserved in many genomes may represent some of these novel functions that so far have eluded functional assays and screens. For example, by these criteria, a broadly conserved uncharacterized gene, ybeB (and to a lesser extent, ybeA), clustered on the chromosome with nadD in more than 50 diverse genomes, seems to constitute a primary suspect. Among other anticipated future developments are metabolic profiling and modeling studies that will provide us with better integration of NAD biogenesis and utilization in a broader context of metabolic and regulatory networks in the cell.
Acknowledgments
I would like to thank my Lab members Oleg Kurnasov, Xiaoqing Li, Dmitryi Rodionov, Irina Rodionova, Leonardo Sorci, Chen Yang, and Ying Zhang for their contribution to our research work in the field of NAD biosynthesis and their direct help with preparation of this chapter. Our work in this field was supported by grants R01AI059146 and R01AI066244 from the National Institute Of Allergy and Infectious Diseases. I am deeply grateful to my long-term collaborators, Hong Zhang (UT Southwestern), Nadia Raffaelli (Università Politecnica delle Marche, Italy), Tadhg Begley (Cornell University), Valerie de Crecy-Lagard (University of Florida), Svetlana Gerdes and Ross Overbeek (Fellowship for Interpretation of Genomes), and to many members of their research groups for their tremendous help and support through all these years. I also want to thank Giulio Magni (Università Politecnica delle Marche, Italy) and John Roth (University of California at Davis) for the most insightful discussions. I am greateful to Cindy Cook for the technical help with a manuscript.
Metabolite Abbreviations used in this chapter
(for the list of functional role abbreviations, see Table 1)
- ADP
Adenosine 5′-diphosphate
- ADPR
ADP-ribose
- AMP
Adenosine 5′-monophosphate
- ATP
Adenosine 5′-triphosphate
- DHAP
Dihydroxyacetone phosphate
- FAD
Flavine Adenine dinucleotide
- IA
Iminoaspartate
- L-Asp
L-Aspartate
- Na
Nicotinic acid
- NaAD
Nicotinic acid adenine dinucleotide (desamido-NAD)
- NAD
Nicotinamide adenine dinucleotide (irrespective of the redox state)
- NAD+
Nicotinamide adenine dinucleotide, oxidized form
- NADH
Nicotinamide adenine dinucleotide, reduced form
- NADP
Nicotinamide adenine dinucleotide phosphate (irrespective of the redox state)
- NADP+
Nicotinamide adenine dinucleotide phosphate, oxidized form
- NADPH
Nicotinamide adenine dinucleotide phosphate, reduced form
- NaMN
Nicotinic acid mononucleotide
- Nm
Nicotinamide
- NMN
Nicotinamide mononucleotide
- PPi
Inorganic pyrophosphate
- PRPP
5-Phosphoribosyl 1-pyrophosphate
- Qa
Quinolinic acid
- RNm
N-Ribosylnicotinamide
References
- 1.Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–25. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aravind L, Anantharaman V, Koonin EV. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: implications for protein evolution in the RNA. Proteins. 2002;48:1–14. doi: 10.1002/prot.10064. [DOI] [PubMed] [Google Scholar]
- 3.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. 2006;2:msb4100050-E1–msb4100050-E11. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Begley TP, Kinsland C, Mehl RA, Osterman A, Dorrestein P. The biosynthesis of nicotinamide adenine dinucleotides in bacteria. Vitam Horm. 2001;61:103–19. doi: 10.1016/s0083-6729(01)61003-3. [DOI] [PubMed] [Google Scholar]
- 5.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–9. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell. 2007;129:473–84. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 7.Bellinzoni M, Buroni S, Pasca MR, Guglierame P, Arcesi F, De Rossi E, Riccardi G. Glutamine amidotransferase activity of NAD+ synthetase from Mycobacterium tuberculosis depends on an amino-terminal nitrilase domain. Res Microbiol. 2005;156:173–7. doi: 10.1016/j.resmic.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Bergthorsson U, Roth JR. Natural isolates of Salmonella enterica serovar Dublin carry a single nadA missense mutation. J Bacteriol. 2005;187:400–3. doi: 10.1128/JB.187.1.400-403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatia R, Calvo KC. The sequencing expression, purification, and steady-state kinetic analysis of quinolinate phosphoribosyl transferase from Escherichia coli. Arch Biochem Biophys. 1996;325:270–8. doi: 10.1006/abbi.1996.0034. [DOI] [PubMed] [Google Scholar]
- 10.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- 11.Bieganowski P, Pace HC, Brenner C. Eukaryotic NAD+ synthetase Qns1 contains an essential, obligate intramolecular thiol glutamine amidotransferase domain related to nitrilase. J Biol Chem. 2003;278:33049–55. doi: 10.1074/jbc.M302257200. [DOI] [PubMed] [Google Scholar]
