

Abstract
Axl, a member of receptor tyrosine kinases (RTKs), has been established as a strong candidate for targeted therapy of cancer. Some reports showed that Axl is a promising therapeutic target to enhance EGFR TKI response in selected EGFR WT NSCLC patients. The present study was aimed to investigate the role of Axl in non-small cell lung carcinoma (NSCLC) drug resistance and the progress of epithelial-to-mesenchymal transition (EMT). MTT was used to detect the cytotoxicity of chemotherapeutic drugs in NSCLC cells, and Western blot to detect the expression of Axl in EGFR wild type NSCLC cell lines. The EMT markers were also determined by Western blot. We found that when downregulating Axl in EGFR WT NSCLC cells, the cells showed a more sensitive response to erlotinib than those overexpressed Axl. The further study showed that when downregulating Axl, the EMT markers E-cadherin was increased while N-cadherin and vimentin were decreased. Those data showed that the inhibition of Axl could reverse the EMT. Combined therapeutic strategies of the inhibitor of Axl and EGFR TKI could be more effective in the treatment of NSCLC drug resistance patients. The EMT signature and Axl might be predictive biomarkers of drug response and therapeutic targets in patients with NSCLC.
Keywords: Axl, E-cadherin, N-cadherin, vimentin, EMT, MDR
Introduction
Lung cancer is the leading cause of cancer morbidity and mortality worldwide. Non-small cell lung carcinoma (NSCLC) is a highly malignant and aggressive neoplasm accounting for approximately 75-80% of all lung cancers reported [1]. The NSCLC is classified as squamous cell carcinoma, large cell carcinoma and adenocarcinoma and characterized by high mortality and poor prognosis [2]. Even though the treatments of NSCLC has been progressed in recent years, the 5-year survival rate of lung cancer is still lowest (15%) [3]. Although surgery is regarded as the best possible treatment for NSCLC, only 20-25% of tumors are suitable for potentially curative resection and despite complete surgical resection, the recurrence rate remains high (30-70%) [4]. Adjuvant chemotherapy may reduce recurrences by eradicating the subclinical and micro-metastatic disease [4]. However, the heterogeneity of NSCLC tumors and resistance to multiple drugs results in a poor response to chemotherapy and reduced survival [4]. Multidrug resistance (MDR) is a multifactorial process and a major obstacle to treatment for NSCLC patients [4].
Epithelial-mesenchymal transition (EMT) plays an important role in multiple physiological and pathological processes of human body. EMT pathway was found to play important roles in regulating the transcription of genes involved in embryonic development [5], inflammatory response [6], tissue regeneration [7], organ fibrosis [8,9], tumor invasion and metastasis. The morphology of EMT was characterized the loose of epithelial cells and loss of cell-cell adhesion. The down-regulation of E-cadherin and cytokeratins, with the up-regulation of mesenchymal proteins like vimentin, fibronectin and N-cadherin were the markers of EMT [10,11]. The previous reports had shown that EGF and resultant EGFR activation could promote EMT by altered the expression and morphology of EMT-associated markers (E-cadherin/β-catenin complex) [11].
Axl is a member of receptor tyrosine kinases (RTK) family [12]. Some research showed that blocking Axl expression by RNAi could lead to a decrease of cells invasion, proliferation and increased cells chemosensitivity through activation of AKT and mitogen-activated protein kinases (MAPK) pathways in solid tumors [13-15]. Besides, over-expression of Axl was initially identified as an oncogene of human leukemia cells [16-18]. The expression of Axl was found to be increased in more than half of NSCLC cell lines [19]. A clinical importance of Axl was also suggested by the data that in 48.3% of lung adenocarcinoma tissues, Axl level was high and proportional to lymph node metastasis as well as disease stage [18]. Furthermore, targeting of Axl with RNA interference or specific monoclonal antibodies has been shown to inhibit proliferation of cancer cells and even tumor growth in mouse xenograft model. Recent studies on NSCLC indicated that increased activation of Axl-induced acquired resistance to epidermal growth factor receptor (EGFR)-targeted therapy [20].
The aim of this study was to investigate the role of Axl in NSCLC drug resistance and the molecular mechanism that was associated with EMT. These novel findings provide strong evidence that Axl may be used as a potential therapeutic target to sensitize cancer cells to chemotherapeutic drugs in NSCLC and provide more evidence for making a new strategy in the treatment of NSCLC patients.
