

Abstract
Non small cell lung cancer (NSCLC) accounts for 85% of all lung cancers and is the most common cause of lung cancer death. Currently, the epidermal growth factor receptor inhibitor gefitinib is widely used for patients with advanced NSCLC. However, drug resistance is a major obstacle. Mig-6 is a feedback inhibitor of EGFR and its down-stream pathway; it has been shown to play a role in gefitinib sensitivity. There is neither systematical research on the relationship between Mig-6 expression and gefitinib sensitivity, nor has the contribution of up-regulated Mig-6 on the gefitinib-resistant cell lines. In the present work, four NSCLC cell lines (H1299, A549, PC-9, and PC-9/AB11) with different sensitivities to gefitinib were subjected to analysis of the expression of Mig-6. We found that Mig-6 is over-expressed in gefitinib-sensitive NSCLC cell lines, but is low in gefitinib-resistant NSCLC cell lines. Further analysis revealed that over-expression of Mig-6 increased cell apoptosis and inhibited proliferation of gefitinib-resistant NSCLC cells treated with gefitinib, whereas lowering the expression of Mig-6 decreased cell apoptosis and promoted cell proliferation after treatment with gefitinib in gefitinib-sensitive NSCLC cell lines. These results suggest that Mig-6 is involved in mediating the response to gefitinib in NSCLC cell lines. Additionally we demonstrated that Mig-6 could reverse gefitinib resistance through inhibition of EGFR/ERK pathway in NSCLC cells. Our work uncovered that Mig-6 may be an effective therapeutic target in gefitinib-resistant lung cancer patients.
Keywords: Mig-6, gefitinib resistance, NSCLC, EGFR signaling
Introduction
Lung cancer is the leading cause of cancer deaths worldwide. Non small cell lung cancer (NSCLC) accounts for 85% of all lung cancers and is the most common cause of lung cancer death. Most of lung cancer patients are diagnosed at an advanced stage with extremely poor prognoses. With the elucidation of the molecular abnormalities in NSCLC, much hope has been laid on epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), such as gefitinib [1]. Thus, gefitinib have been suggested for use as a first line therapy for patients with NSCLC harboring EGFR mutations [2]. Despite gefitinib has improved progression-free and overall survival in patients, drug resistance remained a big problem to affect the patient survival [3].
Previous studies have proposed several aberrant signaling pathways are associated with the sensitivity of NSCLC cells to gefitinib, such as PI3K/Akt/mTOR pathway, Ras/Raf/MAPK pathway [4]. Another study found phosphorylation levels of the EGFR downstream proteins inhibited by gefitinib is large between gefitinib sensitive and resistant cells [5]. These all indicated that inhibiting the down-stream pathway of EGFR could be an effective way to increase the sensitivity of NSCLC cells to gefitinib.
Mitogen-inducible gene 6 (Mig-6), an immediate early response gene, is a specific negative regulator of EGFR. Down-regulation of Mig-6 was found in various tumor tissues [6-8]. Mig-6 binds to EGFR family tyrosine kinase via its EGFR-binding domain thus leading to inhibition of EGFR autophosphorylation and reduced MAPK activity [9,10]. Mig-6 can restore down-regulation of oncogenic EGFR molecules that escape ubiquitylation [11]. Mig-6 can also inhibit the binding of EGFR to Shc, which enhances the synergistic effect of Mig-6 and gefitinib [5]. However, to date there has no systematic evaluation of the role of Mig-6 in gefitinib resistance in NSCLC cells.
In this study, we demonstrated that Mig-6 is overexpressed in gefitinib-sensitive PC-9 cell lines, but not gefitinib resistant NSCLC cell lines. Up-regulated Mig-6 was found to increase the sensitivity of NSCLC cells to gefitinib-induced proliferation, inhibition and apoptosis, while down-regulated Mig-6 led to gefitinib resistance. Moreover, we also determined that Mig-6 mediates gefitinib sensitization by inhibiting EGFR/ERK pathway.