- 12.Boonstra B, French CE, Wainwright I, Bruce NC. The udhA gene of Escherichia coli encodes a soluble pyridine nucleotide transhydrogenase. J Bacteriol. 1999;181:1030–4. doi: 10.1128/jb.181.3.1030-1034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bossi RT, Negri A, Tedeschi G, Mattevi A. Structure of FAD-bound L-aspartate oxidase: insight into substrate specificity and catalysis. Biochemistry. 2002;41:3018–24. doi: 10.1021/bi015939r. [DOI] [PubMed] [Google Scholar]
- 14.Brenner C. Evolution of NAD biosynthetic enzymes. Structure. 2005;13:1239–40. doi: 10.1016/j.str.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Cao H, Pietrak BL, Grubmeyer C. Quinolinate phosphoribosyltransferase: kinetic mechanism for a type II PRTase. Biochemistry. 2002;41:3520–8. doi: 10.1021/bi012148g. [DOI] [PubMed] [Google Scholar]
- 16.Ceciliani F, Caramori T, Ronchi S, Tedeschi G, Mortarino M, Galizzi A. Cloning, overexpression, and purification of Escherichia coli quinolinate synthetase. Protein Expr Purif. 2000;18:64–70. doi: 10.1006/prep.1999.1153. [DOI] [PubMed] [Google Scholar]
- 17.Chappie JS, Canaves JM, Han GW, Rife CL, Xu Q, Stevens RC. The structure of a eukaryotic nicotinic acid phosphoribosyltransferase reveals structural heterogeneity among type II PRTases. Structure. 2005;13:1385–96. doi: 10.1016/j.str.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 18.Cheng W, Roth J. Isolation of NAD cycle mutants defective in nicotinamide mononucleotide deamidase in Salmonella typhimurium. J Bacteriol. 1995;177:6711–7. doi: 10.1128/jb.177.23.6711-6717.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng W, Roth JR. Evidence for two NAD kinases in Salmonella typhimurium. J Bacteriol. 1994;176:4260–8. doi: 10.1128/jb.176.14.4260-4268.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colovos C, Cascio D, Yeates TO. The 1.8 A crystal structure of the ycaC gene product from Escherichia coli reveals an octameric hydrolase of unknown specificity. Structure. 1998;6:1329–37. doi: 10.1016/s0969-2126(98)00132-4. [DOI] [PubMed] [Google Scholar]
- 21.Depping R, Lohaus C, Meyer HE, Ruger W. The mono-ADP-ribosyltransferases Alt and ModB of bacteriophage T4: target proteins identified. Biochem Biophys Res Commun. 2005;335:1217–23. doi: 10.1016/j.bbrc.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 22.Du X, Wang W, Kim R, Yakota H, Nguyen H, Kim SH. Crystal structure and mechanism of catalysis of a pyrazinamidase from Pyrococcus horikoshii. Biochemistry. 2001;40:14166–72. doi: 10.1021/bi0115479. [DOI] [PubMed] [Google Scholar]
- 23.Eads JC, Ozturk D, Wexler TB, Grubmeyer C, Sacchettini JC. A new function for a common fold: the crystal structure of quinolinic acid phosphoribosyltransferase. Structure. 1997;5:47–58. doi: 10.1016/s0969-2126(97)00165-2. [DOI] [PubMed] [Google Scholar]
- 24.Flachmann R, Kunz N, Seifert J, Gutlich M, Wientjes FJ, Laufer A, Gassen HG. Molecular biology of pyridine nucleotide biosynthesis in Escherichia coli. Cloning and characterization of quinolinate synthesis genes nadA and nadB. Eur J Biochem. 1988;175:221–8. doi: 10.1111/j.1432-1033.1988.tb14187.x. [DOI] [PubMed] [Google Scholar]
- 25.Foster JW, Kinney DM, Moat AG. Pyridine nucleotide cycle of Salmonella typhimurium: isolation and characterization of pncA, pncB, and pncC mutants and utilization of exogenous nicotinamide adenine dinucleotide. J Bacteriol. 1979;137:1165–75. doi: 10.1128/jb.137.3.1165-1175.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frothingham R, Meeker-O'Connell WA, Talbot EA, George JW, Kreuzer KN. Identification, cloning, and expression of the Escherichia coli pyrazinamidase and nicotinamidase gene, pncA. Antimicrob Agents Chemother. 1996;40:1426–31. doi: 10.1128/aac.40.6.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabelli SB, Bianchet MA, Bessman MJ, Amzel LM. The structure of ADP-ribose pyrophosphatase reveals the structural basis for the versatility of the Nudix family. Nat Struct Biol. 2001;8:467–72. doi: 10.1038/87647. [DOI] [PubMed] [Google Scholar]
- 28.Gabelli SB, Bianchet MA, Ohnishi Y, Ichikawa Y, Bessman MJ, Amzel LM. Mechanism of the Escherichia coli ADP-ribose pyrophosphatase, a Nudix hydrolase. Biochemistry. 2002;41:9279–85. doi: 10.1021/bi0259296. [DOI] [PubMed] [Google Scholar]
