

Abstract
Endoglin, a 180 kDa disulfide-linked homodimeric, transmembrane receptor protein mostly expressed in tumor-associated endothelial cells, is an endogenous binding partner of GAIP-interacting protein, C terminus (GIPC). Endoglin functions as a co-receptor of TβRII that binds Transforming Growth Factor-β (TGF-β) and is important for vascular development, and consequently has become a compelling target for anti-angiogenic therapies. A few recent studies in Gastrointestinal Stromal Tumor (GIST), breast cancer and ovarian cancer, however, suggest that endoglin is upregulated in tumor cells and is associated with poor prognosis. These findings indicate a broader role of endoglin in tumor biology, beyond anti-angiogenic effects. The goal of our current study is to evaluate the effects of targeting endoglin in pancreatic cancer both in vitro and in vivo. We analyzed the anti-proliferative effect of both RNAi-based and peptide ligand-based inhibition of endoglin in pancreatic cancer cell lines, the latter yielding a GIPC PDZ domain-targeting lipopeptide with notable anti-proliferative activity. We further demonstrated that endoglin inhibition induced a differentiation phenotype in the pancreatic cancer cells and sensitized them against conventional chemotherapeutic drug gemcitabine. Most importantly, we have demonstrated the anti-tumor effect of both RNAi based and competitive inhibitor based blocking of endoglin in pancreatic cancer xenograft models in vivo. To our knowledge, this is the first report exploring the effect of targeting endoglin in pancreatic cancer cells.
Keywords: Endoglin, GIPC, Pancreatic Cancer, Differentiation, Sensitization
INTRODUCTION
Pancreatic cancer is among the most lethal and unforgiving of human cancers, and continues to be a major unsolved health problem (1). It has a five-year survival rate of only 6% and is estimated to have 46420 new cases and cause 39590 deaths in 2014 in the United States, a number that has been steadily increasing for more than a decade (2, 3). Conventional treatment approaches, including surgery, radiation, chemotherapy, or combinations thereof, have had little impact on the course of this aggressive cancer. To address an unmet clinical need for new agents to treat pancreatic cancer, there is a corresponding, urgent requirement to identify novel targets that could lead the way towards therapeutic strategies to combat this disease.
Endoglin (or CD105) is a transmembrane glycoprotein and a component of the Transforming Growth Factor-β (TGF-β) receptor system (4). It is mainly expressed in endothelial cells and promotes their proliferation by modulating TGF-β signaling through its PDZ domain-binding association with GAIP-interacting protein, C terminus (GIPC) (5–7). Mice lacking endoglin die at the mid-gestation stage as a result of defective vascular development, suggesting that endoglin has roles in endothelial proliferation and vascular development i.e. angiogenesis and vasculogenesis (8). Several groups have shown that endoglin is also expressed in peritumoral and intratumoral blood vessels of brain, prostate, breast, colorectal, pancreatic and hepatic cancers (9–15). The specific expression pattern of endoglin in tumoral vessels indicates that it may be a worthy target molecule for anti-angiogenic therapy in cancer. In deed, anti-endoglin therapy is currently being explored as an anti-angiogenic therapeutic in several cancers (16–19).
However, endoglin may serve in a capacity beyond angiogenesis alone. Studies in GIST, breast cancer and ovarian cancer suggest that endoglin is upregulated not only in tumor endothelial cells, but also in tumor cells, and is associated with a poor prognosis (20–22). Additionally, it has been recently shown that while endoglin is rarely expressed in primary ovarian cancer cells, it is frequently expressed in recurrent platinum-resistant tumor cells, as compared with the primary untreated tumor (23). These findings point to a broader role of endoglin in tumor cell biology beyond that of endothelial expression alone.
A recent study reported a wide range of endoglin expression levels in pancreatic cancer cell lines but no detectable levels of endoglin in normal pancreatic epithelial cells (24). Endoglin expression could also be seen in a subset of pancreatic cancer cells obtained from Pancreatic Ductal Adenocarcinoma (PDAC) patients. Interestingly, downregulation of epithelial marker E-cadherin and overexpression of mesenchymal marker vimentin were observed in endoglin-positive pancreatic cancer cells compared with those in endoglin-negative cells. These findings prompted us to evaluate the effects of endoglin inhibition in pancreatic cancer progression.
GIPC was originally identified as a binding partner of the GTPase-activating protein RGS-GAIP (25). The PDZ domain of GIPC binds to several endogenous partner proteins such as RGS-GAIP, APPL1, Glut-1 transporter, Semaphorin-F, neuropilin-1, IGF-1R and endoglin through a direct association with the C-terminal tail of the partner proteins (7, 25–30) and this PDZ interaction has been implicated as a critical player in the biology of normal and malignant cells (30–34). Therefore, it is quite understandable that disruption of this GIPC PDZ-specific interaction with selective inhibitors should inhibit protein-protein interactions within cellular signaling pathways that are required for cancer growth. Previous studies from our group had utilized peptide-based inhibitors for this purpose (30, 34, 35).
In the present study, we have analyzed the anti-proliferative effect of both RNAi-based and competitive, peptide-based inhibition of endoglin, via the PDZ domain of one of its endogenous binding partners, GIPC, in pancreatic cancer cell lines. We have further shown that endoglin inhibition induced a differentiation phenotype in the pancreatic cancer cells and sensitized them against the conventional chemotherapeutic drug gemcitabine. Most importantly, we have shown the anti-proliferative effect of both RNAi-based and molecular competitive peptide-based inhibition of endoglin in pancreatic cancer xenograft models in vivo. To our knowledge, this is the first report exploring the effect of targeting endoglin in pancreatic cancer cells.
