

Abstract
The sea urchin larva is shaped by a calcite endoskeleton. That skeleton is built by 64 primary mesenchyme cells (PMCs) in Lytechinus variegatus. The PMCs originate as micromeres due to an unequal fourth cleavage in the embryo. Micromeres are specified in a well-described molecular sequence and enter the blastocoel at a precise time using a classic epithelial–mesenchymal transition. To make the skeleton, the PMCs receive signaling inputs from the overlying ectoderm, which provides positional information as well as control of the growth of initial skeletal tri-radiates. The patterning of the skeleton is the result both of autonomous inputs from PMCs, including production of proteins that are included in the skeletal matrix, and of non-autonomous dynamic information from the ectoderm. Here, we summarize the wealth of information known about how a PMC contributes to the skeletal structure. The larval skeleton is a model for understanding how information encoded in DNA is translated into a three-dimensional crystalline structure.
Introduction
More than 80% of extant modern sea urchin (euechinoid) species are indirect developers, producing a swimming larva that feeds and later metamorphoses into an adult. The larvae possess a calcite endoskeleton that is produced by a small number of cells called primary mesenchyme cells (PMCs). The skeletogenic cells originate as micromeres during unequal fourth and fifth cleavages at the vegetal pole of the embryo, allowing these cells to be followed throughout their unique developmental trajectory (Fig. 1A). Extensive research has established a comprehensive gene regulatory network (GRN) model for specification of this cell type (Fig. 1B) (Oliveri et al. 2002, 2003, 2008), and many studies have followed this cell type through production of the skeletal calcite crystal.
Fig. 1.
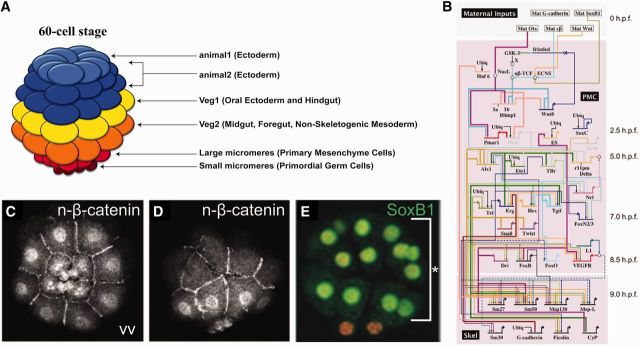
Early specification of micromere/PMC lineage. (A) 60-cell stage fate map. Cleavages are highly stereotypic. Ectoderm arises from tiers of cells at the animal pole, the endomesoderm is below that, and the PMC/micromere lineage originates at the vegetal pole. The unequal fifth cleavage places the primordial germ cells (small micromeres) below the large micromere/PMC lineage. (B) GRN showing specification of the micromere/PMC lineage (Saunders and McClay 2014). (C and D) Vegetal view (VV) and lateral views, respectively, of β-catenin protein staining in vegetal nuclei in the early embryo (Logan et al. 1999). (E) Co-staining for DAPI (vegetal) and SoxB1 (animal) (Kenny et al. 1999).
The PMCs display a wide range of behaviors prior to, and during, skeletogenesis and therefore demonstrate the versatility built into the genome as it instructs cells along their pathway toward differentiation. In order to make the skeleton, the PMCs undergo a controlled series of mitoses and ingress through a basement membrane to become mesenchyme cells via an epithelial–mesenchymal transition (EMT) (Fig. 2). After the EMT, they become motile and move through the blastocoel using lamellapodia and thick filopodia. To sense the spatial environment, they read signals using dynamic thin filopodia and respond to ectodermal growth factors for positional cues (Fig. 3). They organize and assemble in a ring structure in response to those growth factors. Just prior to initiation of skeletogenesis, the PMCs fuse to form a syncytium (Fig. 4). This allows them to collaborate in the deposition of the initial calcite crystal granual. The physical mold of the patterned syncytium continues to regulate subsequent calcium deposits as they transition from amorphous calcium carbonate (ACC) into a crystal of calcite. In this way, the PMCs control skeletal shape (Fig. 5E) and also deposit a variety of matrix proteins that are incorporated in the growing skeletal rods.
Fig. 2.

Control of PMC EMT. (A) GRN showing the TCFs inputs into the morphogenetic events necessary for ingression of PMCs (Saunders and McClay 2014). (B–B″) Staining for two PMC-specific antigens during ingression, demonstrating that these proteins are rapidly exocytosed during EMT. (B) MSP130 is localized to intercellular vesicles prior to ingression, NgCAM is not expressed at this time. (B′) Shortly after the PMCs have ingressed, both antigens are expressed at the cell surface and continue to be expressed. (B″) Migration of PMCs inside the blastocoel (Wu et al. 2007). (C–C′) Time-lapse images of embryos expressing a GFP-cadherin construct demonstrating that cadherin is endocytosed rapidly following EMT. In C, arrow points to cadherin on the membrane of a PMC that has just ingressed into the blastocoel. In C′, the arrow points to the same cell surface, showing that cadherin-GFP signal has been lost form this domain and now has increased in vesicles inside the cell (Miller and McClay 1997). (D) PMCs breaching the basement membrane during EMT. PMCs are stained with a cell-specific marker MSP130, and the basement membrane is stained with laminin. Arrowheads indicate a break in the membrane where PMCs enter the blastocoel (Saunders and McClay 2014). (E) Maximum projection of laminin-staining alone, at the stage shown in D (Saunders and McClay 2014). A vegetal hole has formed and is marked by a dashed line. (F) Diagram of a micromere’s behavior during ingression (Katow and Solursh 1981).
Fig. 3.
