

Abstract
Endotoxin tolerance (ET) is an important phenomenon, which affects inflammation and phagocytosis. Pretreatment with low dose of lipopolysaccharide (LPS) can protect liver injury from various hepatotoxicants such as acetaminophen and pseudomonas aeruginosa exotoxin A. The current study aimed to investigate the protecting mechanisms of endotoxin tolerance in acute liver failure induced by D-galactosamine (D-GalN)/LPS and possible role of toll-like receptors 4 (TLR4) signaling pathway in this phenomenon. Acute liver failure was induced by Injection of D-GalN/LPS. To mimic endotoxin tolerance, male Sprague-Dawley rats were treated with low dose of LPS (0.1 mg/kg once a day intraperitoneally for consecutive five days) before subsequent injection of D-GalN/LPS. Rat survival was determined by survival rate. Liver injury was confirmed by serum biochemical and liver histopathological examination. Inflammatory cytokines were determined by ELISA and nuclear factor-kappa B (NF-κB) (P65), toll-like receptors 4 (TLR4) and Interleukin-1 receptor-associated kinase-1 (IRAK-1) were measured by reverse transcriptase polymerase chain reaction and western blot respectively. Pretreatment of LPS significantly improved rat survival. Moreover, rats pretreated with LPS exhibited lower serum enzyme (ALT, AST and TBiL) level, lower production of inflammatory cytokines and more minor liver histopathological damage than rats without pretreatment of LPS. LPS pretreatment suppressed production of TLR4 and IRAK-1. LPS pretreatment also inhibited activation of hepatic NF-κB. These results indicated that endotoxin tolerance contributed to liver protection against D-GalN/LPS induced acute liver failure through down-regulation of TLR4 and NF-κB pathway.
Keywords: Endotoxin tolerance, D-GalN/LPS, liver failure, TLR4, NF-κB
Introduction
Acute liver failure (ALF) is the result of a catastrophic injury to the liver, which develops coagulopathy and encephalopathy within a short period of time and induces systemic inflammation that involves in multi-organs [1,2]. The causes of ALF are diverse including infectious or metabolic disease, drug toxicity and a variety of other reasons. In general, mechanism to develop ALF is considered to be closely related to the disorder of the body’s immune balance but the specific mechanism is still unclear. However, massive necrosis and apoptosis of liver parenchymal cells (hepatocytes) are believed to release various inflammatory cytokines, which will directly lead to acute liver failure, systemic inflammation and disorder of immune system [3,4]. Moreover, signaling molecules such as toll-like receptors (TLRs) also play an important role in inflammatory necrosis.
Endotoxin, as a gram-negative bacterial lipopolysaccharide (LPS), can stimulate release of a wide variety of inflammatory mediators, which contribute to the development of ALF [5]. Prior exposure of innate immune cells like monocytes/macrophages or organs to small amounts of endotoxin is known to induce tolerance to subsequent endotoxin challenge, a phenomenon called endotoxin tolerance (ET) [6]. Although Dr. Beeson described the phenomenon of ET more than 60 years ago [7], the mechanism of ET is still yet fully understood and more in its clinical application. However, ET state can significantly alleviate severity of polymicrobial sepsis, inflammatory necrosis and function damage of organs with decreased inflammation and increased phagocytosis [8,9].
TLRs are membrane receptor and can recognize pathogen-associated molecular patterns (PAMPs). They participate in the body’s immune response system. LPS is the main component of endotoxin and can bind to TLR4. Recent studies suggest that TLR pathways may mediate interactions among dendritic cells, T lymphocytes and mast cells to modulate allergic immune responses [10]. TLR4 is the major pattern recognition receptors (PRR) involved in the detection of gram-negative bacteria and their associated endotoxins [11]. Once TLR4 is activated, it will activate transcription factor-nuclear factor-kappa B (NF-κB), which is a downstream intracellular molecule of different receptors and involves in activation of different pro-inflammatory genes expression such as pro-inflammatory cytokines. Increased expression of pro-inflammatory cytokines will also activate NF-κB. Therefore, regulation of TLR signaling is important in inflammatory reaction. For example, mutation in TLR4 gene could ameliorate cellular injury through down-regulation of NF-κB activation [12]. Moreover, interleukin-1 receptor-associated kinase-1 (IRAK-1) is an important positive regulator of TLR signaling and studies have found that enhanced expression of IRAK-1 could strongly activate the NF-κB [13] through binding to Toll-interacting protein (Tollip), which suppresses TLR activation and phosphorylation to promote inflammatory reaction termination and maintain resting state of immune cells.
Because of the important role of NF-κB and TLR4 in inflammation, in this study we tested whether small dose of LPS could ameliorate acute liver failure induced by D-GalN/LPS through regulation of TLR4, IRAK-1 and NF-κB (p65). It could explain possible role of ET in protection of inflammatory response induced organ injuries.
