

Abstract
Peripheral biomarkers play an indispensable role in quick and reliable diagnoses of any kind of disease. With the population ageing, the number of people suffering from age-related diseases is expected to rise dramatically over the coming decades. In particular, all types of cognitive deficits, such as Alzheimer’s disease, will increase. Alzheimer’s disease is characterised mainly by coexistence of amyloid plaques and neurofibrillary tangles in brain. Reliable identification of such molecular characteristics antemortem, however, is problematic due to restricted availability of appropriate sample material and definitive diagnosis is only possible postmortem. Currently, the best molecular biomarkers available for antemortem diagnosis originate from cerebrospinal fluid. Though, this is not convenient for routine diagnosis because of the required invasive lumbar puncture. As a consequence, there is a growing demand for additional peripheral biomarkers in a more readily accessible sample material. Blood platelets, due to shared biochemical properties with neurons, can constitute an attractive alternative as discussed here. This review summarises potential platelet Alzheimer’s disease biomarkers, their role, implication, and alteration in the disease. For easy comparison of their performance, the Hedge effect size was calculated whenever data were available.
Keywords: Alzheimer’s disease, Diagnosis, Platelets, Peripheral biomarker, APP, Amyloid beta, Tau, Mao-B, Neurotransmitter, Hedge effect size
Introduction
Alzheimer’s disease
One of the most common forms of dementia is Alzheimer’s disease (AD), which is characterised by a progressive cognitive decline. It is estimated to currently affect about 35 million people worldwide and by 2050 this number is predicted to increase to >115 million, thus reaching over 1% of the total population. Loss of memory is common amongst the aged and once this condition becomes more pronounced, it is termed ‘amnestic mild cognitive impairment’ (aMCI) and is often considered as a very early stage of AD. Early-onset familiar AD (EOFAD) is caused by genetic factors like mutations in genes encoding the amyloid precursor protein (APP) and the subunits of the APP-cleaving enzyme γ-secretase, termed presenilin-1 and −2 (PS1 and PS2). The present review, however, focuses on the more common late-onset AD (LOAD), which is characterised by more heterogeneous conditions [1]. Besides neurofibrillary tangles (NFT) and plaques composed of amyloid beta (Aβ), typical neuropathological characteristics associated with AD are loss of cholinergic neurons in the basal forebrain and reduced acetylcholine concentrations [2]. In addition, neocortical biopsies of AD brains show a severe decrease in the levels of the three monoamine neurotransmitters dopamine, norepinephrine, and serotonin [3]. Further comorbid signs of AD are changes in the metabolome such as vitamin B12 and folate deficiencies, as well as elevated homocysteine levels, potentially providing a functional link to the assumed influence of lifestyle factors on AD development [4].
Despite worldwide efforts at improving AD detection, ‘definite’ AD diagnosis is still restricted to postmortem evaluation of coexisting plaques and NFTs in brain preparations from deceased patients [5]. Antemortem, one of the most reliable diagnostic tools is brain imaging, i.e. visualisation of atrophies most apparent in the temporal lobe [6]. Hippocampal lesions in patients appear to be very important characteristics to determine progression from aMCI to overt AD [7]. However, brain imaging requires exorbitantly expensive equipment and highly trained staff, making these examinations a low-throughput screening method. Therefore, easily accessible molecular markers for early and reliable diagnosis would be of tremendous importance. Moreover, correct diagnosis at the stage of aMCI followed by immediate treatment may be more efficacious than administering medication later on [8, 9].
Biomarker sources for AD
Cerebrospinal fluid (CSF) Aβ1–42, total tau, and phospho-tau181 represent some of the best available AD biomarkers for clinical diagnosis. When combined, they give accuracy in the range of 90% [10]. A significant drawback is the required invasive lumbar puncture, making CSF impractical for initial diagnosis or clinical routine screening. Easily accessible blood, in contrast, is a desired sample material and multiple plasma proteins have already been identified as potential AD biomarker candidates, for example cytokines, acute phase proteins, adhesion molecules, and growth factors [11]. Nevertheless, most of these seem to reflect secondary inflammatory processes of neurodegeneration rather than being causative agents. In addition, many factors are likely to influence the abundance of these plasma markers, e.g. accompanying infection and inflammation, as well as disease progression of AD itself. Because of that, many AD-related plasma profiles are hard to reproduce [12]. Since the mean biological variation of platelet proteins is more stable (coefficient of variation, CV = 17% [13]) as compared to other body fluids such as plasma (CV = 23% [14]), urine (CV = 82% [15]), or CSF (CV = 83% [16]) and since AD-related pathological changes responsible for neuron decay most likely start inside cells, cellular biomarkers may be a more preferable choice for detection of causal AD signs.
Platelets and their implication as peripheral model for neurons
Blood platelets are cell fragments shed from megakaryocytes and released into the blood stream. Therefore, platelets lack a nucleus but contain a number of organelles such as mitochondria for energy generation, dense granules for storage of small molecules (e.g. serotonin, ADP, etc.), and α-granules serving as reservoir for secretory proteins. Upon stimulation, a process required for haemostasis and other physiological functions, platelets undergo a rapid shape change and release a repertoire of substances from mentioned granules [17]. With regards to neurons, they have been described to share many biochemical similarities [18–20] and mirror abnormalities in psychiatric and neuronal disorders [21, 22]. For instance, platelets express high levels of APP [23], in fact highest concentrations of this protein are found in brain and platelets. Additionally, tau protein has recently been detected in their proteome [24, 25]. Both proteins are described to have different isoform patterns in AD [26, 27]. A more obvious advantage of platelets is their fast, easy, and minimally invasive accessibility from whole blood in high numbers [28]. For these reasons, several research teams investigated whether AD-related changes of the brain are also reflected in platelets. This review aims to provide an overview of these studies with particular emphasis on the reliability of potential platelet AD biomarkers summarised in Figure 1. Additionally, in order to enable the comparison of their diagnostic power, we calculated the Hedge effect size (ES) http://www.polyu.edu.hk/mm/effectsizefaqs/calculator/calculator.html [29] for accessible data sets (Table 1). The relative values, either positive or negative (up- or down-regulation), are a measure of the difference of mean values between groups under consideration of their standard deviation. Thus, the higher the ES, the bigger the difference of a parameter with concomitant small standard deviations between the groups analysed, indicating a more powerful biomarker candidate.
Figure 1.
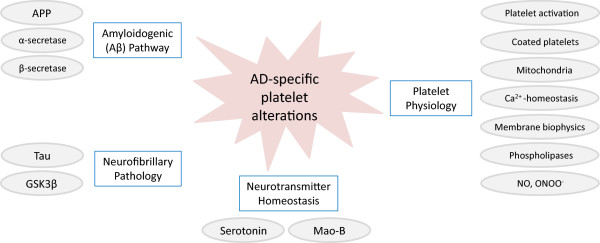
Overview of potential peripheral platelet AD biomarkers. Besides key players involved in the generation of amyloid plaques and neurofibrillary tangles, a good many additional AD-specific platelet alterations have been described. APP, amyloid precursor protein; GSK3β, glycogen synthase kinase 3β; Mao-B, monoamine oxidase B; NO, nitric oxide; ONOO−, peroxynitrite.
Table 1.
Comparison of platelet AD biomarker performance by Hedge effect size
| AD platelet biomarkers | Reg. AD | AD | Hedge ES | Controls | Pubmed ID | ||||
|---|---|---|---|---|---|---|---|---|---|
| n | Age | MMSE | MMSE | Age | n | ||||
| ADAM 10 | ↓ | 10a | 75 ± 8 | 16 ± 5 | −4.4 | 27 ± 2 | 76 ± 7 | 8 | [30] |
| ADAM 10 | ↓ | 11b | 76 ± 8 | 13 ± 5 | −2.6 | 27 ± 2 | 76 ± 7 | 8 | [30] |
| ADAM 10 | ↓ | 9c | 75 ± 8 | 1 ± 2 | −2.0 | 27 ± 2 | 76 ± 7 | 9 | [30] |
| APP total mRNA | ↑ | 20 | 78 ± 5 | 17 ± 4 | 1.8 | 29 ± 1 | 76 ± 5 | 18 | [31] |
| APP KPI mRNA | ↑ | 20 | 78 ± 5 | 17 ± 4 | 1.2 | 29 ± 1 | 76 ± 5 | 18 | [31] |
| APP 115 kDa protein abundance | ↑ | 30 | 67 ± 10 | 25 ± 3 | 1.6 | 29 ± 1 | 68 ± 10 | 23 | [32] |
| APP ratio 130/106-110 kDa ratio | ↓ | 30 | 67 ± 10 | 25 ± 3 | −2.0 | 29 ± 1 | 68 ± 10 | 23 | [32] |
| APP ratio 130/106-110 kDa ratio | ↓ | 12 | 71 ± 2 | 28 ± 2 | −1.2 | 29 ± 2 | 70 ± 6 | 10* | [33] |
| APP ratio 130/106-110 kDa ratio | ↓ | 33 | 68 ± 6 | 18 ± 4 | −2.4 | 29 ± 1 | 63 ± 6 | 26 | [34] |
| APP ratio 130/106-110 kDa ratio | ↓ | 32 | 72 ± 10 | 13 ± 7 | −3.0 | 28 ± 4 | 68 ± 14 | 25 | [35] |
| APP ratio 130/106-110 kDa ratio | ↓ | 85 | 68 ± 0 | 14 ± 7 | −2.4 | 29 ± 2 | n.a. | 24 | [36] |
| APP ratio 130/106-110 kDa ratio | ↓ | 23 | 74 ± 9 | 19 ± 5 | −1.1 | 29 ± 1 | 70 ± 6 | 29 | [37] |
| APP ratio 130/106-110 kDa ratio | ↔ | 66 | 77 ± 10 | 14 ± 8 | −0.1 | 29 ± 1 | 73 ± 11 | 46 | [38] |
| APP ratio 130/106-110 kDa ratio | ↓ | 15 | n.a | n.a | −1.0 | n.a | n.a | 19 | [39] |
| APP ratio 130/106-110 kDa ratio (AChE-inhib.) | ↓ | 20 | 70 ± 10 | 19 ± 4 | −0.4 | 28 ± 2 | 70 ± 10 | 10 | [40] |
| BACE1-β secretase activity | ↑ | 86 | 80 ± 7 | 18 ± 5 | 0.6 | 29 ± 1 | 79 ± 8 | 115 | [41] |
| BACE1-β secretase activity | ↑ | 52* | 76 | 25 ± 1 | 0.7 | 30 | 74 | 75 | [42] |
| BACE1 whole protein abundance | ↓ | 15 | 82 ± 5 | 19 ± 6 | −0.9 | 29 ± 1 | 80 ± 5 | 12 | [43] |
| Calcium intracellular | ↑ | 100 | 68 ± 7 | n.a. | 2.0 | n.a. | 65 ± 9 | 50 | [44] |
| Calcium intracellular female | ↑ | 60 | 72 ± 7 | 18 ± 2 | 1.6 | 29 ± 1 | 70 ± 8 | 25 | [45] |
| Calcium intracellular male | ↑ | 40 | 66 ± 5 | 18 ± 3 | 1.5 | 29 ± 2 | 63 ± 4 | 25 | [45] |
| Coated-platelet levels | ↑ | 10 | n.a. | < 20 | 1.1 | n.a. | n.a. | 19 | [46] |
| Coated-platelet levels | ↑ | 36* | 74 ± 8 | 28 ± 1 | 0.5 | 28 ± 1 | 73 ± 8.8 | 30* | [47] |
| Coated-platelet levels | ↑ | 20 | 72 ± 10 | 23 ± 2 | 0.9 | 30 ± 0 | 74 ± 7 | 40 | [48] |
| Cytochrome c oxidase activity | ↓ | 10 | <60 | n.a. | −0.8 | n.a | 61 ± 3 | 5 | [49] |
| Cytochrome c oxidase activity | ↓ | 10 | >60 | n.a. | −1.0 | n.a | 61 ± 3 | 5 | [49] |
| Cytochrome c oxidase activity | ↓ | 20 | 65 ± 9 | 18 ± 5 | −4.4 | n.a. | 63 ± 9 | 20 | [50] |
| Cytochrome c oxidase activity | ↓ | 22 | 66 ± 9 | 17 ± 8 | −1.5 | 26 ± 3 | 63 ± 9 | 20 | [51] |
| Cytochrome c oxidase activity | ↓ | 6 | n.a. | n.a. | −1.3 | n.a. | n.a. | 8 | [52] |
| Cytochrome c oxidase activity (Complex IV) | ↓ | 8 | 78 ± 7 | 17 ± 7 | −1.4 | 30 ± 1 | 73 ± 5.7 | 7 | [53] |
| Cytochrome c oxidase activity (Complex IV) | ↓ | 5* | 78 ± 10 | 26 ± 2 | −1.4 | 30 ± 1 | 73 ± 5.7 | 7 | [53] |
| GSK3β Ser-9 phosphorylated/total GSK3β | ↓ | 24 | 76 ± 4 | 19 ± 4 | −1.1 | 28 ± 3 | 71 ± 5 | 23 | [54] |
| GSK3β Ser-9 phosphorylated/total GSK3β | ↓ | 22* | 74 ± 7 | 26 ± 2 | −0.6 | 28 ± 3 | 71 ± 5 | 23 | [54] |
| Hyperacidification after activation | ↓ | 19 | 71 ± 5 | n.a. | 4.7 | n.a. | 61 ± 8 | 14 | [55] |
| Immunoglobulin | ↑ | 25 | 78 ± 1 | 21 ± 1 | 2.7 | 28 ± 0 | 71 ± 2 | 26 | [56] |
| Mao-B activity (phenylethylamine) | ↑ | 20 | 81 ± 11 | 5 ± 7 | 1.0 | 28 ± 1 | 80 ± 11 | 9 | [57] |
| Mao-B activity (phenylethylamine) | ↔ | 15 | 68 ± 3 | n.a. | 1.2 | n.a. | 54 ± 2 | 8 | [58] |
| Mao-B activity (benzylamine) | ↑ | 50 | 68 ± 14 | n.a. | 2.1 | n.a. | 64 ± 14 | 50 | [59] |
| Mao-B activity (phenylethylamine) | ↑ | 11 | 65 ± 1 | n.a. | 1.2 | n.a. | 65 | 11 | [60] |
| Mao-B activity (kynuramine) | ↔ | 11 | 64 ± 7 | 14 ± 4 | −0.7 | n.a. | 64 ± 8 | 11 | [61] |
