

Abstract
Background
Two novel avipoxviruses from South Africa have been sequenced, one from a Feral Pigeon (Columba livia) (FeP2) and the other from an African penguin (Spheniscus demersus) (PEPV). We present a purpose-designed bioinformatics pipeline for analysis of next generation sequence data of avian poxviruses and compare the different avipoxviruses sequenced to date with specific emphasis on their evolution and gene content.
Results
The FeP2 (282 kbp) and PEPV (306 kbp) genomes encode 271 and 284 open reading frames respectively and are more closely related to one another (94.4%) than to either fowlpox virus (FWPV) (85.3% and 84.0% respectively) or Canarypox virus (CNPV) (62.0% and 63.4% respectively). Overall, FeP2, PEPV and FWPV have syntenic gene arrangements; however, major differences exist throughout their genomes. The most striking difference between FeP2 and the FWPV-like avipoxviruses is a large deletion of ~16 kbp from the central region of the genome of FeP2 deleting a cc-chemokine-like gene, two Variola virus B22R orthologues, an N1R/p28-like gene and a V-type Ig domain family gene. FeP2 and PEPV both encode orthologues of vaccinia virus C7L and Interleukin 10. PEPV contains a 77 amino acid long orthologue of Ubiquitin sharing 97% amino acid identity to human ubiquitin.
Conclusions
The genome sequences of FeP2 and PEPV have greatly added to the limited repository of genomic information available for the Avipoxvirus genus. In the comparison of FeP2 and PEPV to existing sequences, FWPV and CNPV, we have established insights into African avipoxvirus evolution. Our data supports the independent evolution of these South African avipoxviruses from a common ancestral virus to FWPV and CNPV.
Keywords: Avipoxvirus, Poxvirus, Pigeonpox, Penguinpox, Genome, Vectors
Background
Avipoxviruses have been shown to naturally infect 232 of the approximately 9,000 described species of both wild and domestic birds [1], yet relatively little is known about the host range and genetic diversity of these viruses. There are currently only ten defined- and three tentative avipoxvirus species [2] with species names assigned according to the bird species which they infect or from which they were isolated [1]. This current method of taxonomy and classification has been criticized because it has been found that multiple avian species can be infected by a single viral species and/or multiple viral species can infect a single avian species [3-6]. Phylogenetic relationships of avipoxviruses have been analyzed based on the gene corresponding to vaccinia virus (VACV) P4b (fpv167, VACV A3L) [3-8], indicating that all avipoxvirus strains cluster into 3 major clades, namely, A (Fowlpox (FWPV)-like), B (Canarypox (CNPV)-like) and C (Psittacine). Clade A can be further divided into seven subclades (A1-A7) and Clade B is comprised of three subclades (B1-B3) [6].
Thus far, the genomes of only three avipoxviruses have been published; a pathogenic American strain of fowlpox virus (FPVUS) [9], an attenuated European strain of fowlpox virus derived from European FWPV HP1 passaged over 400 times in chick embryo fibroblasts (FP9) [10], and a virulent canarypox virus isolated from a canary (CNPVATCC VR-111) [11]. Comparison of the CNPV and FWPV genomes reveals overall synteny in genome arrangement with similar genetic complements. They do however, exhibit significant differences in the terminal, variable regions as well as in three internal, variable regions which is in contrast to the conservation of central genomic regions in other Chordopoxviruses (ChPVs). These variable regions within the conserved central region of the genomes occur near the junctions of areas that were identified in FWPV as rearranged relative to other ChPVs and contain genes involved in virus-host interactions [9,11]. Avipoxviruses are considerably divergent from other ChPVs [9,11] and may constitute a separate subfamily within the Poxviridae family [11-13].
FP9, FPVUS and CNPV have large genomes of 258, 280 and 365 kbp encoding 238, 260 and 320 open reading frames (ORFs) respectively [9-11]. 90 genes have been found to be conserved amongst all ChPVs and to comprise the minimum essential genome [14]. The remainder of the genetic component of avian poxviruses is largely made up of immunomodulatory and host specific genes located in the terminal regions of the genome, that have allowed the viruses to take advantage of their unique hosts.
Laidlaw and Skinner [10] compared virulent (FPVUS (American) and HP1 (European)) and attenuated, tissue culture adapted (FP9 (European)) FWPV strains. Instead of the predicted changes in immunomodulators as the mechanism of attenuation, members of multigene families, especially those encoding ankyrin repeat proteins were seen to be the drivers of attenuation [10]. Ankyrin repeat proteins are thought to be involved in poxvirus host-range [15], and have previously been implicated in the attenuation of sheeppox virus [16]. Over 49% (137 genes) of the CNPV genome and 38% (89 genes) of the FWPV genome are comprised of genes belonging to gene families, 51 and 31 of which are ORFs containing ankyrin repeats respectively [9,11]. These and other differences in immunomodulatory gene families encoded by avipoxviruses may account for the extensive variability in virulence, host range and host interaction [11].
FPVUS and CNPV are significantly divergent with amino acid identity between ORF homologues (55-74%) being similar to that observed between different ChPV genera [11]. The CNPV genome contains 39 ORFs not present in FWPV, 29 of which encode unique, hypothetical proteins. CNPV contains ORFs coding for two additional proteins involved in nucleotide metabolism (thymidylate kinase and the small subunit of ribonucleotide reductase), TNFR (cnpv086), an IL-10 like protein (cnpv018), cellular ubiquitin (cnpv096), a protein tyrosine phosphatase (cnpv085), a thioredoxin binding protein (cnpv149), and two Rep like proteins (cnpv153 and cnpv200). FWPV contains 15 ORFs not present in CNPV, 13 of which encode hypothetical proteins. Homologues of fpv217 and fpv250 are notably absent from CNPV, and are similar to ORFs from insect baculoviruses, and from avian herpes- and adenoviruses respectively.
Members of the Columbidae family, consisting of pigeons and doves are susceptible to avipoxvirus infection and cases have been reported in several Columbiform species including the mourning dove [17], laughing dove [18], white-tailed laurel-pigeon [19], rock pigeon [3,5], feral pigeon [5] and others [1]. Due to the host species based approach to avipoxvirus taxonomy, poxviruses infecting the Columbiformes are designated as Pigeonpox viruses (PGPV).
This study focuses on two avipoxviruses, one isolated from a Feral Pigeon (Columba livia) from Port Elizabeth, South Africa (FeP2) in 2011 and the other from an African penguin (Spheniscus demersus) housed at SANCCOB, a seabird rehabilitation centre, following an oil spill off the coast of Cape Town, South Africa (PEPV) in 1988 [8,20]. These locations are 790 km apart.
Isolation and preliminary characterisation of PEPV has been previously described [8,20]. Poxvirus infections have not been observed in wild African penguins living in coastal waters around Cape Town (Personal communication with Nola Parsons and Tertius Gous, SANCCOB). Mosquito netting was introduced at SANCCOB in 2008 to protect housed penguins from contracting avian malaria and there has been no evidence of pox infections since then, suggesting that the initial infections were transmitted by biting insects. It is worth noting that many pigeons nest in the SANCCOB enclosure and could well have been the primary host for this virus, which could have been transmitted to penguins via biting insects. It is therefore possible that the African penguin is not the primary host of PEPV.
When grown on the chorioallantoic membranes of embryonated 10–11 day old chicken eggs, the pigeonpox virus FeP2 caused greater cell proliferation and immune cell infiltration as compared to PEPV, CNPV, FWPV and some of the other avipoxvirus isolates investigated in our laboratory [5]. Furthermore, based on phylogenetic analysis of four conserved genetic regions, FeP2 groups in clade A, subclade A3 and PEPV belongs to clade A, sub-clade A2. FPVUS and FP9 group in clade A1 and CNPV groups in clade B1 [5]. Poxviruses isolated from the African, Magellanic and Humboldt penguins group in Clades A2, A3 (FWPV-like) and B1 (CNPV-like) respectively [6,8]. Columbiformes have been shown to be susceptible to viruses from clade A (Fowlpox-like) [3,5,21,22] and clade B (Canarypox-like) [3,4,23].
In this study we have sequenced the genomes of two novel South African avipoxviruses using the 454 (Roche, Life Sciences) and Ion Torrent (Life Technology) platforms. We present a purpose-designed bioinformatics pipeline for analysis of next generation sequence data of avian poxviruses and compare the different avipoxviruses sequenced to date with particular attention to the viruses isolated from a Feral Pigeon (Port Elizabeth, SA) and African Penguin (Cape Town, SA).
Results
The FeP2 and PEPV genomes were assembled into contiguous sequences of 282,356 bp and 306,862 bp respectively. Because the terminal hairpin loops were not sequenced, for each genome the left most nucleotide was arbitrarily nominated base 1.
Phylogenetic analysis of FeP2 and PEPV
Phylogenetic analysis was performed using the DNA polymerase gene, a concatenated amino acid sequence of 17 translated ORFs and the entire genomic sequence (see Methods). All three analyses produced comparable tree topologies and the tree based on DNA polymerase is given in Figure 1. FeP2 and PEPV are most closely related to one another, grouping on the FWPV branch (Clade A), and separate from CNPV (Clade B).
Figure 1.