Materials and methods
Materials
D-GalN (G0500-1 g) and LPS (L-2880-10 mg) were purchased from Sigma Chemical (St. Louis, MO, USA). Antibodies against TLR4, IRAK-1, P65 and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Murine tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) enzyme-linked immunosorbent assay (ELISA) kits were acquired from the Beyotime institute of biotechnology (Nanjing, China). The mRNA primers (TLR4, IRAK-1, P65 and β-actin) and RNAiso Plus reagent were purchased from the Aidlab Biotechnologies Co., China.
Animals
Male Sprague-Dawley rats weighing 200-220 g were purchased from the Shanghai Laboratory Animal Center (Shanghai, China). The rats were fed with a standard chow diet and water freely. They were housed under normal laboratory conditions (21 ± 2°C, 12 h light-dark cycle). All experiments were conducted under the standard procedure set by the Committee for the Purpose of Control and Supervision of Experiments on Animals and the National Institutes of Health for the specification use of the experimental animals. The research protocol was approved by the Animal Ethics Committee of Wenzhou Medical University, China.
Experimental protocols
D-GalN and LPS were dissolved in sterile 0.9% sodium chloride according to the product description. The rats were divided into three groups randomly: control group, ALF group, ET+ALF group. For ET+ALF model, rats were given 0.1 mg/kg LPS intraperitoneally once every day for five days. In the ALF and control groups, rats were injected with the same volume of sterile 0.9% sodium chloride instead of LPS for the same treatment. After five days, rats in ET+ALF and ALF groups were given one intraperitoneal injection of 500 μL saline containing 800 mg/kg D-GalN (Sigma, USA) and 8 μg LPS (Sigma, USA). Rats in control group were injected with the same volume of sterile 0.9% sodium chloride. All rats were finally sacrificed with chloral hydrate (1.0 g/kg, intraperitoneally (i.p.)) at 2, 6, 12, 24, 48 hours, and blood was withdrawn from the heart. Livers were removed immediately and stored at -80°C for further research. Partial liver specimens were put in 10% neutral formalin before being processed for histopathological analysis.
In a separate experiment, survival rate was monitored for 3 days after injection of D-GalN/LPS both in ALF group (n = 10) and ET+ALF group (n = 10).
Serum tests and liver histological examination
Liver injury was estimated by biochemical serum markers such as ALT, AST and total bilirubin (TBiL) and pathological examination. Blood assays were performed by a biochemical automatic analyzer (Vitros750, Johnson & Johnson, Rochester, USA). Serum levels of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) were assayed by using antibody enzyme-linked immunosorbent assay (ELISA) kits according to the product specifications. Partial liver tissue samples were embedded in paraffin after fixed in formalin. Liver tissues were cut into 4 μm sections, which were then stained with hematoxylin and eosin (H&E) and observed under light microscopy.
RNA isolation and reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from liver tissue by using RNAiso Plus reagent (Aidlab Biote-chnologies Co., China) in accordance with the manufacturer’s description. Reverse transcription was conducted using SYBR ExScript RT-PCR kit (Aidlab Biotechnologies Co., China). The pri-mers of different genes were listed as following: TLR4 (225bp): forward primers: 5’-CAGCAAAGTCCCTGATGACA-3’ and reverse pri-mers: 5’-CCTGGGGAAAAACTCTGGAT-3’. IRAK-1 (103bp): forward primers: 5’-CCCATGACCCA-GAGGCAAAA-3’ and reverse primers: 5’-AGC-AAAGCAGCAGCCCTTTA-3’. P65 (505bp): forward primers: 5’-TTGAGCAGCCCAAGCAGCGG-3’ and reverse primers: 5’-GCAGTGTTGGGG-GCACGGTT-3’. β-actin (149bp): forward primers 5’-CAAGTTCAACGGCACAGTCAA-3’ and reverse primers 5’-TGGTGAAGACGCCAGTAGACTC-3’.
The following protocols were used for amplification: the mixture was first denatured at 94°C for 5 minutes following 35 cycles of denaturing at 94°C for 30 seconds, annealing at 58°C for 30 seconds (P65), 59°C for 30 seconds (IRAK-1), 60°C for 30 seconds (TLR4), 61°C for 30 seconds (β-actin) and extending at 72°C for 20 seconds followed by a final extension step at 72°C for 5 min.
Protein isolation and western blotting
Total intracellular protein was isolated using lysis buffer (RIPA buffer contains sodium deoxycholate, NP-40, SDS and PBS) supplemented with protease inhibitor cocktail. Following heat denaturation at 95°C for 5 min, the samples (15 μg protein each) were subjected to polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto a nitrocellulose membrane. The membrane was then blocked by 5% skim milk in TBS for 90 minutes at room temperature. The primary antibodies against TLR4 (Santa Cruz), IRAK-1 (Santa Cruz), P65 (Santa Cruz) and β-actin (Santa Cruz) were then incubated with the membranes overnight in TBST with 1% skim milk at 4°C, respectively. After being washed with TBST three times, the membrane was incubated with the secondary antibodies at room temperature for 1h. The bands were then observed by enhanced chemiluminescent reagents and exposed to x-ray film.