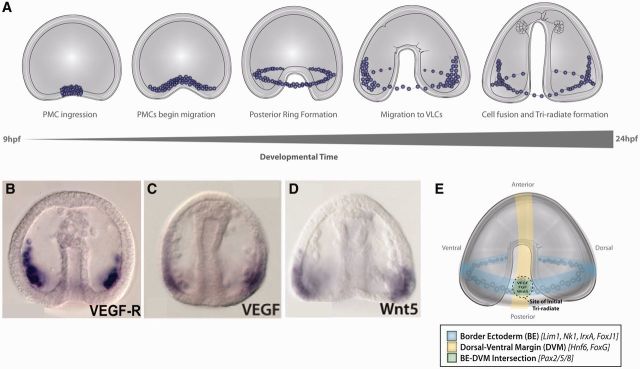
PMCs migrate inside the blastocoel and form the ventrolateral clusters. (A) Diagram of the migration of PMCs inside the blastocoel during gastrulation. After PMCs complete EMT, they migrate along the sides of the blastocoel and within about 2 h arrange themselves into a ventral ring, fuse, and then many of the PMCs coalesce into ventrolateral clusters (VLCs) underneath a patch of ectoderm corresponding to the site where the BE intersects with the DVM. Within the ventrolateral clusters, the rudiment of a tri-radiate spicule is formed. (B) PMCs express the VEGFR. (C) VEGF is expressed in ectoderm at the junction of the BE and DVM. (D) Wnt5 is also expressed in the ectodermal BE/DVM junction. (E) Diagram showing the intersection of the BE and the DVM. Domain-specific markers for these domains are indicated in the legend. B–D are taken from McIntyre et al. (2013). E is taken from McIntyre et al. (2014).
Fig. 4.
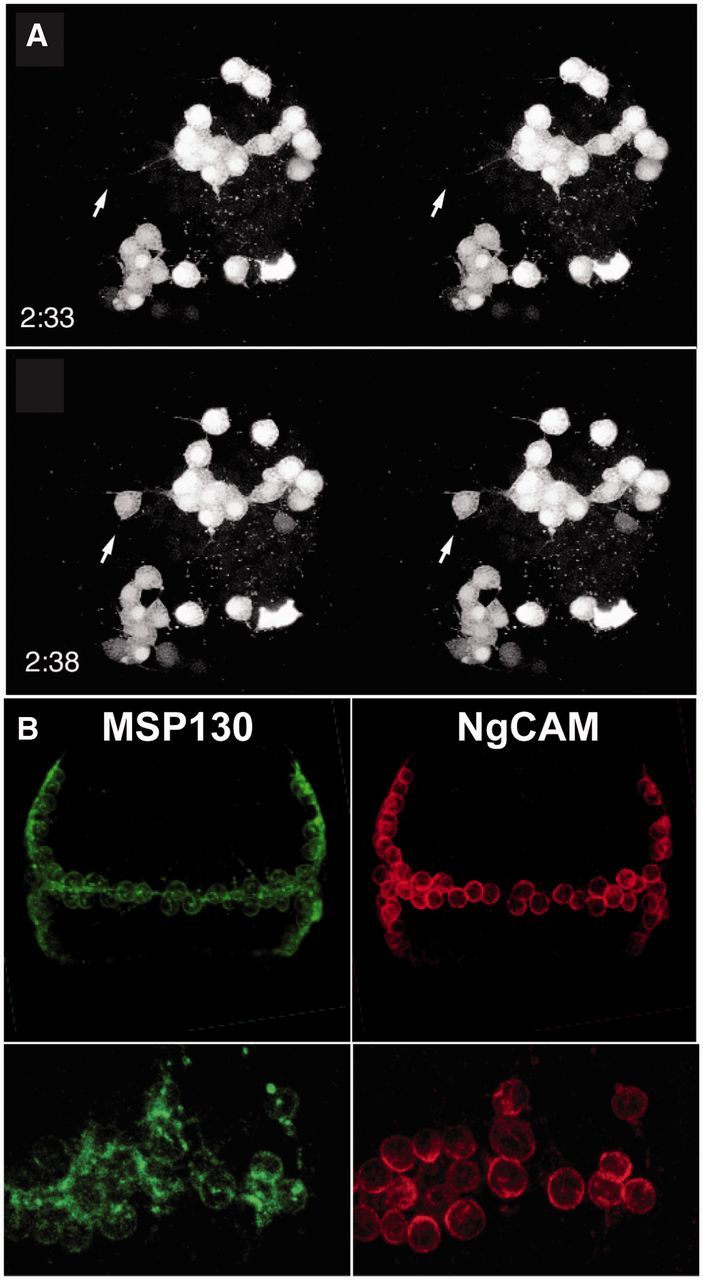
PMCs undergo programmed cell fusion and display membrane microdomains in the syncytium. (A) Stero images of a time-lapse recording of dye-coupled cells during the process of fusion. In the top panel, the arrow points to an unlabeled cell body near a filopodium from a nearby cluster of PMCs. The bottom panel shows the arrow in the same position; 5 min later the dye from the neighboring cell cluster has filled this cell (Hodor and Ettensohn 1998). (B) Staining for MSP130 and NgCAM, two PMC-specific membrane associated proteins. Top panel shows the entire PMC syncytial network at late gastrula, and the bottom panel shows magnified views of a portion of the syncytium. MSP130 is expressed rather evenly all over the PMC cell bodies and connecting membrane, whereas NgCAM is confined to the cell bodies only.
Fig. 5.

PMCs re-arrange themselves as the endoskeleton grows and branches. (A and B) Late gastrula-stage embryo showing the spacing of 64 PMCs in the skeletal rudiment (Hodor and Ettensohn 1998). (C and D) Pluteus stage of embryo showing the 64 PMCs spaced farther apart as the syncytium accommodates the larger, more complex skeleton (Hodor and Ettensohn 1998). (E) Diagram of the growth and branching of the original tri-radiate skeletal rudiment. Modified from Armstrong and McClay (1994). The tri-radiate skeleton begins to form between a tetrad of cells in the middle of the ventrolateral cluster immediately beneath the BE–DVM intersection. Shortly thereafter, one of the branches (#3) of the tri-radiate branches again to produce the post-oral skeletal arm (#5) that grows toward the ventral side, and another rod (#4) that grows toward the dorsal side. Elements #4, #1, (that crosses toward the posterior midline), #2 (that runs anterior-from the initial tri-radiate parallel to the DVM), and #7 (that extends dorsally on the anterior side) provide the framework inside which the body of the pluteus differentiates. The anterior arms (#6) and the postoral arms (#5) along with the anterior–posterior body rod (#2) provide the “catch basin” for larval food that is then streamed into the mouth. Once skeletogenesis is underway, as rod #2, elongates anteriorly, parallel to the DVM, it reaches a site two-third of the way to the animal pole where it branches to extend the ventral arm (#6) that establishes the oral hood of the larva, and the dorsal rod (#7) that provides the anterior skeletal element of the body (Note: anterior is the site closest to the original animal pole whereas posterior is closest to the site of the blastopore). The branchpoint along the DVM is a second site of VEGF production (Duloquin et al. 2007), suggesting that VEGF is secreted and controls skeletal branching (Knapp et al. 2012). In each case, skeletal arms originate and extend from either the BE or from a location along the DVM, and rods encasing the body grow parallel to either the BE or the DVM. The one exception to this is the dorsal extension of rod #7 of the anterior body, suggesting that there is a region of ectoderm patterning that remains to be discovered. A recent publication on ectoderm specification suggests that another band of ectoderm may exist along this location, and if so, it is an excellent candidate for the patterning of band #7 (Li et al. 2014).