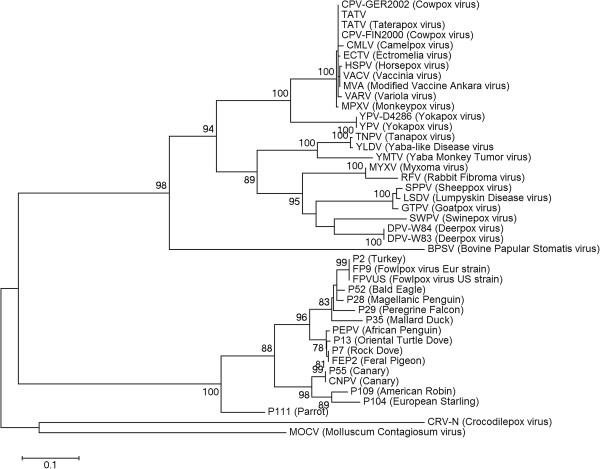
Maximum likelihood phylogenetic trees based on the amino acid sequence alignment of the DNA polymerase genes from various poxviruses. Multiple sequence alignment was performed with MUSCLE and the appropriate phylogenetic model was selected using ProtTest 2.4. The maximum likelihood phylogenetic tree based on the JTT matrix-based model (121) with 100 bootstrap replicates were performed in MEGA 5.10. Discrete Gamma distribution was used to model evolutionary rate differences among sites. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated.
Comparisons of FeP2, PEPV, FWPV and CNPV DNA
The four genomic sequences of FeP2, PEPV, FWPV and CNPV were compared for nucleotide percentage identity. PEPV and FeP2 are more closely related to one another (94.3%) than they are to either FWPV (85.3% and 84.0% respectively) or CNPV (62.0% and 63.4% respectively). Dot plot analysis reveals a large deletion of 16 kbp in FeP2 compared to both of PEPV and FWPV (Figure 2A and B). Like the other avipoxviruses, FeP2 and PEPV have A/T rich genomes, both with 70.5% A + T content.
Figure 2.
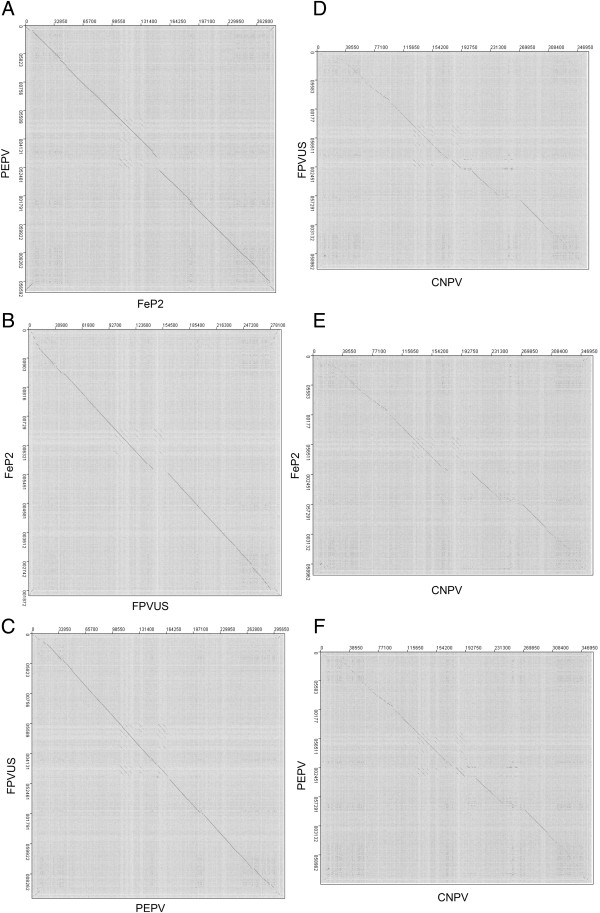
Dotplot comparisons of Pigeonpox virus (FeP2), Penguinpox virus (PEPV), Fowlpox virus (FPVUS) and Canarypox virus (CNPV). A) Dotplot comparing FeP2 (horizontal) and PEPV (vertical). B) Dotplot comparing FPVUS (horizontal) and FeP2 (vertical). C) Dotplot comparing PEPV (horizontal) and FPVUS (vertical). D) Dotplot comparing CNPV (horizontal) and FPVUS (vertical). E) Dotplot comparing CNPV (horizontal) and FeP2 (vertical). F) Dotplot comparing CNPV (horizontal) and PEPV (vertical). Dotplots were constructed using Jdotter (Brodie et al., 2004) [73]. Major deletions can be seen as breaks in the diagonal line. Lines perpendicular to the main diagonal in the top-right and bottom-left corners indicate the inverted terminal repeats (ITRs). The short diagonal lines, parallel to the main diagonal and near the centre of the plots, represent the members of the Variola virus B22R family.
Inverted Terminal Repeats (ITRs)
The FeP2 and PEPV genomes, like most other poxviruses, contain two identical inverted repeated sequences at their termini (ITR). These are 4,682 bp in FeP2 and 5,766 bp in PEPV, both of which are shorter than the ITRs of the other sequenced avipoxviruses (FPVUS: 9,520 bp, FP9: 10,158 bp and CNPV: 6,491 bp). Like FPVUS and CNPV, their A + T content within the ITRs is lower than average - 64.8% for FeP2 and 65.7% for PEPV. At the terminal ends of each ITR in FeP2 exists a 257 bp repeat region with 3.1 and 3.5 copies respectively of a 34 bp and 55 bp tandem repeat. In contrast, PEPV has a 165 bp repeat region containing 3.5 copies of a 47 bp tandem repeat. FPVUS contains a 1.7 kbp region with 42 copies of a 31-32 bp tandem repeat and CNPV contains at least 31, 9, and 7 copies of 17-, 41-, and 48-bp tandem repeats respectively within the terminal regions of each ITR [11]. The repeated sequences are unique for the different avipoxviruses.
Overall arrangement of ORFs in genome
The genomes of FeP2 and PEPV have been compared to FPVUS, FP9 and CNPV in Figure 3. Figure 4 provides a multiple sequence alignment and comparative ORF map of FeP2, PEPV and FPVUS genomes. Clearly, there is conservation amongst the 5 sequenced avipoxviruses, but, at the same time, there are major differences between them. Unlike the orthopoxviruses, the avipoxviruses exhibit variation within the central core region where blocks of avipoxvirus genes have been inverted and/or transposed, suggesting that this genus is the most divergent of the ChPVs [9,11,24]. Comparison of gene orthologues indicate overall genomic synteny between FeP2, PEPV and FPVUS and genomic organisation is similar to that of other sequenced ChPVs [9,25,26]. Additional file 1: Table S1 lists all Open Reading Frames (ORFs) potentially coding for proteins of >30 amino acids for FeP2 and PEPV, and Figures 3 and 4 represent these graphically.
Figure 3.
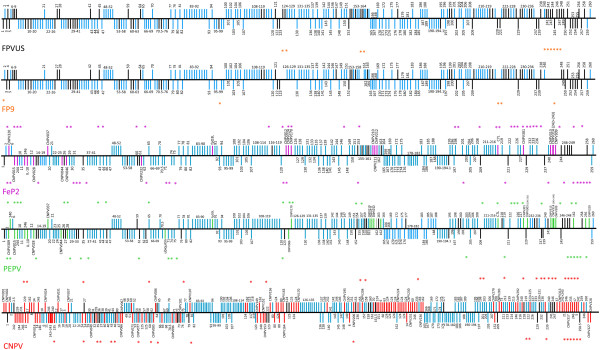
Gene content of the 5 sequenced avipoxvirus genomes. Fowlpox virus United States strain (FPVUS) is used as the reference genome against which other avipoxviruses (fowlpox virus FP9 strain (FP9), pigeonpox virus (FeP2), penguinpox virus (PEPV), canarypox virus (CNPV)) are compared. For each genome, the genes (vertical bars) are upwards when transcribed towards the right and downwards when transcribed towards the left. The genes that are shared amongst all 5 sequences are represented in blue on each genome. Genes shared amongst at least two viruses are represented as black. Differences, as compared to FPVUS, are represented in each genome as the same colour as the virus name. Deletions relative to FPVUS are indicated by an asterisk.
Figure 4.
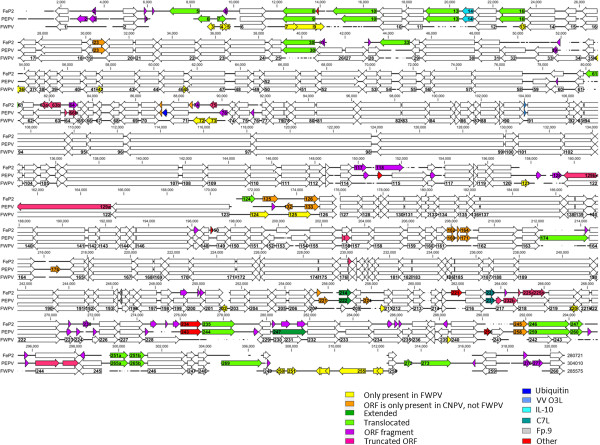
A multiple sequence alignment (MUSCLE) and comparative ORF map of FeP2, PEPV and FPVUS genomes. Blocks represent ORFs, with arrows depicting the direction of transcription. White blocks depict ORF homologues present in all three genomes at the same/similar location and differences between genomes are labelled and shown as coloured blocks in FeP2 and PEPV relative to FPVUS.
FeP2 contains 271 ORFs which have been annotated as putative genes and as orthologues of FPVUS and other genes (Additional file 1: Table S1, Figures 3, 4). These ORFs represent an approximate coding density of 82.5% and encode proteins between 34 and 1,937 amino acids (Additional file 1: Table S1). As published by Hendrickson et al., [27], an ORF was annotated as a whole gene if intact at its 5’ end with at least 80% the length of its orthologue. An ORF intact at its 5’ end but less than 80% of its orthologue length has been annotated as a truncated gene. Any ORF that has been disrupted at its 5’ end has been annotated as a fragmented gene, which is not expected to be transcribed and/or translated into a functional gene product [27]. Of these 271 ORFs, 11 have been annotated as truncated genes (Additional file 1: Table S1). A further 36 of these ORFs are fragmented forms of 28 larger orthologous genes (Additional file 1: Table S1). Therefore a total of 224 ORFs have been annotated as full length putative genes.