| Mao-B protein abundance | ↑ | 34 | 79 ± 8 | 5 ± 4 | 1.4 | 28 ± 2 | 79 ± 9 | 34 | [62] |
| Mean platelet volume | ↑ | 126 | 76 ± 7 | n.a. | 0.3 | n.a. | 75 ± 6 | 286 | [63] |
| Membrane fluidity | ↑ | 12 | 72 ± 4 | 11 ± 2 | 1.0 | n.a. | 68 ± 4.5 | 18 | [64] |
| Membrane fluidity | ↔ | 23 | 74 ± 9 | 19 ± 5 | −0.5 | 29 ± 1 | 70 ± 5.8 | 29 | [37] |
| Membrane fluidity | ↑ | 24 | n.a. | n.a. | −1.6 | n.a. | n.a. | 36 | [65] |
| Membrane fluidity external | ↓ | 100 | 68 ± 7 | n.a. | 0.3 | n.a. | 65 ± 9 | 50 | [44] |
| Membrane fluidity in submitoch. particles | ↓ | 30 | n.a. | n.a. | −2.3 | n.a. | n.a. | 30 | [66] |
| Membrane fluidity internal | ↓ | 100 | 68 ± 7 | n.a. | 0.8 | n.a. | 65 ± 9 | 50 | [44] |
| Na+/K + −ATPase activity | ↓ | 100 | 68 ± 7 | n.a. | −6.0 | n.a. | 65 ± 9 | 50 | [44] |
| Na+/K + −ATPase activity female | ↓ | 60 | 72 ± 7 | 18 ± 2 | −8.5 | 29 ± 1 | 70 ± 8 | 25 | [45] |
| Na+/K + −ATPase activity male | ↓ | 40 | 66 ± 5 | 18 ± 3 | −7.3 | 29 ± 2 | 63 ± 4 | 25 | [45] |
| NO production | ↑ | 100 | 68 ± 7 | n.a. | 6.3 | n.a. | 65 ± 9 | 50 | [44] |
| NO production female | ↑ | 60 | 72 ± 7 | 18 ± 2 | 4.5 | 29 ± 1 | 70 ± 8 | 25 | [45] |
| NO production male | ↑ | 40 | 66 ± 5 | 18 ± 3 | 6.5 | 29 ± 2 | 63 ± 4 | 25 | [45] |
| ONOO − production | ↑ | 100 | 68 ± 7 | n.a. | 6.8 | n.a. | 65 ± 9 | 50 | [44] |
| ONOO − production female | ↑ | 60 | 72 ± 7 | 18 ± 2 | 8.4 | 29 ± 1 | 70 ± 8 | 25 | [45] |
| ONOO − production male | ↑ | 40 | 66 ± 5 | 18 ± 3 | 8.1 | 29 ± 2 | 63 ± 4 | 25 | [45] |
| Phospholipase A2 activity | ↓ | 16 | 70 ± 11 | n.a. | −0.9 | n.a. | 63 ± 10 | 13 | [67] |
| Phospholipase A2 activity | ↓ | 21 | 75 ± 7 | 14 ± 9 | −1.6 | 28 ± 2 | 73 ± 5 | 17 | [68] |
| Phospholipase A2 activity | ↓ | 11* | 73 ± 5 | 25 ± 4 | −0.7 | 28 ± 2 | 73 ± 5 | 17 | [68] |
| Phospholipase A2 activity | ↑ | 37 | 73 ± 6 | 19 ± 5 | 0.3 | n.a. | 72 ± 5 | 27 | [69] |
| Phospholipase A2 activity | ↓ | 44 | 75 ± 7 | 19 ± 5 | −0.6 | 28 ± 4 | 75 ± 7 | 66 | [70] |
| Phospholipase A2 activity | ↓ | 59* | 72 ± 6 | 27 ± 3 | −0.6 | 28 ± 4 | 75 ± 7 | 66 | [70] |
| Phospholipase C delta protein and activity | ↓ | 10 | 81 ± 1 | 1 ± 2 | −3.4 | 1 ± 2 | 80 ± 2 | 10 | [71] |
| Serotonin [c] | ↓ | 22 | n.a. | n.a. | −1.5 | n.a. | n.a. | 20 | [72] |
| Serotonin [c] | ↑ | 57 | n.a. | n.a. | 1.0 | n.a. | n.a. | 20 | [73] |
| Tau high molecular weight/monomeric tau | ↑ | 15 | 81 | 15 | 1.6 | 28 | 68 | 10 | [24] |
| TRAP-induced CD62P expression [%] | ↓ | 23 | 70 ± 8 | n.a. | −1.2 | n.a. | 60 ± 10 | 17 | [74] |
The Hedge effect size was calculated based on published values of mean, standard deviation, standard error of mean, and sample size. Positive ES indicate up-regulation, negative ES down-regulation in AD patients. aMild AD, bModerate AD, cAdvanced AD, *MCI.
Review
Platelet activation
Platelets can become activated by a number of agonists. This introduces shape changes and degranulation of dense- and α-granules [17], redistributing several proteins from α-granules to their surface. An elevated degree of platelet activation in AD patients has been reported by quantification of CD62P (P-selectin) surface expression, platelet aggregates, and platelet leukocytes complexes [75]. Results from another study contradict these findings as these show no difference in surface CD62P expression of unstimulated platelets but significantly lower levels in thrombin receptor activating peptide 6 (TRAP-6)-activated AD platelets [74]. However, this may be a sign of already exhausted platelets in vivo after sustained activation, reflected by the increase of sCD62P plasma levels [74].
Coated platelets
Upon parallel activation with collagen and thrombin, a subset of so called ‘coated’ platelets retains augmented concentrations of pro-coagulant proteins on their cell membrane [76]. Determination of surface fibrinogen levels by flow cytometry is an established method for their quantification [77]. Coated platelet numbers have been found to be significantly increased in early stages of AD as compared to matched controls but decreased in late stages of AD, even below levels of cognitively healthy individuals [48, 78–80]. This group also showed that coated platelets can be used to predict which aMCI patients would rapidly progress to AD [80]. Additionally, this parameter could distinguish amnestic from non-amnestic MCI patients by higher levels of coated platelets in these patients [47]. Similarly, they suggested that these elevated levels could discriminate frontotemporal dementia from AD [48]. Importantly, all these publications originate from the same research group; therefore, and because of the only moderate sample numbers and ES (Table 1), reproduction by other international experts is stringently required.
Serotonin metabolism
The idea of using platelets as an experimental system for neurons was triggered by discovering that platelets’ dense granules (which release their content after stimulation) are the major storage site for serotonin (5-hydroxytryptamin, 5-HT) in blood [81], similar to its vesicular storage in neurons [82]. Consequently, experiments with the antipsychotic drug reserpine were carried out. Reserpine mediates depletion of monoamine neurotransmitters in synapses; the same effect could be simulated in platelets [18]. Since then, many studies confirmed that platelets are an adequate model resembling storage and release of serotonin from serotonergic neurons under both physiological and pathological conditions [83–85]. Moreover, a significant correlation between interindividual 5-HT2A receptor binding characteristics in the brain cortex and in platelets has been observed both in animals [86] and humans [87]. A decrease in serotonin uptake and in the number of transporter binding sites in brain tissue and platelets has been shown to be general molecular evidence for serotonergic abnormalities in depression [85, 88].
Serotonin metabolism in platelets of AD patients
Concentrations of serotonin and 5-hydroxyindoleacetic acid, serotonin uptake, and K+-stimulated release of endogenous serotonin have all been found to be reduced below control values in neocortical biopsy samples from patients with histologically verified AD [89]. A significant reduction in serotonin binding in non-depressed AD patients hints that presynaptic serotonergic function is already affected before development of psychiatric problems such as depression [90]. Further evidence for reflection of cerebral biochemical abnormalities in platelets was the diminished affinity of the platelet 5-HT2A receptor towards its radioactively labelled ligand [3H] LSD in AD cases [91], suggesting decreased serotonin uptake by these peripheral cell bodies. A number of other studies also registered an impaired uptake of serotonin in platelets [92–94]. However, the reliability of this marker is questionable since this finding could not be reproduced in two other investigations [95, 96]. In strong contrast, another study even showed increased accumulation of the neurotransmitter in platelets of female AD patients [97]. Intraplatelet serotonin concentrations reported in literature are also quite inconsistent as reduced [72, 98, 99], as well as increased [73] values in AD patients with delusions have been measured. In the latter observation, there might be a weak parallel to findings of higher serotonin level in AD patients with psychotic features than in inconspicuous subjects [98]. Similarly, in demented patients concentrations of platelet serotonin were higher as compared to controls [73]. Further details about the role of serotonin in depression, ageing, and AD can be found in the review by Meltzer et al. [100] which also compares results from platelets and neurons. Some of the discrepancies may arise from different stages of AD, given a recent study showing that serotonin levels were only reduced in late stage AD [99]. Additionally, diurnal and seasonal variations have been described for intraplatelet serotonin content [101]. The isolation method of easy-to-activate platelets can also influence the measured concentration as this monoamine is taken up from plasma, stored in dense granules of platelets, and released upon stimulation, contributing to an aggregative response [82]. Therefore, by having an effect on the degree of activation and degranulation, the choice of anticoagulant and centrifugation force can strongly influence serotonin concentrations [102]. This might be one reason for the inconsistent results (positive and negative ES in Table 1) for this parameter and great care should be taken if serotonin is considered as AD biomarker.
Monoamine oxidase B
Another neuropharmacological drug target frequently studied in platelets is the neurotransmitter-degrading enzyme monoamine oxidase (Mao, EC 1.4.3.4). The existence of Mao in platelets was first reported in 1964 [103]. This enzyme exists in two isoforms, Mao-A and Mao-B, which are encoded by two genes with exactly the same exon-intron pattern, implying evolvement from a common ancestral gene. Differences do exist though in their primary structures and tissue-specific expression patterns, substrate preferences, and inhibitor sensitivities [104]. Mao-B is a mitochondrial membrane protein that catalyses oxidative deamination of monoamines, including phenethylamine and the neurotransmitters dopamine and aforementioned serotonin. Human platelets exclusively express Mao-B with the same amino acid sequence as brain Mao-B [105]. This enzyme has been proposed to be involved in ageing, as well as the pathogenesis of AD and Parkinson’s disease (PD) through increased generation of reactive oxygen species (ROS) and neurotoxic aldehyde catabolites [106]. The observation that Mao-B activities in brain and platelets correlate positively with ageing [107] has led researchers to suggest a systemic alteration of this potential neurological biomarker. In the course of AD, Mao-B activity was elevated in the temporal lobe and white matter as compared to age-matched controls [108]. These profiles were confirmed at the mRNA level in brain tissue, signifying that the increased activity arises from an enhanced level of transcription and a higher Mao-B concentration rather than from post-translational mechanisms [109]. Though, intraneuronal increase in Mao-B activity in both AD and ageing has been doubted [110, 111] with the hypothesis that because of higher constitutive Mao-B expression by glial cells, elevated brain Mao-B levels may be a consequence of gliosis during AD [108, 110].
Already in 1980, the finding by Adolfsson et al. of a similar increase in Mao-B activity in brain and platelets of AD patients suggested to use platelets as a peripheral diagnostic tool for AD [60]. Higher Mao-B activity in patient samples could only be detected with the Mao-B-specific substrate phenethylamine. These AD-related findings were subsequently confirmed in at least 12 clinical studies from different research groups [57, 59, 73, 112–120]. Five other studies, however, reported no AD-related activity increase [58, 61, 73, 99, 121]. Importantly, instead of phenethylamine, three of these studies used the substrates tyramine or kynuramine [61, 73, 99], both converted by Mao-A and Mao-B [122]. In a recent study, Mao-B activity quantified by kynuramine has been found to be significantly lower in late stage AD patients as compared to healthy controls, whereas in early and middle stage AD Mao-B activity was not changed significantly [99]. This finding stands in strong contradiction to the majority of previous studies, where most AD patients also suffered from moderate (mini–mental state examination (MMSE) score <20) to severe (MMSE score <10) dementia (unless otherwise specified; Table 1). Curiously, there does also not seem to be an agreement in the conclusions drawn by this group, since they additionally demonstrated a significant increase in Mao-B activity in a considerably larger AD patient cohort with an average MMSE of 18.9 [98].