Materials and methods
Cell culture
H1299, A549 cell lines were purchased from ATCC, PC-9 and PC-9 gefitinib resistant (PC-9/AB11) were gifts from Shanghai Pulmonary Hospital of Tongji University, China. Cells are maintained in RPMI 1640 or DMEM medium supplemented with 10% FBS (Hyclone) in a 5% CO2 incubator at 37°C with saturated humidity. Gefitinib was purchased from Astra Zeneca (Britain). One table of gefitinib was dissolved into 22.37 mL DMSO to prepare 25 mM mother solution. It takes 0.1 mL mother solution, by adding 100 mL DMEM medium, to make 25 μM working solution (final concentration of DMSO < 0.1%), which does not cause the cell biological properties change. Add 0.05 μM gefitinib culture to maintain PC9/AB11 drug resistance. Add DMSO into the culture of sensitive cell line PC9. Cells in the exponential growth phase were used for all experiments.
Cell proliferation assay
Cell proliferation assay was performed using Cell Counting Kit-8® solution (Dojindo, Gaithersburg, MD) according to the manufacturer’s protocol. Briefly, cells were seeded at a concentration of 5×103 cells/100 μl/well in 96-well culture plates and treated with 10 μl/well of Cell Counting Kit-8® solution during the last 4 hours of the culture. Optical density of the wells was measured at 450 nm using a microplate reader. Each treatment was assayed in triplicate in the same experiment.
Quantitative real-time PCR (SYBR Green method)
Quantitative real-time PCR was performed using SYBR Green PCR master mix (Applied Biosystems) in a total volume of 20 μl on 7900HT Fast Real-Time PCR System (Applied Biosystems) as follows: 95°C for 30 seconds, 40 cycles of 95°C for 5 seconds, 60°C for 30 seconds. A dissociation step was performed to generate a melting curve to confirm the specificity of the amplification. β-actin was used as the reference gene. The relative levels of gene expression were represented as ΔCt=Ctgene-Ctreference, and the fold change of gene expression was calculated by the 2-ΔΔCt method. The primer sequences are provided in Table 1, Experiments were repeated in triplicate.
Table 1.
Primer sequences
| Name | Primer sequences |
|---|---|
| ACTIN forward | 5’-ATAGCACAGCCTGGATAGCAACGTAC-3’ |
| ACTIN reverse | 5’-CACCTTCTACAATGAGCTGCGTGTG-3’ |
| Mig-6 forward | 5’-TCTTCCACCGTTGCCAATCT-3’ |
| Mig-6 reverse | 5’-TTCGCCTGCCAGGAACATC-3’ |
Western blot analysis
Total proteins from cell lines were extracted in a lysis buffer (Thermo Fisher Scientific, Rockford, IL) and quantified using the Bradford method. Fifty micrograms of protein were separated by SDS-PAGE (12%). After transferring, the polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA) were incubated overnight at 4°C with the following antibodies Mig-6 (1: 1000; protein tech), β-actin (1: 000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-ERK, anti-phospho-EGFR (1: 1000; Cell Signaling Technology, Danvers, MA). After incubation with peroxidase-coupled anti-mouse or rabbit IgG (Santa Cruz Biotechnology) at 37°C for 2 hours, bound proteins were visualized using ECL (Thermo Fisher Scientific) and detected using Chemidoc XRS+ (Bio-rad, USA). The relative protein levels were calculated based on β-actin as the loading control.
Cell apoptosis experiments
Apoptosis was detected using an Annexin V-FITC/PI double staining Kit (KeyGEN, Nanjing, China). Cells were harvested and washed twice with cold PBS by gentle shaking. Cells were then resuspended and added to Binding buffer (1×); cell density was adjusted to 2-5×105/mL. In the dark, 5 μL Annexin V-FITC was added to the cell suspension volume of 195 μL and incubated for 10 min at room temperature before the addition of 190 μL Binding buffer (1×) and 10 μL PI. Ten thousand events per sample were acquired using a FACS-scan flow cytometer (Becton-Dickinson, San Jose, CA, USA) and the percentage of cell apoptosis was analyzed using CellQuest analysis software (Becton-Dickinson).
Statistical analysis
SPSS version 19.0 for Windows was used for all analyses. Student’s t-test was used to compare differences between groups. P-values were based on the two-sided statistical analysis and P < 0.05 was considered to indicate statistical significance.