Material and methods
Cell lines
The human NSCLC cancer cell lines, H460, A549 were obtained from ATCC (Manassas, VA). These cells were cultured in DMEM (Gibco, Carlsbad, CA) supplemented with 10% FBS (Invitrogen Life Technologies, Carlsbad, CA) and 1% penicillin/streptomycin (Gibco). All cells were grown in drug-free culture medium for more than 2 weeks before assay.
Chemicals and reagents
Erlotinib and 3-(4,5-dimethylthiazol-yl)-2,5-diphenyllapatinibrazolium bromide (MTT) were products of Sigma Chemical Co. from Genewindows Co. (Guangzhou, China). Dulbecco’s modified Eagle’s medium (DMEM) and RPMI medium 1640 were products of Gibco BRL. from Genewindows Co. (Guangzhou, China). Axl, E-cadherin, N-cadherin, Vimentin, p-Akt, Akt, p-Erk, Erk and GAPDH antibodies were purchased from Santa Cruz Biotechnology Inc from Genetime Co. (Guangzhou, China). Other routine laboratory reagents were obtained from commercial sources of analytical (Guangzhou, China).
MTT assay
Cell viability was assessed by MTT assay as described previously [13]. Briefly, cells were plated at a density of 3000 cells per well into 96-well plates. At the end of treatment, the supernatant was removed, and 20 μl of the tetrazolium compound, MTT, and 270 ml of fresh DMEM medium were added. After incubation for 4 h at 37°C, 120 μl of DMSO was placed in each well to dissolve the tetrazolium crystals. Finally, the absorbance at a wavelength of 570 nm was recorded using a multi-well plate reader (Tecan, Maennedorf, Switzerland).
Plasmids and siRNAs
Multiple short hairpin RNA (siRNA) probes targeting the human Axl gene and a probe containing a scrambled sequence (control) were designed and constructed into a pU6-mRFP expression vector as described previously [26]. Two siRNA Axl probes, one containing the sequence 5’-AACCTTCAACTCCTGCCTTCTCG-3’ in the coding region, and another with the sequence 5’-CAGCTTCTCCTTCAGCTCTTCAC-3’ in the 3’-UTR region, were verified by Western blot analysis to be effective in silencing the Axl gene.
Western blot analysis
Cell lysates were prepared in RIPA buffer (50 mmol/l Tris-HCl buffer, pH 7.4, 150 mmol/l NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) supplemented with 1×Halt protease inhibitor cocktail and 1×Halt phosphatase inhibitor cocktail (Pierce, Rockford, IL). A Bio-Rad protein assay (Bio-Rad) was used to determine protein concentrations. Proteins were separated on 10-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to PVDF membranes (Whatman, Boston, MA). Membranes were first hybridized with specific primary antibodies and then with HRP-conjugated secondary antibodies (Cell Signaling Technology). Protein bands were visualized using a commercial Immobilon Western Chemiluminescent HRP Substrate detection reagent (Millipore, Billerica, MA). The chemiluminescence of proteins transferred to PVDF membranes was detected with ECL Plus (GE Healthcare Amersham, Piscataway, NJ). Relative protein expression values were quantitatively determined via densitometry with ImageJ software.
Statistical analysis
All findings were analyzed with statistical software package SPSS 16.0 and Graphpad Prism 5. The results were expressed as means ± SD. The statistical significance of the studies was determined by the parametric unpaired Student’s t test. Differences with P < 0.05 are considered significant.
Results
Downregulation of Axl suppresses cell viability of NSCLC
To investigate the role of Axl in NSCLC cell survival, we chose two NSCLC cells for our research: the EGFR wild type A549 cell and H460 cell. We employed A549 and H460 cells with high endogenous Axl expression to stably or transiently down-regulation Axl, respectively (Figure 1A and 1B). Western blot analysis data confirmed the down-regulation of exogenous and endogenous Axl proteins as indicated by the full-length protein (138 kDa) and the short fragment (55 kDa; Figure 1A and 1B). The cell viability assay results demonstrated that the downregulation of Axl expression in A549 and H460 cells significantly inhibited cell viability compared to control cells (Figure 1C and 1D). The data indicated that knockdown of endogenous Axl significantly slow down the cell survival. To assess the effect of Axl inhibition on long-term cellular colony formation potential, control and knockdown shAxl clones were plated in soft agar for 3 weeks. The colony formation data showed that the cell growth in shAxl cells was less than 20% of control in both A549 and H460 cells. These data indicated that inhibition of Axl significantly bock cell survival and colony formation in NSCLC cells.