PEPV contains 284 ORFs which have been annotated here as putative genes (Additional file 1: Table S1) representing an approximate coding density of 85.4% and encoding proteins ranging between 34 and 1,922 amino acids long (Additional file 1: Table S1). A total of 242 of these ORFs have been annotated as whole genes, 14 as truncated genes (Table 1) and 28 as fragments of larger genes (Table 1).
Table 1.
Deletions and disruptions in Pigeonpox virus (FeP2) and Penguinpox virus (PEPV) relative to Fowlpox virus United States Strain (FPVUS)
| FeP2 | PEPV | Function |
|---|---|---|
|
Deleted genes |
|
|
| fpv003 |
fpv003 |
C-type lectin family |
| fpv004 |
fpv004 |
hypothetical protein |
| fpv005 |
fpv005 |
Efc family |
| fpv007 |
fpv007 |
hypothetical protein |
| fpv008 |
fpv008 |
C-type lectin family |
| fpv009 |
fpv009 |
hypothetical protein |
| fpv013 |
fpv013 |
hypothetical protein |
| fpv027 |
I |
G-protein coupled receptor family |
| fpv032 |
I |
Dnase II |
| fpv033 |
I |
α-SNAP |
| fpv036 |
fpv036 |
hypothetical protein |
| fpv042 |
fpv042 |
hypothetical protein |
| fpv045 |
fpv045 |
hypothetical protein |
| F |
fpv072 |
β-NGF |
| T |
fpv073 |
IL-18 binding protein |
| fpv076 |
F |
β-NGF |
| fpv115 |
I |
Ankyrin repeat family |
| fpv121 |
fpv121 |
CC-chemokine family |
| fpv122 |
T |
B22R |
| fpv123 |
I |
B22R |
| fpv124 |
fpv124 |
N1R/p28 family |
| fpv125 |
fpv125 |
V-type Ig Domain |
| fpv152 |
I |
HT-motif |
| I |
fpv153 |
hypothetical protein |
| fpv154 |
I |
hypothetical protein |
| fpv210 |
fpv210 |
hypothetical protein |
| fpv220 |
fpv220 |
hypothetical protein |
| fpv224 |
F |
Ankyrin repeat family |
| fpv225 |
F |
hypothetical protein |
| fpv238 |
fpv238 |
hypothetical protein |
| fpv250 |
fpv250 |
US ORF2 |
| fpv251 |
fpv251 |
Serpin |
| fpv252 |
fpv252 |
hypothetical protein |
| fpv253 |
fpv253 |
C-type lectin family |
| fpv254 |
fpv254 |
hypothetical protein |
| fpv255 |
fpv255 |
C4L/C10L-like family |
| fpv256 |
trans |
Efc family |
| fpv257 |
fpv257 |
hypothetical protein |
|
Disrupted genes |
|
|
| fpv026 fragmented |
I |
ankyrin repeat |
| fpv028 truncated |
I |
hypothetical protein |
| fpv034 fragmented |
fpv034 fragmented |
ankyrin repeat |
| I |
fpv060 fragmented |
cc-chemokine |
| fpv063 truncated |
I |
hypothetical protein |
| fpv064 fragmented |
fpv064 truncated |
glutathione peroxidase |
| fpv072 fragmented |
- |
β-NGF |
| fpv073 truncated |
- |
IL-18 binding protein |
| - |
fpv076 fragmented |
β-NGF |
| - |
fpv122 truncated |
B22R |
| I |
fpv156 truncated |
hypothetical protein |
| fpv162.2 fragmented |
I |
ankyrin repeat |
| fpv177 truncated |
I |
hypothetical protein |
| fpv199 fragmented |
I |
V-Type Ig Domain |
| fpv200 fragmented |
I |
V-Type Ig Domain |
| fpv217 fragmented |
fpv217 truncated |
hypothetical protein |
| fpv218 truncated |
I |
ankyrin repeat |
| fpv223 fragmented |
fpv223 fragmented |
ankyrin repeat |
| - |
fpv224 fragmented |
ankyrin repeat |
| - |
fpv225 fragmented |
ankyrin repeat |
| fpv228 fragmented |
fpv228 fragmented |
ankyrin repeat |
| fpv234 fragmented |
I |
ankyrin repeat |
| fpv239 fragmented |
I |
C-type lectin |
| I |
fpv244 truncated |
ankyrin repeat |
| fpv245 fragmented |
I |
ankyrin repeat |
| I | fpv249 fragmented | hypothetical protein |
Where a gene is deleted or disrupted in only one virus (FeP2/PEPV), then the status of the other is given as either Intact (I), Truncated (T), Fragmented (F) or Translocated (Trans). Putative functions for the deleted genes have been given where possible.
Relative to the FPVUS genome, FeP2 and PEPV have 36 and 25 deleted ORFs respectively (Table 1). FeP2 contains 34 inserted ORFs relative to FPVUS, comprising 12 fragments of larger genes, 2 truncated genes and 20 intact genes (Table 2). Similarly, PEPV contains 42 inserted ORFs, which include 17 gene fragments, 2 truncated genes and 23 intact genes (Table 2). A total of 214 intact ORFs are shared between PEPV and FeP2 throughout the genome and 5 of these do not have any functional orthologue in FPVUS (Additional file 1: Table S1). Amongst PEPV, FeP2 and FWPV, the most conserved ORF is the orthologue of fpv103 (fep105, pep107 and VACV F17R) which encodes the DNA binding virion core phosphoprotein and shares 100% amino acid identity amongst the three viruses.
Table 2.
Insertions in Pigeonpox virus (FeP2) and Penguinpox virus (PEPV) relative to Fowlpox virus United States Strain (FPVUS)
| FeP2 | PEPV | Function |
|---|---|---|
| Fragmented cnpv021 |
- |
ankyrin repeat |
| - |
Fragmented cnpv319 |
ankyrin repeat |
| - |
Fragmented cnpv310 |
ankyrin repeat |
| Fragmented cnpv006 |
Fragmented cnpv006 |
hypothetical protein |
| cnpv320 |
- |
Ig Domain protein |
| Fragmented cnpv309 |
cnpv309 |
ankyrin repeat |
| - |
fpv250 |
hypothetical |
| cnpv015 |
cnpv015 |
ankyrin repeat |
| Fragmented Trichomonas vaginalis |
- |
ankyrin repeat |
| fpv244 |
fpv244 |
ankyrin repeat |
| fpv246 |
fpv246 |
ankyrin repeat |
| IL-10 (Ficedula albicollis) |
IL-10 (Ficedula albicollis) |
IL-10 |
| cnpv028 |
cnpv028 |
ankyrin repeat |
| cnpv037 |
cnpv037 |
hypothetical protein |
| cnpv044 |
cnpv044 |
ankyrin repeat |
| cnpv046 |
- |
ankyrin repeat |
| cnpv232 |
- |
CC-chemokine-like protein |
| cnpv095 |
cnpv095 |
hypothetical protein |
| - |
Ubiquitin |
Ubiquitin |
| Fragmented cnpv098 |
- |
hypothetical protein |
| - |
fragment cnpv012 |
hypothetical protein |
| fp03L |
fp03L |
vaccinia 03 L ortholog |
| Fragmented cnpv011 |
- |
ankyrin repeat |
| Fragmented cnpv004 |
Fragmented cnpv004 |
ankyrin repeat |
| - |
Fragment Pfs, Nacht protein Neosartorya Fischeri |
ankyrin repeat |
| - |
truncated cnpv221 |
N1R/p28-like protein |
| - |
fragmented cnpv165 |
N1R/p28-like protein |
| - |
fragmented cnpv162 |
TGF-β |
| - |
cnpv086 |
TNFR-like protein |
| cnpv012 |
- |
hypothetical protein |
| cnpv224 |
- |
hypothetical protein |
| cnpv170 |
cnpv170 |
thymidilate kinase |
| Fragmented E. Bacterium |
- |
hypothetical |
| cnpv210 |
cnpv210 |
N1R/p28-like protein |
| cnpv211 |
cnpv211 |
hypothetical protein |
| cnpv212 |
cnpv212 |
N1R/p28-like protein |
| - |
Fragmented cnpv041 |
ankyrin repeat |
| Truncated cnpv279 |
Fragmented cnpv279 |
beta-NGF protein |
| - |
fragmented cnpv283 |
CC-chemokine-like protein |
| Fragmented protein (X. tropicalis) |
- |
hypothetical |
| Tanapox 67R |
Tanapox 67R |
Host Range (C7L-like) |
| Fragmented O.tsutsugamushi str |
Fragmented O.tsutsugamushi str |
ankyrin repeat |
| cnpv301 |
cnpv301 |
ankyrin repeat |
| - |
Fragmented GTPV gp138 |
hypothetical |
| cnpv313 |
cnpv313 |
Ig Domain protein |
| - |
cnpv014 |
Ig Domain protein |
| fragment cnpv320 |
fragment cnpv320 |
Ig Domain protein |
| truncated cnpv014 |
truncated cnpv014 |
Ig Domain protein |
| - |
fpv162 |
ankyrin repeat |
| - |
cnpv320 |
Ig Domain protein |
| - |
fragment cnpv006 |
hypothetical protein |
| cnpv021 Frag |
- |
ankyrin repeat |
| - |
cnpv310 Frag |
ankyrin repeat |
| - | cnpv319 Frag | ankyrin repeat |
Inserted genes have been listed if they are present in FeP2 or PEPV in an unexpected site as predicted by genomic synteny with the FPVUS. Where no homology to FPVUS genes is present, the name of the best BlastP hit has been used.