Results
Different expression of Mig-6 in four NSCLC cell lines has relationship with their different sensitivity to gefitinib
H1299, A549, gefitinib-sensitive (PC9) and PC9 gefitinib-resistant (PC9/AB11) cells were treated with different doses of gefitinib (0.01 μM, 0.1 μM, 1 μM, 5 μM, 10 μM, and 20 μM) and cell proliferation rate was detected by the CCK8 assay. The results showed that PC9 cell lines were sensitive to gefitinib whereas H1299, A549 and PC9/AB11 were relatively resistant (Figure 1A). We further examined the expression of Mig-6 in four NSCLC cell lines by Real time-PCR or western blot. Mig-6 was high expression in gefitinib-sensitive cell line PC9. In contrast, Mig-6 expression was significantly lower in the gefitinib-resistant cell lines, H1299, A549 and PC9/AB11 (Figure 1B and 1C).
Figure 1.
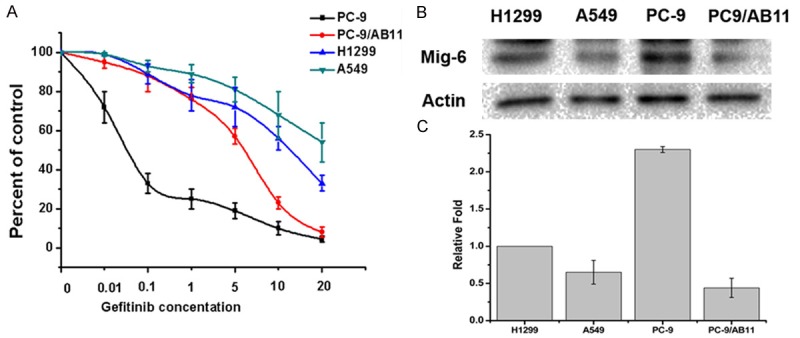
Four NSCLC cell lines exhibited different sensitivity to gefitinib and different expression level of Mig-6. A. H1299, A549, PC9 and PC9/AB11 were treated with different dose of gefitinib, as indicated. Seventy-two hours later cell proliferation rates were detected by CCK-8 assay. The error bar represents the standard deviation (SD); B. The expressions of Mig-6 were detected in H1299, A549, PC9 and PC9/AB11 cells by western blot; C. The expressions of Mig-6 were detected in H1299, A549, PC9 and PC9/AB11 cells by Real-time PCR.
Mig-6 overcomes gefitinib resistant in NSCLC cell lines
To verify the relationship between Mig-6 expression and the sensitivity to gefitinib in different cell lines, Mig-6 over-expression and knockdown experiment were launched. We examined transfection efficiency by protein expression levels after 48h of transfection treatment (Figure 2A). We next analyzed the effect of Mig-6 on the proliferation rates of gefitinib-resistant cells after gefitinib treatment using the CCK8 assay. We found that transfection of H1299, A549, and PC9/AB11 with Mig-6 plasmid significantly promoted gefitinib-mediated growth inhibition to different degrees. At the same time proliferation of cells transfected with siMig-6 was increased in a dose-dependent fashion compared with that of cells transfected with NC after gefitinib treatment in PC9 cell line (Figure 2B). Thus, together these findings suggest that Mig-6 is involved in mediating the response to gefitinib in NSCLC cell lines.
Figure 2.

The sensitivity to gefitinib was changed by up-regulated or down-regulated of Mig-6 in lung cancer cells. A. Western blots analysis of Mig-6 transfection efficiency in H1299, A549 and PC9/AB11 and knock down efficiency of Mig-6 siRNA in the PC9 cell lines; B. After transfection with Mig-6 plasmid or siRNA for 48 h, cell proliferation rates were detected by CCK-8 assay.