Figure 1.
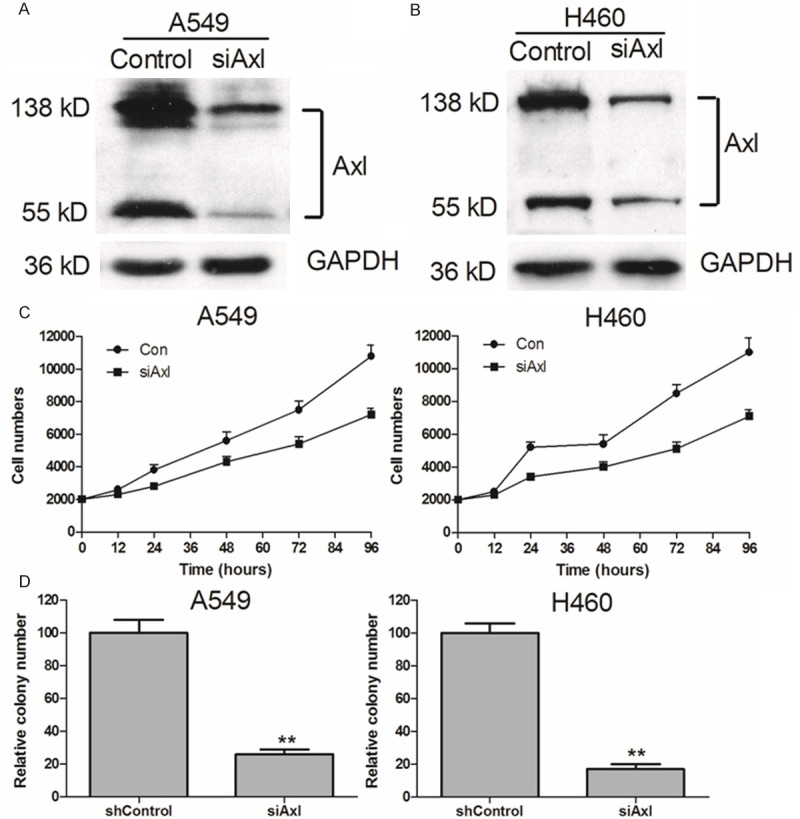
Axl siRNA significantly decreased cell proliferation in EGFR wild type NSCLC cells. Axl siRNA noticeably down regulated expression of Axl protein in A549 and H460 cells (A and B). Scramble and Axl siRNA were transfected into NSCLC cells for 24 h. Relative Axl expressions were detected by Western blot. The cell viabilities were determined using MTT assays (C). A549 and H460 knockdown cells were plated in soft agar and colonies were counted 2 wk later. shControl represents the parental line after transduction of siAxl represents independent constructs against Axl (D). **P < 0.01, in comparison to the control (two-tailed t test). Error bars represent three independent experiments that were done in triplicate.
Downregualtion of Axl abrogates drug resistance in NSCLC cells
To identify the relationship of Axl overexpression in NSCLC cells drug resistance, we knockdown the Axl expression in A549 and H460 cells. The concentrations of chemotherapeutic agents (such as vincristin, doxorubicin and paclitaxel) required for the IC50 values in A549/siRNA and H460/siRNA cells were higher than those in their parental sensitive A549, H460 cells (Table 1). Furthermore, down-regulating Axl could recover the erlotinib sensitivity to A549 and H460 cells (Figure 2). The data unequivocally indicated that knockdown of endogenous Axl significantly enhanced the sensitivity of cells to chemotherapeutic drugs. In addition, we demonstrated that the pro-survival function of Axl was dependent on its kinase activity. These results indicate that knockdown Axl expression can strongly enhance the efficacy of conventional chemotherapeutic agents in NSCLC cells.
Table 1.