Statistical analysis
All data are presented as means ± standard deviation (SD). Statistical significances were analyzed through one-way analysis of variance (ANOVA) or the least significant difference (LSD) test. Survival after injection of D-GalN/LPS was compared using the log-rank method. SPSS19.0 software (IBM, USA) was used for statistical analyses and a value of P < 0.05 was considered statistically significance.
Results
Effect of LPS pretreatment on survival rate of ALF
We first examined the effect of LPS pretreatment on rat survival rate. As shown in Figure 1, after injection of D-GalN/LPS in ten rats of the ALF group, six rats died in total and four of them died in the first 12h. The mortality rate of rats in ALF group was 60% in 3 days. In contrast, all rats survived in LPS pretreated rats in ET+ALF group after injection of D-GalN/LPS. The mortality rate was 0% in 3 days. There was a significant difference in term of survival rate between these two groups (P < 0.05).
Figure 1.
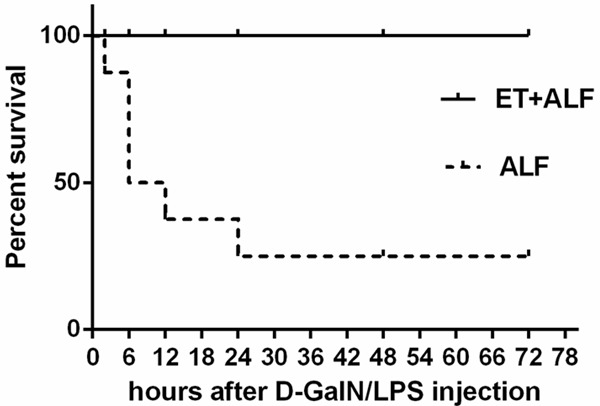
Effect of LPS pretreatment on survival rate of acute liver failure. After injection of D-GalN/LPS, there were significant death of rats in ALF group however, there was no death of rats in ET+ALF group (P < 0.05 by Log-rank test).
Effect of LPS pretreatment on liver injury and cytokines release in ALF
Liver injury and cytokines release were then investigated. Liver histopathological examination was observed after H&E staining, there was a normal histology of the liver from control group (Figure 2). However, liver tissue in ALF group and ET+ALF group shown different levels of injury with significant difference. In ALF group, liver tissue was injured seriously with damaged hepatic lobule, ruined hepatic cords, and a large number of infiltrated inflammatory cells (mostly are monocyte and lymphocyte). Necrosis and apoptosis can also be found (Figure 2). Meanwhile, liver injury in ET+ALF group was not severe. Necrosis of liver cells was less, hepatic lobule structure was visable and there were less infiltrated inflammatory cells (Figure 2). In addition, we examined serum markers of hepatic injury. As shown in Table 1, after intraperitoneal injection of D-GalN/LPS, ALT, AST and TBiL levels were elevated in both ALF and ET+ALF group. However, the increase in ALT, AST and TBiL in ALF group were much higher than that in ET+ALF group (p < 0.05) at every time point of the experiment. Moreover, ALT and AST in both groups reached the peak at 12h and TBiL levels continuously after D-GalN/LPS injection respectively. In addition to release of liver injury, pretreatment of LPS also reduced production of inflammatory mediators (TNF-α and IL-6) (Table 1). Although there were increased expression of both TNF-α and IL-6 after injection of D-GalN/LPS in both ALF and ET+ALF groups, the levels of TNF-α and IL-6 in ET+ALF rats were significantly lower than that in ALF rats (P < 0.05) at every time point of the experiment.
Figure 2.
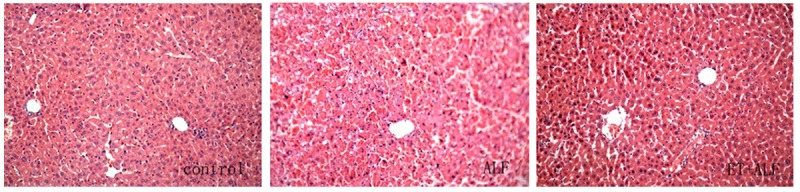
Histopathological examination of liver section. Liver sections from control, ALF and ET+ALF groups were stained with hematoxylin and eosin stain (original magnifications ×200). In control rats the liver tissues appear normal, however, the liver sections from ALF group show significant injury, totally damaged hepatic lobule, ruined hepatic cords, and a large number of inflammatory cells infiltrated. In ET+ALF rats, liver injury was not severe, hepatic lobule structure was visable and there was less necrosis of liver cells.
Table 1.