In an unbiased proteome analysis, our laboratory found a predominant elevation in Mao-B expression in platelets from AD patients which strongly correlated with the enzymatic activity [62]. Nevertheless, concentration of Mao-B also increased with age (55 to 104 years) in healthy subjects. Centenarians exhibited platelet Mao-B levels comparably high to those of AD platelets [62], potentially reflecting the age-dependent cognitive decline indicated by lower MMSE scores. This suggests an age-related increase in Mao-B expression that is more pronounced in AD patients.
Despite a number of studies about excessive platelet and brain Mao-B activity in AD, little is known about the molecular causes underlying these changes. Platelet Mao-B activity is strongly influenced by lifestyle factors such as nutrition, alcohol, and smoking. Norharman, a compound of tobacco smoke, is a specific Mao-B inhibitor [123]. Paradoxically, it was observed that Mao-B inhibition in smokers was accompanied by elevated enzyme concentration in platelets [124]. Smoking induced a hypomethylation of the Mao-B promoter, followed by an increased Mao-B protein expression [124]. Since high homocysteine, as well as low vitamin B12 and folate levels are associated with AD [125], homocysteine-lowering therapies based on these B-vitamins counteract the accelerated atrophy in brains of MCI patients [126]. In good agreement with these observations, a correlation between plasma vitamin B12 levels and platelet Mao-B activity was observed in dementia patients: the lower the vitamin levels, the higher the enzymatic activity. Following vitamin B12 supplementation, platelet Mao-B activity was significantly reduced in these patients to apparently normal levels [127]. Recently, we have shown that a high animal protein diet in healthy young adults decreased platelet Mao-B expression by 26% compared to controls fed with normal protein diet. This was accompanied by improved cognitive function and correlated with increased plasma vitamin B12 levels [128]. Since both increased Mao-B protein expression levels and enzymatic activity (determined with the substrate phenylethylamine) in AD patients have repeatedly been reported by different research groups (Table 1), Mao-B is one of the most reliable and promising AD biomarker candidates.
Nitric oxide and oxidative stress
Nitric oxide (NO) produced by activated astrocytes is thought to contribute to neurodegenerative processes. Reaction of NO with oxygen radicals leads to formation of peroxynitrite (ONOO−). This unstable structural nitrate isomer generates cytotoxic species which oxidise and nitrate proteins. Increased levels of nitrated proteins have been reported in AD brain and CSF [129]. Platelet NO production, as well as ONOO− levels were evaluated in a large AD study cohort and significantly increased in afflicted individuals [44] with one of the highest ES of 6.3 (Table 1). In a subsequent study, elevated NO and ONOO− production was more pronounced in male than female AD patients, but generally higher in males [45].
Defects in the mitochondrion’s electron transport chain are the main source of ROS, the central element of the free radical theory of ageing [130]. Given that ageing is the main risk factor for AD, this may also have an impact on the pathology of AD. Platelets have a high number of mitochondria what makes them attractive to study AD-related systemic malfunctions of the electron transport system. Investigations have revealed that both in isolated platelet and hippocampal mitochondria cytochrome c oxidase (complex IV of the respiratory chain) activity was significantly lower in AD patients as compared to controls [50]. Different research teams have confirmed this finding consistently [49, 51–53, 131, 132]. While one of these studies additionally found reduced adenosine triphosphate (ATP) concentrations in AD platelets [49], another investigation reported no significant differences between platelet ATP levels of patient and control groups [133]. Increased ROS levels in AD platelets have been detected [49], just as an enhanced blood lactate concentration correlating inversely with diminished platelet cytochrome c oxidase activity [51]. Similar findings in adult children of AD-affected mothers imply an exclusively maternal heredity of this biomarker [134]. Taken together, most results point towards the importance of increased oxidative stress in AD. Whether any of the indicators could be used as biomarker remains to be elucidated.
Inflammatory mediators
In the periphery of amyloid plaques and NFTs, pro-inflammatory molecules are highly expressed [135]. One enzyme synthesised by platelets after an inflammatory stimulus is cyclooxygenase-2 (COX-2) that in turn is responsible for the production of prostaglandins. Several studies indicate that treatment with non-steroidal anti-inflammatory drugs and COX-2 inhibitors may reduce the risk of developing AD [136]. Indeed, Western blot analysis revealed a 50% increase in platelet COX-2 in MCI and a 25% increase in AD patients [137]. COX-2 expression profiles in platelets could indicate which patient groups may benefit from a COX-2 inhibitor therapy. Further details can be found in a recent review on inflammatory AD biomarkers in platelets [138].
Amyloid precursor protein
One of the first milestones in understanding the pathogenesis of AD dates back to 1985, when cerebral Aβ deposits in senile and neuritic plaques were recognised as playing a central role [139, 140]. Amyloid plaques are formed by aggregation of Aβ peptides produced by proteolytic cleavage of APP. This single-pass type I membrane protein can be N-terminally hydrolysed by two alternative, differently initiated routes. In the non-amyloidogenic pathway, α-secretase cleaves APP first, releasing the neuro-protective, soluble fragment sAPP-α. In the amyloidogenic pathway, though, β-secretase is the first enzyme to cleave and the resulting soluble fragment is sAPP-β. The remaining carboxyl-terminal fragment derivatives are subsequently cut by γ-secretase to generate either a 3 kDa product or Aβ, respectively. Accordingly, when α-secretase cleaves, Aβ is not produced [141]. It is hypothesised (but still under debate [142]) that Aβ is a causative molecule of AD by inducing neuronal cell death and concomitant disturbance of synaptic function.
Platelets are equipped with α-, β-, and γ-secretases [143]. Studies have shown a reduction of α-secretase protein level in platelets of early stage AD patients [30, 144], in line with attenuated release of sAPP-α from thrombin-activated AD platelets [145]. In addition, an increased activity of β-secretase was indirectly shown by a decreased ratio of its 37/56 kDa fragments from two different groups [143, 144]. Another group determined it directly by an enzymatic assay with a 17% elevation in activity in AD platelets [41] and with an even more pronounced 24% upregulation in MCI platelets [42]. The concentration of β-secretase cleavage products was also found to be significantly increased [145]. On the other hand, the enzymatic activity did not correlate with MMSE scores, signifying that it might be a primary pathophysiological sign and may predict disease onset [41]. A recent study has confirmed altered soluble fragment ratios: while sAPP-α level were unchanged in both MCI and AD patients, there was a strong increase in sAPP-β level. Therefore, an ELISA-based assay to detect this altered β-fragment released by platelets incubated with recombinant BACE1 might be used as diagnostic screening tool [146].
Alternative splicing generates several APP mRNAs with the three major isoforms being APP695, APP751, and APP770 [141]. While the two longer forms (APP751 and APP770) possess a Kunitz-type serine protease inhibitor domain and are found in most tissues, the shorter APP695 is predominantly expressed in neurons [147]. Analysis of brain biopsies is difficult because of the mixture of cells such as astrocytes, glia cells, and neurons. Additionally, protein profiles may be influenced by gliosis (generally occurring in AD), masking neuron-specific expression changes. Microdissection studies would specifically show which proteome changes originate from a single cell type but only few have been conducted from AD brain so far. Therefore, scant and conflicting data are available on abundance and processing of the Aβ parent molecule in AD-affected brains [148–151]: APP751 and APP770 isoform patterns were found to be unchanged but concentrations of total APP and fragment APP695 decreased on mRNA level [150] and protein level [151] with an increase of the Aβ peptide level in samples of the frontal cortex of AD cases [151]. Due to these confusing data on neuronal APP isoform abundance, platelets may be an interesting alternative sample material. At this point it should be mentioned that it is assumed that generation of Aβ is boosted in EOFAD, whereas clearance of Aβ may be diminished in LOAD [152]. Since above mentioned alterations of APP processing and isoform abundance are mainly found in LOAD patients, this suggests that not only clearance of Aβ is affected in LOAD.
In platelets, the two prevalent isoforms are APP751 and APP770 though APP695 is also present [153]. This fact may have significance in haemostasis, since the Kunitz-type domain inhibits certain blood coagulation factors [154–156]. Intraplatelet localisation of the numerous APP isoforms is also different: full-length protein (140–150 kDa) is plasma membrane-bound, the predominant 100–130 kDa species (C-terminally truncated forms in activated platelets) are located in α-granule membranes [157]. APP metabolism has been found to be specifically altered in platelets of LOAD patients: a large number of publications from different groups report a decreased ratio of the two major platelet APP isoforms (130 kDa/110 kDa) in AD patients [26, 33, 35, 36, 39, 143, 158, 159]. One of the studies could also distinguish AD from PD and stroke patients since these values equalled those of controls [158]. Still others correlated the reduced ratio with disease progression [160]. Thus, despite huge variation in the reported values, APP ratio is still the most reproducible and promising AD biomarker in platelets to date (Table 1).
Interesting in this context is a newly identified C-terminally-truncated 115 kDa APP isoform which is not glycosylated and non-releasable upon platelet activation [32]. Experiments of this laboratory suggest that this fragment could represent an easily detectable diagnostic marker as it significantly and inversely correlated with reduced 130/110 kDa isoform ratios of AD patients. From a therapeutic perspective, the acetylcholine (ACh) esterase inhibitor donepezil was found to restore APP metabolism [145] and alter APP ratios in AD patients [40]. At the same time, statin-lowered cholesterol levels inversely correlated with increased APP ratios [158], indicating great potential of the APP isoform ratio or the 115 kDa fragment as a prognostic and surrogate marker for medication efficacy.
Since several smaller isoforms are produced from 130 kDa APP upon platelet activation [161], it can be possible that APP ratios in AD are due to altered platelet reactivity. This might have an important influence on APP biomarker studies as platelets are easily activated during sample collection. On top of that, anticoagulants strongly affect platelet activation and degranulation, as demonstrated previously [28]. Great care is therefore essential to avoid activation during venipuncture, blood collection, and platelet isolation. CTAD blood tubes [162] (containing citrate, theophylline, adenosine, and dipyridamol) are most suitable, as indicated by strongly reduced levels of plasma platelet factor 4 compared to those measured from EDTA or citrate blood tubes [28].
Membrane fluidity and cholesterol
There are indications that low membrane cholesterol levels have a considerable impact on the pathogenesis of AD [163]. Cholesterol is the major lipid constituent of biological membranes and plays a key role in defining their physical state by regulating fluidity in a concentration-dependent manner [164]. An example was provided in a study demonstrating that an initial decrease in cholesterol content of total brain lipid extracts by approximately 5% in vitro also reduced membrane fluidity, whereas further decrease in cholesterol increased fluidity again [165]. Moreover, this study could demonstrate that addition of Aβ1–40 to brain lipid membranes resulted in diminished vesicle fluidity, thereby linking the amyloidogenic pathway with altered membrane fluidity. In line are findings that Aβ aggregates can disturb the structure of brain membranes and that membranes derived from AD-affected hippocampi had lower fluidity [166]. This together with lower cholesterol levels in brain preparations of AD patients challenge the application of cholesterol-lowering statins as therapeutic agents [167].
Literature on AD accompanying changes of platelet membrane fluidity is rather inconsistent. Platelet membrane cholesterol content has been found to be lower in cognitively impaired subjects and β-secretase activity correlated bimodally with these levels: when concentrations were below 50 pmol cholesterol/μg membrane protein, the correlation was negative, above this threshold it was positive [168]. Additionally, this study identified elevated cholesterol levels in platelet membranes of statin-treated versus untreated subjects. Another study has detected reduced fluidity in platelet membranes from AD patients; this effect was even more pronounced in males [45]. Similarly, lower fluidity has been detected in membranes of sub-mitochondrial particles of AD platelets [66]. On the other hand, only insignificant differences in this parameter between LOAD patients and controls have been reported [169]. Despite a weak correlation with the 130 kDa/110 kDa APP ratio, platelet membrane fluidity differed only minimally between AD, MCI, and controls [37]. Others have even reported results pointing to the opposite direction: elevated fluidity in AD platelet membranes [64, 170, 171] and intracellular membranes [172]. This inconsistency on the relationship of Aβ, cholesterol, and AD has been reviewed with the outcome that more data are needed before a definitive conclusion can be drawn on the connection of membrane fluidity and AD pathogenesis [173]. Since AD is an age-related disease, it needs to be emphasised that membrane fluidity is also reduced during ageing, a decline less pronounced in centenarians [174].
Na+/K+-ATPase is another indicator of membrane functionality as the physicochemical properties of the microenvironment have great influence on the activity of this integral membrane protein. The activity of this sodium-potassium pump has been found to be reduced in platelets of AD patients [44]. In agreement are results of significantly diminished levels of both Na+/K+-ATPase enzymatic activity and protein expression in AD brains [175]. A further study compared its activity in platelets from male and female AD patients as well as control subjects. While enzyme activity was generally higher in females, an AD-related decrease persisted within both gender groups [45]. In summary, these results indicate that Na+/K+-ATPase activity could be a promising candidate in AD diagnosis with a high ES of −6 (Table 1).