In NSCLC cell lines, Mig-6 regulate the sensitivity to gefitinib through inhibit EGFR/ERK pathway
We further studied transfection of Mig-6 affecting the proliferation, apoptosis level after gefitinib treatment and the possible pathway. After transfection with Mig-6, as well as added 10 μM gefitinib, cell proliferation rate was determined by CCK-8 assay for 5 days. Compared to the PC group, tansfection of Mig-6 can significant improve gefitinib induced the cell growth inhibiting (Figure 3A). Similar to the proliferation results, gefitinib induced cell apoptosis rates of Mig-6 transfection group is much higher than PC group (H1299 empty vector versus Mig-6 plasmid: 10.34±2.22% versus 33.78± 4.26%, P < 0.05; A549 empty vector versus Mig-6 plasmid: 8.75±2.56% versus 20±3.36%, P < 0.05) (Figure 3B). Mig-6 is the feedback factor of EGFR pathway that has been proved in many studies. Now, we want to explore whether this pathway is contributed to the synergy of Mig-6 and gefitinib. As show in Figure 3C, transfection of Mig-6 can reduce the expression of P-EGFR and P-ERK. At the same time gefitinib-induced P-EGFR and P-ERK inhibitions were significant in Mig-6 transfected cell lines, but not in PC group. Mig-6-induced increase in the gefitinib sensitivity was at least partly by EGFR pathway in NSCLC cell lines.
Figure 3.

Mig-6 prompts H1299, A549 sensitivity to gefitinib by regulating EGFR/ERK pathway. A. H1299 and A549 cells were transfected with pcDNA3 vector-, or pcDNA3-Mig-6 over-expression vector-, for 24 h, then 10 μM gefitinib was added, cell proliferation rates were detected by CCK-8 assay for five days; B. H1299 and A549 cells were transfected with pcDNA3 vector-, or pcDNA3-Mig-6 over-expression vector-, for 24 h, then 10 μM gefitinib were added another 24 h before detected. The percentage of apoptotic cells is expressed as the mean + SD of three independent experiments. *P < 0.05; C. After transfected with pcDNA3 vector-, or pcDNA3-Mig-6 over-expression vector-, for 24 h, and added 10 μM gefitinib another 24 h, levels of p-EGFR and p-ERK were determined by using Western blot analysis.
Mig-6 reverses gefitinib resistance through inhibition of EGFR/ERK pathway in NSCLC cells
Next, we employed Mig-6 over-expression study in PC-9 and PC-9/AB11 cell lines. Before transfection of Mig-6 plasmid, the proliferation rate of PC-9/AB11 was much higher that PC-9 after 1 μM gefitinib treatment. In PC-9/AB11 the levels of P-EGFR and P-ERK were also much higher than PC-9, which indicated the activation of EGFR/ERK pathway. After transfection of Mig-6 plasmid, the proliferation rate of PC-9/AB11 was decreased as well as the level of P-EGFR and P-ERK. After transfection of Mig-6, the cell proliferation rate and the activity of EGFR/ERK pathway in PC-9/AB11 with the treatment of gefitinib became the same as gefitinib sensitive cell line PC-9 (Figure 4).
Figure 4.

Mig-6 reverses gefitinib-resistance through inhibiting EGFR/ERK pathway. A. PC9/AB11 was transfected with PC (pcDNA3 vector) or Mig-6, after 24 h, cells were treated with 1 μM gefitinib, cell proliferation rates were measured by CCK8 assay 24 h later; B. PC9/AB11 was transfected with PC (pcDNA3 vector) or Mig-6, after 24 h, cells were treated with 1 μM gefitinib, levels of p-EGFR and p-ERK were measured by western blot 24 h later.
Discussion
Gefitinib is a tyrosine kinase inhibitor selective for the EGFR patients [12]. EGFR mutation-positive is concerned suitable for targeted therapy [13]. Despite promising progress was made in gefitinib treatment in NSCLC. A series of problem remains challenging: (1) Only a subgroup of patients had EGFR mutation-positive [14]. (2) Not all patients with activating EGFR mutations, such as delE746-A750 (exon 19) and L858R (exon 21), have demonstrated high clinical response to gefitinib [1,15,16]. (3) Almost all patients develop acquired resistance to gefitinib in 9 to 11 months [17]. How to overcome primary and secondary drug resistant to gefitinib in NSCLC is an urgent problem need to be solved.
The mechanism of gefitinib resistant had been studied recent years, including: Mutation in codon 790 (T790M) in EGFR exon 20 [18], MET amplication /overexpression [19], HGF overexpression [20], and the activation of downstream signaling pathways of EGFR such as the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways [21]. Mig-6 can also effectively inhibit the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways in many type of cancer [8,22,23]. These reports indicated Mig-6 may influence tumor cell sensitivity to gefitinib, but no such studies launched.