MATERIALS AND METHODS
Reagents
The antibodies against phospho-Smad 1/5, Smad 1, phospho-Smad 2 and Smad 2/3 were purchased from Cell Signaling Technology. Antibodies against Id-1, Ki67 and CD31 were purchased from Santa Cruz Biotechnology. Anti-β-actin and Anti-E-cadherin were from BD Biosciences. Anti-endoglin antibody was purchased from Sigma-Aldrich. Anti-GIPC antibody was purchased from Pierce. Immunohistochemistry was performed using the IHC Select HRP/DAB kit from Millipore.
Cell culture
Pancreatic cancer cell lines ASPC-1, MiaPaca-2 and cells isolated from pancreatic cancer patient-derived xenografts (MCPAN002, 5160-1, MCPAN014, 4866-1, 5647-1 and 4482-1) were used in the present study. ASPC-1 (CRL-1682; purchased on January, 2006) and MiaPaca-2 (CRL-1420; purchased on November, 2005) cells were from American Type Culture Collection (ATCC). No authentification of the cell lines was done by the authors. The procedure for isolation of cells from pancreatic cancer patient-derived xenografts is described below. ASPC-1 cell line was maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. MiaPaca-2 cell line was maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS and 1% penicillin-streptomycin. Cells isolated from patients were cultured in DMEM-F12 medium supplemented with 10% FBS and 1% Antimycotic/Antibiotic (Invitrogen). Cells from a culture of 70–80% confluence were used in experiments.
Isolation of cells from pancreatic cancer patient xenografts
The human primary pancreatic cell lines were developed from successful patient-derived xenografts. To generate our xenografts, we obtained human pancreatic tumor tissue from Mayo Clinic’s surgical pathology department. All tissues were obtained and used in accordance to the Mayo Clinic Institutional Review Board and the Institutional Animal Care and Use Committee. The tumor tissue was maintained under sterile conditions and was washed twice with PBS. It was then minced with a razor blade and mixed with high growth factor Matrigel (BD Biosciences) to create a slurry. Approximately 0.1 ml slurry was injected subcutaneously into the right flank of SCID mice. Tumors were grown to a spherical diameter of approximately 1.7cm, if permitted by the health of the animal. Animals were then euthanized and xenografts were harvested. The harvested xenograft was washed twice with HBSS supplemented with glucose (0.9 g/L) and 1% Antibiotic-Antimycotic. The tissue was then cut into 1–2 mm pieces and digested with Collagenase V (Sigma). After a 20 minute incubation in a 37°C water bath, the tissue was washed three times with the HBSS glucose solution. The tissue was further dissociated with TrypLE (Life Technologies) for 5 mins at room temperature and then washed with complete culture medium. The sample was finally resuspended in the culture medium and plated on rat tail collagen-coated 60 mm plates (BD Biosciences) and incubated at 37°C with 5% CO2/95% relative humidity. The plates were then trypsinized over a series of weeks to remove non-tumor cells. Tumor cells were replated on rat tail collagen-coated plates for expansion when needed. Cells were maintained in DMEM-F12 medium (Invitrogen) supplemented with 10% FBS and 1% Antimycotic/Antibiotic (Invitrogen).
Quantitative RT-PCR
RNA was isolated from pancreatic cancer cell lines using RNAEasy plus mini kit (Qiagen), and was used to prepare cDNA using iScript cDNA synthesis kit (Bio-Rad); both procedures followed the manufacturers’ protocols. They were subjected to a quantitative PCR to evaluate the expression levels of Endoglin using RT2 qPCR Primer Assay (SABioscience). The values were normalized against β-actin.
Western blot analysis and immunoprecipitation
Whole cell lysates were prepared in NP-40 lysis buffer supplemented with protease inhibitor cocktail (Sigma) and Halt phosphatase inhibitor (Thermo Scientific). Supernatant was collected by centrifugation at 12,000 rpm for 10 minutes at 4 °C. Thereafter, samples were subjected to SDS-Page and then transferred to PVDF membranes. The membranes were then immunoblotted and antibody-reactive bands detected by using the Supersignal West Pico substrate (Thermo Scientific). Immunoprecipitation experiments were performed as previously described (30).
siRNA transfection
Transient knockdown of endoglin was performed in ASPC-1 and MiaPaca-2 cell lines with two different anti-endoglin siRNA (Qiagen, Target sequence si 1: 5′-CGCCATGACCCTGGTACTAAA-′, Target sequence si 2: 5′-CAGCAATGAGGCGGTGGTCAA-3′) using Dharmafect-4 (Dharmacon). A control siRNA (Qiagen) lacking known human or mouse targets was used as a non-target control. RNA or protein was isolated from the cells 72–96 hours after the siRNA transfection and endoglin downregulation was determined by quantitative RT-PCR and Western blot analysis.
Lentiviral shRNA transduction
Stable knockdown of endoglin was performed in ASPC-1 cells using lentivirus shRNA transduction method. Two different Endoglin shRNA (Sigma TRC1.5, Target sequence for sh1: GTCTTGCAGAAACAGTCCATT, Target sequence for sh2: CCACTTCTACACAGTACCCAT) were used. The lentivirus particles were prepared using 293T cells following standard methods. ASPC-1 cells were transduced with the prepared lentivirus particles and stable colonies were isolated after selection with puromycin (1 μg/ml).
Design and synthesis of peptides
Peptides were designed based on the native eight-residue C-terminal sequence of endoglin (PCSTSSMA), with varying degrees of N-terminal lipidation to enhance cellular membrane permeability (Table 1). In addition, two positions were independently mutated to Lys residues, in which the side chain was benzoylated (AP1035, myristoyl-PCSTSS[K(εN-benzoyl)]A, and AP1036, myristoyl-PCST[K(εN-benzoyl)]SMA). The details of the synthesis procedures are given in the supplementary materials and methods.
Table 1.