Both PEPV and FeP2 lack sequences similar to the reticuloendotheliosis virus (REV) observed in FPVUS and other avipoxvirus strains [9,28,29].Although the FeP2 and PEPV genomes are closely related and syntenic to the FWPV genome overall (Figure 2), when compared to each other, and to FWPV, these viruses display differences in gene content throughout their genomes. Of particular note is the large 16 kbp deletion in FeP2 relative to both PEPV (Figure 2A) and FPVUS (Figure 2B).
Conserved genes
There are 179 genes that are conserved amongst all five sequenced avipoxviruses, which have been highlighted in blue in Figure 3. These include genes that lie within the more variable terminal regions of the genome. These avipoxvirus core genes were defined as such if intact orthologues of the genes were present at least once in all five avipoxvirus sequences. The 179 APV core genes include the 90 core genes conserved in all ChPVs which are involved in essential functions such as replication, transcription and virion assembly [24,30] (indicated in bold and italic in Additional file 1: Table S1). There are an additional 89 genes conserved in all avipoxviruses with a variety of different functions (indicated in bold in Additional file 1: Table S1).
Gene families
Avipoxviruses contain extensive gene families, which vary greatly between different species. In CNPV, these comprise over 49% of the genome, whereas they encompass 38% of the FWPV genome [9,11]. In CNPV and FWPV, these gene families account for much of the variation between the two genomes [11]. Intact members of gene families comprise 33% of the FeP2 and PEPV genomes. Table 3 summarises the differences in the number of intact gene family proteins found in the five sequenced avipoxvirus genomes.
Table 3.
A summary of the numbers of intact gene family proteins found in different poxviruses
| FeP2 | PEPV | FPVUS | FP9 | CNPV | VACV | |
|---|---|---|---|---|---|---|
| Ankyrin repeat proteins |
26 |
33 |
31 |
22 |
51 |
17 |
| B22R |
4 |
5 |
6 |
5 |
6 |
1 |
| N1R/p28 |
11 |
11 |
10 |
8 |
26 |
0 |
| C4L/C10L |
2 |
2 |
3 |
3 |
3 |
3 |
| CC chemokine |
4 |
1 |
4 |
4 |
5 |
2 |
| C-type Lectin |
4 |
7 |
9 |
6 |
11 |
2 |
| G protein-coupled receptor gene family |
2 |
3 |
3 |
2 |
4 |
0 |
| HT Motif |
4 |
5 |
6 |
6 |
5 |
0 |
| Ig-like domain protein |
4 |
6 |
5 |
4 |
9 |
3 |
| Serpin |
4 |
4 |
5 |
5 |
5 |
2 |
| Efc family |
1 |
1 |
3 |
2 |
2 |
0 |
| TGF-β |
1 |
1 |
1 |
1 |
5 |
0 |
| β-NGF |
0 |
0 |
2 |
2 |
2 |
0 |
| interleukin 18 (IL-18)-binding protein | 0 | 1 | 1 | 1 | 3 | 1 |
A summary of the numbers of intact gene family proteins in pigeonpox virus (FeP2), penguinpox virus (PEPV), fowlpox virus US strain (FPVUS), fowlpox virus FP9 strain (FP9), canarypox virus (CNPV) and vaccinia virus (VACV). “0” is given where no member of the gene family is present in the respective virus.
Gene translocations and duplications
Analysis of the FeP2 and PEPV genomes revealed several occurances of gene duplication and translocation relative to FWPV. In some instances the postion of the gene was closer to that observed in CNPV than FWPV. A second copy of the Ankyrin repeat family gene, fpv244 orthologue (fep010; pepv010; cnpv009) is found in the equivalent place in the left hand region of each African avipoxvirus genome.
Disrupted and deleted genes
Relative to FPVUS, FeP2 contains 5 truncated FPVUS genes and the fragmented forms of 13 FPVUS orthologues. PEPV contains 5 truncated genes and the ORF fragments of 8 FPVUS orthologues (Additional file 1: Table S1). Both FeP2 and PEPV contain truncated or fragmented remains of ORFs with similarity to CNPV (Table 2).
Relative to the FPVUS genome, FeP2 and PEPV have 36 and 25 deleted ORFs respectively (Table 1) that are absent from any potential coding regions of their genomes.
Inserted genes
Relative to FWPV, the PEPV and FeP2 genomes both contain several inserted genes (Table 2). PEPV, but not FeP2 or FWPV [9], has orthologues of cnpv086 (pepv132) which encodes a protein similar to Tumour necrosis factor receptor (TNFR) [11] and a 77 amino acid long orthologue of Ubiquitin (pepv074) which is most similar to human ubiquitin, S5a ubiquitin-interacting motif-1 (UIM-1) (Accession: 1YX5_B) [31]. Both PEPV (pepv133) and FeP2 (fep126) contain orthologues of thymidylate kinase (TMPK) (cnpv170; VACV A48R) (Table 2). FeP2 and PEPV both encode a putative Interleukin-10 (IL-10) gene with 80.9% amino acid identity between them (fep014, pepv014) and limited similarity to the CNPV orthologue (22.1% and 23.3% aa identity respectively) (not shown). Amino acid identity between all available avian IL-10 genes and PEPV and FeP2 fall between 32.5% and 27.1%, however for CNPV the identities are significantly lower (23.1%-19.0%). The PEPV, FeP2 and CNPV IL-10 genes are most similar to that of a Collared Flycatcher (Ficedula albicollis) sharing 32.5%, 30.2% and 23.1% amino acid identity respectively (not shown). FWPV does not encode an IL-10 gene [9]. The respective positions of FeP2, PEPV and CNPV IL-10 orthologues were analysed to determine the possibility of these genes existing in the three genomes as a result of independent horizontal gene transfer events (Figure 5). Based on this analysis, it is possible that there were two independent transfer events into avian poxviruses, one into CNPV and another into the progenitor of PEPV and FeP2.
Figure 5.
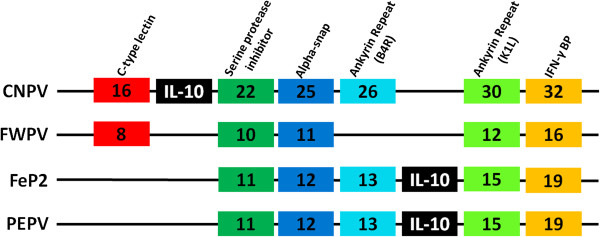
Analysis of Interleukin-10 (IL-10) in Avipoxviruses. Gene synteny analysis depicting syntenic ORFs (that are present in at least 2 of the depicted genomes) highlighting the location of IL-10.
PEPV and FeP2 both encode homologues of Tanapox virus (TANV) and Yaba-Like Disease virus (YLDV) 67R which are orthologues of VACV C7L. These C7L-like genes (pepv231 and fep223) are in the equivalent genome positions, between orthologues of fpv216 (pepv230; fep222) and fpv217 (pepv232 [truncated]; fep224 [fragmented]) (Additional file 1: Table S1, Figures 3 and 4).
Comparison of FeP2 and PEPV to attenuation and lineage specific mutations
In order to determine lineage specific mutations, Laidlaw and Skinner [10] determined the sequence of HP1 (European strain), the virulent parent of FP9, at all loci where FP9 differed from FPVUS (American strain) [10]. Here we compared nucleotide changes in PEPV and FeP2 to 44 genomic positions influencing amino acid composition of 25 open reading frames between European and American strains as previously identified [10]. In 15 and 17 of the places, FeP2 and PEPV were the same as HP1 (European lineage) respectively. In 18 and 17 of the places, FeP2 and PEPV were the same as FPVUS (American lineage) respectively. PEPV and FeP2 differ from both HP1 and FPVUS in 9 and 10 places respectively.
Discussion
Genomic sequences have been determined for two novel South African avipoxviruses. The DNA sequences of PEPV and FeP2 are significantly different from those of CNPV (approx. 63% identity) and FWPV (approx. 85% identity). In Orthopoxviruses (OPVs), the internal region of different species share at least 96% identity when compared at the nucleotide level, while different strains from the same species share at least 99% nucleotide sequence identity [27-33]. These identities have been listed as criteria for establishing poxvirus taxonomy [33]. Therefore, according to the identities between PEPV, FeP2 and FPVUS genomes (94.4% between PEPV and FeP2, 85.3% between PEPV and FPVUS and 84.0% between FeP2 and FPVUS), as well as the differences in phylogenetics and growth characteristics between these two viruses [5], one can deduce that PEPV and FeP2 are separate species of APV.
Laidlaw and Skinner [10] identified geographical lineage-specific mutations which distinguished European fowlpox viruses, FP9 and HP1 from the American strain FPVUS. A comparison of PEPV and FeP2 with the published FWPV sequences at these sites shows that PEPV and FeP2 share lineage-specific mutations with both HP1 and FPVUS. The South African viruses are no more closely related to either the American or the European virus. These results suggest that FeP2 and PEPV originate from a common ancestor that diverged relatively recently from a FWPV-like progenitor which was more distantly related to the CNPV-like progenitor. It is important to note that the divergence between avipoxvirus species is comparable to that between some poxvirus genera and the different species within the OPV genus are highly similar.