Effect of chemotherapeutic agents on the growth of NSCLC cells transfected with Axl siRNA
| Compounds | IC50 ± SDs μM (fold-reverse) | |
|
|
||
| A549 | A549/siAxl | |
|
| ||
| Doxorubicin | 1.1004 ± 0.1022 | 0.7004 ± 0.0902 (1.57)* |
| Vincristine | 0.0066 ± 0.0053 | 0.0018 ± 0.0012 (3.67)** |
| Paclitaxel | 0.0017 ± 0.0134 | 0.0007 ± 0.0821 (2.43)** |
|
| ||
| H460 | H460/siAxl | |
|
| ||
| Doxorubicin | 12.0003 ± 1.0243 | 4.0124 ± 0.3002 (2.99)** |
| Vincristine | 0.0014 ± 0.0091 | 0.0006 ± 0.0034 (2.33)** |
| Paclitaxel | 0.0030 ± 0.0230 | 0.0011 ± 0.0149 (2.73)** |
Cell survival was determined by MTT assays as described in “Materials and Methods”. Data are the means ± SDs of at least three independent experiments.
P < 0.05, for values versus that obtained in the parent cells;
P < 0.01, for values versus that obtained in the parent cells.
Figure 2.
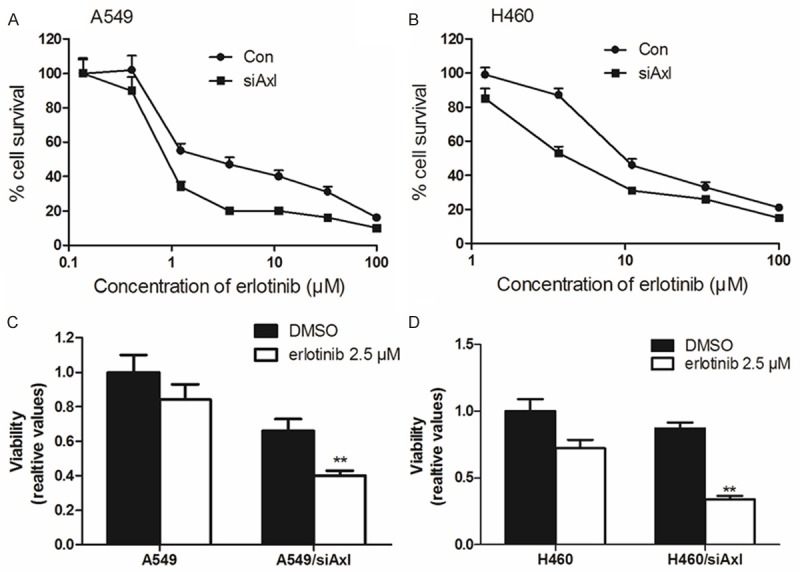
Down-regulation Axl could reverse drug resistance in A549 and H460 cells. Down-regulation Axl could significantly reverse erlotinib resistance in A549 and H460 cells (A, B). The MTT assays showed that knockdown Axl could decreased cell viabilities in NSCLC cells (C, D).
Inhibition of Axl blocks the phosphorylation of AKT and ERK1/2
Previous studies have shown that the inhibition of the AKT and ERK1/2 pathways may decrease the resistance to antineoplastic drugs in cancer cells. Consequently, we determined the effect of Axl inhibitor and siRNA on the levels of total and phosphorylated forms of AKT and ERK1/2 in all cell lines. As shown in Figure 3, the incubation of cells with Axl siRNA for 48 hours did significantly alter the total and phosphorylated forms of AKT and ERK1/2. This suggested that the reversal effect of Axl inhibition in NSCLC cells is dependent on the inhibition of AKT and ERK1/2 phosphorylation.
Figure 3.
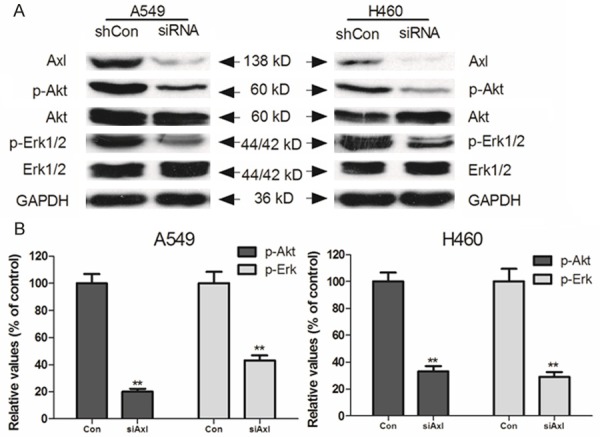
Effect of Axl on blockage of AKT and ERK1/2 phosphorylation. A549 and H460 cells were treated with Axl siRNA for 48 h. Equal amounts of protein were loaded for Western blot analysis as described in “Materials and Methods”. Independent experiments were performed at least three times and results from a representative experiment were shown.