List of peptides designed on the basis of the native eight-residue C-terminal sequence of endoglin (PCSTSSMA), with varying degrees of N-terminal lipidation.
| Compound code | Lipidated peptide | N-acyl C length |
|---|---|---|
| AP1030 | Acetyl-PCSTSSMA | 2 |
| AP1031 | Lauroyl-PCSTSSMA | 12 |
| AP1013 | Myristoyl-PCSTSSMA | 14 |
| AP1032 | Palmitoyl-PCSTSSMA | 16 |
| AP1033 | Stearoyl-PCSTSSMA | 18 |
| AP1063 | Palmitoyl-SMTSCAPS | 16 |
| AP1035 | Myristoyl-PCSTSS[K(Bn)]A | 16 |
| AP1036 | Myristoyl-PCST[K(Bn)]SMA | 16 |
Surface Plasmon Resonance (SPR)
The affinity of N-myristoyl-PCSTSSMA (AP1013) for recombinant GIPC PDZ was measured by surface plasmon resonance, using a Biacore X100 (GE Healthcare Life Sciences). After pH scouting was performed, determining an optimal pH of 5.5, GIPC PDZ was immobilized on a CM5 sensor chip via amine coupling reaction using N-hydroxysuccinimide (NHS) and N-ethyl-N′-(dimethylaminopropyl) carbodiimide (EDC). Solutions of AP1013 were prepared using HBS-EP buffer, in dilutions ranging from 10×Kd to 0.1×Kd. SPR experiments were conducted using the kinetic/affinity measurement workflow of the instrument. Collected data from the sensorgrams were analyzed using the Biacore evaluation software included with the instrument.
Thymidine incorporation assay
~2×104 cells were seeded in 24-well plates and cultured for indicated time in presence of siRNA or peptides. Then, 1 mCi of [3H] thymidine was added to each well and incubated for 4 hours. The cells were then washed with ice-cold PBS, fixed with chilled methanol, and lysed with 0.1N NaOH. The lysates were collected for measurement of trichloroacetic acid-precipitable radioactivity.
Drug sensitivity assay
~5×103 Cells were seeded in 96-well plates and treated with Endoglin siRNA or peptides for 24 hours followed by gemcitabine for 48 hours. At the end of the treatment period, cell viability was measured using Celltiter 96 Aqueous One Solution Cell Proliferation Assay (Promega) as per the manufacturer’s protocol. Half maximal inhibitory concentration (IC50) values were calculated as concentrations corresponding to a 50% reduction of cell viability.
In vivo tumor progression analysis
6–8 weeks old male SCID mice were obtained from NIH and housed in the institutional animal facilities. All animal work was performed under protocols approved by Mayo Clinic Institutional Animal Care and Use Committee. 1×106 of either control or Endoglin shRNA treated cells suspended in 50 μl PBS were injected orthotopically into the pancreas of 6–8 weeks old male SCID mice (5 mice in each group). Tumors were allowed to grow for three weeks. After three weeks, mice were sacrificed and tumor growth was analyzed. In another set of experiments, 5×106 ASPC-1 cells suspended in 100 μl PBS were injected subcutaneously into the right flanks of 6–8 weeks old male SCID mice (7 mice in each group). After 9 days, mice were randomized and either AP1063 or AP1032 dissolved in PBS containing 80% DMSO were injected intratumorally everyday for three weeks (500μg/mouse/day). After three weeks of treatment, mice were sacrificed and tumor growth was analyzed. Tumor volumes were calculated using the formula: V=0.5×a×b2, where ‘a’ is the longest tumor axis and ‘b’ is the shortest tumor axis.
Histological study
Tumors were removed and fixed in neutral buffered 10% formalin at room temperature for 24 hours prior to embedding in paraffin and sectioning. Sections were deparaffinized and then subjected to different immunohistochemical staining according to manufacturer’s instructions (DAB 150, Millipore). Stable diaminobenzidine was used as a chromogen substrate and the sections were counterstained with a hematoxylin solution. Images were acquired using Zeiss Axioplan 2 Microscope.
Statistical analysis
The independent-samples t-test was used to test the probability of significant differences between groups. Statistical significance was defined as p<0.05 (*) and statistical high significance was defined as p<0.01 (**). Error bars are given on the basis of calculated SD values.
RESULTS
Endoglin downregulation inhibits cell proliferation
Endoglin expression could be seen in both the pancreatic cancer cell lines tested (e.g. ASPC-1, MiaPaca-2). It was also expressed in several cell lines isolated from pancreatic cancer patient-derived xenografts such as 5160-1, MCPAN014, 5647-1 and 4482-1 (Figures 1A & 1B). However, the expression levels were varied among the cell lines. To check if the expression of endoglin is important for pancreatic cancer growth, downregulation of endoglin was performed in two different cell lines with different amount of endoglin expression (ASPC-1 with lower expression and MiaPaca-2 with higher expression). Two different siRNAs (ENG si 1 and ENG si 2) and shRNAs (ENG sh1 and ENG sh2) effectively reduced the endoglin expression at the mRNA and protein levels (Figures 1C, 1D & 1E). Both siRNAs inhibited cell proliferation in ASPC-1 and MiaPaca-2 cell lines (Figure 1F). Similarly, both shRNAs showed significant inhibition of proliferation in ASPC-1 (Figure 1G). Overall, these observations suggest that endoglin plays a significant role in proliferation.
Figure 1. Endoglin downregulation inhibits cell proliferation.

A & B. Endoglin mRNA and protein expression in pancreatic cancer cell lines as well as cells isolated from patients. C, D & E. Estimation of endoglin downregulation in ASPC-1 and MiaPaca-2 cell lines by endoglin siRNAs and shRNAs. F. Inhibition of cell proliferation in ASPC-1 and MiaPaca-2 after siRNA-mediated endoglin downregulation. G. Inhibition of cell proliferation in ASPC-1 after shRNA-mediated endoglin downregulation. ** denotes p<0.01 compared to respective controls.