In Lytechinus variegatus, the species described here, the entire skeleton is built by 64 PMCs. Each PMC is capable of building any part of the skeleton: if any PMC in the blastocoel is moved to any position in the ring, it still contributes to patterning a normal skeleton. For this reason, each PMC can be considered equivalent. Diverse parts of the skeleton (branches, bends, rods, spikes, or baskets) are built by PMCs based on local interpretation of the spatial information they receive. They respond to produce a skeleton that is genotypically correct. How they accomplish that feat is the subject of this essay.
Because all PMCs are essentially identical, we describe the developmental sequence from the point of view of a single micromere/PMC; what it experiences, what it does, and how it works. Even within the syncytium, each nucleus operates semi-independently to instruct the cell in production of the local skeletal pattern. The story is remarkable in that a single cell type engages in an amazingly diverse array of functions in order to assemble the larval endoskeleton. As such, it serves as a model for the functional diversity of a single cell type.
Fourth cleavage to EMT
The first six to seven cleavages of the sea urchin embryo are highly stereotypic, so that each group of cells has a known ancestry (Fig. 1A; McClay 2011; Lyons et al. 2012). The micromere-PMC lineage is born with an unequal fourth cleavage in which the cleavage furrow at the vegetal pole of the embryo is offset to produce four macromeres and four micromeres with a 95–5% distribution of cytoplasm to the two daughter cells. This unequal distribution of cytoplasm contributes mechanistically to the initial specification of the micromeres as described below. The fourth cleavage is followed one cell division later by yet another unequal cleavage, this time with the large micromere-PMC lineage receiving more than 80% of the remaining cytoplasm of the micromere whereas the small micromere sister blastomeres receive 20% or less of the cytoplasm (Fig. 1C–E). The small micromeres divide only once more prior to the larval-feeding stage and make the primordial germ cells of the next generation in normal development (Pehrson and Cohen 1986; Yajima and Wessel 2011). The large micromere progeny of that second unequal cleavage become the PMCs. Most micromeres divide three more times prior to their EMT and then once again just following EMT to produce an average of 64 PMCs. Those 64 PMCs are the entire complement of cells that will build the initial four-armed skeleton of the pluteus larva because the PMCs do not divide again until after the larva begins to feed a day or so after the four-armed skeleton is built.
During the fourth cleavage, all maternal transcription factors (TCFs) are distributed throughout the cytoplasm. The maternal TCFs return to the nuclei only when assembly of the nuclear membrane is established in the daughter cells. As a consequence, Soxb1, a TCF produced maternally in abundance, is asymmetrically distributed to the nuclei of macromeres versus micromeres (more than 20-fold higher amount of protein in the nuclei of macromeres relative to those of micromeres) (Fig. 1E; Kenny et al. 1999). In addition, the micromeres activate the Wnt pathway due to asymmetrically localized pathway components (i.e., Dsh), and as a result, the micromeres are the first cells to accumulate nuclear β-catenin (Logan et al. 1999; Weitzel et al. 2004) (Fig. 1C, D). Additionally, a third TCF, Otx, becomes localized to nuclei of micromeres at the 16-cell stage (Chuang et al. 1996). As a consequence of these asymmetries, micromeres begin a specification trajectory that is unique in the embryo.
Specification of micromeres uses β-catenin and maternal Otx to activate a repressor called Pmar1 (Fig. 1B; Oliveri et al. 2002, 2003). Twenty minutes later, Pmar1 represses transcription of a TCF called HesC in the micromeres. Elsewhere in the embryo, HesC is activated because there is no Pmar1 to repress its expression, and as a result, HesC represses the micromere GRN in non-micromere cells. Soxb1, the protein that was unequally distributed between macromeres and micromeres, is abundant in nuclei everywhere but the micromeres and contributes to specification of macromeres and mesomeres (Fig. 1E; Kenny et al. 1999). Soxb1 is thought to compete with β-catenin for binding to the TCF, so in the micromeres, where Soxb1 is sparse, nuclear β-catenin is free to activate transcription through the binding of TCF. Ubiquitous activators in the micromeres begin specification of this unique cell type in the absence of HesC expression (Fig. 2). Maternal Ets1 contributes to activation of Alx1 and Tbr. These three TCFs then initiate expression of a cascade of 10 TCFs that specify the micromeres (Fig. 2).
Included in the micromere specification sequence is the expression of Tel, Erg, Hex, and TGIF that act in a feed-forward loop, thereby assuring that micromere development moves forward and relinquishes the earlier requirement for Pmar1 repression of HesC. In fact, Pmar1 is active for only a few hours until the feed-forward regulatory loop is established. At that point, the fate of the micromeres is established, and they are committed to production of skeletogenic cells. Unlike other cells of the embryo, they are unable to reprogram beyond this point.
The micromeres differentiate into PMCs beginning at about 9 h after fertilization (at 23°C). To prepare for that event, each early micromere divides three times, expresses a suite of 10 TCFs, and initiates transcription of a number of effector genes that will be used in the initial differentiated state. A number of those differentiation genes are expressed prior to EMT by 1–2 h. As an example, MSP130, a membrane glycoprotein on PMCs, is transcribed, beginning 2 h prior to EMT (Fig. 2F). Its protein product is collected into secretory vesicles and stored until EMT. Glycosylation of the stored MSP130 is completed just prior to release of the vesicles to the surface of the PMCs. The appearance of MSP130 on the membranes of the PMCs provides a marker to show how dramatically these cells change in a 45-min period (Fig. 2F). At EMT, the PMCs lose adhesiveness to the epithelium of the blastula (Fink and McClay 1985), become motile, move basally, breach the basement membrane (Fig. 2B, C), and move to the inside of the blastocoel (Fig. 2F).