- 29.Garavaglia S, Galizzi A, Rizzi M. Allosteric regulation of Bacillus subtilis NAD kinase by quinolinic acid. J Bacteriol. 2003;185:4844–50. doi: 10.1128/JB.185.16.4844-4850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem. 2007;282:30239–45. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- 31.Gerasimova AV, Gelfand MS. Evolution of the NadR regulon in Enterobacteriaceae. J Bioinform Comput Biol. 2005;3:1007–19. doi: 10.1142/s0219720005001387. [DOI] [PubMed] [Google Scholar]
- 32.Gerdes SY, Kurnasov OV, Shatalin K, Polanuyer B, Sloutsky R, Vonstein V, Overbeek R, Osterman AL. Comparative genomics of NAD biosynthesis in cyanobacteria. J Bacteriol. 2006;188:3012–23. doi: 10.1128/JB.188.8.3012-3023.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerdes SY, Scholle MD, Campbell JW, Balazsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D'Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabasi AL, Oltvai ZN, Osterman AL. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol. 2003;185:5673–84. doi: 10.1128/JB.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerdes SY, Scholle MD, D'Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, Campbell JW, Anantha S, Shatalin KY, Chowdhury SA, Fonstein MY, Osterman AL. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J Bacteriol. 2002;184:4555–72. doi: 10.1128/JB.184.16.4555-4572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grose JH, Bergthorsson U, Roth JR. Regulation of NAD synthesis by the trifunctional NadR protein of Salmonella enterica. J Bacteriol. 2005;187:2774–82. doi: 10.1128/JB.187.8.2774-2782.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grose JH, Bergthorsson U, Xu Y, Sterneckert J, Khodaverdian B, Roth JR. Assimilation of nicotinamide mononucleotide requires periplasmic AphA phosphatase in Salmonella enterica. J Bacteriol. 2005;187:4521–30. doi: 10.1128/JB.187.13.4521-4530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grose JH, Joss L, Velick SF, Roth JR. Evidence that feedback inhibition of NAD kinase controls responses to oxidative stress. Proc Natl Acad Sci U S A. 2006;103:7601–6. doi: 10.1073/pnas.0602494103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gross J, Rajavel M, Segura E, Grubmeyer C. Energy coupling in Salmonella typhimurium nicotinic acid phosphoribosyltransferase: identification of His-219 as site of phosphorylation. Biochemistry. 1996;35:3917–24. doi: 10.1021/bi9517906. [DOI] [PubMed] [Google Scholar]
- 39.Gross JW, Rajavel M, Grubmeyer C. Kinetic mechanism of nicotinic acid phosphoribosyltransferase: implications for energy coupling. Biochemistry. 1998;37:4189–99. doi: 10.1021/bi972014w. [DOI] [PubMed] [Google Scholar]
- 40.Grubmeyer CT, Gross JW, Rajavel M. Energy coupling through molecular discrimination: nicotinate phosphoribosyltransferase. Methods Enzymol. 1999;308:28–48. doi: 10.1016/s0076-6879(99)08004-0. [DOI] [PubMed] [Google Scholar]
- 41.Haferkamp I, Schmitz-Esser S, Linka N, Urbany C, Collingro A, Wagner M, Horn M, Neuhaus HE. A candidate NAD+ transporter in an intracellular bacterial symbiont related to Chlamydiae. Nature. 2004;432:622–5. doi: 10.1038/nature03131. [DOI] [PubMed] [Google Scholar]
- 42.Han S, Forman MD, Loulakis P, Rosner MH, Xie Z, Wang H, Danley DE, Yuan W, Schafer J, Xu Z. Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Staphyloccocus aureus: structural basis for NaAD interaction in functional dimer. J Mol Biol. 2006;360:814–25. doi: 10.1016/j.jmb.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 43.Hochmann H, Pust S, von Figura G, Aktories K, Barth H. Salmonella enterica SpvB ADP-ribosylates actin at position arginine-177-characterization of the catalytic domain within the SpvB protein and a comparison to binary clostridial actin-ADP-ribosylating toxins. Biochemistry. 2006;45:1271–7. doi: 10.1021/bi051810w. [DOI] [PubMed] [Google Scholar]
- 44.Huang N, Sorci L, Zhang X, Brautigam CA, Li X, Raffaelli N, Magni G, Grishin NV, Osterman AL, Zhang H. Bifunctional NMN Adenylyltransferase/ADP-Ribose Pyrophosphatase: Structure and Function in Bacterial NAD Metabolism. Structure. 2008;16:196–209. doi: 10.1016/j.str.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hughes KT, Dessen A, Gray JP, Grubmeyer C. The Salmonella typhimurium nadC gene: sequence determination by use of Mud-P22 and purification of quinolinate phosphoribosyltransferase. J Bacteriol. 1993;175:479–86. doi: 10.1128/jb.175.2.479-486.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes KT, Ladika D, Roth JR, Olivera BM. An indispensable gene for NAD biosynthesis in Salmonella typhimurium. J Bacteriol. 1983;155:213–21. doi: 10.1128/jb.155.1.213-221.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes KT, Olivera BM, Roth JR. Structural gene for NAD synthetase in Salmonella typhimurium. J Bacteriol. 1988;170:2113–20. doi: 10.1128/jb.170.5.2113-2120.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hughes KT, Roth JR, Olivera BM. A genetic characterization of the nadC gene of Salmonella typhimurium. Genetics. 1991;127:657–70. doi: 10.1093/genetics/127.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jauch R, Humm A, Huber R, Wahl MC. Structures of Escherichia coli NAD synthetase with substrates and products reveal mechanistic rearrangements. J Biol Chem. 2005;280:15131–40. doi: 10.1074/jbc.M413195200. [DOI] [PubMed] [Google Scholar]