Endoglin downregulation inhibits tumor growth
When endoglin-downregulated ASPC-1 cells were injected orthotopically into the pancreas of 6–8 week old SCID mice (5 mice in each group), and the resulting tumors were allowed to grow for 3 weeks, they were significantly smaller compared to the tumors arising from control shRNA treated cells (Figure 2A & 2B). The tumor volumes were 416.94±125.24 mm3 in control shRNA group versus 232.97±102.4 mm3 and 215.34±63.4 mm3 in ENG sh1 and ENG sh2 groups respectively. The tumor weights were 436.72±76.21 mg in control shRNA group versus 232.97±102.4 mg and 215.34±63.4 mg in ENG sh1 and ENG sh2 groups respectively. The proliferation of tumor cells was significantly lower in tumors from ENG sh1 and ENG sh2 groups compared to control shRNA group, as evident from Ki67 staining of the tumor tissue sections (Figure 2C). The abundance of Ki67 positive nuclei was 50.86±4.58% in control shRNA group versus 25.3±4.39% and 21.24±3.7% in ENG sh1 and ENG sh2 groups respectively (Figure 2D). This is in accord with our hypothesis that endoglin plays an important role in cancer progression, which is affected by endoglin downregulation, thus causing a slower tumor growth. However, no anti-angiogenic effects such as reduced microvessel density (MVD) or increased necrosis were observed in tumor tissue sections obtained from endoglin-downregulated ASPC-1 cells (Supplementary Figure S1). This was not surprising, considering these orthotopic tumors were mostly non-vascular. This also confirmed that the observed effect was due to the inhibition of tumor-cell specific endoglin rather than an anti-angiogenic effect.
Figure 2. Endoglin downregulation inhibits tumor growth.

A & B. 1×106 of control or endoglin shRNA treated ASPC-1 cells were injected orthotopically into the pancreas of 6–8 week old male SCID mice and tumors were allowed to grow for 3 weeks. Tumors arising from endoglin downregulated ASPC-1 cells were significantly smaller. * denotes p<0.05 compared to control. C. H&E and Ki67 immunohistochemical staining in the tumor tissues. Scale= 200 μm. D. Quantification of Ki67 stained nuclei in control and Endoglin shRNA-treated tumor tissue sections respectively. ** denotes p<0.01 compared to control.
Endoglin-GIPC interaction is necessary for proliferation
Endoglin has been shown to act in endothelial cells through its association with GIPC through a PDZ domain-binding motif (7). However, little has been known about the importance of endoglin-GIPC association in cancer cells. To this end, we designed an octapeptide (PCSTSSMA) mimicking the C-terminus of endoglin, a PDZ domain-binding motif, with the intent of selectively blocking endoglin-GIPC association in a competitive manner. A biochemical binding assay, using SPR under equilibrium analysis mode, demonstrated that N-myristoyl-PCSTSSMA (AP1013) binds to recombinant GIPC PDZ with a binding constant of Kd = 7.1±2.8 μM.
Derivatives in which the amino terminus of the peptide was acylated with fatty acids other than myristic (i.e., lauric, palmitic and stearic), as well as simple acetic acid, were prepared to serve as a form of control for the effect of lipidation (Table 1). Two additional N-myristoylated peptides were synthesized in which one of two native residues was sequentially replaced with Lys, in which the side chain had been modified with benzoyl moieties. We screened all these lipidated peptides in ASPC-1 cells and monitored their anti-proliferative activities, with the intent of selecting the analog with maximal activity. Only myristoyl, palmitoyl and stearoyl derivatives showed significant anti-proliferative activity, with a noticeable dose- response relationship (Figure 3A). The N-acetyl analog (AP1030), lacking a sufficiently long aliphatic tail that might enhance cell membrane penetration and permeability, was, unsurprisingly, both ineffective and lacked a dose-response effect.
Figure 3. Endoglin-GIPC interaction is necessary for proliferation.

A. Screening of endoglin PDZ domain-binding sequence peptides N-acylated with acetic and fatty acids in ASPC-1 cells. Palmitoyl-PCSTSSMA (AP1032) was selected for further experiments. B. AP1032 effectively blocked Endoglin-GIPC interaction but palmitoyl-SMTSCAPS (control peptide, AP1063) could not, as seen from the immunoprecipitation experiment. C & D. AP1032 showed greater inhibition of proliferation in ASPC-1 cells compared to MiaPaca-2 cells. AP1063 was used as a control. E. AP1032 also inhibited proliferation in cells isolated from patient-derived xenografts. Remarkably, AP1032 was more effective in cells with a lower level of endoglin expression.
We selected palmitoyl-PCSTSSMA (AP1032) for further studies, since it exhibited the highest anti-proliferative activity. We also synthesized a scrambled peptide with the same residues (SMTSCAPS), with N-terminal palmitoylation, to use as a negative control (AP1063). Treatment with AP1032 effectively inhibited endoglin-GIPC interaction in both ASPC-1 and MiaPaca-2 cell lines, compared to control AP1063 (Figure 3B). AP1032 inhibited proliferation in ASPC-1 (Figure 3C) and MiaPaca-2 (Figure 3D) cell lines. Interestingly, the peptide AP1032 is more effective in ASPC-1, which has lower endoglin expression, than in MiaPaca-2, with higher endoglin expression. To check if this observation can be generalized, two sets of cell lines isolated from patient-derived xenografts were chosen. 5647-1 and 4482-1 express comparatively lower amount of endoglin whereas 5160-1 and MCPAN014 express higher amount of endoglin (Figure 1A). As we presumed, the peptide AP1032 is more effective in 5647-1 and 4482-1, thus corroborating our model of competitive inhibition (Figure 3E).