Thus, from the fourth cleavage at 2.5 hpf (hours post-fertilization), the micromeres employ an asymmetric cleavage mechanism to inherit less than 5% of the cytoplasm of the egg and, therefore, the asymmetrical distribution of proteins necessary for specification. They become specified, part of which generates a feed-forward regulatory mechanism for the GRN, and they generate a number of sub-circuits for differentiation and for the different components of the EMT. Finally, they invade the blastocoel to become the PMCs that go on to build the skeleton.
EMT to tri-radiate skeleton
Thirty-two PMCs ingress into the blastocoel beginning at 9 hpf. Three TCFs control de-adhesion (Alx1, Snail, and Twist), which occurs rather late in the EMT sequence (Fig. 2A, D). When that de-adhesion occurs, each PMC releases itself from the adherens junction and initiates a rapid endocytosis of the cadherin on the membrane’s surface (Fig. 2E). Once endocytosed, the cadherin is quickly broken down in lysosomes (Miller and McClay 1997; Wu and McClay 2007; Wu et al. 2008; Saunders and McClay 2014). Another component of the EMT is motility, which, along with change in the polarity and shape of the cell, is responsible for movement of the cell out of the epithelium and into the blastocoel. When these EMT properties are quantified after perturbations of each single TCF, a different set of TCFs are found to control the directional movement of the PMCs into the blastocoel (Hex, FoxN2/3, FoxO, Tel, Erg, TGIF, and FoxB). Each of these TCFs, when perturbed, causes a slightly different PMC ingression behavior defect, but in each knockdown, the outcome is a deficit in the ability of the PMC to move directionally into the blastocoel (Fig. 3).
The third component of the EMT is passage through the basement membrane. At the time the PMCs invade the blastocoel, laminin staining reveals a hole in the basement membrane the size of the patch of ingressing PMCs (Fig. 2B, C). Knockdown of three TCFs eliminates the hole (Tbr, Dri, and Hex). These, and additional data, show that up to 12 TCFs operate in sub-circuits of the PMC GRN to control the EMT (Fig. 2D). The mechanism is coordinated, so that EMT occurs in a 45-min period, but those sub-circuits are somewhat independent of each other. For example, FoxN2/3 knockdown blocks the motility phase of EMT but does not block formation of the hole in the basement membrane. As a result, a hole appears, but the PMCs cannot take advantage of it since they cannot move without FoxN2/3 function (Saunders and McClay 2014).
Once a PMC enters the blastocoel, its behavior changes dramatically. At the time the cadherin is endocytosed, an entirely new set of glycoproteins is exocytosed, providing the PMC with the ability to adhere to the blastocoelar substrate (Fig. 2F). Also, in preparation for EMT, a number of genes are activated, and their protein products accumulate, so that at the time of ingression, the PMC is ready to assume its new migratory role. EMT itself is coincident with activation of a number of additional genes. For example, the PMCs activate the vegfr and fgfr genes at EMT (Duloquin et al. 2007). They also activate L1 (NgCAM), a glycoprotein that has been associated with adhesion in a number of systems (Fig. 2F). Once in the blastocoel, the PMCs begin migrating up the wall of the blastocoel (Fig. 3A; Malinda and Ettensohn 1994; Malinda et al. 1995; Peterson and McClay 2003). Their motile behavior uses lamellapodia and thick filopodia to transit up the blastocoelar wall, often beyond the equator (Fig. 3A). The PMCs then reverse direction and organize into a ring around the posterior floor of the blastocoel. Ring formation is now thought to be a consequence of VEGF–VEGFR recognition (Fig. 3B–E) (see below for the mechanism). During their migratory phase, the PMCs begin to extend long thin filopodia (Miller et al. 1995; McClay 1999). Observation of the thin (ca. 250 nm in diameter) filopodia under control and experimental conditions suggests that they are used as sensors for ectodermal cues. Consequently, as the PMCs initiate ring formation, each PMC can be seen extending a number of thin filopodia toward the substrate below it.
Initially, the distribution of VEGF from ectoderm is uniform around the ring (judging from the in situ pattern only). Later, production of VEGF becomes stronger at two lateral sites of ectoderm, and consequently, many of the PMCs, initially distributed equidistant from one another around the ring, begin converging beneath the two sites of higher ectodermal production of VEGF (Fig. 3B–E). As VEGF production increases at those two sites, it declines and disappears from the remainder of ectoderm. The PMCs respond to the localized secretion of VEGF, and a majority of them collect beneath the two lateral sites on the ectoderm. Those lateral sites are called the ventrolateral clusters of PMCs. As a test of the hypothesis that VEGF signaling is responsible for the formation of ventrolateral clusters, when VEGF or VEGFR is knocked down, formation of clusters does not occur (Duloquin et al. 2007; Adomako-Ankomah and Ettensohn 2013; McIntyre et al. 2013).
Coincident with formation of the ring and ventrolateral clusters, the 64 PMCs form a syncytium (Fig. 4A). Although the molecular details of how cell–cell fusions occur are not known, there are several observations about the process worth noting. When formation of the ring is underway, about 2 h past EMT, cellular extensions of the PMCs make contact with one another, and these contacts often initiate a fusion (Hodor and Ettensohn 1998). By the time ring formation is complete and the lateral PMCs collect into the ventrolateral clusters, the entire complement of PMCs on each side of the embryo is contained within a single syncytium as demonstrated by dye-transfer experiments (Fig. 4A; Hodor and Ettensohn 1998). Despite being part of a syncytium, some proteins within the syncytium are distributed regionally, and the pattern of the skeleton emerges locally, as will be seen below (Fig. 4B). The syncytium allows the skeleton to form as one crystal, since it grows within one continuous space provided by the folded cellular extensions between the nuclei of the syncytium. The membranes of the cellular extensions wrap entirely around the growing skeleton, thus, it is possible that the space in which the skeleton grows is extracellular, but the extensions of the syncytium surround the growing skeleton in a way that is reminiscent of the way a Schwann cell ensheaths a neuron.