- 50.Johansson T, Oswald C, Pedersen A, Tornroth S, Okvist M, Karlsson BG, Rydstrom J, Krengel U. X-ray structure of domain I of the proton-pumping membrane protein transhydrogenase from Escherichia coli. J Mol Biol. 2005;352:299–312. doi: 10.1016/j.jmb.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 51.Katoh A, Uenohara K, Akita M, Hashimoto T. Early steps in the biosynthesis of NAD in Arabidopsis start with aspartate and occur in the plastid. Plant Physiol. 2006;141:851–7. doi: 10.1104/pp.106.081091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawai S, Mori S, Mukai T, Hashimoto W, Murata K. Molecular characterization of Escherichia coli NAD kinase. Eur J Biochem. 2001;268:4359–65. doi: 10.1046/j.1432-1327.2001.02358.x. [DOI] [PubMed] [Google Scholar]
- 53.Kawai S, Mori S, Mukai T, Suzuki S, Yamada T, Hashimoto W, Murata K. Inorganic Polyphosphate/ATP-NAD kinase of Micrococcus flavus and Mycobacterium tuberculosis H37Rv. Biochem Biophys Res Commun. 2000;276:57–63. doi: 10.1006/bbrc.2000.3433. [DOI] [PubMed] [Google Scholar]
- 54.Khan JA, Tao X, Tong L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat Struct Mol Biol. 2006;13:582–8. doi: 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- 55.Kim MK, Kang GB, Song WK, Eom SH. The role of Phe181 in the hexamerization of Helicobacter pylori quinolinate phosphoribosyltransferase. Protein J. 2007;26:517–21. doi: 10.1007/s10930-007-9093-0. [DOI] [PubMed] [Google Scholar]
- 56.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–9. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 57.Kornberg A. The participation of inorganic pyrophosphate in the reversible enzymatic synthesis of diphosphopyridine nucleotide. J Biol Chem. 1948;176:1475. [PubMed] [Google Scholar]
- 58.Kurnasov O, Goral V, Colabroy K, Gerdes S, Anantha S, Osterman A, Begley TP. NAD biosynthesis: identification of the tryptophan to quinolinate pathway in bacteria. Chem Biol. 2003;10:1195–204. doi: 10.1016/j.chembiol.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 59.Kurnasov OV, Polanuyer BM, Ananta S, Sloutsky R, Tam A, Gerdes SY, Osterman AL. Ribosylnicotinamide Kinase Domain of NadR Protein: Identification and Implications in NAD Biosynthesis. J Bacteriol. 2002;184:6906–17. doi: 10.1128/JB.184.24.6906-6917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labesse G, Douguet D, Assairi L, Gilles AM. Diacylglyceride kinases, sphingosine kinases and NAD kinases: distant relatives of 6-phosphofructokinases. Trends Biochem Sci. 2002;27:273–5. doi: 10.1016/s0968-0004(02)02093-5. [DOI] [PubMed] [Google Scholar]
- 61.Liu H, Woznica K, Catton G, Crawford A, Botting N, Naismith JH. Structural and kinetic characterization of quinolinate phosphoribosyltransferase (hQPRTase) from homo sapiens. J Mol Biol. 2007;373:755–63. doi: 10.1016/j.jmb.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu J, Lou Y, Yokota H, Adams PD, Kim R, Kim SH. Crystal structures of an NAD kinase from Archaeoglobus fulgidus in complex with ATP, NAD, or NADP. J Mol Biol. 2005;354:289–303. doi: 10.1016/j.jmb.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 63.Loiseau L, Ollagnier-de Choudens S, Lascoux D, Forest E, Fontecave M, Barras F. Analysis of the heteromeric CsdA-CsdE cysteine desulfurase, assisting Fe-S cluster biogenesis in Escherichia coli. J Biol Chem. 2005;280:26760–9. doi: 10.1074/jbc.M504067200. [DOI] [PubMed] [Google Scholar]
- 64.Maggio-Hall LA, Escalante-Semerena JC. In vitro synthesis of the nucleotide loop of cobalamin by Salmonella typhimurium enzymes. Proc Natl Acad Sci U S A. 1999;96:11798–803. doi: 10.1073/pnas.96.21.11798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Structure and function of nicotinamide mononucleotide adenylyltransferase. Curr Med Chem. 2004;11:873–85. doi: 10.2174/0929867043455666. [DOI] [PubMed] [Google Scholar]
- 66.Magni G, Amici A, Emanuelli M, Raffaelli N, Ruggieri S. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol. 1999;73:135–82. doi: 10.1002/9780470123195.ch5. [DOI] [PubMed] [Google Scholar]