Inhibition of Endoglin-GIPC interaction inhibits tumor growth
Intratumoral injections of AP1032 in a subcutaneous ASPC-1 tumor model developed in 6–8 week old SCID mice (7 mice in each group) showed significant inhibition of tumor growth compared to control peptide AP1063 (Figure 4A & 4B). The tumor volumes were 385.96±94.95 mm3 in the AP1063 treated group versus 153.95±27.13 mm3 in the AP1032 treated group. The tumor weights were 333.57±47.53 mg in the AP1063 treated group versus 166.7±29.49 mg in the AP1032 treated group. The proliferation of tumor cells was significantly lower in the AP1032-treated group as evident from Ki67 staining of the tumor tissue sections (Figure 4C). The abundance of Ki67 positive nuclei was 44.44±3.94% in the AP1063 treated group versus 11.22±2.25% in the AP1032 treated group (Figure 4D). This suggests that endoglin-GIPC interaction plays an important role in cancer progression. Interestingly, treatment with the peptide AP1032 resulted in a significant decrease in MVD and increased necrotic area compared to AP1063 in the subcutaneous model suggesting that the peptide AP1032 was also affecting the tumor vasculature (Supplementary Figure S2).
Figure 4. Inhibition of Endoglin-GIPC interaction inhibits tumor growth.

A & B. 5×106 ASPC-1 cells were injected subcutaneously into the right flanks of 6–8 week old male SCID mice and tumors were allowed to grow. After 9 days mice were randomized into two groups (n=7) and intra-tumoral injections of AP1032 or AP1063 (dissolved in PBS containing 80% DMSO) at a dosage of 500 μg/day/mouse were started. The daily treatment continued for 3 weeks, after which the mice were sacrificed, and the tumors were collected and analyzed. Tumors obtained from AP1032-treated group were significantly smaller compared to AP1063-treated group. C. H&E and Ki67 immunohistochemical staining in the tumor tissues. Scale= 200 μm. D. Quantification of Ki67 stained nuclei in AP1063-treated and AP1032-treated tumor tissue sections respectively. ** denotes p<0.01 compared to AP1063 treated group.
Endoglin downregulation does not affect Smad phophorylation but inhibition of Endoglin-GIPC interaction does
Since endoglin is a co-receptor of TGF-β signaling pathway, we thought to see whether the canonical Smad signaling is affected due to endoglin downregulation. Surprisingly, endoglin downregulation did not have significant effect on Smad phosphorylation (Figure 5A). We then checked whether blocking endoglin-GIPC interaction acts in the same manner. However, treatment with AP1032 clearly inhibits Smad-1/5 phosphorylation, which is involved in signaling pathways responsible for cell proliferation (Figure 5B). Expressions of Smad1 and Smad 2 were also inhibited due to the treatment with AP1032. From these result, we may conclude that downregulation of endoglin and blocking endoglin-GIPC interaction acts differently in pancreatic cancer cells.
Figure 5. Endoglin downregulation does not affect Smad phophorylation but inhibition of Endoglin-GIPC interaction does.
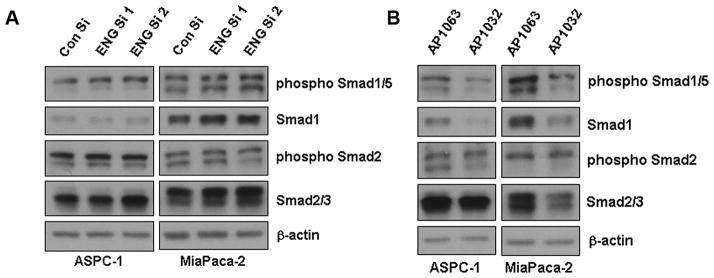
A. Smad phosphorylation was not significantly affected by endoglin downregulation. B. Inhibition of endoglin-GIPC interaction with treatment of AP1032 showed inhibition of Smad1/5 phosphorylation.
Both Endoglin downregulation and inhibition of endoglin-GIPC interaction induce differentiation
Since both endoglin downregulation and inhibition of endoglin-GIPC interaction inhibited tumor growth, we thought to check whether they induce differentiation in the pancreatic cancer cells. Endoglin downregulation increased E-cadherin expression in ASPC-1 and inhibited Id-1 expression in both ASPC-1 and MiaPaca-2 (Figure 6A). Treatment with AP1032 did not significantly increase E-cadherin but inhibited Id-1 expression in both ASPC-1 and MiaPaca-2 (Figure 6B). Therefore, both endoglin downregulation and inhibition of endoglin-GIPC interaction induce a differentiation phenotype to pancreatic cancer cells.
Figure 6. Both endoglin downregulation and inhibition of Endoglin-GIPC interaction induce differentiation and sensitize pancreatic cancer cells towards gemcitabine.

A. Endoglin downregulation increased E-cadherin expression in ASPC-1 and inhibited Id-1 in both ASPC-1 and MiaPaca-2 cell lines. B. Treatment with 100μmol/L AP1032 inhibited Id-1 in both ASPC-1 and MiaPaca-2 cell lines. C & D. Endoglin downregulation sensitized both ASPC-1 and MiaPaca-2 cell lines towards gemcitabine. E & F. Treatment with 50μmol/L AP1032 sensitized both ASPC-1 and MiaPaca-2 cell lines towards gemcitabine.
Both Endoglin downregulation and inhibition of endoglin-GIPC interaction sensitizes pancreatic cancer cells towards gemcitabine
Since both endoglin downregulation and inhibition of endoglin-GIPC interaction induced differentiation in pancreatic cancer cells, we explored whether they could sensitize pancreatic cancer cells towards standard chemotherapeutic drug gemcitabine. Our results show that endoglin downregulation decreases the IC50 value of gemcitabine from 44.92 nmol/L to 18.14 nmol/L (ENG si 1) and 24.54 nmol/L (ENG si 2) in ASPC-1 cells (Figure 6C). Similarly, the IC50 values for MiaPaca-2 cell lline are 52.08 nmol/L (con si), 15.73 nmol/L (ENG si 1) and 19.99 nmol/L (ENG si 2) (Figure 6D). Inhibition of endoglin-GIPC interaction also decreases the IC50 value from 47.37 nmol/L (AP1063) to 9.59 nmol/L (AP1032) in ASPC-1 (Figure 6E) and from 42.73 nmol/L (AP1063) to 15.83 nmol/L (AP1032) in MiaPaca-2 (Figure 6F). So, in both ASPC-1 and MiaPaca-2 cell lines, either genetic inhibition of endoglin or inhibition of endoglin-GIPC interaction elicited a significantly greater drug response compared to control, when treated with gemcitabine.