FeP2 and PEPV are most closely related to FWPV. Despite this, relative to FWPV, the PEPV and FeP2 genomes both contain several genes that are more closely related to CNPV throughout their genomes. FeP2 and PEPV encode 16 and 15 intact CNPV orthologues respectively (Table 3), five of which are not in FWPV, including a TNFR-like protein (cnpv086, pepv132) and a TMPK protein (cnpv170, fep126, pepv133) (Additional file 1: Table S1 and Table 3). The remainder are more similar to CNPV orthologues but are present in FWPV. Rather than being acquired through horizontal gene transfer or recombination, these genes are most probably from a common ancestor. Their existence suggests that FeP2 and PEPV viruses diverged prior to the present day FWPV, somewhere between the CNPV and FWPV branches. Several of these CNPV-like genes exist as gene fragments (Table 2), which is indicative of their gradual loss through progressive mutation, rearrangements and/or deletions over many rounds of virus replication.
There are several instances of gene duplication and translocation in FeP2 and PEPV relative to FWPV (Figures 3 and 4). Most of these involve genes containing ankyrin repeats. Virulent FWPV contains 31 ankyrin repeat containing genes and attenuated FP9 contains 22 of these genes. PEPV contains 33 ankyrin repeat-containing genes whereas FeP2 contains only 26 (Additional file 1: Table S1; Table 3). It is postulated that loss of these genes may be responsible for the attenuation of the virus [9,10]. Thus the reduced numbers of intact ankyrin repeat genes observed in FeP2 compared to PEPV would suggest that this virus is less virulent. On the other hand, the expansion of the number of ankyrin repeat genes in certain avipoxvirus strains could represent the forming of adaptive genomic accordions, similar to that of the K3L gene in OPV, the formation of which has been described to play an adaptive role in these viruses [34].
The most striking difference between FeP2 and the FWPV-like avipoxviruses, is a large deletion of ~16 kbp from the central, usually conserved, region of the genome. ORFs corresponding to fpv121 (cc-chemokine family), fpv122 and fpv123 (VAR B22R family), fpv124 (N1R/p28 family) and fpv125 (V-type Ig domain gene family) are deleted in FeP2 at this site. The existence of gene fragments and insertions on the borders of this deletion, as well as in the equivalent regions in FWPV and PEPV, suggests that this is a “hotspot” of genetic change, the mechanism of which is uncertain.
Relative to the FPVUS genome, FeP2 and PEPV have 36 and 25 deleted ORFs respectively (Table 1). Most of the genes deleted in PEPV and FeP2 are members of multi-gene families, the disruption of which has been implicated in the attenuation of poxviruses [10], however there are several other deletions of significant ORFs. Of interest, the orthologue of fpv032 encoding a DNase II has been deleted in FeP2 but is in intact in PEPV. In FWPV, fpv032 represents the large subunit of cellular DNase II [9] and cellular DNase II is thought to function in DNA catabolism during apoptosis [35]. Additionally, the orthologue of fpv033 encoding one of the two α-SNAP genes present in FWPV, is absent from FeP2 but intact in PEPV (pepv038). Eukaryotic α-type soluble NSF attachment proteins (α-SNAP) are involved in vesicular transport through the Golgi apparatus and for exocytosis [36]. The fpv033 gene has been shown to be conserved in FWPV strains but non-essential to viral replication and it is thought to be involved in virus-host interactions [37]. Although the fpv033 gene orthologue has been deleted in FeP2, a second α-SNAP-like gene (fpv011, pepv012) exists in the genome which exhibits 34.0% and 33.8% amino acid identity with fpv033 and pepv038 respectively.
FWPV contains two genes with homology to cellular β-nerve growth factor (β-NGF) (fpv072 and fpv076). In FeP2, the orthologue of fpv076 is completely deleted and the orthologue of fpv072 (fep074) is fragmented (Table 1). Conversely, in PEPV, the orthologue of fpv072 is completely deleted and the orthologue of fpv076 (pep079) is fragmented (Table 1). An additional β-NGF-like gene is observed in a truncated form in FeP2 (fep213) and as a fragmented gene in PEPV (pepv221) (Table 2). These are most similar to the CNPV gene cnpv279, which exists as a fragment in FWPV [11]. The FeP2 ORF, fep213 is truncated to 67 amino acids by the introduction of a premature stop codon, where the intact cnpv279 orthologue is 169 amino acids. It is unknown whether this protein would be functional in FeP2 but it is unlikely that PEPV encodes a functional β-NGF protein. β-NGF has proinflammatory properties and is produced by cells in response to infection, injury and stress [38]. FPV encoded β-NGF is thought to be involved in promoting survival in infected cells and could have a role in inhibiting antiviral immune responses in the host [9]. Previously, it was speculated that the absence of a β-NGF gene in an avipoxvirus genome could be partly responsible for the decreased inflammatory response observed in PEPV-infected CAMs as compared to FeP2 [5].
An orthologue of fpv073 encoding an IL-18 binding protein has been deleted in PEPV. In FeP2 this ORF (fep075) has been truncated due to a 1 bp deletion resulting in a premature stop codon. IL-18 is a multifunctional pro-inflammatory cytokine that induces interferon gamma (IFN-γ) production, Th-1 responses and NK cell activity [39]. It is modulated in vivo by IL-18 binding protein (IL18BP) via a negative feedback mechanism [40]. IL18BP homologues are encoded by many poxviruses [9,11,41,42] and have been found to inhibit IL-18-dependent IFN-γ production [43]. In FWPV, fpv073 is thought to have an anti-inflammatory function [9].
PEPV contains an orthologue of cnpv086 (pepv132) which encodes a protein similar to TNF receptor [11]. This ORF is absent from FeP2 (Additional file 1: Table S1) and FWPV [9]. Tumour Necrosis Factor (TNF) is a pro-inflammatory cytokine which is involved in induction of cytokines, cell proliferation, differentiation, necrosis and apoptosis [44]. TNF -induced cellular responses are mediated by one of two types of TNF receptors [44]. Several poxviruses encode TNFR homologues which function to modulate TNF-induced anti-viral responses [11,45-47].
PEPV (pepv133) and FeP2 (fep126) both contain an orthologue of thymidylate kinase (TMPK) (cnpv170; VACV A48R), which is absent in FWPV [9]. This is the only difference found with respect to the complement of nucleotide metabolism genes. Interestingly, despite being within the conserved central region of the genome, the TMPK gene orthologues occur within a region of PEPV and FeP2 that is highly variable compared to FWPV. In addition, the CNPV orthologue insertions that occur within this particular variable region in FeP2 and PEPV occur in different sites across the CNPV genome. The higher than average amino acid identity shared between FeP2 and PEPV (90.8%) and the CNPV genomes (81.6% and 80.2% respectively), suggests that this TMPK gene is necessary and conserved in these viruses. CNPV, FeP2 and PEPV are the only poxvirus species outside of the OPV genus to contain a TMPK homologue. VACV encodes TMPK (A48R) that is 42% identical to human TMPK [48]. TMPK catalyzes an important step in the biosynthesis of (deoxy) thymidine triphosphate and is essential for cell metabolism [49]. In CNPV, FeP2 and PEPV, the presence of a TMPK gene suggests a different optimization of cellular nucleotide pools as compared to FWPV and probably affects cell and tissue tropism in the different viruses [11].
PEPV contains a 77 amino acid long orthologue of Ubiquitin (pepv074) which is most similar to human ubiquitin (97% BLASTp identity), S5a ubiquitin-interacting motif-1 (UIM-1)(Accession: 1YX5_B). CNPV also contains an intact ubiquitin orthologue (cnpv096) (86.1% amino acid identity to pepv074) [11]. FeP2 (nt81,771-81,622) and FPVUS (nt74,550-74,220) encode the fragmented remains of an ubiquitin gene but no open reading frame is observed in either genome [9]. In all four genomes these Ubiquitin ORFs or sequence remains are in the same location (between orthologues of fpv070.5 and fpv071). Ubiquitin is a highly conserved 76 amino acid protein with diverse cellular functions effected by means of marking proteins for intracellular signalling [50]. Ubiquitin proteins are thought to affect the diverse ubiquitination-mediated cellular functions, which may impact on CNPV and PEPV virulence and host range [11]. Other than CNPV and PEPV, ubiquitin genes have only been found in two insect poxviruses, Melanoplus sanguinipes (MSEV) [51] and Amsacta moorei (AMEV) [52]. Virus-encoded ubiquitin genes have also been identified in the Baculoviridae, including in the Autographa californica nuclear polyhedrosis virus (AcNPV) [53]. In AcNPV, disruption of the ubiquitin gene was shown to have no effect on virus viability but to cause a decrease in virion budding and total infectious particle production [53]. The ubiquitin encoding genes in PEPV and CNPV may have an effect on virus production and budding but this remains to be determined.
FeP2 and PEPV both encode a putative Interleukin-10 (IL-10) gene (Figures 3 and 4). Putative orthologues of IL-10 are also found in ORF virus, Bovine Papular Stomatitis Virus (BPSV) [54], Lumpy skin disease virus (LSDV) [55] and Yaba-Like disease virus (YLDV) [56]. IL-10 is a cytokine that has both immunostimulatory and immunosuppressive functions [57] and the ORF virus IL-10 orthologue has been shown to be immunomodulatory in function [58]. The IL-10 genes encoded by PEPV and FeP2 may also be involved immune evasion, however, because of the divergence from host IL-10 proteins (20-31% amino acid identities to various avian IL-10 genes), like cnpv018, these genes may have a novel biological function [11].