- 67.Magni G, Orsomando G, Raffaelli N. Structural and functional properties of NAD kinase, a key enzyme in NADP biosynthesis. Mini Rev Med Chem. 2006;6:739–46. doi: 10.2174/138955706777698688. [DOI] [PubMed] [Google Scholar]
- 68.Martin PR, Shea RJ, Mulks MH. Identification of a plasmid-encoded gene from Haemophilus ducreyi which confers NAD independence. J Bacteriol. 2001;183:1168–74. doi: 10.1128/JB.183.4.1168-1174.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mattevi A, Tedeschi G, Bacchella L, Coda A, Negri A, Ronchi S. Structure of L-aspartate oxidase: implications for the succinate dehydrogenase/fumarate reductase oxidoreductase family. Structure. 1999;7:745–56. doi: 10.1016/s0969-2126(99)80099-9. [DOI] [PubMed] [Google Scholar]
- 70.McLennan AG. The Nudix hydrolase superfamily. Cell Mol Life Sci. 2006;63:123–43. doi: 10.1007/s00018-005-5386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McMurry J, Begley T. The Organic Chemistry of Biological Pathways. Roberts and Co. publishers; Greenwood Village, CO: 2005. [Google Scholar]
- 72.Mehl RA, Kinsland C, Begley TP. Identification of the Escherichia coli nicotinic acid mononucleotide adenylyltransferase gene. J Bacteriol. 2000;182:4372–4. doi: 10.1128/jb.182.15.4372-4374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Merdanovic M, Sauer E, Reidl J. Coupling of NAD+ Biosynthesis and Nicotinamide Ribosyl Transport: Characterization of NadR Ribonucleotide Kinase Mutants of Haemophilus influenzae. J Bacteriol. 2005;187:4410–20. doi: 10.1128/JB.187.13.4410-4420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Messner KR, Imlay JA. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J Biol Chem. 2002;277:42563–71. doi: 10.1074/jbc.M204958200. [DOI] [PubMed] [Google Scholar]
- 75.Meuller J, Rydstrom J. The membrane topology of proton-pumping Escherichia coli transhydrogenase determined by cysteine labeling. J Biol Chem. 1999;274:19072–80. doi: 10.1074/jbc.274.27.19072. [DOI] [PubMed] [Google Scholar]
- 76.Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin AS, Ciecko A, Feldblyum TV, White O, Paulsen IT, Nierman WC, Lee J, Szczypinski B, Fraser CM. Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. J Bacteriol. 2003;185:5220–33. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moran-Zorzano MT, Viale AM, Munoz FJ, Alonso-Casajus N, Eydallin GG, Zugasti B, Baroja-Fernandez E, Pozueta-Romero J. Escherichia coli AspP activity is enhanced by macromolecular crowding and by both glucose-1,6-bisphosphate and nucleotide-sugars. FEBS Lett. 2007;581:1035–40. doi: 10.1016/j.febslet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 78.Mori S, Yamasaki M, Maruyama Y, Momma K, Kawai S, Hashimoto W, Mikami B, Murata K. NAD-binding mode and the significance of intersubunit contact revealed by the crystal structure of Mycobacterium tuberculosis NAD kinase-NAD complex. Biochem Biophys Res Commun. 2005;327:500–8. doi: 10.1016/j.bbrc.2004.11.163. [DOI] [PubMed] [Google Scholar]
- 79.Mortarino M, Negri A, Tedeschi G, Simonic T, Duga S, Gassen HG, Ronchi S. L-aspartate oxidase from Escherichia coli. I. Characterization of coenzyme binding and product inhibition. Eur J Biochem. 1996;239:418–26. doi: 10.1111/j.1432-1033.1996.0418u.x. [DOI] [PubMed] [Google Scholar]
- 80.Mushegian A. The Purloined Letter: bacterial orthologs of archaeal NMN adenylyltransferase are domains within multifunctional transcription regulator NadR. J Mol Microbiol Biotechnol. 1999;1:127–8. [PubMed] [Google Scholar]
- 81.Nandakumar J, Nair PA, Shuman S. Last stop on the road to repair: structure of E. coli DNA ligase bound to nicked DNA-adenylate. Mol Cell. 2007;26:257–71. doi: 10.1016/j.molcel.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 82.Nasu S, Wicks FD, Gholson RK. L-Aspartate oxidase, a newly discovered enzyme of Escherichia coli, is the B protein of quinolinate synthetase. J Biol Chem. 1982;257:626–32. [PubMed] [Google Scholar]
- 83.Ollagnier-de Choudens S, Loiseau L, Sanakis Y, Barras F, Fontecave M. Quinolinate synthetase, an iron-sulfur enzyme in NAD biosynthesis. FEBS Lett. 2005;579:3737–43. doi: 10.1016/j.febslet.2005.05.065. [DOI] [PubMed] [Google Scholar]
- 84.Ollagnier-de-Choudens S, Sanakis Y, Abdel-Ghany SE, Rousset C, Ye H, Fontecave M, Pilon-Smits EA, Pilon M. Characterization of Arabidopsis thaliana SufE2 and SufE3: functions in chloroplast iron-sulfur cluster assembly and Nad synthesis. J Biol Chem. 2007;282:18254–64. doi: 10.1074/jbc.M701428200. [DOI] [PubMed] [Google Scholar]