Chemistry changes and cytokines production after D-GalN/LPS injection at various time points
| Group | ALT (U/L) | AST (U/L) | TBiL (U/L) | IL-6 (A vaule) | TNF-α (A vaule) |
|---|---|---|---|---|---|
| Control | 32.9 ± 7.9 | 37.1 ± 10.0 | 7.6 ± 2.6 | 0.020 ± 0.005 | 0.023 ± 0.005 |
| ALF2h | 87.0 ± 9.5* | 359.0 ± 54.7* | 11.0 ± 2.6 | 0.136 ± 0.018* | 0.791 ± 0.451* |
| ALF6h | 760.8 ± 160.8* | 981.8 ± 93.6* | 17.8 ± 3.0* | 0.204 ± 0.025* | 0.945 ± 0.448* |
| ALF12h | 3474.8 ± 456.2* | 3339.5 ± 432.3 | 32.5 ± 6.8* | 0.286 ± 0.027* | 1.099 ± 0.474* |
| ALF24h | 1790.4 ± 127.4* | 1878.2 ± 215.9* | 65.3 ± 25.6* | 0.611 ± 0.178* | 1.429 ± 0.631* |
| ALF48h | 1618.6 ± 234.3* | 2039.4 ± 165.6* | 78.3 ± 15.5* | 0.394 ± 0.024* | 1.223 ± 0.516* |
| ET+ALF2h | 65.8 ± 10.8 | 251.3 ± 49.0*,# | 10.4 ± 2.2 | 0.056 ± 0.008 | 0.402 ± 0.075*,# |
| ET+ALF6h | 184.1 ± 39.8*,# | 323.9 ± 82.8*,# | 12.2 ± 2.9*,∆ | 0.077 ± 0.007*,# | 0.514 ± 0.074*,# |
| ET+ALF12h | 388.4 ± 53.6*,# | 385.2 ± 78.8*,# | 20.6 ± 6.7*,# | 0.097 ± 0.010*,# | 0.635 ± 0.122*,# |
| ET+ALF24h | 387.0 ± 130.5*,# | 488.1 ± 65.7*,# | 40.1 ± 20.9* | 0.251 ± 0.017*,# | 0.895 ± 0.152*,# |
| ET+ALF48h | 140.4 ± 30.4*,# | 136.8 ± 46.9*,# | 50.8 ± 17.3*,# | 0.181 ± 0.027*,# | 0.757 ± 0.137*,# |
Serum concentration of ALT, AST and TBiL; Cytokines production of IL-6 level and TNF-α level. Data shown are mean ± SD of 6 samples at each time point.
P < 0.05 versus control group;
P < 0.01 versus ALF group;
P < 0.05 versus ALF group.
Effect of LPS pretreatment on expression of P65, TLR4 and IRAK-1 in ALF
To elucidate the effect of LPS pretreatment on survival rate and liver injury, we further examined the mechanisms that may be involved in cytokine release specially NF-κB activation. The mRNA and protein levels of P65, TLR4 and IRAK-1 at 2h, 6h, 12h, 24h, 48h following injection of D-GalN/LPS were examined by RT-PCR and western blot respectively. As shown in Figure 3, substantial increases in P65, TLR4 and IRAK-1 expression were observed both in ALF and ET+ALF group compared to that in control group. However, LPS pretreatment significantly reduced the mRNA abundances of P65, TLR4 and IRAK-1 in rat liver of ET+ALF group (P < 0.05). The protein levels of P65, TLR4 and IRAK-1 in liver tissues were further documented by Western Blotting in Figure 4. Compared to control group, protein levels of P65, TLR4 and IRAK-1 were significantly increased in ALF and ET+ALF group after D-GalN/LPS injection. Moreover, after pretreatment of LPS, the protein abundance of P65, TLR4 and IRAK-1 were significantly reduced at the most time points of experiment respectively (P < 0.05, Figure 4).
Figure 3.
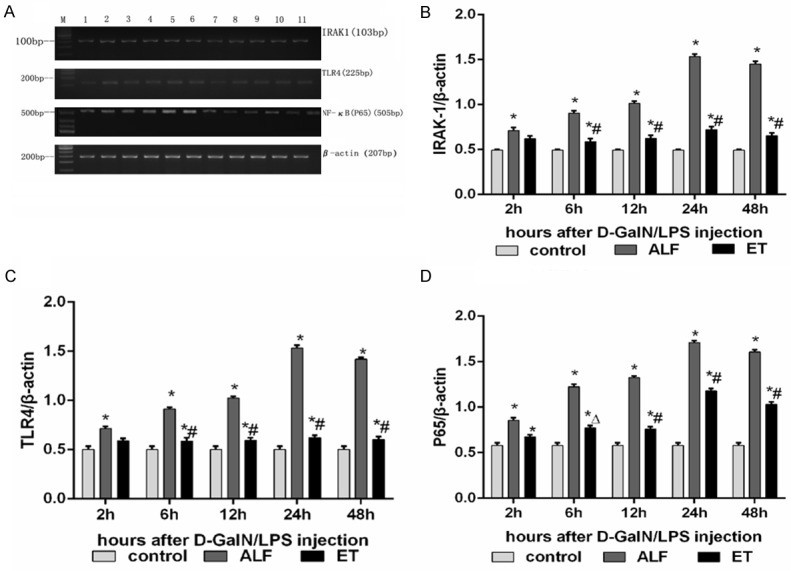
Inhibitory effects of LPS pretreatment on mRNA abundance of NF-κB pathway in the liver of acute liver failure. A displays typical pictures of mRNA abundance in the liver. (B-D) shows the quantitative levels of IRAK-1, TLR4 and P65 mRNA measured by RT-PCR respectively. Label M = mark; Lane 1 as control rat; Lane 2-6 represent rat liver of ET+ALF group at 2h, 6h, 12h, 24h, 48h after injection of D-GalN/LPS; Lane 7-11 represent rat liver of ALF group at 2h, 6h, 12h, 24h, 48h after injection of D-GalN/LPS; Levels of IRAK-1 (B), TLR4 (C) and P65 (D) were standardized to β-actin content. All data were expressed as mean ± SD of six rats at every point of time. *Represents P < 0.05 versus control group; ∆indicates P < 0.05 versus ALF group, #indicates P < 0.01 versus ALF group.