In this study, we evaluated the expression of Mig-6 in four different NSCLC cell lines and their sensitivity to gefitinib. Mig-6 was overexpression in gefitinib-sensitive NSCLC cell lines PC-9 but was low in gefitinib-resistant NSCLC cell lines. Further study show that up-regulate Mig-6 can increase the proliferation inhibiting and apoptosis treated by gefitinib, however, down-regulate of Mig-6 can decrease the proliferation inhibiting and apoptosis treated by gefitinib in NSCLC cell lines. Mig-6 also inhibiting the activity of EGFR/ERK pathway in gefitinib treated NSCLC cell lines. Mig-6 overcomes gefitinib resistant in NSCLC via EGFR/ERK pathway in gefitinib-resistant cell line PC-9/AB11. The combination of Mig-6 and gefitinib has a synergistic effect in inhibiting cell proliferation and increase cell apoptosis.
Clinically, EGFR exon 19 deletions (delE746-A750) is one of the most robust predictive biomarker for symptom improvement when gefitinib is used for patients with advanced NSCLC [24]. Previous study show that both PC-9 and PC-9/AB11 harbor EGFR exon 19 deletions [25]. However, there is a great difference between the two cells lines for sensitivity to gefitinib. This indicated there are other factors influence cancer cell sensitivity to gefitinib. In this research, in gefitinib sensitive PC-9 cell line, the Mig-6 level is higher than those three gefitinib resistant cell lines. H1299 is relative sensitive to gefitinib in three gefitinib resistant cell lines, and its Mig-6 expression level is much higher than other two cell lines. Taken together the Mig-6 expression may have a positive relationship with the sensitivity to gefitinib in NSCLC, which needed to be further investigation.
In conclusion, our work suggests that the level of Mig-6 may affect the cell sensitivity to gefitinib in NSCLC. In gefitinib resistant cell line up-regulation of Mig-6 may reverses gefitinib resistant through inhibiting EGFR/ERK pathway. Mig-6 may become potential predictors of tumor cell sensitivity to gefitinib.
Acknowledgements
We thank Dr. Oreste Segatto (Regina Elena Cancer Institute, Via Delle Messi d’Oro, 156, Rome 00158, Italy) for kindly providing the pcDNA3 vector- and pcDNA3-Mig-6 overexpression vector. This work was supported by Liaoning S&T Project (2013225585), National Natural Science Foundation of China (No. 30972967), Specialized Research Fund for the Doctoral Program of Higher Education (No. 20092104110018) and Program for Liaoning Excellent Talents in University (LR2011021).
Disclosure of conflict of interest
None.
References
- 1.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T Asami K, Katakami N, Takada M, Yoshioka H, Shibata K, Kudoh S, Shimizu E, Saito H, Toyooka S, Nakagawa K, Fukuoka M West Japan Oncology Group. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 2.Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–8923. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 3.Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J. Clin. Oncol. 2007;25:587–595. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 4.Gadgeel SM, Wozniak A. Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clin Lung Cancer. 2013;14:322–332. doi: 10.1016/j.cllc.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Naruo Y, Nagashima T, Ushikoshi-Nakayama R, Saeki Y, Nakakuki T, Naka T, Tanaka H, Tsai SF, Okada-Hatakeyama M. Epidermal growth factor receptor mutation in combination with expression of MIG6 alters gefitinib sensitivity. BMC Syst Biol. 2011;5:29. doi: 10.1186/1752-0509-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeong JW, Lee HS, Lee KY, White LD, Broaddus RR, Zhang YW, Vande Woude GF, Giudice LC, Young SL, Lessey BA, Tsai SY, Lydon JP, DeMayo FJ. Mig-6 modulates uterine steroid hormone responsiveness and exhibits altered expression in endometrial disease. Proc Natl Acad Sci U S A. 2009;106:8677–8682. doi: 10.1073/pnas.0903632106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin CI, Du J, Shen WT, Whang EE, Donner DB, Griff N, He F, Moore FD Jr, Clark OH, Ruan DT. Mitogen-inducible gene-6 is a multifunctional adaptor protein with tumor suppressor-like activity in papillary thyroid cancer. J Clin Endocrinol Metab. 2011;96:E554–565. doi: 10.1210/jc.2010-1800. [DOI] [PubMed] [Google Scholar]