Skeletogenesis begins in the two ventrolateral clusters (Figs. 3A and 5). The first sign of a skeleton is seen as a small birefringent granule within each cluster at mid gastrula to late gastrula. That granule rapidly grows into a tri-radiate spicule, and then each arm grows in a stereotypic pattern to eventually form the skeleton of the pluteus (Fig. 5).
Inputs of ectodermal patterning into skeletogenesis
Although the skeleton is produced by the PMCs, they must obtain positional information so the pattern of the skeleton can be oriented correctly with respect to the remainder of the embryo. That patterning is a consequence of signaling from the ectoderm. To understand how skeletal patterning works, it is necessary to understand how the ectoderm contributes to that elaborate pattern.
Recent work established that patterning information for skeletogenesis comes not from the entire ectoderm, but from two bands of ectoderm that provide the necessary patterning signals for production of the skeleton (Fig. 3E; McIntyre et al. 2013, 2014). The first band is the “dorsal–ventral margin” (DVM) of ectoderm, established by Nodal and BMP signaling, which separates the dorsal ectoderm and ventral ectoderm and later becomes part of the ciliary band (yellow band modeled in Fig. 3E). The second is the “border ectoderm” (BE), a band about four cells wide immediately adjacent to the endoderm (blue band in Fig. 3E). It surrounds the embryo just below the equator prior to gastrulation and is established by Wnt5 signaling from the endoderm. The BE and DVM are orthogonal to each other and intersect at two sites, one on the right side and the other on the left side (Fig. 3E). Those intersections are the sites where the production of VEGF is highest, leading to formation of the ventrolateral clusters of PMCs just beneath. As skeletogenesis continues, these two bands of ectoderm continue to provide inputs to patterning, so it is worthwhile taking time to understand how the DVM and BE form.
The DVM is largely a consequence of Nodal and BMP signaling, along with their inhibitors Lefty and Chordin (Duboc et al. 2004, 2008; Bradham et al. 2009; Saudemont et al. 2010). Each of these four proteins is synthesized in the ventral ectoderm, and Nodal expression is required for the other three. Nodal is synthesized first in response to a cascade of earlier signals that sets up a dorsal–ventral asymmetry (Bradham and McClay 2006; Coffman et al. 2014). Once activated, Nodal signaling feeds back to neighboring cells of the ventral ectoderm in a community effect to continue its own expression. Nodal activates expression of its own inhibitor, Lefty, and expression of BMP and Chordin (Duboc et al. 2004; Duboc and Lepage 2008; Bradham et al. 2009). Lefty is modeled to diffuse more readily than Nodal and thus limits the extent of Nodal signaling (Range et al. 2007; Duboc et al. 2008). BMP is produced by the ventral ectoderm and diffuses to the dorsal ectoderm where it activates a group of TCFs that specify the dorsal ectoderm. Chordin, a BMP inhibitor, is also expressed in the ventral ectoderm where it prevents BMP from signaling. Chordin likely serves as a chaperone that carries BMP to the dorsal half of the embryo before being released, allowing BMP to signal (Shimmi et al. 2005; Bradham et al. 2009). As a consequence of these two signals, distinct domains of ventral and dorsal ectoderm become specified. At the boundary between these domains, the DVM cells become uniquely specified. For example, this region expresses OneCut (Hnf6) and is thought to somehow exclude both Nodal and BMP signaling, but an understanding of its specification is incomplete. Those cells provide patterning information for a portion of the skeleton; they will become the ciliary band of the larva, and the band will also harbor neurons and neurites of the larval nervous system (Bradham et al. 2009; Li et al. 2014).
The BE was recently discovered as a distinct territory of ectodermal cells. It is uniquely activated by short-range Wnt5 signaling from the adjacent endoderm prior to the mesenchyme blastula stage (McIntyre et al. 2013). Several TCFs in the BE are activated by Wnt5 signaling. Ectodermal Nodal and BMP signaling restricts the expression of the BE TCFs, so that some are expressed in the dorsal half of the BE due to Nodal repression in the ventral half, and others are restricted in their expression to the ventral half by BMP signaling (McIntyre et al. 2013). The DVM intersection with the BE on either side is the site where VEGF and FGF are expressed by the ectoderm (Fig. 3E). As discussed below, the DVM, the BE, and their intersection are important regions of ectoderm for patterning the skeleton.
Skeletogenesis
The synthesis and assembly of the calcite crystalline skeleton is a remarkable feat of construction performed by the 64 PMCs. This section covers several known components of that complex process. As mentioned above, the intersection between the DVM and the BE is the location of production of VEGF and FGF, and that is the site where skeletogenesis begins (Rottinger et al. 2008; McIntyre et al. 2013).
Although the ectoderm provides the spatial signaling that positions the skeleton in space, it has also been known for some time that each PMC contributes to the detailed structure of the local piece of skeleton. Perhaps the best experimental evidence for this is the collaboration between PMCs of two species in the same embryo (Armstrong and McClay 1994). PMCs of L. variegatus produce simple smooth rods for their skeletons whereas the post-oral rod of Tripneustes esculentus is fenestrated. If one adds a few T. esculentus PMCs to a L. variegatus blastocoel, the PMCs of the two species collaborate to form a syncytium and produce a skeleton. The informative aspect of this collaboration is seen when a T. esculentus PMC lands in the group of cells that produces the post-oral rod. For a short distance, that rod is fenestrated, coincident with the placement of the T. esculentus PMC, and on either side of that stretch, the rod is simple, in keeping with the patterning output of L. variegatus PMCs. This simple experiment illustrates several principles of skeletal patterning in larvae of sea urchins. First, there are autonomous patterning elements provided by the PMCs themselves. Since each PMC can be moved to any location in the blastocoel prior to formation of the syncytium and still contribute a correctly patterned piece of the skeleton, the local pattern produced by each PMC must be based on instructions received locally by that PMC. At least some of those instructions come from the ectodermal patterning cells as described above. The possibility exists (without current evidence, however) that additional instructions originate from neighboring PMCs. After reception of positional information, each PMC patterns a local region of skeletal growth. The changes in pattern revealed by the two-species experiment show that each PMC is responsible for the portion of the skeleton up to a line roughly midway between that PMC and its neighbor. The question then becomes how patterning molds the skeletal element that results.