Figure 4.
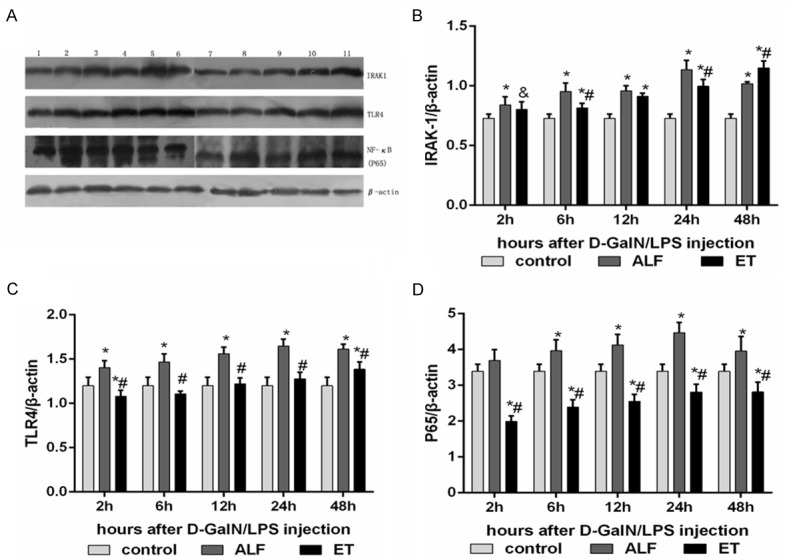
Inhibitory effects of LPS pretreatment on protein abundance NF-κB pathway in the liver of acute liver failure. A displays typical pictures of mRNA abundance in the liver. (B-D) shows the quantitative levels of IRAK-1, TLR4 and P65 protein measured by Western blot respectively. Label M = mark; Lane 1 as control rat; Lane 2-6 represent rat liver of ET+ALF group at 2h, 6h, 12h, 24h, 48h after injection of D-GalN/LPS; Lane 7-11 represent rat liver of ALF group at 2h, 6h, 12h, 24h, 48h after injection of D-GalN/LPS. Levels of IRAK-1 (B), TLR4 (C) and P65 (D) were standardized to β-actin content. All data were expressed as mean ± SD of six rats at every point of time. *Represents P < 0.05 versus control group; ∆indicates P < 0.05 versus ALF group, #indicates P < 0.01 versus ALF group.
Discussion
Currently, ALF in liver clinical still remains an extremely poor prognosis and results in high mortality. Therefore, mechanism research is urgently needed for better treatment. When liver failure occurs, hepatic function damaged seriously accompanied with intestinal endotoxemia. It has been confirmed that endotoxin (i.e., LPS)-mediated macrophage activation and inflammatory responses played a significant role in acute and chronic liver diseases [14].
D-GalN/LPS-induced ALF in mice has been used as a promising animal model for simulating the formation of ALF in human and illuminating the exact pathogenesis mechanism [15]. D-GalN can expend uridine monophosphate (UMP) in hepatic cells directly, resulting in reduction of nucleic acid, glycoprotein and lipid, ruining organelle, damaging hepatic cell structure and function and causing liver apoptosis [16,17]. Liver injury induced by LPS was reported to be related to activation of hepatic sinus gap endothelial cells and kupffer cells [18]. Therefore, D-GalN with LPS can extend liver injury involving biomacromolecule synthesis, hepatocyte apoptosis stimulation, free radical generation, lipid peroxidation and other various mechanisms. In our research, animal model of liver injury with the changes of serum ALT, AST, TBiL and liver pathology were consistent of clinical observation of patients with ALF. Our findings are also kept in line with other group using D-GalN/LPS as ALF model.