Phospholipase A2
Another enzyme family involved in membrane physiology are the phospholipases A2 (PLA2, EC 3.1.1.4) which play an important role especially in lipid metabolism. Catalysing the hydrolysis of membrane glycerophospholipid ester bonds, free fatty acids (e.g. arachidonic acid) and lysophospholipids are generated [176, 177]. Their exact role in neurodegeneration is still to be explored as earlier data showed ambiguity regarding the consequences of altered PLA2 activity. On the one hand, studies have indicated that increased PLA2 activity alters membrane integrity which (especially via Ca2+-induced lipolysis) ultimately leads to cell structure disruption and thus to neurodegeneration [178]. On the other hand, in animal experiments inhibition of PLA2 impaired learning and spatial memory, similar signs occurring in the earliest phases of AD [179]. Additionally, PLA2 inhibition led to reduced fatty acyl chain flexibility, linking PLA2 with AD via altered membrane fluidity [180]. In line, membrane phospholipid metabolism has been described to be reduced in the prefrontal cortex of mildly and moderately demented AD patients [181]. These findings of PLA2-dependent reduction in membrane hydrophobic core fluidity correspond with the recent observation of diminished membrane fluidity in platelets of AD patients [45]. Moreover, PLA2 activity was significantly reduced in parietal and frontal cortices of AD patients; this enzyme deficiency could be connected with earlier onset and severity of the disease, as well as a worse outcome [182]. A further hint of a link between PLA2 and AD was provided by differentially processed and secreted APP after inhibition or activation of PLA2 [183]. An increase in both sAPPα secretion and membrane fluidity, as well as a decrease in Aβ1–42 levels was detected when neuronal cells were exposed to sPLA2 [184].
The search for peripheral surrogates of cerebral changes in AD encouraged several groups to investigate PLA2 activity in platelet membranes. In 1996, a study demonstrated significant reduction in enzymatic PLA2 activity in platelets of AD patients [67]. In a subsequent publication, this group reported a correlation of lower platelet PLA2 activity in MCI and AD patients with severity of cognitive decline. Accordingly, MCI individuals had mean PLA2 activity levels between those of AD patients and controls [68]. After a one-month long cognitive training period, platelet PLA2 activity increased in healthy elderly subjects [185]. In contrast stands a significantly increased activity of PLA2 in platelets of AD patients reported by another team [69]. They assumed that the differences of these results may be due to different severity of the disease since their AD patients were at an earlier stage. Finally, a 4-year follow-up study of MCI subjects detected that patients with a low baseline platelet PLA2 activity had a higher risk of progressing to AD, suggesting that low platelet PLA2 activity may be an AD risk marker in such patients [70]. As indicated in Table 1, only few data sets originating from different publications are available. Together with the low sample numbers and inconsistent ES, we would not recommend PLA2 as trustworthy AD platelet biomarker until other researchers confirm a reduced platelet PLA2 expression in AD and MCI patients.
Tau
A second hallmark of AD is represented by intracellular NFT that consist of hyperphosphorylated tau protein [186, 187]. Tau is mostly found in axons where it attaches to and stabilizes microtubules, crucial for anterograde and retrograde axonal transport. Hyperphosphorylation of tau causes it to dissociate from microtubules, thereby greatly reducing their stability and ultimately leading to cell death. Free hyperphosphorylated tau tends to aggregate into helical filaments which result in formation of the aforementioned NFT [188, 189].
Apart from hyperphosphorylation, aberrations in tau splicing are further regulations directly causing neurodegenerative diseases [190]. High molecular weight tau protein (130 kDa) was detected in tangles of AD brains already in 1992 [191]. Several years later, a 110 kDa tau variant was also found in peripheral tissues such as human oral epithelium [192] and rat muscle [193]. Expression of tau protein in human platelets has been reported [56] together with an elevated ratio of high molecular weight (>80 kDa) oligomeric isoform variants to monomeric tau in AD patients [24]. In a follow-up study, this group identified tau modifications also in healthy subjects, without any age dependency. Nevertheless, the above described ratio seemed to correlate with the cognitive status of AD patients [27]. Other studies analysed platelet tau quantities in AD patients and control subjects but could not detect any disease-specific differences [25, 56]. However, C-terminal end tau protein levels of MCI subjects were significantly different from normal ones. Additionally, they detected elevated total tau levels in older AD patients as compared to both younger AD patients and healthy controls, concluding that tau might be a diagnostic marker for the detection of the onset of the disease [25]. Together, results on platelet tau as AD biomarker are sparse, unsettled, and originate from two research groups only. Thus, interpretation should be done with great care and more data are required to define its diagnostic value, ideally by conducting clinical studies.
Glycogen synthase kinase 3β
One of the most important tau kinases in neurons is glycogen synthase kinase 3β (GSK3β) which is suspected to play a central role in AD [194]. Its activity has been found to be increased in specific regions of the AD brain [195–197], thereby promoting hyperphosphorylation of tau and formation of NFTs. Normally, GSK3β is constitutively expressed in all cells and primarily inhibited via phosphorylation of serine-9 [198]. Lithium can increase GSK3β phosphorylation indirectly [199], thereby reducing its tau hyperphosphorylation activity and stabilising cognitive ability in AD patients [200]. Comparison of the ratio of Ser-9-phosphorylated GSK3β versus total GSK3β in platelets of AD patients and controls revealed a significant reduction in AD and MCI samples, indicating seriously enhanced enzyme activity [54]. This ratio correlated with cognitive scores and might therefore represent an interesting diagnostic target in platelets. Nevertheless, these findings need to be reproduced by others before the diagnostic value can be defined.
Conclusion
Due to increased ageing of the world population and concomitantly elevated prevalence of AD, a vast number of publications have been produced during the last decades, deepening our understanding about AD and accompanying molecular changes. Nevertheless, to date there is no alternative for diagnosis of definite AD than postmortem brain autopsy. However, findings that blood platelets could serve as a source of efficient biomarkers to assist early AD detection are promising.
Here, we compiled AD platelet biomarker candidates with available information to calculate their Hedge ES as a measure for comparability in a tabular form (Table 1). AD-related alterations (and standard deviations) of various molecules and physicochemical platelet changes from different studies are quite heterogeneous and data therefore difficult to compare. In order to offer a more standardised factor, the Hedge ES is provided as useful tool to determine the separating power of a given biomarker. It is interesting to observe that highest ES were calculated for potential biomarkers that are indicators of either oxidative stress or membrane integrity. This supports the idea that inflammation and oxidative stress play crucial roles in the pathogenesis of AD. The quite large ES of sub-mitochondrial platelet membrane fluidity implies that these structures become extremely sensitive to oxidative stress and may be involved in initiating disease progression. At the same time, it is important to note that ROS production is considered to be a function of ageing, which in turn is the most significant risk factor of LOAD. However, the large effect sizes of NO metabolism and membrane fluidity together with changed Na+/K + −ATPase activity have only been shown by one laboratory so far [44] and need to be validated by other research facilities.
Conflicting data on some potential AD biomarkers are highlighted by inconsistent ES calculated for e.g. platelet membrane fluidity and serotonin concentration (Table 1). These contradictions are perhaps due to difficulties analysts face when examining heterogeneous and hard-to-identify target populations such as suspected AD patients and their appropriate controls. Furthermore, it is crucial to avoid activation in the case of platelets, thus the choice of both the anticoagulant and the protein extraction method can modify the outcome of any blood-based study. Complications also arise from the lack of standardised sample collection and isolation methods, which have a dramatic impact on the results. These latter shortcomings are not restricted to platelets either but generally apply to biomarker studies in all media, e.g. CSF or plasma, which have similar drawbacks. Accordingly, some of the less obvious candidates like membrane fluidity have been found to be both increased [170] and decreased [44] in AD patients. On the other hand, some of the molecular marker candidates indeed showed great reliability, such as decreased APP isoform ratio and APP-processing proteases ADAM10 and BACE1. Elevated levels of multimer tau, the second hallmark of AD, have been reported, though available data are limited to two publications from the same group [24, 27] and a contradictory report from another [25]. Mao-B, a further highly promising candidate, has uniformly been found to be up-regulated with quite stable ES, at least when the substrate phenethylamine was used. We suggest that Mao-B and APP isoform ratios are currently the two top AD biomarker candidates in platelets due to consistent results from several different groups.
Finally, we can conclude that platelets represent a promising peripheral surrogate to detect AD-related molecular changes and provide crucial data necessary for taking the next step towards development of a diagnostic and/or therapy-predictive tool for AD. However, as none of the individual markers described is powerful enough to meet the required levels of sensitivity and specificity for routine AD diagnosis [201], it may be useful to exploit several of these biomarker candidates contemporaneously. It remains to be seen whether a combination would be robust enough to expose molecular changes occurring at the early phase of AD so as to distinguish between healthy and affected individuals. Future work into AD platelet biomarkers might shift the focus towards a proteomic approach in order to identify the best combination of biomarkers with the intention of designing diagnostic multiplex devices.
Acknowledgements
SB Guterres thanks the São Paulo Research Foundation FAPESP for financial support (2008/04050-4).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Michael Veitinger, Email: michael.veitinger@meduniwien.ac.at.
Balazs Varga, Email: balazs.varga@meduniwien.ac.at.
Sheila B Guterres, Email: sheilaguterres@gmail.com.
Maria Zellner, Email: maria.zellner@meduniwien.ac.at.
References
- 1.Kennedy JL, Farrer LA, Andreasen NC, Mayeux R, St George-Hyslop P. The genetics of adult-onset neuropsychiatric disease: complexities and conundra? Science. 2003;302:822–826. doi: 10.1126/science.1092132. [DOI] [PubMed] [Google Scholar]
- 2.Schliebs R, Arendt T. The significance of the cholinergic system in the brain during aging and in Alzheimer's disease. J Neural Transm. 2006;113:1625–1644. doi: 10.1007/s00702-006-0579-2. [DOI] [PubMed] [Google Scholar]