- 85.Olland AM, Underwood KW, Czerwinski RM, Lo MC, Aulabaugh A, Bard J, Stahl ML, Somers WS, Sullivan FX, Chopra R. Identification, characterization, and crystal structure of Bacillus subtilis nicotinic acid mononucleotide adenylyltransferase. J Biol Chem. 2002;277:3698–707. doi: 10.1074/jbc.M109670200. [DOI] [PubMed] [Google Scholar]
- 86.Osterman A, Overbeek R. Missing genes in metabolic pathways: a comparative genomics approach. Curr Opin Chem Biol. 2003;7:238–51. doi: 10.1016/s1367-5931(03)00027-9. [DOI] [PubMed] [Google Scholar]
- 87.Osterman AL, Begley TP. A subsystems-based approach to the identification of drug targets in bacterial pathogens. Prog Drug Res. 2007;64:131, 133–70. doi: 10.1007/978-3-7643-7567-6_6. [DOI] [PubMed] [Google Scholar]
- 88.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Ruckert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ozment C, Barchue J, DeLucas LJ, Chattopadhyay D. Structural study of Escherichia coli NAD synthetase: overexpression, purification, crystallization, and preliminary crystallographic analysis. J Struct Biol. 1999;127:279–82. doi: 10.1006/jsbi.1999.4152. [DOI] [PubMed] [Google Scholar]
- 90.Penfound T, Foster JW. Biosynthesis and Recycling of NAD. In: Neihardt F, editor. Escherichia Coli and Salmonella. American Society for Microbiology; Washington, D.C: 1996. pp. 721–730. ed. ASM. [Google Scholar]
- 91.Penfound T, Foster JW. NAD-dependent DNA-binding activity of the bifunctional NadR regulator of Salmonella typhimurium. J Bacteriol. 1999;181:648–55. doi: 10.1128/jb.181.2.648-655.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Poncet-Montange G, Assairi L, Arold S, Pochet S, Labesse G. NAD kinases use substrate-assisted catalysis for specific recognition of NAD. J Biol Chem. 2007;282:33925–34. doi: 10.1074/jbc.M701394200. [DOI] [PubMed] [Google Scholar]
- 93.Preiss J, Handler P. Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. J Biol Chem. 1958;233:493–500. [PubMed] [Google Scholar]
- 94.Prunier AL, Schuch R, Fernandez RE, Maurelli AT. Genetic structure of the nadA and nadB antivirulence loci in Shigella spp. J Bacteriol. 2007;189:6482–6. doi: 10.1128/JB.00525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Prunier AL, Schuch R, Fernandez RE, Mumy KL, Kohler H, McCormick BA, Maurelli AT. nadA and nadB of Shigella flexneri 5a are antivirulence loci responsible for the synthesis of quinolinate, a small molecule inhibitor of Shigella pathogenicity. Microbiology. 2007;153:2363–72. doi: 10.1099/mic.0.2007/006916-0. [DOI] [PubMed] [Google Scholar]
- 96.Raffaelli N, Finaurini L, Mazzola F, Pucci L, Sorci L, Amici A, Magni G. Characterization of Mycobacterium tuberculosis NAD kinase: functional analysis of the full-length enzyme by site-directed mutagenesis. Biochemistry. 2004;43:7610–7. doi: 10.1021/bi049650w. [DOI] [PubMed] [Google Scholar]
- 97.Raffaelli N, Lorenzi T, Amici A, Emanuelli M, Ruggieri S, Magni G. Synechocystis sp. slr0787 protein is a novel bifunctional enzyme endowed with both nicotinamide mononucleotide adenylyltransferase and ‘Nudix’ hydrolase activities. FEBS Lett. 1999;444:222–6. doi: 10.1016/s0014-5793(99)00068-x. [DOI] [PubMed] [Google Scholar]
- 98.Raffaelli N, Lorenzi T, Mariani PL, Emanuelli M, Amici A, Ruggieri S, Magni G. The Escherichia coli NadR regulator is endowed with nicotinamide mononucleotide adenylyltransferase activity. J Bacteriol. 1999;181:5509–11. doi: 10.1128/jb.181.17.5509-5511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biol. 2003;4:R54. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rizzi M, Bolognesi M, Coda A. A novel deamido-NAD+-binding site revealed by the trapped NAD-adenylate intermediate in the NAD+ synthetase structure. Structure. 1998;6:1129–40. doi: 10.1016/s0969-2126(98)00114-2. [DOI] [PubMed] [Google Scholar]
- 101.Rizzi M, Schindelin H. Structural biology of enzymes involved in NAD and molybdenum cofactor biosynthesis. Curr Opin Struct Biol. 2002;12:709–20. doi: 10.1016/s0959-440x(02)00385-8. [DOI] [PubMed] [Google Scholar]
- 102.Rodionov DA, Li X, Rodionova IA, Yang C, Sorci L, Dervyn E, Martynowski D, Zhang H, Gelfand MS, Osterman AL. Transcriptional regulation of NAD metabolism in bacteria: genomic reconstruction of NiaR (YrxA) regulon. Nucleic Acids Res. 2008;36:2032–46. doi: 10.1093/nar/gkn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–34. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 104.Rossolillo P, Marinoni I, Galli E, Colosimo A, Albertini AM. YrxA is the transcriptional regulator that represses de novo NAD biosynthesis in Bacillus subtilis. J Bacteriol. 2005;187:7155–60. doi: 10.1128/JB.187.20.7155-7160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rowe JJ, Lemmon RD, Tritz GJ. Nicotinic acid transport in Escherichia coli. Microbios. 1985;44:169–84. [PubMed] [Google Scholar]