Down-regulation of Axl could inhibit the progress of EMT
The acquisition of mesenchymal cell characteristics by epithelial cells, in particular the ability to migrate as single cells and invade the extracellular matrix, is the functional hallmark of EMT (3). Therefore, we investigated whether EMT markers expression is associated with Axl in human lung cancer cells. After treated the cells with Axl siRNA for 48 hours, we observed an EMT phenotype characterized by down-regulation of the mesenchymal markers N-cadherin and vimentin and up-regulation of the epithelial marker E-cadherin in A549 and H460 cells (Figure 4). These results suggested that downexpression of Axl could abrogate EMT program induction in NSCLC cells.
Figure 4.
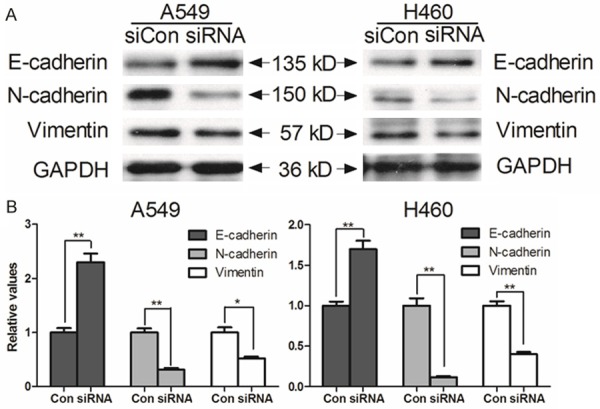
Effect of Axl on the progress of EMT. A549 and H460 cells were treated with Axl siRNA for 48 h. Western blot findings of vimentin, E-cadherin, N-cadherin and GAPDH served as the loading control and results are representative of three independent.
Discussion
In this study, we explored the role of Axl in drug resistance and EMT progress of NSCLC cells. Our data was similar to the data published in previous reports, we observed that downregulation of Axl could inhibit the cell survival and overcame drug resistance in vitro. Recently, we demonstrated that increased Axl expression could contribute to drug resistance in cell lines. Altered Axl-related signaling was also observed in approximately 20% of patients with acquired resistance to EGFR-TKI [21], although it remains unknown whether these patients could benefit from Axl inhibition. Axl could activate the downstream signals such as Akt and Erk to motivate cell survival and growth in EGFR-TKI resistance cells. Therefore, combined treatment with EGFR and Axl inhibitors might effectively abrogate the growth of tumor cells. A similar phenomenon can be observed in MET-mediated resistance, as shown in a previous report [22].
NSCLC could be classified into 3 groups in the terms of EGFR gene status: EGFR wild type, EGFR mutant and UN-EGFR-GS [23]. The patients with EGFR mutations in NSCLC can benefit from EGFR tyrosine kinase inhibitor (TKI) therapy and several TKIs was used as a first-line treatment for patients with EGFR mutations, Compared with the traditional chemotherapy, the EGFR TKI can make longer progression-free survival (PFS) and fewer side effects but the overall survival (OS) was not extended [24]. The EGFR TKI gefitinib and erlotinib had been approved for the treatment of advanced NSCLC in 2007 [25-27]. However, the drug resistance to TKI is an obstacle for the treatment in NSCLC patients [28]. The mutations in exon 21 missense point mutation L858R and frame deletions exon 19 deletion are highly response to EGFR-TKI [29]. In addition, the EGFR T790M mutation and MET amplification are responsible for the acquired resistance in most patients [30,31]. The challenge of tumor drug resistance therefore represents a barrier that confounds the ultimate goal of cure or long-term control of NSCLC [32].
We tested whether the downregulation of Axl can inhibit the phosphorylation of Akt or Erk1/2. As shown in Figure 3A, inhibiting Axl significantly blocked the phosphorylation of Akt and Erk1/2 in any of the two cell lines. This result suggested that Axl induced the resistance of chemotherapeutic agents in EGFR WT cells may be due to its activation of EGFR receptors. In addition, the EMT markers E-cadherin was increased wile N-cadherin and vimentin were decreased after downregulated Axl. These results indicated clinical significance in the design of therapies targeting Axl and EMT signaling. Some reports pointed that the effect of dual treatment with Axl and EGFR inhibitors may be more reliant on Axl or EMT for downstream signaling than EGFR itself, and that the effects of increased EGFR abundance may be in large part manifested by activation of Axl or EMT. Consequently, blocking Axl or EMT signal may be more effective that blocking signal from EGFR and that inhibitors of their kinase activity or treatments that reduce receptor abundance may be more effective [33-36]. In some carcinomas, EMT and the expression of Axl and c-MET amplification have been identified as a mechanism of acquired resistance to EGFR-TKI therapies [37,38]. After EGFR-TKI treatment, the cross talk between cells is halted due to RTK-mediated secondary resistance and may provide qualitatively distinct signaling through new receptors, though in response to the original activating ligand. Consequently, upon withdrawal of treatment, cells could respond to the original ligand with a more invasive or aggressive phenotype as a result of the dynamic network rewiring that created resistance [39,40].