- 3.Trillo L, Das D, Hsieh W, Medina B, Moghadam S, Lin B, Dang V, Sanchez MM, De MZ, Ashford JW, Salehi A. Ascending monoaminergic systems alterations in Alzheimer's disease. Translating basic science into clinical care. Neurosci Biobehav Rev. 2013;37:1363–1379. doi: 10.1016/j.neubiorev.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Fuso A, Scarpa S. One-carbon metabolism and Alzheimer's disease: is it all a methylation matter? Neurobiol Aging. 2011;32:1192–1195. doi: 10.1016/j.neurobiolaging.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 5.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chincarini A, Bosco P, Calvini P, Gemme G, Esposito M, Olivieri C, Rei L, Squarcia S, Rodriguez G, Bellotti R, Cerello P, De Mitri I, Retico A, Nobili F. Local MRI analysis approach in the diagnosis of early and prodromal Alzheimer's disease. Neuroimag. 2011;58:469–480. doi: 10.1016/j.neuroimage.2011.05.083. [DOI] [PubMed] [Google Scholar]
- 7.Jack CR, Jr, Wiste HJ, Vemuri P, Weigand SD, Senjem ML, Zeng G, Bernstein MA, Gunter JL, Pankratz VS, Aisen PS, Weiner MW, Petersen RC, Shaw LM, Trojanowski JQ, Knopman DS. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease. Brain. 2010;133:3336–3348. doi: 10.1093/brain/awq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker RE, Greig NH, Giacobini E. Why do so many drugs for Alzheimer's disease fail in development? Time for new methods and new practices? J Alzheimers Dis. 2008;15:303–325. doi: 10.3233/jad-2008-15213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jelic V, Winblad B. Treatment of mild cognitive impairment: rationale, present and future strategies. Acta Neurol Scand Suppl. 2003;179:83–93. doi: 10.1034/j.1600-0404.107.s179.12.x. [DOI] [PubMed] [Google Scholar]
- 10.Maddalena A, Papassotiropoulos A, Muller-Tillmanns B, Jung HH, Hegi T, Nitsch RM, Hock C. Biochemical diagnosis of Alzheimer disease by measuring the cerebrospinal fluid ratio of phosphorylated tau protein to beta-amyloid peptide42. Arch Neurol. 2003;60:1202–1206. doi: 10.1001/archneur.60.9.1202. [DOI] [PubMed] [Google Scholar]
- 11.Lista S, Faltraco F, Prvulovic D, Hampel H. Blood and plasma-based proteomic biomarker research in Alzheimer's disease. Prog Neurobiol. 2013;101–102:1–17. doi: 10.1016/j.pneurobio.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Bjorkqvist M, Ohlsson M, Minthon L, Hansson O. Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer's disease. PLoS One. 2012;7:e29868. doi: 10.1371/journal.pone.0029868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veitinger M, Umlauf E, Baumgartner R, Badrnya S, Porter J, Lamont J, Gerner C, Gruber CW, Oehler R, Zellner M. A combined proteomic and genetic analysis of the highly variable platelet proteome: from plasmatic proteins and SNPs. J Proteomics. 2012;75:5848–5860. doi: 10.1016/j.jprot.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 14.Corzett TH, Fodor IK, Choi MW, Walsworth VL, Turteltaub KW, McCutchen-Maloney SL, Chromy BA. Statistical analysis of variation in the human plasma proteome. J Biomed Biotechnol. 2010;2010:258494. doi: 10.1155/2010/258494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nolen BM, Orlichenko LS, Marrangoni A, Velikokhatnaya L, Prosser D, Grizzle WE, Ho K, Jenkins FJ, Bovbjerg DH, Lokshin AE. An extensive targeted proteomic analysis of disease-related protein biomarkers in urine from healthy donors. PLoS One. 2013;8:e63368. doi: 10.1371/journal.pone.0063368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoop MP, Coulier L, Rosenling T, Shi S, Smolinska AM, Buydens L, Ampt K, Stingl C, Dane A, Muilwijk B, Luitwieler RL, Sillevis Smitt PA, Hintzen RQ, Bischoff R, Wijmenga SS, Hankemeier T, van Gool AJ, Luider TM. Quantitative proteomics and metabolomics analysis of normal human cerebrospinal fluid samples. Mol Cell Proteomics. 2010;9:2063–2075. doi: 10.1074/mcp.M110.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.George JN. Platelets. Lancet. 2000;355:1531–1539. doi: 10.1016/S0140-6736(00)02175-9. [DOI] [PubMed] [Google Scholar]
- 18.Pletscher A, Laubscher A. Blood platelets as models for neurons: uses and limitations. J Neural Transm Suppl. 1980;16:7–16. doi: 10.1007/978-3-7091-8582-7_2. [DOI] [PubMed] [Google Scholar]
- 19.Catricala S, Torti M, Ricevuti G. Alzheimer disease and platelets: how's that relevant. Immun Ageing. 2012;9:20. doi: 10.1186/1742-4933-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talib LL, Joaquim HP, Forlenza OV. Platelet biomarkers in Alzheimer's disease. World J Psychiatry. 2012;2:95–101. doi: 10.5498/wjp.v2.i6.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freson K, Labarque V, Thys C, Wittevrongel C, Geet CV. What's new in using platelet research? To unravel thrombopathies and other human disorders. Eur J Pediatr. 2007;166:1203–1210. doi: 10.1007/s00431-007-0543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behari M, Shrivastava M. Role of platelets in neurodegenerative diseases: a universal pathophysiology. Int J Neurosci. 2013;123:287–299. doi: 10.3109/00207454.2012.751534. [DOI] [PubMed] [Google Scholar]
- 23.Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem. 1990;265:15977–15983. [PubMed] [Google Scholar]
- 24.Neumann K, Farias G, Slachevsky A, Perez P, Maccioni RB. Human platelets tau: a potential peripheral marker for Alzheimer's disease. J Alzheimers Dis. 2011;25:103–109. doi: 10.3233/JAD-2011-101641. [DOI] [PubMed] [Google Scholar]
- 25.Mukaetova-Ladinska EB, Abdell-All Z, Andrade J, da Silva JA, Boksha I, Burbaeva G, Kalaria RJ, O'Brien JT. Platelet Tau protein as a potential peripheral biomarker in Alzheimer's disease: an explorative study. Curr Alzheimer Res. 2013;10:1–8. doi: 10.2174/1567205015666180404165915. [DOI] [PubMed] [Google Scholar]
- 26.Di Luca M, Pastorino L, Cattabeni F, Zanardi R, Scarone S, Racagni G, Smeraldi E, Perez J. Abnormal pattern of platelet APP isoforms in Alzheimer disease and down syndrome. Arch Neurol. 1996;53:1162–1166. doi: 10.1001/archneur.1996.00550110114019. [DOI] [PubMed] [Google Scholar]
- 27.Farias G, Perez P, Slachevsky A, Maccioni RB. Platelet tau pattern correlates with cognitive status in Alzheimer's disease. J Alzheimers Dis. 2012;31:65–69. doi: 10.3233/JAD-2012-120304. [DOI] [PubMed] [Google Scholar]
- 28.Zellner M, Oehler R. Platelet Proteomics. New Jersey: John Wiley & Sons, Inc; 2011. Sample preparation variables in platelet proteomics for biomarker research; pp. 67–86. [Google Scholar]
- 29.Ellis PD: Effect Size Calculators. http://www.polyu.edu.hk/mm/effectsizefaqs/calculator/calculator.html
- 30.Manzine PR, de Franca Bram JM, Barham EJ, do Vale FA, Selistre-de-Araujo HS, Cominetti MR, Iost Pavarini SC. ADAM10 as a biomarker for Alzheimer's disease: a study with Brazilian elderly. Dement Geriatr Cogn Disord. 2013;35:58–66. doi: 10.1159/000345983. [DOI] [PubMed] [Google Scholar]
- 31.Vignini A, Morganti S, Salvolini E, Sartini D, Luzzi S, Fiorini R, Provinciali L, Di PR, Mazzanti L, Emanuelli M. Amyloid precursor protein expression is enhanced in human platelets from subjects with Alzheimer's disease and frontotemporal lobar degeneration: a real-time PCR study. Exp Gerontol. 2013;48:1505–1508. [PubMed] [Google Scholar]
- 32.Jelic V, Hagman G, Yamamoto NG, Teranishi Y, Nishimura T, Winblad B, Pavlov PF. Abnormal platelet amyloid-beta protein precursor (AbetaPP) metabolism in Alzheimer's disease: identification and characterization of a new AbetaPP isoform as potential biomarker. J Alzheimers Dis. 2013;35:285–295. doi: 10.3233/JAD-122122. [DOI] [PubMed] [Google Scholar]
- 33.Borroni B, Colciaghi F, Caltagirone C, Rozzini L, Broglio L, Cattabeni F, Di Luca M, Padovani A. Platelet amyloid precursor protein abnormalities in mild cognitive impairment predict conversion to dementia of Alzheimer type: a 2-year follow-up study. Arch Neurol. 2003;60:1740–1744. doi: 10.1001/archneur.60.12.1740. [DOI] [PubMed] [Google Scholar]
- 34.Colciaghi F, Borroni B, Pastorino L, Marcello E, Zimmermann M, Cattabeni F, Padovani A, Di LM. [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med. 2002;8:67–74. [PMC free article] [PubMed] [Google Scholar]
- 35.Di Luca M, Pastorino L, Bianchetti A, Perez J, Vignolo LA, Lenzi GL, Trabucchi M, Cattabeni F, Padovani A. Differential level of platelet amyloid beta precursor protein isoforms: an early marker for Alzheimer disease. Arch Neurol. 1998;55:1195–1200. doi: 10.1001/archneur.55.9.1195. [DOI] [PubMed] [Google Scholar]
- 36.Padovani A, Pastorino L, Borroni B, Colciaghi F, Rozzini L, Monastero R, Perez J, Pettenati C, Mussi M, Parrinello G, Cottini E, Lenzi GL, Trabucchi M, Cattabeni F, Di LM. Amyloid precursor protein in platelets: a peripheral marker for the diagnosis of sporadic AD. Neurology. 2001;57:2243–2248. doi: 10.1212/wnl.57.12.2243. [DOI] [PubMed] [Google Scholar]
- 37.Zainaghi IA, Forlenza OV, Gattaz WF. Abnormal APP processing in platelets of patients with Alzheimer's disease: correlations with membrane fluidity and cognitive decline. Psychopharmacology (Berl) 2007;192:547–553. doi: 10.1007/s00213-007-0748-5. [DOI] [PubMed] [Google Scholar]
- 38.Liu HC, Wang HC, Ko SY, Wang PN, Chi CW, Hong CJ, Lin KN, Liu TY. Correlation between platelet amyloid precursor protein isoform ratio and cognition in Alzheimer's disease. J Alzheimers Dis. 2007;11:77–84. doi: 10.3233/jad-2007-11111. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg RN, Baskin F, Fosmire JA, Risser R, Adams P, Svetlik D, Honig LS, Cullum CM, Weiner MF. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch Neurol. 1997;54:139–144. doi: 10.1001/archneur.1997.00550140019007. [DOI] [PubMed] [Google Scholar]
- 40.Borroni B, Colciaghi F, Pastorino L, Pettenati C, Cottini E, Rozzini L, Monastero R, Lenzi GL, Cattabeni F, Di Luca M, Padovani A. Amyloid precursor protein in platelets of patients with Alzheimer disease: effect of acetylcholinesterase inhibitor treatment. Arch Neurol. 2001;58:442–446. doi: 10.1001/archneur.58.3.442. [DOI] [PubMed] [Google Scholar]
- 41.Johnston JA, Liu WW, Coulson DT, Todd S, Murphy S, Brennan S, Foy CJ, Craig D, Irvine GB, Passmore AP. Platelet beta-secretase activity is increased in Alzheimer's disease. Neurobiol Aging. 2008;29:661–668. doi: 10.1016/j.neurobiolaging.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Liu WW, Todd S, Craig D, Passmore AP, Coulson DT, Murphy S, Irvine GB, Johnston JA. Elevated platelet beta-secretase activity in mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;24:464–468. doi: 10.1159/000110739. [DOI] [PubMed] [Google Scholar]
- 43.Decourt B, Walker A, Gonzales A, Malek-Ahmadi M, Liesback C, Davis KJ, Belden CM, Jacobson SA, Sabbagh MN. Can platelet BACE1 levels be used as a biomarker for Alzheimer's disease? Proof-of-concept study. Platelets. 2013;24:235–238. doi: 10.3109/09537104.2012.688899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vignini A, Nanetti L, Moroni C, Tanase L, Bartolini M, Luzzi S, Provinciali L, Mazzanti L. Modifications of platelet from Alzheimer disease patients: a possible relation between membrane properties and NO metabolites. Neurobiol Aging. 2007;28:987–994. doi: 10.1016/j.neurobiolaging.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Vignini A, Giusti L, Raffaelli F, Giulietti A, Salvolini E, Luzzi S, Provinciali L, Mazzanti L, Nanetti L. Impact of gender on platelet membrane functions of Alzheimer's disease patients. Exp Gerontol. 2013;48:319–325. doi: 10.1016/j.exger.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 46.Prodan CI, Szasz R, Vincent AS, Ross ED, Dale GL. Coated-platelets retain amyloid precursor protein on their surface. Platelets. 2006;17:56–60. doi: 10.1080/09537100500181913. [DOI] [PubMed] [Google Scholar]
- 47.Prodan CI, Ross ED, Vincent AS, Dale GL. Coated-platelets are higher in amnestic versus nonamnestic patients with mild cognitive impairment. Alzheimer Dis Assoc Disord. 2007;21:259–261. doi: 10.1097/WAD.0b013e31811ec11f. [DOI] [PubMed] [Google Scholar]
- 48.Prodan CI, Ross ED, Vincent AS, Dale GL. Differences in coated-platelet production between frontotemporal dementia and Alzheimer disease. Alzheimer Dis Assoc Disord. 2009;23:234–237. doi: 10.1097/WAD.0b013e318199dd1a. [DOI] [PubMed] [Google Scholar]