- 106.Saitoh M, Tanaka K, Nishimori K, Makino S, Kanno T, Ishihara R, Hatama S, Kitano R, Kishima M, Sameshima T, Akiba M, Nakazawa M, Yokomizo Y, Uchida I. The artAB genes encode a putative ADP-ribosyltransferase toxin homologue associated with Salmonella enterica serovar Typhimurium DT104. Microbiology. 2005;151:3089–96. doi: 10.1099/mic.0.27933-0. [DOI] [PubMed] [Google Scholar]
- 107.Sakuraba H, Kawakami R, Ohshima T. First archaeal inorganic polyphosphate/ATP-dependent NAD kinase, from hyperthermophilic archaeon Pyrococcus horikoshii: cloning, expression, and characterization. Appl Environ Microbiol. 2005;71:4352–8. doi: 10.1128/AEM.71.8.4352-4358.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sakuraba H, Tsuge H, Yoneda K, Katunuma N, Ohshima T. Crystal structure of the NAD biosynthetic enzyme quinolinate synthase. J Biol Chem. 2005;280:26645–8. doi: 10.1074/jbc.C500192200. [DOI] [PubMed] [Google Scholar]
- 109.Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E. The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J Biol Chem. 2004;279:6613–9. doi: 10.1074/jbc.M311657200. [DOI] [PubMed] [Google Scholar]
- 110.Sauve AA, Celic I, Avalos J, Deng H, Boeke JD, Schramm VL. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry. 2001;40:15456–63. doi: 10.1021/bi011858j. [DOI] [PubMed] [Google Scholar]
- 111.Schneider BL, Reitzer LJ. Salmonella typhimurium nit is nadE: defective nitrogen utilization and ammonia-dependent NAD synthetase. J Bacteriol. 1998;180:4739–41. doi: 10.1128/jb.180.17.4739-4741.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schwarzenbacher R, Jaroszewski L, von Delft F, Abdubek P, Ambing E, Biorac T, Brinen LS, Canaves JM, Cambell J, Chiu HJ, Dai X, Deacon AM, DiDonato M, Elsliger MA, Eshagi S, Floyd R, Godzik A, Grittini C, Grzechnik SK, Hampton E, Karlak C, Klock HE, Koesema E, Kovarik JS, Kreusch A, Kuhn P, Lesley SA, Levin I, McMullan D, McPhillips TM, Miller MD, Morse A, Moy K, Ouyang J, Page R, Quijano K, Robb A, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, Vincent J, Wang X, West B, Wolf G, Xu Q, Hodgson KO, Wooley J, Wilson IA. Crystal structure of a type II quinolic acid phosphoribosyltransferase (TM1645) from Thermotoga maritima at 2.50 A resolution. Proteins. 2004;55:768–71. doi: 10.1002/prot.20029. [DOI] [PubMed] [Google Scholar]
- 113.Seifert J, Kunz N, Flachmann R, Laufer A, Jany KD, Gassen HG. Expression of the E. coli nadB gene and characterization of the gene product L-aspartate oxidase. Biol Chem Hoppe Seyler. 1990;371:239–48. [PubMed] [Google Scholar]
- 114.Sharma V, Grubmeyer C, Sacchettini JC. Crystal structure of quinolinic acid phosphoribosyltransferase from Mmycobacterium tuberculosis: a potential TB drug target. Structure. 1998;6:1587–99. doi: 10.1016/s0969-2126(98)00156-7. [DOI] [PubMed] [Google Scholar]
- 115.Shin DH, Oganesyan N, Jancarik J, Yokota H, Kim R, Kim SH. Crystal structure of a nicotinate phosphoribosyltransferase from Thermoplasma acidophilum. J Biol Chem. 2005;280:18326–35. doi: 10.1074/jbc.M501622200. [DOI] [PubMed] [Google Scholar]
- 116.Singh P, Mishra AK, Malonia SK, Chauhan DS, Sharma VD, Venkatesan K, Katoch VM. The paradox of pyrazinamide: an update on the molecular mechanisms of pyrazinamide resistance in Mycobacteria. J Commun Dis. 2006;38:288–98. [PubMed] [Google Scholar]
- 117.Singh SK, Kurnasov OV, Chen B, Robinson H, Grishin NV, Osterman AL, Zhang H. Crystal structure of Haemophilus influenzae NadR protein. A bifunctional enzyme endowed with NMN adenyltransferase and ribosylnicotinimide kinase activities. J Biol Chem. 2002;277:33291–9. doi: 10.1074/jbc.M204368200. [DOI] [PubMed] [Google Scholar]
- 118.Starai VJ, Escalante-Semerena JC. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J Mol Biol. 2004;340:1005–12. doi: 10.1016/j.jmb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 119.Starai VJ, Takahashi H, Boeke JD, Escalante-Semerena JC. A link between transcription and intermediary metabolism: a role for Sir2 in the control of acetyl-coenzyme A synthetase. Curr Opin Microbiol. 2004;7:115–9. doi: 10.1016/j.mib.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 120.Symersky J, Devedjiev Y, Moore K, Brouillette C, DeLucas L. NH3-dependent NAD+ synthetase from Bacillus subtilis at 1 A resolution. Acta Crystallogr D Biol Crystallogr. 2002;58:1138–46. doi: 10.1107/s0907444902006698. [DOI] [PubMed] [Google Scholar]