Our work more broadly raises the implication that modulation of RTK expression may benefit the treatment of NSCLC. Thus, cancer therapies targeting Axl may be circumvented by activation in inhibitor resistance that may take place by amplification of signaling through other receptors.
Acknowledgements
This work was supported by Educational Commission of Jiangxi Province of China (No. GJJ13036).
Disclosure of conflict of interest
None.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira CG, Huisman C, Giaccone G. Novel approaches to the treatment of non-small cell lung cancer. Crit Rev Oncol Hematol. 2002;41:57–77. doi: 10.1016/s1040-8428(01)00197-4. [DOI] [PubMed] [Google Scholar]
- 3.Xu MM, Mao GX, Liu J, Li JC, Huang H, Liu YF, Liu JH. Low Expression of the FoxO4 Gene may Contribute to the Phenomenon of EMT in Non-small Cell Lung Cancer. Asian Pac J Cancer Prev. 2014;15:4013–4018. doi: 10.7314/apjcp.2014.15.9.4013. [DOI] [PubMed] [Google Scholar]
- 4.Arriagada R, Auperin A, Burdett S, Higgins JP, Johnson DH, Le Chevalier T, Le Pechoux C, Parmar MK, Pignon JP, Souhami RL, Stephens RJ, Stewart LA, Tierney JF, Tribodet H, van Meerbeeck J. Adjuvant chemotherapy, with or without postoperative radiotherapy, in operable non-small-cell lung cancer: two meta-analyses of individual patient data. Lancet. 2010;375:1267–1277. doi: 10.1016/S0140-6736(10)60059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denlinger CE, Ikonomidis JS, Reed CE, Spinale FG. Epithelial to mesenchymal transition: the doorway to metastasis in human lung cancers. J Thorac Cardiovasc Surg. 2010;140:505–513. doi: 10.1016/j.jtcvs.2010.02.061. [DOI] [PubMed] [Google Scholar]
- 6.Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S, Baba H, Mori M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010;101:293–299. doi: 10.1111/j.1349-7006.2009.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796:75–90. doi: 10.1016/j.bbcan.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Cannito S, Novo E, di Bonzo LV, Busletta C, Colombatto S, Parola M. Epithelial-mesenchymal transition: from molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal. 2010;12:1383–1430. doi: 10.1089/ars.2009.2737. [DOI] [PubMed] [Google Scholar]
- 10.Chow G, Tauler J, Mulshine JL. Cytokines and growth factors stimulate hyaluronan production: role of hyaluronan in epithelial to mesenchymal-like transition in non-small cell lung cancer. J Biomed Biotechnol. 2010;2010:485468. doi: 10.1155/2010/485468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 13.Valverde P, Obin MS, Taylor A. Role of Gas6/Axl signaling in lens epithelial cell proliferation and survival. Exp Eye Res. 2004;78:27–37. doi: 10.1016/j.exer.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Goruppi S, Ruaro E, Schneider C. Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3T3 fibroblasts. Oncogene. 1996;12:471–480. [PubMed] [Google Scholar]
- 15.Goruppi S, Ruaro E, Varnum B, Schneider C. Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol Cell Biol. 1997;17:4442–4453. doi: 10.1128/mcb.17.8.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Donnell K, Harkes IC, Dougherty L, Wicks IP. Expression of receptor tyrosine kinase Axl and its ligand Gas6 in rheumatoid arthritis: evidence for a novel endothelial cell survival pathway. Am J Pathol. 1999;154:1171–1180. doi: 10.1016/S0002-9440(10)65369-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen JW, Schulz AS, Steenvoorden AC, Schmidberger M, Strehl S, Ambros PF, Bartram CR. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene. 1991;6:2113–2120. [PubMed] [Google Scholar]