- 49.Cardoso SM, Proenca MT, Santos S, Santana I, Oliveira CR. Cytochrome c oxidase is decreased in Alzheimer's disease platelets. Neurobiol Aging. 2004;25:105–110. doi: 10.1016/s0197-4580(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 50.Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, Murri L, Rapoport SI, Solaini G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol Aging. 2002;23:371–376. doi: 10.1016/s0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 51.Mancuso M, Filosto M, Bosetti F, Ceravolo R, Rocchi A, Tognoni G, Manca ML, Solaini G, Siciliano G, Murri L. Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Exp Neurol. 2003;182:421–426. doi: 10.1016/s0014-4886(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 52.Parker WD, Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 53.Valla J, Schneider L, Niedzielko T, Coon KD, Caselli R, Sabbagh MN, Ahern GL, Baxter L, Alexander G, Walker DG, Reiman EM. Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion. 2006;6:323–330. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forlenza OV, Torres CA, Talib LL, de Paula VJ, Joaquim HP, Diniz BS. Increased platelet GSK3B activity in patients with mild cognitive impairment and Alzheimer's disease. J Psychiatr Res. 2011;45:220–224. doi: 10.1016/j.jpsychires.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 55.Davies TA, Long HJ, Sgro K, Rathbun WH, McMenamin ME, Seetoo K, Tibbles H, Billingslea AM, Fine RE, Fishman JB, Levesque CA, Smith SJ, Wells JM, Simons ER. Activated Alzheimer disease platelets retain more beta amyloid precursor protein. Neurobiol Aging. 1997;18:147–153. doi: 10.1016/s0197-4580(97)00013-4. [DOI] [PubMed] [Google Scholar]
- 56.Mukaetova-Ladinska EB, Abdel-All Z, Dodds S, Andrade J, Alves da Silva J, Kalaria RN, O'Brien JT. Platelet immunoglobulin and amyloid precursor protein as potential peripheral biomarkers for Alzheimer's disease: findings from a pilot study. Age Ageing. 2012;41:408–412. doi: 10.1093/ageing/afr171. [DOI] [PubMed] [Google Scholar]
- 57.Fischer P, Gotz ME, Ellinger B, Streifler M, Riederer P, Danielczyk W. Platelet monoamine oxidase B activity and vitamin B12 in dementia. Biol Psychiatry. 1994;35:772–774. doi: 10.1016/0006-3223(94)91138-x. [DOI] [PubMed] [Google Scholar]
- 58.Mann JJ, Stanley M, Neophytides A, de Leon MJ, Ferris SH, Gershon S. Central amine metabolism in Alzheimer's disease: in vivo relationship to cognitive deficit. Neurobiol Aging. 1981;2:57–60. doi: 10.1016/0197-4580(81)90060-9. [DOI] [PubMed] [Google Scholar]
- 59.Bongioanni P, Gemignani F, Boccardi B, Borgna M, Rossi B. Platelet monoamine oxidase molecular activity in demented patients. Ital J Neurol Sci. 1997;18:151–156. doi: 10.1007/BF02048483. [DOI] [PubMed] [Google Scholar]
- 60.Adolfsson R, Gottfries CG, Oreland L, Wiberg A, Winblad B. Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci. 1980;27:1029–1034. doi: 10.1016/0024-3205(80)90025-9. [DOI] [PubMed] [Google Scholar]
- 61.Konings CH, Scheltens P, Kuiper MA, Wolters EC. No evidence for abnormalities in kinetics of platelet monoamine oxidase in Alzheimer's disease. Clin Chim Acta. 1995;240:99–102. doi: 10.1016/0009-8981(95)06130-4. [DOI] [PubMed] [Google Scholar]
- 62.Zellner M, Baureder M, Rappold E, Bugert P, Kotzailias N, Babeluk R, Baumgartner R, Attems J, Gerner C, Jellinger K, Roth E, Oehler R, Umlauf E. Comparative platelet proteome analysis reveals an increase of monoamine oxidase-B protein expression in Alzheimer's disease but not in non-demented Parkinson's disease patients. J Proteomics. 2012;75:2080–2092. doi: 10.1016/j.jprot.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 63.Yesil Y, Kuyumcu ME, Cankurtaran M, Uz B, Kara A, Kilic MK, Halil M, Ulger Z, Yavuz BB, Haznedaroglu IC, Ariogul S. Increased mean platelet volume (MPV) indicating the vascular risk in Alzheimer's disease (AD) Arch Gerontol Geriatr. 2012;55:257–260. doi: 10.1016/j.archger.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 64.Kozubski W, Swiderek M, Kloszewska I, Gwozdzinski K, Watala C. Blood platelet membrane fluidity and the exposition of membrane protein receptors in Alzheimer disease (AD) patients–preliminary Study. Alzheimer Dis Assoc Disord. 2002;16:52–54. doi: 10.1097/00002093-200201000-00009. [DOI] [PubMed] [Google Scholar]
- 65.Zubenko GS, Cohen BM, Boller F, Malinakova I, Keefe N, Chojnacki B. Platelet membrane abnormality in Alzheimer's disease. Ann Neurol. 1987;22:237–244. doi: 10.1002/ana.410220208. [DOI] [PubMed] [Google Scholar]
- 66.Ortiz GG, Pacheco-Moises F, El Hafidi M, Jimenez-Delgado A, Macias-Islas MA, Rosales Corral SA, de la Rosa AC, Sanchez-Gonzalez VJ, Arias-Merino ED, Velazquez-Brizuela IE. Detection of membrane fluidity in submitochondrial particles of platelets and erythrocyte membranes from Mexican patients with Alzheimer disease by intramolecular excimer formation of 1,3 dipyrenylpropane. Dis Markers. 2008;24:151–156. doi: 10.1155/2008/642120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gattaz WF, Cairns NJ, Levy R, Forstl H, Braus DF, Maras A. Decreased phospholipase A2 activity in the brain and in platelets of patients with Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci. 1996;246:129–131. doi: 10.1007/BF02189113. [DOI] [PubMed] [Google Scholar]
- 68.Gattaz WF, Forlenza OV, Talib LL, Barbosa NR, Bottino CM. Platelet phospholipase A(2) activity in Alzheimer's disease and mild cognitive impairment. J Neural Transm. 2004;111:591–601. doi: 10.1007/s00702-004-0142-y. [DOI] [PubMed] [Google Scholar]
- 69.Krzystanek E, Krzystanek M, Opala G, Trzeciak HI, Siuda J, Malecki A. Platelet phospholipase A2 activity in patients with Alzheimer's disease, vascular dementia and ischemic stroke. J Neural Transm. 2007;114:1033–1039. doi: 10.1007/s00702-007-0669-9. [DOI] [PubMed] [Google Scholar]
- 70.Gattaz WF, Talib LL, Schaeffer EL, Diniz BS, Forlenza OV. Low platelet iPLA2 activity predicts conversion from mild cognitive impairment to Alzheimer's disease: a 4-year follow-up study. J Neural Transm. 2014;121:193–200. doi: 10.1007/s00702-013-1088-8. [DOI] [PubMed] [Google Scholar]
- 71.Matsushima H, Shimohama S, Kawamata J, Fujimoto S, Takenawa T, Kimura J. Reduction of platelet phospholipase C-delta1 activity in Alzheimer's disease associated with a specific apolipoprotein E genotype (epsilon3/epsilon3) Int J Mol Med. 1998;1:91–93. doi: 10.3892/ijmm.1.1.91. [DOI] [PubMed] [Google Scholar]
- 72.Kumar AM, Sevush S, Kumar M, Ruiz J, Eisdorfer C. Peripheral serotonin in Alzheimer's disease. Neuropsychobiology. 1995;32:9–12. doi: 10.1159/000119205. [DOI] [PubMed] [Google Scholar]
- 73.Meszaros Z, Borcsiczky D, Mate M, Tarcali J, Szombathy T, Tekes K, Magyar K. Platelet MAO-B activity and serotonin content in patients with dementia: effect of age, medication, and disease. Neurochem Res. 1998;23:863–868. doi: 10.1023/a:1022458928442. [DOI] [PubMed] [Google Scholar]
- 74.Jaremo P, Milovanovic M, Buller C, Nilsson S, Winblad B. P-selectin paradox and dementia of the Alzheimer type: circulating P-selectin is increased but platelet-bound P-selectin after agonist provocation is compromised. Scand J Clin Lab Invest. 2013;73:170–174. doi: 10.3109/00365513.2013.764572. [DOI] [PubMed] [Google Scholar]
- 75.Sevush S, Jy W, Horstman LL, Mao WW, Kolodny L, Ahn YS. Platelet activation in Alzheimer disease. Arch Neurol. 1998;55:530–536. doi: 10.1001/archneur.55.4.530. [DOI] [PubMed] [Google Scholar]
- 76.Dale GL. Coated-platelets: an emerging component of the procoagulant response. J Thromb Haemost. 2005;3:2185–2192. doi: 10.1111/j.1538-7836.2005.01274.x. [DOI] [PubMed] [Google Scholar]
- 77.Dale GL, Friese P, Batar P, Hamilton SF, Reed GL, Jackson KW, Clemetson KJ, Alberio L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 2002;415:175–179. doi: 10.1038/415175a. [DOI] [PubMed] [Google Scholar]
- 78.Prodan CI, Ross ED, Vincent AS, Dale GL. Rate of progression in Alzheimer's disease correlates with coated-platelet levels–a longitudinal study. Transl Res. 2008;152:99–102. doi: 10.1016/j.trsl.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 79.Prodan CI, Ross ED, Vincent AS, Dale GL. Coated-platelets correlate with disease progression in Alzheimer disease. J Neurol. 2007;254:548–549. doi: 10.1007/s00415-006-0323-8. [DOI] [PubMed] [Google Scholar]
- 80.Prodan CI, Ross ED, Stoner JA, Cowan LD, Vincent AS, Dale GL. Coated-platelet levels and progression from mild cognitive impairment to Alzheimer disease. Neurology. 2011;76:247–252. doi: 10.1212/WNL.0b013e3182074bd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tyce GM. Origin and metabolism of serotonin. J Cardiovasc Pharmacol. 1990;16(Suppl 3):S1–S7. [PubMed] [Google Scholar]
- 82.Jonnakuty C, Gragnoli C. What do we know about serotonin? J Cell Physiol. 2008;217:301–306. doi: 10.1002/jcp.21533. [DOI] [PubMed] [Google Scholar]
- 83.Hergovich N, Aigner M, Eichler HG, Entlicher J, Drucker C, Jilma B. Paroxetine decreases platelet serotonin storage and platelet function in human beings. Clin Pharmacol Ther. 2000;68:435–442. doi: 10.1067/mcp.2000.110456. [DOI] [PubMed] [Google Scholar]
- 84.Castermans D, Volders K, Crepel A, Backx L, De VR, Freson K, Meulemans S, Vermeesch JR, Schrander-Stumpel CT, De RP, Del-Favero J, Van Geet C, Van De VEN WJ, Steyaert JG, Devriendt K, Creemers JW. SCAMP5, NBEA and AMISYN: three candidate genes for autism involved in secretion of large dense-core vesicles. Hum Mol Genet. 2010;19:1368–1378. doi: 10.1093/hmg/ddq013. [DOI] [PubMed] [Google Scholar]
- 85.Jackman H, Luchins D, Meltzer HY. Platelet serotonin levels in schizophrenia: relationship to race and psychopathology. Biol Psychiatry. 1983;18:887–902. [PubMed] [Google Scholar]
- 86.Ostrowitzki S, Rao ML, Redei J, Andres AH. Concurrence of cortex and platelet serotonin2 receptor binding characteristics in the individual and the putative regulation by serotonin. J Neural Transm Gen Sect. 1993;93:27–35. doi: 10.1007/BF01244935. [DOI] [PubMed] [Google Scholar]
- 87.Andres AH, Rao ML, Ostrowitzki S, Entzian W. Human brain cortex and platelet serotonin2 receptor binding properties and their regulation by endogenous serotonin. Life Sci. 1993;52:313–321. doi: 10.1016/0024-3205(93)90223-p. [DOI] [PubMed] [Google Scholar]
- 88.Owens MJ, Nemeroff CB. Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem. 1994;40:288–295. [PubMed] [Google Scholar]
- 89.Palmer AM, Francis PT, Benton JS, Sims NR, Mann DM, Neary D, Snowden JS, Bowen DM. Presynaptic serotonergic dysfunction in patients with Alzheimer's disease. J Neurochem. 1987;48:8–15. doi: 10.1111/j.1471-4159.1987.tb13120.x. [DOI] [PubMed] [Google Scholar]
- 90.Ouchi Y, Yoshikawa E, Futatsubashi M, Yagi S, Ueki T, Nakamura K. Altered brain serotonin transporter and associated glucose metabolism in Alzheimer disease. J Nucl Med. 2009;50:1260–1266. doi: 10.2967/jnumed.109.063008. [DOI] [PubMed] [Google Scholar]
- 91.Spigset O, Wilhelmsson C, Mjorndal T, Eriksson S. Serotonin 5-HT2A receptor binding in platelets from patients with Alzheimer's disease or vascular dementia. Int Psychogeriatr. 2000;12:537–545. doi: 10.1017/s1041610200006645. [DOI] [PubMed] [Google Scholar]
- 92.Koren P, Diver-Haber A, Adunsky A, Rabinowitz M, Hershkowitz M. Uptake of serotonin into platelets of senile dementia of the Alzheimer type patients. J Gerontol. 1993;48:B93–B96. doi: 10.1093/geronj/48.3.b93. [DOI] [PubMed] [Google Scholar]
- 93.Suranyi-Cadotte BE, Gauthier S, Lafaille F, DeFlores S, Dam TV, Nair NP, Quirion R. Platelet 3H-imipramine binding distinguishes depression from Alzheimer dementia. Life Sci. 1985;37:2305–2311. doi: 10.1016/0024-3205(85)90022-0. [DOI] [PubMed] [Google Scholar]
- 94.Inestrosa NC, Alarcon R, Arriagada J, Donoso A, Alvarez J. Platelet of Alzheimer patients: increased counts and subnormal uptake and accumulation of [14C]5-hydroxytryptamine. Neurosci Lett. 1993;163:8–10. doi: 10.1016/0304-3940(93)90216-8. [DOI] [PubMed] [Google Scholar]
- 95.Tukiainen E, Wikstrom J, Kilpelainen H. Uptake of 5-hydroxytryptamine by blood platelets in Huntington's chorea and Alzheimer type of presenile dementia. Med Biol. 1981;59:116–120. [PubMed] [Google Scholar]
- 96.Arora RC, Emery OB, Meltzer HY. Serotonin uptake in the blood platelets of Alzheimer's disease patients. Neurology. 1991;41:1307–1309. doi: 10.1212/wnl.41.8.1307. [DOI] [PubMed] [Google Scholar]
- 97.Kumar AM, Kumar M, Sevush S, Ruiz J, Eisdorfer C. Serotonin uptake and its kinetics in platelets of women with Alzheimer's disease. Psychiatry Res. 1995;59:145–150. doi: 10.1016/0165-1781(95)02786-6. [DOI] [PubMed] [Google Scholar]