DISCUSSION
The primary canonical role of endoglin is as a TGF-β co-receptor (4). It is primarily expressed in endothelial cells (5, 6). It is also found in solid tumor vasculature and is a reliable marker of angiogenesis (9, 11–15). However, to date, only a few studies address the expression of endoglin on tumor cells and its potential role in cancer progression (20–22). A recent study reported the expression of endoglin in pancreatic cancer cells and cells isolated from PDAC patients (24). Here we have shown that endoglin is expressed in several pancreatic cancer cells and RNAi based downregulation of endoglin inhibits in vitro pancreatic cancer cell proliferation. In vivo tumorigenic property of pancreatic cancer cell line ASPC-1 was also significantly reduced upon endoglin downregulation. In addition, endoglin downregulation sensitized the cells towards conventional chemotherapeutic drugs possibly through the induction of differentiation as indicated from increased E-cadherin expression and reduced Id-1 expression. Surprisingly, endoglin downregulation had no significant effect on Smad phosphorylation which suggests that other TGF-β co-receptors are possibly compensating for the downregulation of endoglin. Nonetheless, from these results it appears that targeting endoglin in pancreatic cancer may prove to be useful.
In regards to therapeutic development in cancer patients, delivery of siRNA constructs has the potential to offer long duration of target inhibition as well as reduced toxicity compared to other approaches. However, development of a delivery modality for siRNA constructs remains the rate-limiting step in translational research. This is the reason we chose a peptide based competitive inhibition model. We designed an octapeptide based on the C-terminal PDZ domain-binding sequence of endoglin (PCSTSSMA), in order to competitively inhibit the association of endoglin with GIPC. Our SPR studies with AP1013 demonstrated binding to recombinantly-produced PDZ domain derived from the full-length GIPC gene, with a dissociation constant of Kd = 7.1±2.8 μM. This value is in line with binding affinities of peptides for other PDZ domains, which, biologically, are believed to be principally engaged in transient interactions not intended to bind their endogenous partner proteins with high affinity. It is possible, however, through judicious selection of standard (36) or nonstandard residues (37) or through chemical modification (38), to enhance the affinity of a peptide for a PDZ domain, and even selectivity, plasma stability and cellular permeability properties (39). These and other approaches highlight the significant amount of activity that has arisen around the design and development of molecular agents to target of PDZ domains (40).
In order to promote cell permeability, we adopted and expanded upon two strategies we had employed previously (35). For the first, we synthesized derivatives in which the amino terminus of the peptide was acylated with natural fatty acids of increasing carbon length (lauric, myristic, palmitic and stearic), as well as simple acetate, to serve as a form of control for the effect of lipidation (Table 1). Since the mode of cell membrane uptake and transmigration may be a function not only on the length of the fatty acid (i.e., its lipophilicity), but on the influence by the peptide sequence itself; that is, the overall length and combined contribution of polar and nonpolar residues. Thus, with different peptide sequences, it is prudent to explore lipid appendages of varying length, since it may not be possible to predict in advance which fatty acid would prove to be the best lipid conjugate. Addition of moieties at the N-terminal region of peptide ligands for PDZ domains is generally considered unobtrusive, since the primary recognition and binding energetics for PDZ domain-mediated interactions often seems to reside in 4 to 6 residues at the C-terminus.
The second influence from our earlier work (35) inspired us to chemically modify the side chain in each of two different positions within the endoglin peptide sequence. Sequentially substituting Lys for the endogenous Met and Ser residues at the “−1” and “−3” positions, respectively, we acylated the side chain amine with benzoate. This lysine(εN-benzoyl) unit serves as a ‘probe moiety’, to gauge the extent to which that binding position will not only tolerate such a substitution, but also potentially enhance binding affinity and, consequently, biological efficacy. In the absence of structural or other binding data, the chances that a single substitution would improve potency are limited, but the primary motivation would be, as just stated, to test for tolerance. If efficacy is either maintained, or at least not irredeemably compromised, this would provide a degree of justification for pursuing a larger scale chemical library approach, in which hundreds of different organic acids, many of which would be aromatic acids with different functionality, would be placed at one or both position, and each tested for affinity for GIPC PDZ. We have employed this strategy for a different PDZ domain, and were able to generate chemically-modified peptide ligands with enhanced binding affinity (38). Although the modified analogs AP1035 and AP1036 were not superior to the corresponding non-modified AP1013, the parent from which they derived, they did exhibit a degree of efficacy and dose-response that could justify a chemical library approach, as described above. This could prove to be the desired approach for devising next-generation compounds with improved potency over that of the native endoglin sequence.
Such peptide-based inhibitors will be important for targeting the PDZ domain-containing protein GIPC, a central player in various signaling pathways. Previously it has been shown that endoglin modulates TGF-β signaling through its association with GIPC in endothelial cells (7). To the best of our knowledge, however, there have been no reports exploring the role of endoglin-GIPC association in cancer cells. In this study we have shown that blocking the interaction of endoglin and GIPC, by means of targeting the PDZ domain of the latter with a lipidated peptide inhibitor, reduced pancreatic cancer cell proliferation. The in vivo tumor progression was also significantly reduced by this peptide. In addition, this peptide sensitized the cells towards gemcitabine possibly through the induction of differentiation as indicated from reduced Id-1 expression. Interestingly, Smad 1/5 phopsphorylation was inhibited here, which could not be observed in RNAi-based inhibition. We presume the reason behind this difference was due to the similarity between the PDZ-domain binding sequences of endoglin and other TGF-β co-receptors; allowing them to bind to GIPC possibly through the same PDZ domain. As a result, blocking that domain by treatment with AP1032 may block other interactions, thus inhibiting the downstream signaling pathway. However, downregulation of endoglin should have no effect on the binding of other co-receptors with GIPC. So, in the absence of endoglin, other co-receptors may still bind to GIPC and activate the downstream signaling pathway. Taken together, our data suggests that PDZ domain-targeting peptide-based inhibitors may have a better therapeutic potential than that of RNAi-based inhibition.