The IL-10 gene is likely to have been incorporated into poxvirus genomes via horizontal gene transfer (HGT) [59]. Bratke and McLysacht [59] made use of comparative genomic information and synteny conservation to investigate HGT in pox genomes [59]. Here they found that the IL-10 orthologue encoded by CNPV was in an unexpected location as compared to other poxviruses (yatapox and capripox) but because of the lack of conservation at this region, no conclusion could be drawn about the transfer event into CNPV. An analysis of the genome synteny of avipoxviruses in the region of the IL-10 gene shows that the CNPV IL-10 is in a different location despite being in a similar region to FeP2 and PEPV (Figure 5). Although this region is not well conserved between genera, it appears to be relatively well conserved between avian poxviruses making it possible that there were two independent transfer events into avian poxviruses, one into CNPV and one into the common ancestor of FeP2 and PEPV.
PEPV and FeP2 both encode a gene with homology to Tanapox virus (TANV) and Yaba-Like Disease virus (YLDV) 67R which are orthologues of VACV C7L and not found in FWPV or CNPV. The poxvirus C7L family of host range genes functions by mediating poxvirus host range and antagonising the host defence system [60]. VACV C7L orthologues are found in all orthopoxviruses and most mammalian poxviruses, with the exception of molluscum contagiosum virus and parapoxviruses [61]. This is the first report of a C7L-like gene in avipoxviruses. In mammalian cells, C7L has been shown to inhibit apoptosis [62], antagonise the anti-viral effects of type 1 interferons (IFNs) and Interferon Regulatory Factor 1 (IRF-1) [63] and can antagonize the dsRNA-activated protein kinase (PKR) pathway by inhibiting the phosphorylation of eIF2a [64]. It has previously been suggested that C7L orthologues are an important adaptation of mammalian poxviruses for replication in mammalian hosts [63]. The presence of C7L orthologues in PEPV and FeP2 confounds these previous observations but the function of this gene in these two avipoxviruses remains to be determined. The limited amino acid identity may suggest a novel function of these genes as compared to other C7L orthologues. However, YLDV 67 L shows only 28-30% identity to the VACV C7L protein but has been shown to function equivalently in supporting VACV replication in mammalian hosts [61].
Conclusions
The genome sequences of FeP2 and PEPV have greatly added to the limited repository of genomic information available for the Avipoxvirus genus. In the comparison of FeP2 and PEPV to existing sequences, FWPV and CNPV, we have established insights into African avipoxvirus evolution. Although FeP2 and PEPV are more closely related to FWPV, the presence of whole or disrupted genes with similarity to CNPV genes that are absent in FWPV, suggests that FeP2 and PEPV originate from the common ancestor of CNPV and FWPV. The presence of an intact gene in CNPV, FeP2 and PEPV where the FWPV counterpart is fragmented into two ORFs further supports this theory as fragmented genes represent the gradual loss of genetic information during the process of virus evolution. Additional genome sequences of avipoxviruses would help to define avipoxvirus evolution as a whole.
The ongoing search for an ideal vaccine vector makes this work relevant. Future work could focus on how these two avipoxviruses differ from the well characterized FWPV and CNPV with respect to immune activation and foreign gene expression.
Methods
Viral DNA isolation
FeP2 and PEPV were grown on the chorioallantoic membranes (CAMS) of embryonated 10–11 day old chicken eggs and virus purified as described previously [5].
For 454 sequencing, genomic DNA was extracted using a method described previously [5] with an additional incubation with DNase (25 U/100 ul virus) at 55°C for 1 hr and subsequent inactivation of DNase at 80°C for 30 minutes, prior to treatment with proteinase K. Also, following the first phenol extraction, the virus preparation was treated with RNAse (100 μg/μl) and incubated at 37°C for 1 hour.
For Ion Torrent sequencing, special care was taken to prevent host (chicken) chromosomal and mitochondrial DNA contamination of the viral DNA preparation. Before DNA extraction virus preparations were treated with equal volumes of Vertrel® (1,1,1,2,3,4,4,5,5,5-decafluoropentane) (DuPont) which is a Freon substitute that has been shown to separate virus from infected cellular debris in some viral purification methods [65]. The virus preparations were then freeze/thawed three times to lyse any remaining cells, and then treated with DNAse prior to lysis of the virions. DNA extraction was then performed as described previously [5].
DNA sequencing and bioinformatics
High quality viral DNA was sequenced using a Roche 454 GS Junior system as per manufacturer’s instructions. Primary sequence analysis was performed using the GS Junior Software version 2.5p1 and de novo assembly was done using GS De Novo Assembler software and CLC Genomics Workbench. For Ion Torrent sequencing [66], DNA was sequenced using a 316 chip on an Ion Torrent Personal Genome Machine (PGM) (Life Technologies) at the Central Analytical Facilities, Stellenbosch University. The Covaris S2 system was used for physical shearing.
To offset possible remaining host DNA contamination, we made use of a customized bioinformatics pipeline as described here (Additional file 2). For 454 data, the SFF file was converted to FastQ in Galaxy, and filtered such that only reads shorter than 500 nt and with a mean QC > 20 were included. For the Ion torrent data, primary analysis was performed in Torrent Suite version 3.2.1. Firstly, the Raw SFF file was submitted to SFFTrim (Torrent Suite 3.2.1) and reads were trimmed when the average base quality values in a window size of 10 were less than 25 (Q-value of 25). Reads shorter than 50 nt were discarded. Reads were trimmed of adaptor sequences and filtered to remove polyclonal reads and trimmed to remove poor quality bases at the 3’ end of long reads.
These two datasets were then mapped to the Chicken (Gallus gallus) genome (WASHUC2 assembly) with Newbler 2.6 to filter out possible host contamination. Read IDs that did not map to the Chicken genome were obtained from the Newbler output and these unmapped reads were extracted from the original SFF files using Linux commandline tools and SFFFile that forms part of the 454 NGS data analysis software. The resulting SFF files were used as input data for de novo assembly. De novo assembly was performed in Newbler 2.6 using default parameters, as well as in Mira (Version 3.4.0) where de novo assembly was assisted by a reference genome (FPVUS).
For the FeP2 data, the Roche 454 sequencing resulted in 93,654 reads with an average length of 404 bp. Following filtering, only 2,369 (2.5%) of these reads did not map to the chicken genome and met the quality requirements. Ion Torrent sequencing resulted in a total of 3,239,283 reads with a mean read length of 191 bp. The filtered data set contained 1,068,645 reads or 32.9% of the total raw reads. Newbler assembly resulted in 7 contigs greater than 500 bp (278,380 bases total). MIRA assembly also resulted in 7 contigs greater than 500 bp (279,145 bases total). The assemblies were merged using the Genome Assembler, Reconciliation and Merging (GARM) version 0.70 meta assembler pipeline which resulted in 3 contigs and the remaining gaps were closed by PCR and Sanger sequencing and visual inspection. Raw PGM reads were then mapped back to the draft sequence. A total of 3,142,379 raw reads mapped to the draft sequence (97.0% of total) resulting in an average coverage of 2,129.9 X. Remaining gaps between contigs were closed by PCR and Sanger sequencing and raw reads were mapped back to the draft sequence using CLC Genomics workbench 4.7.2 and TMap as part of the Torrent Suite (Version 3.2.1) software.
For the PEPV data, Ion Torrent sequencing resulted in 3,197,371 reads with a mean read length of 203 bp. The filtered data set contained 1, 119,080 reads or 35.0% of the total raw reads. MIRA assembly resulted in one contig of 301,453 bp. PCR and Sanger sequencing were used to find and confirm the location of the inverted terminal repeats.
Open reading frames (ORFs) longer than 30 amino acids with a methionine start codon (ATG) and less than 50% overlap to other ORFs were called using the CLC Genomics Workbench (CLC) ORF analysis tool as well as with an Integrated Services for Genomic Analysis (ISGA) pipeline which makes use of GLIMMER3 [67], BLAST, HMMer2 [68] and other protein coding sequence and annotation software described by Hemmerich, et al. [69]. Similarity searches including nucleotide (BLASTn) and Protein (BLASTp) BLAST analyses were performed on every ORF and ORFs were annotated as potential genes if they shared significant sequence similarity to known viral or cellular genes (BLAST E value ≤ e-5) or contained a putative conserved domain as predicted by BLASTp. ORFs were numbered from left to right, with alphabetic sub-ordering used to indicate multiple potential fragments of larger avipoxvirus ORFs.
Promoters described by Afonso et al. [9] were predicted using CLC motif search tool and tandem repeats were identified using TandemRepeatsFinder [70]. Multiple sequence alignments were performed with progressiveMauve [71] and Base-By-Base v2 [72], and dotplots were done in Jdotter [73].
Phylogenetic analysis
Phylogenetic analyses were performed on representative amino acid sequences of DNA polymerase as well as on the concatenated amino acid sequences of 17 conserved proteins from each ChPV species, as previously described [24]. Amino acid sequences were obtained from Genbank. For the 17 conserved proteins, the individual sequences of each protein were first aligned using MUSCLE [74]. Gblocks.091b [75] was then used to remove any gaps or divergent blocks according to specified criteria [75]. All 17 protein sequences for each virus were then extracted from the Gblocks output and concatenated manually. For the DNA polymerase and concatenated amino acid sequences, multiple sequence alignments were performed with MUSCLE [74]. An additional analysis was performed using the whole genome nucleotide sequences of CNPV, FPVUS, FeP2 and PEPV, which were aligned with ClustalW in Galaxy. Appropriate phylogenetic models were selected using ProtTest 2.4 [76], and phylogenetic analysis was performed in MEGA 5.10 [77] using both maximum parsimony and maximum likelihood methods with 100 bootstrap replicates.