According to our findings, TNF-α and IL-6 could play an important role in D-GalN/LPS induced ALF, which is similar to reports that described D-GalN/LPS induced liver apoptosis is mainly mediated by TNF-α [19]. Moreover, TNF-α level in GalN/LPS induced ALF was correlated with rat survival and apoptosis of hepatocytes [20]. Previous clinical studies have shown that patients with ALF in critical condition often resulted in further liver injury because of sustained severe immune and inflammatory responses [21]. In this study, expression of inflammatory cytokines (TNF-α and IL-6) was closely correlated with liver injury. Moreover, these inflammatory cytokines (TNF-α and IL-6) also play an important role in immunization.
Immune tolerance can be affected by several factors such as deletion of reactive cells, receptor down-regulation, signal retardation and suppression of cytokines [22]. Endotoxin tolerance has been reported to have the liver protection by preventing hepatic mononuclear cell infiltration, reducing the production of pro-inflammatory cytokines and inducing the production of endogenous anti-inflammatory cytokines [23,24]. Our experimental data demonstrated that liver injury induced by D-GalN/LPS in rats was effectively attenuated by pretreatment of non-toxic dose of LPS for few days. This protective effect of LPS was believed to be related to the reduction of inflammatory cytokines, including TNF-α and IL-6. However, some studies have reported that over-expression of certain cytokines such as IL-10 may be related with endotoxin tolerance [25,26]. In this study, we found that LPS pretreatment significantly decreased serum levels of TNF-α and IL-6 in D-GalN/LPS-treated rats. Since TNF-α plays an important role in liver failure and IL-6 is one of the most important molecules involving in inflammation responses, the protective role of LPS pretreatment against D-GalN/LPS-induced liver failure could be due to alleviation of inflammation responses such as inhibition of cytokines production and their signaling pathway.
NF-κB pathway is a critical signal to trigger expression of inflammatory cytokines and exposure to LPS has been shown to induce NF-κB translocation from cytoplasm to the nucleus and bind to NF-κB promoter sites on DNA to activate transcription of TNF-α [27]. Moreover, prevention of NF-κB activation has been shown to reduce inflammation and apoptosis [15,28]. TLR4 is one of the most important signal-transducing receptors for structurally diverse microbial molecules (LPS) and can activate NF-κB to regulate the expression of inflammatory cytokines and immune reaction. Our results in this study are consistent with previous reports that D-GalN/LPS induced expression of TLR4 significantly, and at the same time, D-GalN/LPS activated NF-κB signal and induced the inflammatory cytokines expression, which all lead to liver injury. These findings are in line with TLR4 knock-out study, which indicated reduced inflammatory response and liver injury in TLR4ko mice with decreased NF-κB activation and reduced TNF-α level [29]. Moreover, silencing of TLR4 expression has a consequential correlation to inhibition of cytokine IL-6 and IL-8 production in response to LPS [30]. Therefore, our study confirmed that LPS pretreatment was capable of improving animal survival and alleviating liver injury induced by D-GalN/LPS. We suspected that LPS pretreatment protected the liver from D-GalN/LPS induced injury by reducing the TLR4 expression and suppressing activation of NF-κB.
In addition, IRAK-1, as a key molecule in LPS-mediated TLR4 pathway, can interact with TLR4 on cell membrane and activate LPS signal, which is similar to the signal transduction activated by Myeloid differentiation factor 88 (MyD88). Inhibition of IRAK-1 could alleviate inflammatory reactions via down-regulating the expression of inflammatory mediators [31,32]. Our results was corresponding to the study that IRAK-1 expression was significantly decreased in condition of LPS pretreatment. It is worthy of attention that signaling pathways can often be regulated bidirectionally. In a recent study, inhibition of TNF-α is able to suppress the activation of TLR4 and NF-κB signaling pathway [33]. Therefore, LPS pretreatment may have an important role in interaction of signal pathways that regulate immunologic balance. Moreover, pretreatment with LPS may have an impact on autophosphorylation of signal molecules such as IRAK-1 and TLR4 leading to integration of complex signaling pathways. Or regulation of signaling proteins expression may be the key point in protecting liver tissues from being injured by excessive immune reactions. More importantly, understanding of the specific mechanisms of endotoxin tolerance will improve our knowledge of ALF pathophysiology and contribute to the possibility of clinical therapy in future.
In conclusion, LPS pretreatment significantly improved survival and protected liver injury from D-GalN/LPS induced acute liver failure. The mechanism may be related to effect of LPS pretreatment on inhibiting inflammatory cytokines production through TLR4 pathway such as NF-κ and IRAK-1. Although time of LPS pretreatment could be critical in acute liver failure, current study at lease provides new targets and treatment options for this disease.
Acknowledgements
This work was supported by a grant from the Zhejiang Provincial Natural Science Foundation of China (LY12H03002).
Disclosure of conflict of interest
None.