To understand how PMCs use patterning information to build the skeleton, it is necessary to understand how biomineralization works. To form the biomineral, PMCs accumulate ACC into intracellular vesicles and secrete the ACC as granules into the syncytial space provided by the cellular extensions (Wilt et al. 2008). The ACC then contributes to the growth of the calcite crystal (Gong et al. 2012). Along with accumulating an intracellular concentration of calcium carbonate, the PMCs must somehow keep the ACC from crystallizing prematurely. It has been proposed that it is the proteins produced by the PMCs and incorporated into the crystalline matrix that provide that control (Wilt et al. 2008; Gong et al. 2012). In a recent analysis in which it was possible to distinguish ACC from calcite in high resolution, investigators observed patches of ACC deposited early and then transformed into calcite as developmental time progressed, but even in advanced skeletons, there were patches of ACC that coated the outside of the spicule’s calcite (Gong et al. 2012).
The skeleton grows in length and in girth. Cross-sections of the skeleton show that the calcite is deposited sequentially since growth rings can be discerned. At the same time, however, when the ACC transitions to calcite, it joins and becomes part of the single crystal. It has been suggested that the matrix proteins confer a variety of properties to the ACC and the growing crystal. Since more than 50 proteins are known to be associated with the skeleton, and little is known about their specific function (Livingston et al. 2006; Rafiq et al. 2012). There are many different patterning functions that could be controlled by variation in the kind of protein, or its concentration, produced by any one of the PMCs. The signaling input received by each PMC dictates the complexity of matrix protein output as a means of controlling local variation in skeletal shape. At present, however, much remains to be learned about the role of matrix proteins before the process can be modeled in any detail.
The template produced by the syncytial folds of PMCs constitutes the mold in which the skeleton grows. Figure 5E shows the sequence of skeletal assembly. During this phase of skeletogenesis, from initiation of the tri-radiate spicule to the four-armed pluteus stage, only 64 PMCs are present. As crystals begin to grow, most PMCs are in the two ventrolateral clusters. To form the four arms and the entire anterior skeleton, the PMCs must migrate from the ring to their final destination. The post-oral arm on each side has about seven PMCs (Fig. 5D). Those PMCs begin their journey into the post-oral arm from the ventrolateral cluster, and as the rod elongates, one PMC after the other drops off the advancing tip of the arm. When the arm reaches an intermediate length, most of the cells have dropped off the advancing front. In Fig. 5D, the two post-oral arms are of different lengths, both have the same number of PMCs, yet the PMCs on the shorter arm are closer together. As the arm continues to grow, the distance between adjacent PMCs along the arm also grows. That means the PMC nuclei redistribute themselves, and, at the same time, the syncytium locally associated with those nuclei contributes to the growing girth of the skeletal arm. The post-oral arm (#5 in Fig. 5E) grows outward from the body, beginning at a point source (the BE–DVM intersection), and, unlike the body rods, grows parallel to the ectoderm and stretches it as it grows outward. That local source for outgrowth of the arm likely provides a signal telling the PMCs to produce the arm at that particular location. As the arm grows, PMCs remaining at the growing tip express FGF, and it is likely that the FGF produced by the PMCs and the overlying ectoderm together establish a positive reinforcing signaling circuit that stimulates and coordinates outgrowth of the arm.
Many other details have accumulated through time-lapse analysis of skeletogenesis. Growth of the skeleton involves a diverse set of functions for patterning and for biomineralization. As more is understood about the details underlying growth, it will be possible to discern how evolution led to changes in skeletal patterns. With so many inputs into pattern, alteration in any one of them could lead to dramatic differences between two species. For example, if one process in the patterning was to simply be delayed it could have rather significant consequences.
Scaling
A fascinating component of skeletogenesis is scaling. A famous experiment by Driesch showed that half-sized embryos produce a half-sized skeleton with half the number of PMCs (Marcus 1979; Ettensohn and McClay 1986; Ettensohn and Malinda 1993). Furthermore, a quarter-sized skeleton is produced by an isolated blastomere from the four-celled stage (with 16 PMCs). If one adds PMCs to quarter-sized embryos, the skeletons do not get bigger (Ettensohn and McClay 1986; Ettensohn 1990; Ettensohn and Sweet 2000). We now know the reason for this is that the ectoderm dictates the general shape and size of the skeleton. Sixty-four PMCs in a half-sized ectoderm (instead of 32) still make a half-sized skeleton even though there are enough PMCs for a full-sized skeleton. Thus, the total length of each rod is dictated by the ectoderm. In those cases in which extra PMCs were added, all participated in skeletogenesis, but the space between PMC nuclei decreased as progressively more PMCs were added. This suggests that within the PMC spacing framework, there is an ongoing conversation between adjacent PMCs in the syncytium. Indeed, rather than being programmed to make an element of a precise length, each PMC integrates information from ectoderm locally, to produce a portion of the skeleton that smoothly transitions to the neighboring PMC.
As stated in the beginning of this essay, much, if not all, of the information controlling normal development of the skeleton in the context of the sea urchin embryo is encoded in the DNA. As seen here, the readout involves signaling, timing, spacing, production, and secretion of matrix proteins, and synthesis and deposition of calcium carbonate, all done according to scale. And to think, the sea urchin larval skeleton is simple relative to the skeletons of many animals!
Funding
Support for this research was provided by NIH RO1 HD14483 and PO1 HD037105 [to D.R.M.].