Currently, anti-endoglin therapy is being explored as a therapeutic in several cancers as an anti-angiogenic agent (16–19). The present study shows that endoglin-targeted therapies in pancreatic cancer may offer an additional advantage by targeting not only the vasculature but also the tumor cells expressing endoglin. Their effects on differentiation will also contribute to drug sensitivity, so a combination therapy may prove more efficacious in chemotherapy-resistant tumors. Overall, our data strongly suggests endoglin-targeted therapy as a double-speared approach to improve drug sensitivity through induction of differentiation in tumor cells in addition to anti-angiogenic therapy. This approach should be actively explored as a potential therapy for the treatment of pancreatic cancer.
Supplementary Material
Acknowledgments
We thank the Mayo Clinic Histology Core Facility and Optical Morphology Core Facility for their assistance with this work. We also thank Dr. Thomas C. Smyrk for providing kind and helpful suggestions with histological analysis.
Financial Support: This work is supported by NIH grants CA78383 and CA150190 (D. Mukhopadhyay).
Footnotes
Conflicts of interest: None.
References
- 1.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–57. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Smith BD, Smith GL, Hurria A, Hortobagyi GN, Buchholz TA. Future of cancer incidence in the United States: burdens upon an aging, changing nation. J Clin Oncol. 2009;27:2758–65. doi: 10.1200/JCO.2008.20.8983. [DOI] [PubMed] [Google Scholar]
- 4.Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999;274:584–94. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- 5.Gougos A, Letarte M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem. 1990;265:8361–4. [PubMed] [Google Scholar]
- 6.Bernabeu C, Conley BA, Vary CP. Novel biochemical pathways of endoglin in vascular cell physiology. J Cell Biochem. 2007;102:1375–88. doi: 10.1002/jcb.21594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee NY, Ray B, How T, Blobe GC. Endoglin promotes transforming growth factor beta-mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J Biol Chem. 2008;283:32527–33. doi: 10.1074/jbc.M803059200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, et al. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–7. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- 9.Behrem S, Zarkovic K, Eskinja N, Jonjic N. Endoglin is a better marker than CD31 in evaluation of angiogenesis in glioblastoma. Croat Med J. 2005;46:417–22. [PubMed] [Google Scholar]
- 10.Munson J, Bonner M, Fried L, Hofmekler J, Arbiser J, Bellamkonda R. Identifying new small molecule anti-invasive compounds for glioma treatment. Cell Cycle. 2013;12:2200–9. doi: 10.4161/cc.25334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Gohary YM, Silverman JF, Olson PR, Liu YL, Cohen JK, Miller R, et al. Endoglin (CD105) and vascular endothelial growth factor as prognostic markers in prostatic adenocarcinoma. Am J Clin Pathol. 2007;127:572–9. doi: 10.1309/X6NXYE57DLUE2NQ8. [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Esquer F, Agudo D, Martinez-Arribas F, Nunez-Villar MJ, Schneider J. mRNA expression of the angiogenesis markers VEGF and CD105 (endoglin) in human breast cancer. Anticancer Res. 2004;24:1581–5. [PubMed] [Google Scholar]
- 13.Saad RS, Liu YL, Nathan G, Celebrezze J, Medich D, Silverman JF. Endoglin (CD105) and vascular endothelial growth factor as prognostic markers in colorectal cancer. Mod Pathol. 2004;17:197–203. doi: 10.1038/modpathol.3800034. [DOI] [PubMed] [Google Scholar]
- 14.Yoshitomi H, Kobayashi S, Ohtsuka M, Kimura F, Shimizu H, Yoshidome H, et al. Specific expression of endoglin (CD105) in endothelial cells of intratumoral blood and lymphatic vessels in pancreatic cancer. Pancreas. 2008;37:275–81. doi: 10.1097/mpa.0b013e3181690b97. [DOI] [PubMed] [Google Scholar]
- 15.Yang LY, Lu WQ, Huang GW, Wang W. Correlation between CD105 expression and postoperative recurrence and metastasis of hepatocellular carcinoma. BMC Cancer. 2006;6:110. doi: 10.1186/1471-2407-6-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi N, Haba A, Matsuno F, Seon BK. Antiangiogenic therapy of established tumors in human skin/severe combined immunodeficiency mouse chimeras by anti-endoglin (CD105) monoclonal antibodies, and synergy between anti-endoglin antibody and cyclophosphamide. Cancer Res. 2001;61:7846–54. [PubMed] [Google Scholar]
- 17.Uneda S, Toi H, Tsujie T, Tsujie M, Harada N, Tsai H, et al. Anti-endoglin monoclonal antibodies are effective for suppressing metastasis and the primary tumors by targeting tumor vasculature. Int J Cancer. 2009;125:1446–53. doi: 10.1002/ijc.24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosen LS, Hurwitz HI, Wong MK, Goldman J, Mendelson DS, Figg WD, et al. A phase I first-in-human study of TRC105 (Anti-Endoglin Antibody) in patients with advanced cancer. Clin Cancer Res. 2012;18:4820–9. doi: 10.1158/1078-0432.CCR-12-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Starr MD, Brady JC, Dellinger A, Pang H, Adams B, et al. Modulation of circulating protein biomarkers following TRC105 (anti-endoglin antibody) treatment in patients with advanced cancer. Cancer Med. 2014;3:580–91. doi: 10.1002/cam4.