Nucleotide sequence accession number
The sequences of the South African Feral Pigeonpox virus (FeP2) and Penguinpox virus (PEPV) have been deposited in the NCBI database under GenBank accession numbers: [FeP2 Genbank: KJ801920] and [PEPV Genbank: KJ859677].
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KO, OC, ND and ALW designed the study. KO and OC performed the experiments. KO, OC and APV performed the bioinformatics and data analysis. KO wrote the inital manuscript. All authors read, edited and approved the final manuscript.
Supplementary Material
FeP2 and PEPV Open Reading Frames.
Flow diagram depicting the customized bioinformatics pipeline used to analyse FeP2 and PEPV sequencing data.
Contributor Information
Kristy Offerman, Email: kristy.offerman@uct.ac.za.
Olivia Carulei, Email: olivia.carulei@uct.ac.za.
Anelda Philine van der Walt, Email: anelda.vanderwalt@uct.ac.za.
Nicola Douglass, Email: niki.douglass@uct.ac.za.
Anna-Lise Williamson, Email: Anna-Lise.Williamson@uct.ac.za.
Acknowledgements
This work is based on the research supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation (NRF), South Africa. Any opinion, finding and conclusion or recommendation expressed in this material is that of the authors and the NRF does not accept any liability in this regard. Financial support was also provided by the Clinical Infectious Diseases Research Initiative (CIDRI) and the Carnegie Corporation of New York.
References
- Bolte AL, Meurer J, Kaleta EF. Avian host spectrum of avipoxviruses. Avian Pathol. 1999;28(5):415–432. doi: 10.1080/03079459994434. [DOI] [PubMed] [Google Scholar]
- ICTV 9th Report. Virus taxonomy: classification and nomenclature of viruses. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editor. Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier; 2012. [Google Scholar]
- Jarmin S, Manvell R, Gough RE, Laidlaw SM, Skinner MA. Avipoxvirus phylogenetics: identification of a PCR length polymorphism that discriminates between the two major clades. J Gen Virol. 2006;87(8):2191–2201. doi: 10.1099/vir.0.81738-0. [DOI] [PubMed] [Google Scholar]
- Manarolla G, Pisoni G, Sironi G, Rampin T. Molecular biological characterization of avian poxvirus strains isolated from different avian species. Vet Microbiol. 2010;140(1–2):1–8. doi: 10.1016/j.vetmic.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Offerman K, Carulei O, Gous TA, Douglass N, Williamson A. Phylogenetic and histological variation in avipoxviruses isolated in South Africa. J Gen Virol. 2013;94(Pt 10):2338–2351. doi: 10.1099/vir.0.054049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyuranecz M, Foster JT, Dán Á, Ip HS, Egstad KF, Parker PG, Higashiguchi JM, Skinner MA, Höfle U, Kreizinger Z, Dorrestein GM, Solt S, Sós E, Kim YJ, Uhart M, Pereda A, González-Hein G, Hidalgo H, Blanco J, Erdélyi K. Worldwide phylogenetic relationship of Avian Poxviruses. J Virol. 2013;87(9):4938–4951. doi: 10.1128/JVI.03183-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LH, Lee KH. Application of the polymerase chain reaction for the diagnosis of fowl poxvirus infection. J Virol Methods. 1997;63(1–2):113–119. doi: 10.1016/s0166-0934(96)02119-2. [DOI] [PubMed] [Google Scholar]
- Carulei O, Douglass N, Williamson AL. Phylogenetic analysis of three genes of Penguinpox virus corresponding to Vaccinia virus G8R (VLTF-1), A3L (P4b) and H3L reveals that it is most closely related to Turkeypox virus, Ostrichpox virus and Pigeonpox virus. Virol J. 2009;6:52–422X. doi: 10.1186/1743-422X-6-52. 6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonso CL, Tulman ER, Lu Z, Zsak L, Kutish GF, Rock DL. The genome of Fowlpox Virus. J Virol. 2000;74(8):3815–3831. doi: 10.1128/JVI.74.8.3815-3831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw SM, Skinner MA. Comparison of the genome sequence of FP9, an attenuated, tissue culture-adapted European strain of Fowlpox virus, with those of virulent American and European viruses. J Gen Virol. 2004;85(2):305–322. doi: 10.1099/vir.0.19568-0. [DOI] [PubMed] [Google Scholar]
- Tulman ER, Afonso CL, Lu Z, Zsak L, Kutish GF, Rock DL. The genome of Canarypox Virus. J Virol. 2004;78(1):353–366. doi: 10.1128/JVI.78.1.353-366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle DB. In: Poxviruses; Birkhäuser Advances in Infectious Diseases. Mercer A, Schmidt A, Weber O, editor. Basel: Birkhäuser Verlag; 2007. Genus Avipoxvirus; pp. 217–251. [Google Scholar]
- Amano H, Morikawa S, Shimizu H, Shoji I, Kurosawa D, Matsuura Y, Miyamura T, Ueda Y. Identification of the canarypox virus thymidine kinase gene and insertion of foreign genes. Virology. 1999;256(2):280–290. doi: 10.1006/viro.1999.9648. [DOI] [PubMed] [Google Scholar]
- Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. Poxvirus Orthologous clusters: toward defining the minimum essential Poxvirus genome. J Virol. 2003;77(13):7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkus ME, Goebel SJ, Davis SW, Johnson GP, Limbach K, Norton EK, Paoletti E. Vaccinia virus host range genes. Virology. 1990;179(1):276–286. doi: 10.1016/0042-6822(90)90296-4. [DOI] [PubMed] [Google Scholar]
- Tulman ER, Afonso CL, Lu Z, Zsak L, Sur J, Sandybaev NT, Kerembekova UZ, Zaitsev VL, Kutish GF, Rock DL. The genomes of Sheeppox and Goatpox Viruses. J Virol. 2002;76(12):6054–6061. doi: 10.1128/JVI.76.12.6054-6061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pledger A. Avian pox virus infection in a mourning dove. Can Vet J. 2005;46(12):1143–1145. [PMC free article] [PubMed] [Google Scholar]
- Tageldin MH, Johnson EH, Al-Amri IS, Aisha A. Cutaneous tumor-like Lesions associated with Poxvirus infection in laughing doves (Streptopelia senegalensis) J Avian Med Surg. 2006;20(2):94–96. doi: 10.1647/2005-006RR.1. [DOI] [Google Scholar]
- Medina F, RamÃrez GA, Hernández A. Avian Pox in white-tailed Laurel-pigeons from the Canary Islands. J Wildl Dis. 2004;40(2):351–355. doi: 10.7589/0090-3558-40.2.351. [DOI] [PubMed] [Google Scholar]
- Stannard LM, Marais D, Kow D, Dumbell KR. Evidence for incomplete replication of a penguin poxvirus in cells of mammalian origin. J Gen Virol. 1998;79(7):1637–1646. doi: 10.1099/0022-1317-79-7-1637. [DOI] [PubMed] [Google Scholar]
- Pawar R, Bhushan S, Poornachandar A, Lakshmikantan U, Shivaji S. Avian pox infection in different wild birds in India. Eur J Wildl Res. 2011;57(4):785–793. doi: 10.1007/s10344-010-0488-4. [DOI] [Google Scholar]
- Lüschow D, Hoffmann T, Hafez HM. Differentiation of Avian Poxvirus Strains on the basis of nucleotide sequences of 4b gene fragment. Avian Dis. 2004;48(3):453–462. doi: 10.1637/7111. [DOI] [PubMed] [Google Scholar]
- Weli SC, Traavik T, Tryland M, Coucheron DH, Nilssen Ã. Analysis and comparison of the 4b core protein gene of avipoxviruses from wild birds: Evidence for interspecies spatial phylogenetic variation. Arch Virol. 2004;149(10):2035–2046. doi: 10.1007/s00705-004-0357-0. [DOI] [PubMed] [Google Scholar]
- Gubser C, Hué S, Kellam P, Smith GL. Poxvirus genomes: a phylogenetic analysis. J Gen Virol. 2004;85(1):105–117. doi: 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- Goebel SJ, Johnson GP, Perkus ME, Davis SW, Winslow JP, Paoletti E. The complete DNA sequence of vaccinia virus. Virology. 1990;179(1):247–266. doi: 10.1016/0042-6822(90)90294-2. [DOI] [PubMed] [Google Scholar]
- Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology. 1997;233(1):19–42. doi: 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- Hendrickson RC, Wang C, Hatcher EL, Lefkowitz EJ. Orthopoxvirus genome evolution: the role of gene loss. Viruses. 2010;2(9):1933–1967. doi: 10.3390/v2091933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Schnitzlein WM, Tripathy DN. Reticuloendotheliosis virus sequences within the genomes of field strains of fowlpox virus display variability. J Virol. 2003;77(10):5855–5862. doi: 10.1128/JVI.77.10.5855-5862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KM, Davis JR, Sato T, Yasuda A. Reticuloendotheliosis virus (REV) long terminal repeats incorporated in the genomes of commercial fowl poxvirus vaccines and pigeon poxviruses without indication of the presence of infectious REV. Avian Dis. 2000;44(4):827–841. doi: 10.2307/1593055. [DOI] [PubMed] [Google Scholar]
- Lefkowitz EJ, Wang C, Upton C. Poxviruses: past, present and future. Virus Res. 2006;117(1):105–118. doi: 10.1016/j.virusres.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Wang Q, Young P, Walters KJ. Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. J Mol Biol. 2005;348(3):727–739. doi: 10.1016/j.jmb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Chen N, Li G, Liszewski MK, Atkinson JP, Jahrling PB, Feng Z, Schriewer J, Buck C, Wang C, Lefkowitz EJ, Esposito JJ, Harms T, Damon IK, Roper RL, Upton C, Buller RML. Virulence differences between monkeypox virus isolates from West Africa and the Congo basin. Virology. 2005;340(1):46–63. doi: 10.1016/j.virol.2005.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MA, Buller RM, Damon IK, Lefkowits EJ, McFadden G, McInnes CJ, Mercer AA, Moyer RW, Upton C. In: Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. King AQ, Lefkowitz E, Adams MJ, Carstens EB, editor. USA: Elsevier; 2011. Poxviridae; pp. 290–309. [Google Scholar]
- Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP, Malik HS. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell. 2012;150(4):831–841. doi: 10.1016/j.cell.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torriglia A, Perani P, Brossas JY, Chaudun E, Treton J, Courtois Y, Counis M. L-DNase II, a molecule that links proteases and endonucleases in apoptosis, derives from the ubiquitous serpin leukocyte elastase inhibitor. Mol Cell Biol. 1998;18(6):3612–3619. doi: 10.1128/mcb.18.6.3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A, Burgoyne RD. A role for soluble NSF attachment proteins (SNAPs) in regulated exocytosis in adrenal chromaffin cells. EMBO J. 1995;14(2):232–239. doi: 10.1002/j.1460-2075.1995.tb06996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw SM, Anwar MA, Thomas W, Green P, Shaw K, Skinner MA. Fowlpox virus encodes nonessential homologs of cellular Alpha-SNAP, PC-1, and an orphan human homolog of a secreted nematode protein. J Virol. 1998;72(8):6742–6751. doi: 10.1128/jvi.72.8.6742-6751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloe L, Simone MD, Properzi F. Nerve growth factor: a neurotrophin with activity on cells of the immune system. Microsc Res Tech. 1999;45(4–5):285–291. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<285::AID-JEMT12>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, Akira S. Defective NK cell activity and Th1 response in IL-18–deficient mice. Immunity. 1998;8(3):383–390. doi: 10.1016/S1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- Novick D, Kim S, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10(1):127–136. doi: 10.1016/S1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- Krumm B, Meng X, Wang Z, Xiang Y, Deng J. A unique bivalent binding and inhibition mechanism by the Yatapoxvirus Interleukin 18 binding protein. PLoS Pathog. 2012;8(8):e1002876. doi: 10.1371/journal.ppat.1002876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonso CL, Delhon G, Tulman ER, Lu Z, Zsak A, Becerra VM, Zsak L, Kutish GF, Rock DL. Genome of Deerpox Virus. J Virol. 2005;79(2):966–977. doi: 10.1128/JVI.79.2.966-977.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading PC, Smith GL. Vaccinia virus Interleukin-18-binding protein promotes virulence by reducing gamma interferon production and natural killer and t-cell activity. J Virol. 2003;77(18):9960–9968. doi: 10.1128/JVI.77.18.9960-9968.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Hu F, Smith CA, Pickup DJ. Cowpox virus contains two copies of an early gene encoding a soluble secreted form of the type II TNF receptor. Virology. 1994;204(1):343–356. doi: 10.1006/viro.1994.1539. [DOI] [PubMed] [Google Scholar]
- Upton C, Macen JL, Schreiber M, McFadden G. Myxoma virus expresses a secreted protein with homology to the tumor necrosis factor receptor gene family that contributes to viral virulence. Virology. 1991;184(1):370–382. doi: 10.1016/0042-6822(91)90853-4. [DOI] [PubMed] [Google Scholar]
- Saraiva M, Alcami A. CrmE, a novel soluble tumor necrosis factor receptor encoded by poxviruses. J Virol. 2001;75(1):226–233. doi: 10.1128/JVI.75.1.226-233.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillat C, Topalis D, Agrofoglio LA, Pochet S, Balzarini J, Deville-Bonne D, Meyer P. Crystal structure of poxvirus thymidylate kinase: an unexpected dimerization has implications for antiviral therapy. Proc Natl Acad Sci. 2008;105(44):16900–16905. doi: 10.1073/pnas.0804525105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SJ, Johnston LH, de Carlos A, Smith GL. Vaccinia virus encodes an active thymidylate kinase that complements a cdc8 mutant of Saccharomyces cerevisiae. J Biol Chem. 1991;266(30):20103–20109. [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Afonso CL, Tulman ER, Lu Z, Oma E, Kutish GF, Rock DL. The genome of Melanoplus sanguinipes Entomopoxvirus. J Virol. 1999;73(1):533–552. doi: 10.1128/jvi.73.1.533-552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawden AL, Glassberg KJ, Diggans J, Shaw R, Farmerie W, Moyer RW. Complete genomic sequence of the Amsacta moorei Entomopoxvirus: analysis and comparison with other poxviruses. Virology. 2000;274(1):120–139. doi: 10.1006/viro.2000.0449. [DOI] [PubMed] [Google Scholar]
- Reilly LM, Guarino LA. The viral ubiquitin gene of Autographa californica Nuclear Polyhedrosis Virus is not essential for viral replication. Virology. 1996;218(1):243–247. doi: 10.1006/viro.1996.0185. [DOI] [PubMed] [Google Scholar]
- Delhon G, Tulman ER, Afonso CL, Lu Z, de la Concha-Bermejillo A, Lehmkuhl HD, Piccone ME, Kutish GF, Rock DL. Genomes of the Parapoxviruses Orf Virus and Bovine Papular Stomatitis Virus. J Virol. 2004;78(1):168–177. doi: 10.1128/JVI.78.1.168-177.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulman ER, Afonso CL, Lu Z, Zsak L, Kutish GF, Rock DL. Genome of Lumpy Skin Disease Virus. J Virol. 2001;75(15):7122–7130. doi: 10.1128/JVI.75.15.7122-7130.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Essani K, Smith GL. The genome sequence of Yaba-like Disease Virus, a Yatapoxvirus. Virology. 2001;281(2):170–192. doi: 10.1006/viro.2000.0761. [DOI] [PubMed] [Google Scholar]
- Moore KW, de WM, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19(1):683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Chan A, Baird M, Mercer AA, Fleming SB. Maturation and function of human dendritic cells are inhibited by orf virus-encoded interleukin-10. J Gen Virol. 2006;87(11):3177–3181. doi: 10.1099/vir.0.82238-0. [DOI] [PubMed] [Google Scholar]
- Bratke KA, McLysaght A. Identification of multiple independent horizontal gene transfers into poxviruses using a comparative genomics approach. BMC Evol Biol. 2008;8:67–2148. doi: 10.1186/1471-2148-8-67. -8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Rothenburg S, McFadden G. The poxvirus C7L host range factor superfamily. Curr Opin Virol. 2012;2(6):764–772. doi: 10.1016/j.coviro.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Chao J, Xiang Y. Identification from diverse mammalian poxviruses of host-range regulatory genes functioning equivalently to vaccinia virus C7L. Virology. 2008;372(2):372–383. doi: 10.1016/j.virol.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nájera JL, Gómez CE, Domingo-Gil E, Gherardi MM, Esteban M. Cellular and biochemical differences between two attenuated poxvirus vaccine candidates (MVA and NYVAC) and role of the C7L gene. J Virol. 2006;80(12):6033–6047. doi: 10.1128/JVI.02108-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Schoggins J, Rose L, Cao J, Ploss A, Rice CM, Xiang Y. C7L family of poxvirus host range genes inhibits antiviral activities induced by Type 1 interferons and interferon regulatory factor 1. J Virol. 2012;86(8):4538–4547. doi: 10.1128/JVI.06140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes S, Sperling KM, Zwilling J, Gasteiger G, Ludwig H, Kremmer E, Schwantes A, Staib C, Sutter G. Viral host-range factor C7 or K1 is essential for modified vaccinia virus Ankara late gene expression in human and murine cells, irrespective of their capacity to inhibit protein kinase R-mediated phosphorylation of eukaryotic translation initiation factor 2α. J Gen Virol. 2010;91(2):470–482. doi: 10.1099/vir.0.015347-0. [DOI] [PubMed] [Google Scholar]
- Mendez II, Hermann LL, Hazelton PR, Coombs KM. A comparative analysis of Freon substitutes in the purification of reovirus and calicivirus. J Virol Methods. 2000;90(1):59–67. doi: 10.1016/S0166-0934(00)00217-2. [DOI] [PubMed] [Google Scholar]
- Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, Hoon J, Simons JF, Marran D, Myers JW, Davidson JF, Branting A, Nobile JR, Puc BP, Light D, Clark TA, Huber M, Branciforte JT, Stoner IB, Cawley SE, Lyons M, Fu Y, Homer N, Sedova M, Miao X, Reed B. et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23(6):673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23(1):205–211. [PubMed] [Google Scholar]
- Hemmerich C, Buechlein A, Podicheti R, Revanna KV, Dong Q. An Ergatis-based prokaryotic genome annotation web server. Bioinformatics. 2010;26(8):1122–1124. doi: 10.1093/bioinformatics/btq090. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillary W, Lin S, Upton C. Base-By-Base version 2: single nucleotide-level analysis of whole viral genome alignments. Microb Inform Experimentation. 2011;1(1):1–6. doi: 10.1186/2042-5783-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie R, Roper RL, Upton C. JDotter: a Java interface to multiple dotplots generated by dotter. Bioinformatics. 2004;20(2):279–281. doi: 10.1093/bioinformatics/btg406. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21(9):2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FeP2 and PEPV Open Reading Frames.
Flow diagram depicting the customized bioinformatics pipeline used to analyse FeP2 and PEPV sequencing data.