- 98.Mimica N, Muck-Seler D, Pivac N, Mustapic M, Dezeljin M, Stipcevic T, Presecki P, Radonic E, Folnegovic-Smalc V. Platelet serotonin and monoamine oxidase in Alzheimer's disease with psychotic features. Coll Antropol. 2008;32(Suppl 1):119–122. [PubMed] [Google Scholar]
- 99.Muck-Seler D, Presecki P, Mimica N, Mustapic M, Pivac N, Babic A, Nedic G, Folnegovic-Smalc V. Platelet serotonin concentration and monoamine oxidase type B activity in female patients in early, middle and late phase of Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1226–1231. doi: 10.1016/j.pnpbp.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 100.Meltzer CC, Smith G, DeKosky ST, Pollock BG, Mathis CA, Moore RY, Kupfer DJ, Reynolds CF., III Serotonin in aging, late-life depression, and Alzheimer's disease: the emerging role of functional imaging. Neuropsychopharmacology. 1998;18:407–430. doi: 10.1016/S0893-133X(97)00194-2. [DOI] [PubMed] [Google Scholar]
- 101.Wirz-Justice A, Lichtsteiner M, Feer H. Diurnal and seasonal variations in human platelet serotonin in man. J Neural Transm. 1977;41:7–15. doi: 10.1007/BF01252961. [DOI] [PubMed] [Google Scholar]
- 102.Beck O, Wallen NH, Broijersen A, Larsson PT, Hjemdahl P. On the accurate determination of serotonin in human plasma. Biochem Biophys Res Commun. 1993;196:260–266. doi: 10.1006/bbrc.1993.2243. [DOI] [PubMed] [Google Scholar]
- 103.Paasonen MK, Solatunturi E, Kivalo E. Monoamine oxidase activity of blood platelets and their ability to store 5-hydroxytryptamine in some mental deficiencies. Psychopharmacologia. 1964;6:120–124. doi: 10.1007/BF00413224. [DOI] [PubMed] [Google Scholar]
- 104.Grimsby J, Chen K, Wang LJ, Lan NC, Shih JC. Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc Natl Acad Sci U S A. 1991;88:3637–3641. doi: 10.1073/pnas.88.9.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen K, Wu HF, Shih JC. The deduced amino acid sequences of human platelet and frontal cortex monoamine oxidase B are identical. J Neurochem. 1993;61:187–190. doi: 10.1111/j.1471-4159.1993.tb03554.x. [DOI] [PubMed] [Google Scholar]
- 106.Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev. 2008;60:1527–1533. doi: 10.1016/j.addr.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Robinson DS. Changes in monoamine oxidase and monoamines with human development and aging. Fed Proc. 1975;34:103–107. [PubMed] [Google Scholar]
- 108.Gulyas B, Pavlova E, Kasa P, Gulya K, Bakota L, Varszegi S, Keller E, Horvath MC, Nag S, Hermecz I, Magyar K, Halldin C. Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem Int. 2011;58:60–68. doi: 10.1016/j.neuint.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 109.Emilsson L, Saetre P, Balciuniene J, Castensson A, Cairns N, Jazin EE. Increased monoamine oxidase messenger RNA expression levels in frontal cortex of Alzheimer's disease patients. Neurosci Lett. 2002;326:56–60. doi: 10.1016/s0304-3940(02)00307-5. [DOI] [PubMed] [Google Scholar]
- 110.Nakamura S, Kawamata T, Akiguchi I, Kameyama M, Nakamura N, Kimura H. Expression of monoamine oxidase B activity in astrocytes of senile plaques. Acta Neuropathol. 1990;80:419–425. doi: 10.1007/BF00307697. [DOI] [PubMed] [Google Scholar]
- 111.Gottfries CG. Neurochemical aspects on aging and diseases with cognitive impairment. J Neurosci Res. 1990;27:541–547. doi: 10.1002/jnr.490270415. [DOI] [PubMed] [Google Scholar]
- 112.Grunblatt E, Schlosser R, Fischer P, Fischer MO, Li J, Koutsilieri E, Wichart I, Sterba N, Rujescu D, Moller HJ, Adamcyk W, Dittrich B, Muller F, Oberegger K, Gatterer G, Jellinger KJ, Mostafaie N, Jungwirth S, Huber K, Tragl KH, Danielczyk W, Riederer P. Oxidative stress related markers in the "VITA" and the centenarian projects. Neurobiol Aging. 2005;26:429–438. doi: 10.1016/j.neurobiolaging.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 113.Soto J, Ulibarri I, Jauregui JV, Ballesteros J, Meana JJ. Dissociation between I2-imidazoline receptors and MAO-B activity in platelets of patients with Alzheimer's type dementia. J Psychiatr Res. 1999;33:251–257. doi: 10.1016/s0022-3956(98)00065-x. [DOI] [PubMed] [Google Scholar]
- 114.Gotz ME, Fischer P, Gsell W, Riederer P, Streifler M, Simanyi M, Muller F, Danielczyk W. Platelet monoamine oxidase B activity in dementia. A 4-year follow-up. Dement Geriatr Cogn Disord. 1998;9:74–77. doi: 10.1159/000017026. [DOI] [PubMed] [Google Scholar]
- 115.Parnetti L, Reboldi GP, Santucci C, Santucci A, Gaiti A, Brunetti M, Cecchetti R, Senin U. Platelet MAO-B activity as a marker of behavioural characteristics in dementia disorders. Aging (Milano ) 1994;6:201–207. doi: 10.1007/BF03324240. [DOI] [PubMed] [Google Scholar]
- 116.Bonuccelli U, Piccini P, Marazziti D, Cassano GB, Muratorio A. Increased platelet 3H-imipramine binding and monoamine oxidase B activity in Alzheimer's disease. J Neural Transm Park Dis Dement Sect. 1990;2:139–147. doi: 10.1007/BF02260901. [DOI] [PubMed] [Google Scholar]
- 117.Danielczyk W, Streifler M, Konradi C, Riederer P, Moll G. Platelet MAO-B activity and the psychopathology of Parkinson's disease, senile dementia and multi-infarct dementia. Acta Psychiatr Scand. 1988;78:730–736. doi: 10.1111/j.1600-0447.1988.tb06412.x. [DOI] [PubMed] [Google Scholar]
- 118.Alexopoulos GS, Young RC, Lieberman KW, Shamoian CA. Platelet MAO activity in geriatric patients with depression and dementia. Am J Psychiatry. 1987;144:1480–1483. doi: 10.1176/ajp.144.11.1480. [DOI] [PubMed] [Google Scholar]
- 119.Schneider LS, Severson JA, Chui HC, Pollock VE, Sloane RB, Fredrickson ER. Platelet tritiated imipramine binding and MAO activity in Alzheimer's disease patients with agitation and delusions. Psychiatry Res. 1988;25:311–322. doi: 10.1016/0165-1781(88)90101-1. [DOI] [PubMed] [Google Scholar]
- 120.Smith RC, Ho BT, Kralik P, Vroulis G, Gordon J, Wolff J. Platelet monamine oxidase in Alzheimer's disease. J Gerontol. 1982;37:572–574. doi: 10.1093/geronj/37.5.572. [DOI] [PubMed] [Google Scholar]
- 121.Ahlskog JE, Uitti RJ, Tyce GM, O'Brien JF, Petersen RC, Kokmen E. Plasma catechols and monoamine oxidase metabolites in untreated Parkinson's and Alzheimer's diseases. J Neurol Sci. 1996;136:162–168. doi: 10.1016/0022-510x(95)00318-v. [DOI] [PubMed] [Google Scholar]
- 122.Kalgutkar AS, Castagnoli N., Jr Selective inhibitors of monoamine oxidase (MAO-A and MAO-B) as probes of its catalytic site and mechanism. Med Res Rev. 1995;15:325–388. doi: 10.1002/med.2610150406. [DOI] [PubMed] [Google Scholar]
- 123.Rommelspacher H, Meier-Henco M, Smolka M, Kloft C. The levels of norharman are high enough after smoking to affect monoamineoxidase B in platelets. Eur J Pharmacol. 2002;441:115–125. doi: 10.1016/s0014-2999(02)01452-8. [DOI] [PubMed] [Google Scholar]
- 124.Launay JM, Del PM, Chironi G, Callebert J, Peoc'h K, Megnien JL, Mallet J, Simon A, Rendu F. Smoking induces long-lasting effects through a monoamine-oxidase epigenetic regulation. PLoS One. 2009;4:e7959. doi: 10.1371/journal.pone.0007959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Malouf R, Grimley EJ. Folic acid with or without vitamin B12 for the prevention and treatment of healthy elderly and demented people. Cochrane Database Syst Rev. 2008;8:CD004514. doi: 10.1002/14651858.CD004514.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.de Jager CA, Oulhaj A, Jacoby R, Refsum H, Smith AD. Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: a randomized controlled trial. Int J Geriatr Psychiatry. 2012;27:592–600. doi: 10.1002/gps.2758. [DOI] [PubMed] [Google Scholar]
- 127.Regland B, Gottfries CG, Oreland L. Vitamin B12-induced reduction of platelet monoamine oxidase activity in patients with dementia and pernicious anaemia. Eur Arch Psychiatry Clin Neurosci. 1991;240:288–291. doi: 10.1007/BF02189542. [DOI] [PubMed] [Google Scholar]
- 128.Zellner M, Babeluk R, Jakobsen LH, Gerner C, Umlauf E, Volf I, Roth E, Kondrup J. A proteomics study reveals a predominant change in MaoB expression in platelets of healthy volunteers after high protein meat diet: relationship to the methylation cycle. J Neural Transm. 2011;118:653–662. doi: 10.1007/s00702-011-0617-6. [DOI] [PubMed] [Google Scholar]
- 129.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 130.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 131.Burbaeva GS, Boksha IS, Savushkina OK, Turishcheva MS, Tereshkina EB, Starodubtseva LI, Gavrilova SI, Fedorova I, Zhuravin IA. Platelet cytochrome c-oxidase and glutamine synthetase-like protein in patients with mild cognitive impairment. Zh Nevrol Psikhiatr Im S S Korsakova. 2012;112:55–58. [PubMed] [Google Scholar]
- 132.Parker WD, Jr, Mahr NJ, Filley CM, Parks JK, Hughes D, Young DA, Cullum CM. Reduced platelet cytochrome c oxidase activity in Alzheimer's disease. Neurology. 1994;44:1086–1090. doi: 10.1212/wnl.44.6.1086. [DOI] [PubMed] [Google Scholar]
- 133.Shi C, Guo K, Yew DT, Yao Z, Forster EL, Wang H, Xu J. Effects of ageing and Alzheimer's disease on mitochondrial function of human platelets. Exp Gerontol. 2008;43:589–594. doi: 10.1016/j.exger.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 134.Mosconi L, de Leon M, Murray J, EL, Lu J, Javier E, McHugh P, Swerdlow RH. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer's disease. J Alzheimers Dis. 2011;27:483–490. doi: 10.3233/JAD-2011-110866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Szekely CA, Zandi PP. Non-steroidal anti-inflammatory drugs and Alzheimer's disease: the epidemiological evidence. CNS Neurol Disord Drug Targets. 2010;9:132–139. doi: 10.2174/187152710791012026. [DOI] [PubMed] [Google Scholar]
- 137.Bermejo P, Martin-Aragon S, Benedi J, Susin C, Felici E, Gil P, Ribera JM, Villar AM. Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer's disease. Immunol Lett. 2008;117:198–202. doi: 10.1016/j.imlet.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 138.Casoli T, Di Stefano G, Balietti M, Solazzi M, Giorgetti B, Fattoretti P. Peripheral inflammatory biomarkers of Alzheimer's disease: the role of platelets. Biogerontology. 2010;11:627–633. doi: 10.1007/s10522-010-9281-8. [DOI] [PubMed] [Google Scholar]
- 139.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 141.O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Attems J, Jellinger KA. Amyloid and tau: neither chicken nor egg but two partners in crime! Acta Neuropathol. 2013;126:619–621. doi: 10.1007/s00401-013-1167-9. [DOI] [PubMed] [Google Scholar]
- 143.Colciaghi F, Marcello E, Borroni B, Zimmermann M, Caltagirone C, Cattabeni F, Padovani A, Di Luca M. Platelet APP, ADAM 10 and BACE alterations in the early stages of Alzheimer disease. Neurology. 2004;62:498–501. doi: 10.1212/01.wnl.0000106953.49802.9c. [DOI] [PubMed] [Google Scholar]
- 144.Tang K, Hynan LS, Baskin F, Rosenberg RN. Platelet amyloid precursor protein processing: a bio-marker for Alzheimer's disease. J Neurol Sci. 2006;240:53–58. doi: 10.1016/j.jns.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zimmermann M, Borroni B, Cattabeni F, Padovani A, Di Luca M. Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol Dis. 2005;19:237–242. doi: 10.1016/j.nbd.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 146.Marksteiner J, Humpel C. Platelet-Derived Secreted Amyloid-Precursor Protein-beta as a Marker for Diagnosing Alzheimer's Disease. Curr Neurovasc Res. 2013;10:297–303. doi: 10.2174/15672026113109990022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nalivaeva NN, Turner AJ. The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 2013;587:2046–2054. doi: 10.1016/j.febslet.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 148.Moir RD, Lynch T, Bush AI, Whyte S, Henry A, Portbury S, Multhaup G, Small DH, Tanzi RE, Beyreuther K, Masters CL. Relative increase in Alzheimer's disease of soluble forms of cerebral Abeta amyloid protein precursor containing the Kunitz protease inhibitory domain. J Biol Chem. 1998;273:5013–5019. doi: 10.1074/jbc.273.9.5013. [DOI] [PubMed] [Google Scholar]