References
- Adomako-Ankomah A, Ettensohn CA. Growth factor-mediated mesodermal cell guidance and skeletogenesis during sea urchin gastrulation. Development. 2013;140:4214–25. doi: 10.1242/dev.100479. [DOI] [PubMed] [Google Scholar]
- Armstrong N, McClay DR. Skeletal pattern is specified autonomously by the primary mesenchyme cells in sea urchin embryos. Dev Biol. 1994;162:329–38. doi: 10.1006/dbio.1994.1090. [DOI] [PubMed] [Google Scholar]
- Bradham CA, McClay DR. p38 MAPK is essential for secondary axis specification and patterning in sea urchin embryos. Development. 2006;133:21–32. doi: 10.1242/dev.02160. [DOI] [PubMed] [Google Scholar]
- Bradham CA, Oikonomou C, Kuhn A, Core AB, Modell JW, McClay DR, Poustka AJ. Chordin is required for neural but not axial development in sea urchin embryos. Dev Biol. 2009;328:221–33. doi: 10.1016/j.ydbio.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CK, Wikramanayake AH, Mao CA, Li X, Klein WH. Transient appearance of Strongylocentrotus purpuratus Otx in micromere nuclei: cytoplasmic retention of SpOtx possibly mediated through an alpha-actinin interaction. Dev Genet. 1996;19:231–7. doi: 10.1002/(SICI)1520-6408(1996)19:3<231::AID-DVG6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Coffman JA, Wessels A, Deschiffart C, Rydlizky K. Oral–aboral axis specification in the sea urchin embryo, IV: Hypoxia radializes embryos by preventing the initial spatialization of nodal activity. Dev Biol. 2014;386:302–7. doi: 10.1016/j.ydbio.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc V, Lepage T. A conserved role for the nodal signaling pathway in the establishment of dorso–ventral and left–right axes in deuterostomes. J Exp Zool B Mol Dev Evol. 2008;310:41–53. doi: 10.1002/jez.b.21121. [DOI] [PubMed] [Google Scholar]
- Duboc V, Lapraz F, Besnardeau L, Lepage T. Lefty acts as an essential modulator of Nodal activity during sea urchin oral–aboral axis formation. Dev Biol. 2008;320:49–59. doi: 10.1016/j.ydbio.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Duboc V, Rottinger E, Besnardeau L, Lepage T. Nodal and BMP2/4 signaling organizes the oral–aboral axis of the sea urchin embryo. Dev Cell. 2004;6:397–410. doi: 10.1016/s1534-5807(04)00056-5. [DOI] [PubMed] [Google Scholar]
- Duloquin L, Lhomond G, Gache C. Localized VEGF signaling from ectoderm to mesenchyme cells controls morphogenesis of the sea urchin embryo skeleton. Development. 2007;134:2293–302. doi: 10.1242/dev.005108. [DOI] [PubMed] [Google Scholar]
- Ettensohn CA. The regulation of primary mesenchyme cell patterning. Dev Biol. 1990;150:261–71. doi: 10.1016/0012-1606(90)90076-u. [DOI] [PubMed] [Google Scholar]
- Ettensohn CA, Malinda KM. Size regulation and morphogenesis: a cellular analysis of skeletogenesis in the sea urchin embryo. Development. 1993;119:155–67. doi: 10.1242/dev.119.1.155. [DOI] [PubMed] [Google Scholar]
- Ettensohn CA, McClay DR. The regulation of primary mesenchyme cell migration in the sea urchin embryo: transplantations of cells and latex beads. Dev Biol. 1986;117:380–91. doi: 10.1016/0012-1606(86)90307-6. [DOI] [PubMed] [Google Scholar]
- Ettensohn CA, Sweet HC. Patterning the early sea urchin embryo. Curr Top Dev Biol. 2000;50:1–44. doi: 10.1016/s0070-2153(00)50002-7. [DOI] [PubMed] [Google Scholar]
- Fink RD, McClay DR. Three cell recognition changes accompany the ingression of sea urchin primary mesenchyme cells. Dev Biol. 1985;107:66–74. doi: 10.1016/0012-1606(85)90376-8. [DOI] [PubMed] [Google Scholar]
- Gong YU, Killian CE, Olson IC, Appathurai NP, Amasino AL, Martin MC, Holt LJ, Wilt FH, Gilbert PU. Phase transitions in biogenic amorphous calcium carbonate. Proc Natl Acad Sci USA. 2012;109:6088–93. doi: 10.1073/pnas.1118085109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodor PG, Ettensohn CA. The dynamics and regulation of mesenchymal cell fusion in the sea urchin embryo. Dev Biol. 1998;199:111–24. doi: 10.1006/dbio.1998.8924. [DOI] [PubMed] [Google Scholar]
- Katow H, Solursh M. Ultrastructural and time-lapse studies of primary mesenchyme cell behavior in normal and sulfate-deprived sea urchin embryos. Exp Cell Res. 1981;136:233–45. doi: 10.1016/0014-4827(81)90001-x. [DOI] [PubMed] [Google Scholar]
- Kenny AP, Kozlowski D, Oleksyn DW, Angerer LM, Angerer RC. SpSoxB1, a maternally encoded transcription factor asymmetrically distributed among early sea urchin blastomeres. Development. 1999;126:5473–83. doi: 10.1242/dev.126.23.5473. [DOI] [PubMed] [Google Scholar]
- Knapp RT, Wu CH, Mobilia KC, Joester D. Recombinant sea urchin vascular endothelial growth factor directs single-crystal growth and branching in vitro. J Am Chem Soc. 2012;134:17908–11. doi: 10.1021/ja309024b. [DOI] [PubMed] [Google Scholar]
- Li E, Cui M, Peter IS, Davidson EH. Encoding regulatory state boundaries in the pregastrular oral ectoderm of the sea urchin embryo. Proc Natl Acad Sci USA. 2014;111:E906–13. doi: 10.1073/pnas.1323105111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston BT, Killian CE, Wilt F, Cameron A, Landrum MJ, Ermolaeva O, Sapojnikov V, Maglott DR, Buchanan AM, Ettensohn CA. A genome-wide analysis of biomineralization-related proteins in the sea urchin Strongylocentrotus purpuratus. Dev Biol. 2006;300:335–48. doi: 10.1016/j.ydbio.2006.07.047. [DOI] [PubMed] [Google Scholar]
- Logan CY, Miller JR, Ferkowicz MJ, McClay DR. Nuclear beta-catenin is required to specify vegetal cell fates in the sea urchin embryo. Development. 1999;126:345–57. doi: 10.1242/dev.126.2.345. [DOI] [PubMed] [Google Scholar]
- Lyons DC, Kaltenbach SL, McClay DR. Morphogenesis in sea urchin embryos: linking cellular events to gene regulatory network states. Wiley Interdiscip Rev Dev Biol. 2012;1:231–52. doi: 10.1002/wdev.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinda KM, Ettensohn CA. Primary mesenchyme cell migration in the sea urchin embryo: distribution of directional cues. Dev Biol. 1994;164:562–78. doi: 10.1006/dbio.1994.1224. [DOI] [PubMed] [Google Scholar]