References
- 1.O’Grady JG. Acute liver failure. Postgrad Med J. 2005;81:148–154. doi: 10.1136/pgmj.2004.026005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt LE, Larsen FS. Prognostic implications of hyperlactatemia, multiple organ failure, and systemic inflammatory response syndrome in patients with acetaminophen-induced acute liver failure. Crit Care Med. 2006;34:337–343. doi: 10.1097/01.ccm.0000194724.70031.b6. [DOI] [PubMed] [Google Scholar]
- 3.Galle PR. Apoptosis in liver disease. J Hepatol. 1997;27:405–412. doi: 10.1016/s0168-8278(97)80189-4. [DOI] [PubMed] [Google Scholar]
- 4.Nakama T, Hirono S, Moriuchi A, Hasuike S, Nagata K, Hori T, Ido A, Hayashi K, Tsubouchi H. Etoposide prevents apoptosis in mouse liver with D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure resulting in reduction of lethality. Hepatology. 2001;33:1441–1450. doi: 10.1053/jhep.2001.24561. [DOI] [PubMed] [Google Scholar]
- 5.Ogushi I, Iimuro Y, Seki E, Son G, Hirano T, Hada T, Tsutsui H, Nakanishi K, Morishita R, Kaneda Y, Fujimoto J. Nuclear factor kappa B decoy oligodeoxynucleotides prevent endotoxin-induced fatal liver failure in a murine model. Hepatology. 2003;38:335–344. doi: 10.1053/jhep.2003.50298. [DOI] [PubMed] [Google Scholar]
- 6.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Beeson PB. Development of tolerance to typhoid bacterial pyrogen and its abolition by reticulo-endothelial blockade. Proc Soc Exp Biol Med. 1946;61:248–250. doi: 10.3181/00379727-61-15291p. [DOI] [PubMed] [Google Scholar]
- 8.Shi DW, Zhang J, Jiang HN, Tong CY, Gu GR, Ji Y, Summah H, Qu JM. LPS pretreatment ameliorates multiple organ injuries and improves survival in a murine model of polymicrobial sepsis. Inflamm Res. 2011;60:841–849. doi: 10.1007/s00011-011-0342-5. [DOI] [PubMed] [Google Scholar]
- 9.Virca GD, Kim SY, Glaser KB, Ulevitch RJ. Lipopolysaccharide induces hyporesponsiveness to its own action in RAW 264.7 cells. J Biol Chem. 1989;264:21951–21956. [PubMed] [Google Scholar]
- 10.Bellou A, Schaub B, Ting L, Finn PW. Toll receptors modulate allergic responses: interaction with dendritic cells, T cells and mast cells. Curr Opin Allergy Clin Immunol. 2003;3:487–494. doi: 10.1097/00130832-200312000-00011. [DOI] [PubMed] [Google Scholar]
- 11.Tissieres P, Dunn-Siegrist I, Schappi M, Elson G, Comte R, Nobre V, Pugin J. Soluble MD-2 is an acute-phase protein and an opsonin for Gram-negative bacteria. Blood. 2008;111:2122–2131. doi: 10.1182/blood-2007-06-097782. [DOI] [PubMed] [Google Scholar]
- 12.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 13.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nolan JP. The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology. 2010;52:1829–1835. doi: 10.1002/hep.23917. [DOI] [PubMed] [Google Scholar]
- 15.Park JH, Kim KH, Lee WR, Han SM, Park KK. Protective effect of melittin on inflammation and apoptosis in acute liver failure. Apoptosis. 2012;17:61–69. doi: 10.1007/s10495-011-0659-0. [DOI] [PubMed] [Google Scholar]
- 16.Chen JC, Ng CJ, Chiu TF, Chen HM. Altered neutrophil apoptosis activity is reversed by melatonin in liver ischemia-reperfusion. J Pineal Res. 2003;34:260–264. doi: 10.1034/j.1600-079x.2003.t01-1-00031.x. [DOI] [PubMed] [Google Scholar]
- 17.Tsutsui S, Hirasawa K, Takeda M, Itagaki S, Kawamura S, Maeda K, Mikami T, Doi K. Apoptosis of murine hepatocytes induced by high doses of galactosamine. J Vet Med Sci. 1997;59:785–790. doi: 10.1292/jvms.59.785. [DOI] [PubMed] [Google Scholar]
- 18.Blatteis CM, Li S, Li Z, Perlik V, Feleder C. Signaling the brain in systemic inflammation: the role of complement. Front Biosci. 2004;9:915–931. doi: 10.2741/1297. [DOI] [PubMed] [Google Scholar]
- 19.Josephs MD, Bahjat FR, Fukuzuka K, Ksontini R, Solorzano CC, Edwards CK 3rd, Tannahill CL, MacKay SL, Copeland EM 3rd, Moldawer LL. Lipopolysaccharide and D-galactosamine-induced hepatic injury is mediated by TNF-alpha and not by Fas ligand. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1196–1201. doi: 10.1152/ajpregu.2000.278.5.R1196. [DOI] [PubMed] [Google Scholar]