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gromova P, Rubin BP, Thys A, Cullus P, Erneux C, Vanderwinden JM. ENDOGLIN/CD105 is expressed in KIT positive cells in the gut and in gastrointestinal stromal tumours. J Cell Mol Med. 2012;16:306–17. doi: 10.1111/j.1582-4934.2011.01315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidson B, Stavnes HT, Forsund M, Berner A, Staff AC. CD105 (Endoglin) expression in breast carcinoma effusions is a marker of poor survival. Breast. 2010;19:493–8. doi: 10.1016/j.breast.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Ziebarth AJ, Nowsheen S, Steg AD, Shah MM, Katre AA, Dobbin ZC, et al. Endoglin (CD105) contributes to platinum resistance and is a target for tumor-specific therapy in epithelial ovarian cancer. Clin Cancer Res. 2013;19:170–82. doi: 10.1158/1078-0432.CCR-12-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res. 2012;18:869–81. doi: 10.1158/1078-0432.CCR-11-2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujiwara K, Ohuchida K, Ohtsuka T, Mizumoto K, Shindo K, Ikenaga N, et al. Migratory activity of CD105+ pancreatic cancer cells is strongly enhanced by pancreatic stellate cells. Pancreas. 2013;42:1283–90. doi: 10.1097/mpa.0b013e318293e7bd. [DOI] [PubMed] [Google Scholar]
- 25.De Vries L, Lou X, Zhao G, Zheng B, Farquhar MG. GIPC, a PDZ domain containing protein, interacts specifically with the C terminus of RGS-GAIP. Proc Natl Acad Sci U S A. 1998;95:12340–5. doi: 10.1073/pnas.95.21.12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varsano T, Dong MQ, Niesman I, Gacula H, Lou X, Ma T, et al. GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol Cell Biol. 2006;26:8942–52. doi: 10.1128/MCB.00305-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wieman HL, Horn SR, Jacobs SR, Altman BJ, Kornbluth S, Rathmell JC. An essential role for the Glut1 PDZ-binding motif in growth factor regulation of Glut1 degradation and trafficking. Biochem J. 2009;418:345–67. doi: 10.1042/BJ20081422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang LH, Kalb RG, Strittmatter SM. A PDZ protein regulates the distribution of the transmembrane semaphorin, M-SemF. J Biol Chem. 1999;274:14137–46. doi: 10.1074/jbc.274.20.14137. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Mukhopadhyay D, Xu X. C terminus of RGS-GAIP-interacting protein conveys neuropilin-1-mediated signaling during angiogenesis. Faseb J. 2006;20:1513–5. doi: 10.1096/fj.05-5504fje. [DOI] [PubMed] [Google Scholar]
- 30.Muders MH, Vohra PK, Dutta SK, Wang E, Ikeda Y, Wang L, et al. Targeting GIPC/synectin in pancreatic cancer inhibits tumor growth. Clin Cancer Res. 2009;15:4095–103. doi: 10.1158/1078-0432.CCR-08-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muders MH, Dutta SK, Wang L, Lau JS, Bhattacharya R, Smyrk TC, et al. Expression and regulatory role of GAIP-interacting protein GIPC in pancreatic adenocarcinoma. Cancer Res. 2006;66:10264–8. doi: 10.1158/0008-5472.CAN-06-2321. [DOI] [PubMed] [Google Scholar]
- 32.Kedlaya RH, Bhat KM, Mitchell J, Darnell SJ, Setaluri V. TRP1 interacting PDZ-domain protein GIPC forms oligomers and is localized to intracellular vesicles in human melanocytes. Arch Biochem Biophys. 2006;454:160–9. doi: 10.1016/j.abb.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirikoshi H, Katoh M. Expression of human GIPC1 in normal tissues, cancer cell lines, and primary tumors. Int J Mol Med. 2002;9:509–13. [PubMed] [Google Scholar]
- 34.Wang L, Lau JS, Patra CR, Cao Y, Bhattacharya S, Dutta S, et al. RGS-GAIP-interacting protein controls breast cancer progression. Mol Cancer Res. 2010;8:1591–600. doi: 10.1158/1541-7786.MCR-10-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patra CR, Rupasinghe CN, Dutta SK, Bhattacharya S, Wang E, Spaller MR, et al. Chemically modified peptides targeting the PDZ domain of GIPC as a therapeutic approach for cancer. ACS Chem Biol. 2012;7:770–9. doi: 10.1021/cb200536r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saro D, Li T, Rupasinghe C, Paredes A, Caspers N, Spaller MR. A thermodynamic ligand binding study of the third PDZ domain (PDZ3) from the mammalian neuronal protein PSD-95. Biochemistry. 2007;46:6340–52. doi: 10.1021/bi062088k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Memic A, Spaller MR. How do halogen substituents contribute to protein-binding interactions? A thermodynamic study of peptide ligands with diverse aryl halides. Chembiochem. 2008;9:2793–5. doi: 10.1002/cbic.200800572. [DOI] [PubMed] [Google Scholar]
- 38.Udugamasooriya DG, Sharma SC, Spaller MR. A chemical library approach to organic-modified peptide ligands for PDZ domain proteins: a synthetic, thermodynamic and structural investigation. Chembiochem. 2008;9:1587–9. doi: 10.1002/cbic.200800126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bach A, Eildal JN, Stuhr-Hansen N, Deeskamp R, Gottschalk M, Pedersen SW, et al. Cell-permeable and plasma-stable peptidomimetic inhibitors of the postsynaptic density-95/N-methyl-D-aspartate receptor interaction. J Med Chem. 2011;54:1333–46. doi: 10.1021/jm1013924. [DOI] [PubMed] [Google Scholar]
- 40.Grillo-Bosch D, Choquet D, Sainlos M. Inhibition of PDZ domain-mediated interactions. Drug Discov Today Technol. 2013;10:e531–40. doi: 10.1016/j.ddtec.2012.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.