- Malinda KM, Fisher GW, Ettensohn CA. Four-dimensional microscopic analysis of the filopodial behavior of primary mesenchyme cells during gastrulation in the sea urchin embryo. Dev Biol. 1995;172:552–66. doi: 10.1006/dbio.1995.8044. [DOI] [PubMed] [Google Scholar]
- Marcus NH. Developmental aberrations associated with twinning in laboratory-reared sea urchins. Dev Biol. 1979;70:274–7. doi: 10.1016/0012-1606(79)90025-3. [DOI] [PubMed] [Google Scholar]
- McClay DR. The role of thin filopodia in motility and morphogenesis. Exp Cell Res. 1999;253:296–301. doi: 10.1006/excr.1999.4723. [DOI] [PubMed] [Google Scholar]
- McClay DR. Evolutionary crossroads in developmental biology: sea urchins. Development. 2011;138:2639–48. doi: 10.1242/dev.048967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre DC, Lyons DC, Martik ML, McClay DR. Branching out: origins of the sea urchin larval skeleton in development and evolution. Genesis. 2014;52:173–85. doi: 10.1002/dvg.22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre DC, Seay NW, Croce JC, McClay DR. Short-range Wnt5 signaling initiates specification of sea urchin posterior ectoderm. Development. 2013;140:4881–9. doi: 10.1242/dev.095844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, McClay DR. Characterization of the role of cadherin in regulating cell adhesion during sea urchin development. Dev Biol. 1997;192:323–39. doi: 10.1006/dbio.1997.8740. [DOI] [PubMed] [Google Scholar]
- Miller J, Fraser SE, McClay D. Dynamics of thin filopodia during sea urchin gastrulation. Development. 1995;121:2501–11. doi: 10.1242/dev.121.8.2501. [DOI] [PubMed] [Google Scholar]
- Oliveri P, Carrick DM, Davidson EH. A regulatory gene network that directs micromere specification in the sea urchin embryo. Dev Biol. 2002;246:209–28. doi: 10.1006/dbio.2002.0627. [DOI] [PubMed] [Google Scholar]
- Oliveri P, Davidson EH, McClay DR. Activation of pmar1 controls specification of micromeres in the sea urchin embryo. Dev Biol. 2003;258:32–43. doi: 10.1016/s0012-1606(03)00108-8. [DOI] [PubMed] [Google Scholar]
- Oliveri P, Tu Q, Davidson EH. Global regulatory logic for specification of an embryonic cell lineage. Proc Natl Acad Sci USA. 2008;105:5955–62. doi: 10.1073/pnas.0711220105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrson JR, Cohen LH. The fate of the small micromeres in sea urchin development. Dev Biol. 1986;113:522–6. doi: 10.1016/0012-1606(86)90188-0. [DOI] [PubMed] [Google Scholar]
- Peterson RE, McClay DR. Primary mesenchyme cell patterning during the early stages following ingression. Dev Biol. 2003;254:68–78. doi: 10.1016/s0012-1606(02)00025-8. [DOI] [PubMed] [Google Scholar]
- Rafiq K, Cheers MS, Ettensohn CA. The genomic regulatory control of skeletal morphogenesis in the sea urchin. Development. 2012;139:579–90. doi: 10.1242/dev.073049. [DOI] [PubMed] [Google Scholar]
- Range R, Lapraz F, Quirin M, Marro S, Besnardeau L, Lepage T. Cis-regulatory analysis of nodal and maternal control of dorsal–ventral axis formation by Univin, a TGF-beta related to Vg1. Development. 2007;134:3649–64. doi: 10.1242/dev.007799. [DOI] [PubMed] [Google Scholar]
- Rottinger E, Saudemont A, Duboc V, Besnardeau L, McClay D, Lepage T. FGF signals guide migration of mesenchymal cells, control skeletal morphogenesis [corrected] and regulate gastrulation during sea urchin development. Development. 2008;135:353–65. doi: 10.1242/dev.014282. [DOI] [PubMed] [Google Scholar]
- Saudemont A, Haillot E, Mekpoh F, Bessodes N, Quirin M, Lapraz F, Duboc V, Rottinger E, Range R, Oisel A, et al. Ancestral regulatory circuits governing ectoderm patterning downstream of Nodal and BMP2/4 revealed by gene regulatory network analysis in an echinoderm. PLoS Genet. 2010;6:e1001259. doi: 10.1371/journal.pgen.1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, McClay DR. Sub-circuits of a regulatory network control epithelial-mesenchymal transition. Development. 2014;141:1503. doi: 10.1242/dev.101436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimmi O, Umulis D, Othmer H, O’Connor MB. Facilitated transport of Dpp/Scw heterodimer by Sog/Tsg leads to robust patterning of the Drosophila blastoderm embryo. Cell. 2005;120:873–86. doi: 10.1016/j.cell.2005.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzel HE, Illies MR, Byrum CA, Xu R, Wikramanayake AH, Ettensohn CA. Differential stability of β-catenin along the animal–vegetal axis of the sea urchin embryo mediated by dishevelled. Development. 2004;131:2947–56. doi: 10.1242/dev.01152. [DOI] [PubMed] [Google Scholar]
- Wilt FH, Killian CE, Hamilton P, Croker L. The dynamics of secretion during sea urchin embryonic skeleton formation. Exp Cell Res. 2008;314:1744–52. doi: 10.1016/j.yexcr.2008.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, McClay DR. The Snail repressor is required for PMC ingression in the sea urchin embryo. Development. 2007;134:1061–70. doi: 10.1242/dev.02805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Ferkowicz M, McClay DR. Ingression of primary mesenchyme cells of the sea urchin embryo: a precisely timed epithelial mesenchymal transition. Birth Defects Res C Embryo Today. 2007;81:241–52. doi: 10.1002/bdrc.20113. [DOI] [PubMed] [Google Scholar]
- Wu SY, Yang YP, McClay DR. Twist is an essential regulator of the skeletogenic gene regulatory network in the sea urchin embryo. Dev Biol. 2008;319:406–15. doi: 10.1016/j.ydbio.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima M, Wessel GM. Small micromeres contribute to the germline in the sea urchin. Development. 2011;138:237–43. doi: 10.1242/dev.054940. [DOI] [PMC free article] [PubMed] [Google Scholar]