- 8.Reschke M, Ferby I, Stepniak E, Seitzer N, Horst D, Wagner EF, Ullrich A. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology. 2010;51:1383–1390. doi: 10.1002/hep.23428. [DOI] [PubMed] [Google Scholar]
- 9.Xu D, Makkinje A, Kyriakis JM. Gene 33 is an endogenous inhibitor of epidermal growth factor (EGF) receptor signaling and mediates dexamethasone-induced suppression of EGF function. J Biol Chem. 2005;280:2924–2933. doi: 10.1074/jbc.M408907200. [DOI] [PubMed] [Google Scholar]
- 10.Anastasi S, Baietti MF, Frosi Y, Alema S, Segatto O. The evolutionarily conserved EBR module of RALT/MIG6 mediates suppression of the EGFR catalytic activity. Oncogene. 2007;26:7833–7846. doi: 10.1038/sj.onc.1210590. [DOI] [PubMed] [Google Scholar]
- 11.Frosi Y, Anastasi S, Ballaro C, Varsano G, Castellani L, Maspero E, Polo S, Alema S, Segatto O. A two-tiered mechanism of EGFR inhibition by RALT/MIG6 via kinase suppression and receptor degradation. J Cell Biol. 2010;189:557–571. doi: 10.1083/jcb.201002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riely GJ, Politi KA, Miller VA, Pao W. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res. 2006;12:7232–7241. doi: 10.1158/1078-0432.CCR-06-0658. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 14.Sebastian M, Schmittel A, Reck M. First-line treatment of EGFR-mutated nonsmall cell lung cancer: critical review on study methodology. Eur Respir Rev. 2014;23:92–105. doi: 10.1183/09059180.00008413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 16.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T North-East Japan Study Group. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 17.Xu C, Zhou Q, Wu YL. Can EGFR-TKIs be used in first line treatment for advanced non-small cell lung cancer based on selection according to clinical factors? - A literature-based meta-analysis. J Hematol Oncol. 2012;5:62. doi: 10.1186/1756-8722-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson KW, Sandler AB. The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist. 2013;18:115–122. doi: 10.1634/theoncologist.2012-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, Mitsudomi T, Tanaka H, Kimura T, Kudoh S, Nokihara H, Ohe Y, Yokota J, Uramoto H, Yasumoto K, Kiura K, Higa- shiyama M, Oda M, Saito H, Yoshida J, Kondoh K, Noguchi M. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6:2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 21.Niu FY, Wu YL. Novel agents and strategies for overcoming EGFR TKIs resistance. Exp Hematol Oncol. 2014;3:2. doi: 10.1186/2162-3619-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, Sommergruber W, Kraut N, Ullrich A, Fässler R, Klein R. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006;12:568–573. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 23.Ying H, Zheng H, Scott K, Wiedemeyer R, Yan H, Lim C, Huang J, Dhakal S, Ivanova E, Xiao Y, Zhang H, Hu J, Stommel JM, Lee MA, Chen AJ, Paik JH, Segatto O, Brennan C, Elferink LA, Wang YA, Chin L, DePinho RA. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc Natl Acad Sci U S A. 2010;107:6912–6917. doi: 10.1073/pnas.0914930107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerber DE, Gandhi L, Costa DB. Management and future directions in non-small cell lung cancer with known activating mutations. Am Soc Clin Oncol Educ Book. 2014;34:e353–365. doi: 10.14694/EdBook_AM.2014.34.e353. [DOI] [PubMed] [Google Scholar]
- 25.Wei W, Fan Y, Liu H, Wu Z, Wan H, Yan Z, Xu K, Zhou Q. Relationship between Mutations of the Epidermal Growth Factor Receptor Gene and Drug-Resistance to Gefitinib in Human Lung Cancer in vitro. Zhongguo Fei Ai Za Zhi. 2009;12:28–32. doi: 10.3779/j.issn.1009-3419.2009.01.004. [DOI] [PubMed] [Google Scholar]