- 149.Causevic M, Farooq U, Lovestone S, Killick R. beta-Amyloid precursor protein and tau protein levels are differently regulated in human cerebellum compared to brain regions vulnerable to Alzheimer's type neurodegeneration. Neurosci Lett. 2010;485:162–166. doi: 10.1016/j.neulet.2010.08.088. [DOI] [PubMed] [Google Scholar]
- 150.Tharp WG, Lee YH, Greene SM, Vincellete E, Beach TG, Pratley RE. Measurement of altered AbetaPP isoform expression in frontal cortex of patients with Alzheimer's disease by absolute quantification real-time PCR. J Alzheimers Dis. 2012;29:449–457. doi: 10.3233/JAD-2011-111337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen J, Wang M, Turko IV. Quantification of amyloid precursor protein isoforms using quantification concatamer internal standard. Anal Chem. 2013;85:303–307. doi: 10.1021/ac3033239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Goate A, Hardy J. Twenty years of Alzheimer's disease-causing mutations. J Neurochem. 2012;120(Suppl 1):3–8. doi: 10.1111/j.1471-4159.2011.07575.x. [DOI] [PubMed] [Google Scholar]
- 153.Schlossmacher MG, Ostaszewski BL, Hecker LI, Celi A, Haass C, Chin D, Lieberburg I, Furie BC, Furie B, Selkoe DJ. Detection of distinct isoform patterns of the beta-amyloid precursor protein in human platelets and lymphocytes. Neurobiol Aging. 1992;13:421–434. doi: 10.1016/0197-4580(92)90117-g. [DOI] [PubMed] [Google Scholar]
- 154.Henry A, Li QX, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Cappai R. Inhibition of platelet activation by the Alzheimer's disease amyloid precursor protein. Br J Haematol. 1998;103:402–415. doi: 10.1046/j.1365-2141.1998.01005.x. [DOI] [PubMed] [Google Scholar]
- 155.Schmaier AH, Dahl LD, Rozemuller AJ, Roos RA, Wagner SL, Chung R, Van Nostrand WE. Protease nexin-2/amyloid beta protein precursor. A tight-binding inhibitor of coagulation factor IXa. J Clin Invest. 1993;92:2540–2545. doi: 10.1172/JCI116863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mahdi F, Van Nostrand WE, Schmaier AH. Protease nexin-2/amyloid beta-protein precursor inhibits factor Xa in the prothrombinase complex. J Biol Chem. 1995;270:23468–23474. doi: 10.1074/jbc.270.40.23468. [DOI] [PubMed] [Google Scholar]
- 157.Li QX, Berndt MC, Bush AI, Rumble B, Mackenzie I, Friedhuber A, Beyreuther K, Masters CL. Membrane-associated forms of the beta A4 amyloid protein precursor of Alzheimer's disease in human platelet and brain: surface expression on the activated human platelet. Blood. 1994;84:133–142. [PubMed] [Google Scholar]
- 158.Baskin F, Rosenberg RN, Iyer L, Hynan L, Cullum CM. Platelet APP isoform ratios correlate with declining cognition in AD. Neurology. 2000;54:1907–1909. doi: 10.1212/wnl.54.10.1907. [DOI] [PubMed] [Google Scholar]
- 159.Borroni B, Agosti C, Marcello E, Di Luca M, Padovani A. Blood cell markers in Alzheimer Disease: Amyloid Precursor Protein form ratio in platelets. Exp Gerontol. 2010;45:53–56. doi: 10.1016/j.exger.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 160.Zainaghi IA, Talib LL, Diniz BS, Gattaz WF, Forlenza OV. Reduced platelet amyloid precursor protein ratio (APP ratio) predicts conversion from mild cognitive impairment to Alzheimer's disease. J Neural Transm. 2012;119:815–819. doi: 10.1007/s00702-012-0807-x. [DOI] [PubMed] [Google Scholar]
- 161.Smith RP, Broze GJ., Jr Characterization of platelet-releasable forms of beta-amyloid precursor proteins: the effect of thrombin. Blood. 1992;80:2252–2260. [PubMed] [Google Scholar]
- 162.Yokota M, Tatsumi N, Tsuda I, Nishioka T, Takubo T. CTAD as a universal anticoagulant. J Autom Methods Manag Chem. 2003;25:17–20. doi: 10.1155/S1463924603000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pensalfini A, Zampagni M, Liguri G, Becatti M, Evangelisti E, Fiorillo C, Bagnoli S, Cellini E, Nacmias B, Sorbi S, Cecchi C. Membrane cholesterol enrichment prevents Abeta-induced oxidative stress in Alzheimer's fibroblasts. Neurobiol Aging. 2011;32:210–222. doi: 10.1016/j.neurobiolaging.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 164.Wood WG, Schroeder F, Avdulov NA, Chochina SV, Igbavboa U. Recent advances in brain cholesterol dynamics: transport, domains, and Alzheimer's disease. Lipids. 1999;34:225–234. doi: 10.1007/s11745-999-0357-9. [DOI] [PubMed] [Google Scholar]
- 165.Yip CM, Elton EA, Darabie AA, Morrison MR, McLaurin J. Cholesterol, a modulator of membrane-associated Abeta-fibrillogenesis and neurotoxicity. J Mol Biol. 2001;311:723–734. doi: 10.1006/jmbi.2001.4881. [DOI] [PubMed] [Google Scholar]
- 166.Eckert GP, Cairns NJ, Maras A, Gattaz WF, Muller WE. Cholesterol modulates the membrane-disordering effects of beta-amyloid peptides in the hippocampus: specific changes in Alzheimer's disease. Dement Geriatr Cogn Disord. 2000;11:181–186. doi: 10.1159/000017234. [DOI] [PubMed] [Google Scholar]
- 167.Biondi E. Statin-like drugs for the treatment of brain cholesterol loss in Alzheimer's disease. Curr Drug Saf. 2007;2:173–176. doi: 10.2174/157488607781668927. [DOI] [PubMed] [Google Scholar]
- 168.Liu WW, Todd S, Coulson DT, Irvine GB, Passmore AP, McGuinness B, McConville M, Craig D, Johnston JA. A novel reciprocal and biphasic relationship between membrane cholesterol and beta-secretase activity in SH-SY5Y cells and in human platelets. J Neurochem. 2009;108:341–349. doi: 10.1111/j.1471-4159.2008.05753.x. [DOI] [PubMed] [Google Scholar]
- 169.Fernandes MA, Proenca MT, Nogueira AJ, Oliveira LM, Santiago B, Santana I, Oliveira CR. Effects of apolipoprotein E genotype on blood lipid composition and membrane platelet fluidity in Alzheimer's disease. Biochim Biophys Acta. 1999;1454:89–96. doi: 10.1016/s0925-4439(99)00030-7. [DOI] [PubMed] [Google Scholar]
- 170.Zubenko GS, Cohen BM, Reynolds CF, 3rd, Boller F, Malinakova I, Keefe N. Platelet membrane fluidity in Alzheimer's disease and major depression. Am J Psychiatry. 1987;144:860–868. doi: 10.1176/ajp.144.7.860. [DOI] [PubMed] [Google Scholar]
- 171.Hajimohammadreza I, Brammer MJ, Eagger S, Burns A, Levy R. Platelet and erythrocyte membrane changes in Alzheimer's disease. Biochim Biophys Acta. 1990;1025:208–214. doi: 10.1016/0005-2736(90)90099-a. [DOI] [PubMed] [Google Scholar]
- 172.Piletz JE, Sarasua M, Whitehouse P, Chotani M. Intracellular membranes are more fluid in platelets of Alzheimer's disease patients. Neurobiol Aging. 1991;12:401–406. doi: 10.1016/0197-4580(91)90064-q. [DOI] [PubMed] [Google Scholar]
- 173.Wood GW, Eckert GP, Igbavboa U, Muller WE. Amyloid beta-protein interactions with membranes and cholesterol: causes or casualties of Alzheimer's disease. Biochim Biophys Acta. 2003;1610:281–290. doi: 10.1016/s0005-2736(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 174.Rabini RA, Vignini A, Martarelli D, Nanetti L, Salvolini E, Rizzo MR, Ragno E, Paolisso G, Franceschi C, Mazzanti L. Evidence for reduction of pro-atherosclerotic properties in platelets from healthy centenarians. Exp Gerontol. 2003;38:367–371. doi: 10.1016/s0531-5565(02)00268-1. [DOI] [PubMed] [Google Scholar]
- 175.Hattori N, Kitagawa K, Higashida T, Yagyu K, Shimohama S, Wataya T, Perry G, Smith MA, Inagaki C. CI-ATPase and Na+/K(+)-ATPase activities in Alzheimer's disease brains. Neurosci Lett. 1998;254:141–144. doi: 10.1016/s0304-3940(98)00654-5. [DOI] [PubMed] [Google Scholar]
- 176.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 177.Six DA, Dennis EA. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 178.Farooqui AA, Yang HC, Horrocks L. Involvement of phospholipase A2 in neurodegeneration. Neurochem Int. 1997;30:517–522. doi: 10.1016/s0197-0186(96)00122-2. [DOI] [PubMed] [Google Scholar]
- 179.Fujita S, Ikegaya Y, Nishiyama N, Matsuki N. Ca2 + −independent phospholipase A2 inhibitor impairs spatial memory of mice. Jpn J Pharmacol. 2000;83:277–278. doi: 10.1254/jjp.83.277. [DOI] [PubMed] [Google Scholar]
- 180.Schaeffer EL, Skaf HD, Novaes BA, da Silva ER, Martins BA, Joaquim HD, Gattaz WF. Inhibition of phospholipase A(2) in rat brain modifies different membrane fluidity parameters in opposite ways. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1612–1617. doi: 10.1016/j.pnpbp.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 181.Forlenza OV, Wacker P, Nunes PV, Yacubian J, Castro CC, Otaduy MC, Gattaz WF. Reduced phospholipid breakdown in Alzheimer's brains: a 31P spectroscopy study. Psychopharmacology (Berl) 2005;180:359–365. doi: 10.1007/s00213-005-2168-8. [DOI] [PubMed] [Google Scholar]
- 182.Gattaz WF, Maras A, Cairns NJ, Levy R, Forstl H. Decreased phospholipase A2 activity in Alzheimer brains. Biol Psychiatry. 1995;37:13–17. doi: 10.1016/0006-3223(94)00123-K. [DOI] [PubMed] [Google Scholar]
- 183.Emmerling MR, Moore CJ, Doyle PD, Carroll RT, Davis RE. Phospholipase A2 activation influences the processing and secretion of the amyloid precursor protein. Biochem Biophys Res Commun. 1993;197:292–297. doi: 10.1006/bbrc.1993.2474. [DOI] [PubMed] [Google Scholar]
- 184.Yang X, Sheng W, He Y, Cui J, Haidekker MA, Sun GY, Lee JC. Secretory phospholipase A2 type III enhances alpha-secretase-dependent amyloid precursor protein processing through alterations in membrane fluidity. J Lipid Res. 2010;51:957–966. doi: 10.1194/jlr.M002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Talib LL, Yassuda MS, Diniz BS, Forlenza OV, Gattaz WF. Cognitive training increases platelet PLA2 activity in healthy elderly subjects. Prostaglandins Leukot Essent Fatty Acids. 2008;78:265–269. doi: 10.1016/j.plefa.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 186.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 187.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 189.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 190.Niblock M, Gallo JM. Tau alternative splicing in familial and sporadic tauopathies. Biochem Soc Trans. 2012;40:677–680. doi: 10.1042/BST20120091. [DOI] [PubMed] [Google Scholar]
- 191.Gache Y, Guilleminot J, Ricolfi F, Theiss G, Nunez J. A tau-related protein of 130 kDa is present in Alzheimer brain. J Neurochem. 1992;58:2005–2010. doi: 10.1111/j.1471-4159.1992.tb10940.x. [DOI] [PubMed] [Google Scholar]
- 192.Hattori H, Matsumoto M, Iwai K, Tsuchiya H, Miyauchi E, Takasaki M, Kamino K, Munehira J, Kimura Y, Kawanishi K, Hoshino T, Murai H, Ogata H, Maruyama H, Yoshida H. The tau protein of oral epithelium increases in Alzheimer's disease. J Gerontol A Biol Sci Med Sci. 2002;57:M64–M70. doi: 10.1093/gerona/57.1.m64. [DOI] [PubMed] [Google Scholar]
- 193.Nagao SI, Kumamoto T, Masuda T, Ueyama H, Toyoshima I, Tsuda T. Tau expression in denervated rat muscles. Muscle Nerve. 1999;22:61–70. doi: 10.1002/(sici)1097-4598(199901)22:1<61::aid-mus10>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 194.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Elyaman W, Terro F, Wong NS, Hugon J. In vivo activation and nuclear translocation of phosphorylated glycogen synthase kinase-3beta in neuronal apoptosis: links to tau phosphorylation. Eur J Neurosci. 2002;15:651–660. doi: 10.1046/j.1460-9568.2002.01899.x. [DOI] [PubMed] [Google Scholar]
- 196.Gandy JC, Melendez-Ferro M, Bijur GN, Van LF, Roche JK, Lechat B, Devijver H, Demedts D, Perez-Costas E, Roberts RC. Glycogen synthase kinase-3beta (GSK3beta) expression in a mouse model of Alzheimer's disease: a light and electron microscopy study. Synapse. 2013;67:313–327. doi: 10.1002/syn.21642. [DOI] [PubMed] [Google Scholar]
- 197.Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- 198.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 199.Hampel H, Ewers M, Burger K, Annas P, Mortberg A, Bogstedt A, Frolich L, Schroder J, Schonknecht P, Riepe MW, Kraft I, Gasser T, Leyhe T, Moller HJ, Kurz A, Basun H. Lithium trial in Alzheimer's disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922–931. [PubMed] [Google Scholar]
- 200.Nunes MA, Viel TA, Buck HS. Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer's disease. Curr Alzheimer Res. 2013;10:104–107. doi: 10.2174/1567205011310010014. [DOI] [PubMed] [Google Scholar]
- 201.Consensus report of the Working Group on "Molecular and biochemical markers of Alzheimer's disease". The Ronald and Nancy Reagan research institute of the Alzheimer's association and the national institute on aging working group. Neurobiol Aging. 1998;19:109–116. [PubMed] [Google Scholar]