- 20.Mignon A, Rouquet N, Fabre M, Martin S, Pages JC, Dhainaut JF, Kahn A, Briand P, Joulin V. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Respir Crit Care Med. 1999;159:1308–1315. doi: 10.1164/ajrccm.159.4.9712012. [DOI] [PubMed] [Google Scholar]
- 21.Meloni C, Morosetti M, Turani F, Palombo G, Meschini L, Zupancich E, Taccone-Gallucci M, Di Giulio S, Casciani CU. Cardiac function and oxygen balance in septic patients during continuous hemofiltration. Blood Purif. 1998;16:140–146. doi: 10.1159/000014327. [DOI] [PubMed] [Google Scholar]
- 22.Albrecht V, Hofer TP, Foxwell B, Frankenberger M, Ziegler-Heitbrock L. Tolerance induced via TLR2 and TLR4 in human dendritic cells: role of IRAK-1. BMC Immunol. 2008;9:69. doi: 10.1186/1471-2172-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishikage T, Seki S, Toyabe S, Abo T, Kagata Y, Iwai T, Hiraide H. Inhibition of concanavalin A-induced hepatic injury of mice by bacterial lipopolysaccharide via the induction of IL-6 and the subsequent reduction of IL-4: the cytokine milieu of concanavalin A hepatitis. J Hepatol. 1999;31:18–26. doi: 10.1016/s0168-8278(99)80159-7. [DOI] [PubMed] [Google Scholar]
- 24.Colletti LM, Remick DG, Campbell DA Jr. LPS pretreatment protects from hepatic ischemia/reperfusion. J Surg Res. 1994;57:337–343. doi: 10.1006/jsre.1994.1152. [DOI] [PubMed] [Google Scholar]
- 25.del Fresno C, Garcia-Rio F, Gomez-Pina V, Soares-Schanoski A, Fernandez-Ruiz I, Jurado T, Kajiji T, Shu C, Marin E, Gutierrez del Arroyo A, Prados C, Arnalich F, Fuentes-Prior P, Biswas SK, Lopez-Collazo E. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J Immunol. 2009;182:6494–6507. doi: 10.4049/jimmunol.0803350. [DOI] [PubMed] [Google Scholar]
- 26.Chiu CC, Huang YT, Wang YC, Chang YC, Ching YH, Chen HH, Chuang HL. Pretreatment with lipopolysaccharide ameliorates Pseu-domonas exotoxin A-induced hepatotoxicity in rats. Immunopharmacol Immunotoxicol. 2013;35:296–303. doi: 10.3109/08923973.2013.764503. [DOI] [PubMed] [Google Scholar]
- 27.Lin M, Rippe RA, Niemela O, Brittenham G, Tsukamoto H. Role of iron in NF-kappa B activation and cytokine gene expression by rat hepatic macrophages. Am J Physiol. 1997;272:G1355–1364. doi: 10.1152/ajpgi.1997.272.6.G1355. [DOI] [PubMed] [Google Scholar]
- 28.Watson MR, Wallace K, Gieling RG, Manas DM, Jaffray E, Hay RT, Mann DA, Oakley F. NF-kappaB is a critical regulator of the survival of rodent and human hepatic myofibroblasts. J Hepatol. 2008;48:589–597. doi: 10.1016/j.jhep.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Ben Ari Z, Avlas O, Pappo O, Zilbermints V, Cheporko Y, Bachmetov L, Zemel R, Shainberg A, Sharon E, Grief F, Hochhauser E. Reduced hepatic injury in Toll-like receptor 4-deficient mice following D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure. Cell Physiol Biochem. 2012;29:41–50. doi: 10.1159/000337585. [DOI] [PubMed] [Google Scholar]
- 30.Wang AC, Ma YB, Wu FX, Ma ZF, Liu NF, Gao R, Gao YS, Sheng XG. TLR4 induces tumor growth and inhibits paclitaxel activity in MyD88-positive human ovarian carcinoma. Oncol Lett. 2014;7:871–877. doi: 10.3892/ol.2013.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joh EH, Jeong JJ, Kim DH. Kalopanaxsaponin B inhibits LPS-induced inflammation by inhibiting IRAK1 Kinase. Cell Immunol. 2012;279:103–108. doi: 10.1016/j.cellimm.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Joh EH, Kim DH. Kalopanaxsaponin A ameliorates experimental colitis in mice by inhibiting IRAK-1 activation in the NF-kappaB and MAPK pathways. Br J Pharmacol. 2011;162:1731–1742. doi: 10.1111/j.1476-5381.2010.01195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang F, Li X, Wang LK, Wang LW, Han XQ, Zhang H, Gong ZJ. Inhibitions of NF-kappaB and TNF-alpha result in differential effects in rats with acute on chronic liver failure induced by d-Gal and LPS. Inflammation. 2014;37:848–857. doi: 10.1007/s10753-013-9805-x. [DOI] [PubMed] [Google Scholar]