- 121.Tedeschi G, Negri A, Ceciliani F, Mattevi A, Ronchi S. Structural characterization of l-aspartate oxidase and identification of an interdomain loop by limited proteolysis. Eur J Biochem. 1999;260:896–903. doi: 10.1046/j.1432-1327.1999.00234.x. [DOI] [PubMed] [Google Scholar]
- 122.Tedeschi G, Negri A, Mortarino M, Ceciliani F, Simonic T, Faotto L, Ronchi S. L-aspartate oxidase from Escherichia coli. II. Interaction with C4 dicarboxylic acids and identification of a novel L-aspartate: fumarate oxidoreductase activity. Eur J Biochem. 1996;239:427–33. doi: 10.1111/j.1432-1033.1996.0427u.x. [DOI] [PubMed] [Google Scholar]
- 123.Tempel W, Rabeh WM, Bogan KL, Belenky P, Wojcik M, Seidle HF, Nedyalkova L, Yang T, Sauve AA, Park HW, Brenner C. Nicotinamide riboside kinase structures reveal new pathways to NAD+ PLoS Biol. 2007;5:e263. doi: 10.1371/journal.pbio.0050263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tomkinson AE, Vijayakumar S, Pascal JM, Ellenberger T. DNA ligases: structure, reaction mechanism, and function. Chem Rev. 2006;106:687–99. doi: 10.1021/cr040498d. [DOI] [PubMed] [Google Scholar]
- 125.Tritz G. NAD biosynthesis and recycling. In: Neihardt F, Ingraham J, Low K, Magasanik B, Schaechter M, Umbarger H, editors. Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. Vol. 1 American Society for Microbiology; Washington, D.C: 1987. pp. 557–563. [Google Scholar]
- 126.Tsang AW, Escalante-Semerena JC. CobB, a new member of the SIR2 family of eucaryotic regulatory proteins, is required to compensate for the lack of nicotinate mononucleotide:5,6-dimethylbenzimidazole phosphoribosyltransferase activity in cobT mutants during cobalamin biosynthesis in Salmonella typhimurium LT2. J Biol Chem. 1998;273:31788–94. doi: 10.1074/jbc.273.48.31788. [DOI] [PubMed] [Google Scholar]
- 127.Vinitsky A, Grubmeyer C. A new paradigm for biochemical energy coupling. Salmonella typhimurium nicotinate phosphoribosyltransferase. J Biol Chem. 1993;268:26004–10. [PubMed] [Google Scholar]
- 128.Vinitsky A, Teng H, Grubmeyer CT. Cloning and nucleic acid sequence of the Salmonella typhimurium pncB gene and structure of nicotinate phosphoribosyltransferase. J Bacteriol. 1991;173:536–40. doi: 10.1128/jb.173.2.536-540.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wilkinson A, Day J, Bowater R. Bacterial DNA ligases. Mol Microbiol. 2001;40:1241–8. doi: 10.1046/j.1365-2958.2001.02479.x. [DOI] [PubMed] [Google Scholar]
- 130.Willison JC, Tissot G. The Escherichia coli efg gene and the Rhodobacter capsulatus adgA gene code for NH3-dependent NAD synthetase. J Bacteriol. 1994;176:3400–2. doi: 10.1128/jb.176.11.3400-3402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wubbolts MG, Terpstra P, van Beilen JB, Kingma J, Meesters HA, Witholt B. Variation of cofactor levels in Escherichia coli. Sequence analysis and expression of the pncB gene encoding nicotinic acid phosphoribosyltransferase. J Biol Chem. 1990;265:17665–72. [PubMed] [Google Scholar]
- 132.Yang Z, Savchenko A, Yakunin A, Zhang R, Edwards A, Arrowsmith C, Tong L. Aspartate dehydrogenase, a novel enzyme identified from structural and functional studies of TM1643. J Biol Chem. 2003;278:8804–8. doi: 10.1074/jbc.M211892200. [DOI] [PubMed] [Google Scholar]
- 133.Yoon HJ, Kim HL, Mikami B, Suh SW. Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Pseudomonas aeruginosa in its Apo and substrate-complexed forms reveals a fully open conformation. J Mol Biol. 2005;351:258–65. doi: 10.1016/j.jmb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 134.Zalkin H. Structure, function, and regulation of amidophosphoribosyltransferase from prokaryotes. Adv Enzyme Regul. 1983;21:225–37. doi: 10.1016/0065-2571(83)90016-x. [DOI] [PubMed] [Google Scholar]
- 135.Zerez CR, Moul DE, Gomez EG, Lopez VM, Andreoli AJ. Negative modulation of Escherichia coli NAD kinase by NADPH and NADH. J Bacteriol. 1987;169:184–8. doi: 10.1128/jb.169.1.184-188.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang H, Zhou T, Kurnasov O, Cheek S, Grishin NV, Osterman A. Crystal structures of E. coli nicotinate mononucleotide adenylyltransferase and its complex with deamido-NAD. Structure (Camb) 2002;10:69–79. doi: 10.1016/s0969-2126(01)00693-1. [DOI] [PubMed] [Google Scholar]
- 137.Zhao K, Chai X, Marmorstein R. Structure and substrate binding properties of cobB, a Sir2 homolog protein deacetylase from Escherichia coli. J Mol Biol. 2004;337:731–41. doi: 10.1016/j.jmb.2004.01.060. [DOI] [PubMed] [Google Scholar]