- 18.O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, Espinosa R, Le Beau MM, Earp HS, Liu ET. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11:5016–5031. doi: 10.1128/mcb.11.10.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shieh YS, Lai CY, Kao YR, Shiah SG, Chu YW, Lee HS, Wu CW. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia. 2005;7:1058–1064. doi: 10.1593/neo.05640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6:ra66. doi: 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hata A, Katakami N, Yoshioka H, Takeshita J, Tanaka K, Nanjo S, Fujita S, Kaji R, Imai Y, Monden K, Matsumoto T, Nagata K, Otsuka K, Tachikawa R, Tomii K, Kunimasa K, Iwasaku M, Nishiyama A, Ishida T, Nishimura Y. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: Comparison between T790M mutation-positive and mutation-negative populations. Cancer. 2013;119:4325–4332. doi: 10.1002/cncr.28364. [DOI] [PubMed] [Google Scholar]
- 22.Qi J, McTigue MA, Rogers A, Lifshits E, Christensen JG, Janne PA, Engelman JA. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 2011;71:1081–1091. doi: 10.1158/0008-5472.CAN-10-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y, Fang W, Deng J, Zhao P, Xu N, Zhou J. Sequential treatment of icotinib after first-line pemetrexed in advanced lung adenocarcinoma with unknown EGFR gene status. J Thorac Dis. 2014;6:958–964. doi: 10.3978/j.issn.2072-1439.2014.07.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paz-Ares L, Soulieres D, Moecks J, Bara I, Mok T, Klughammer B. Pooled analysis of clinical outcome for EGFR TKI-treated patients with EGFR mutation-positive NSCLC. J Cell Mol Med. 2014;18:1519–39. doi: 10.1111/jcmm.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Araya T, Kasahara K, Demura Y, Matsuoka H, Nishitsuji M, Nishi K. Successful treatment with erlotinib of severe neutropenia induced by gefitinib in a patient with advanced non-small cell lung cancer. Lung Cancer. 2013;80:344–346. doi: 10.1016/j.lungcan.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Kang XH, Xu ZY, Gong YB, Wang LF, Wang ZQ, Xu L, Cao F, Liao MJ. Bufalin Reverses HGF-Induced Resistance to EGFR-TKIs in EGFR Mutant Lung Cancer Cells via Blockage of Met/PI3k/Akt Pathway and Induction of Apoptosis. Evid Based Complement Alternat Med. 2013;2013:243859. doi: 10.1155/2013/243859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakai K, Horiike A, Irwin DL, Kudo K, Fujita Y, Tanimoto A, Sakatani T, Saito R, Kaburaki K, Yanagitani N, Ohyanagi F, Nishio M, Nishio K. Detection of epidermal growth factor receptor T790M mutation in plasma DNA from patients refractory to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 2013;104:1198–1204. doi: 10.1111/cas.12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y, Chen Y, Mei Q, Chen Y, Yu S, Xia S. Combined inhibition of the EGFR and mTOR pathways in EGFR wild-type non-small cell lung cancer cell lines with different genetic backgrounds. Oncol Rep. 2013;29:2486–2492. doi: 10.3892/or.2013.2357. [DOI] [PubMed] [Google Scholar]
- 29.Kim HJ, Oh SY, Kim WS, Kim SJ, Yoo GH, Kim WD, Lee KY. Clinical investigation of EGFR mutation detection by pyrosequencing in lung cancer patients. Oncol Lett. 2013;5:271–276. doi: 10.3892/ol.2012.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shih JY, Gow CH, Yang PC. EGFR mutation conferring primary resistance to gefitinib in non-small-cell lung cancer. N Engl J Med. 2005;353:207–208. doi: 10.1056/NEJM200507143530217. [DOI] [PubMed] [Google Scholar]
- 31.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 32.Shen H, Zhu F, Liu J, Xu T, Pei D, Wang R, Qian Y, Li Q, Wang L, Shi Z, Zheng J, Chen Q, Jiang B, Shu Y. Alteration in Mir-21/PTEN Expression Modulates Gefitinib Resistance in Non-Small Cell Lung Cancer. PLoS One. 2014;9:e103305. doi: 10.1371/journal.pone.0103305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerchia L, Esposito CL, Camorani S, Rienzo A, Stasio L, Insabato L, Affuso A, de Franciscis V. Targeting Axl with an high-affinity inhibitory aptamer. Mol Ther. 2012;20:2291–2303. doi: 10.1038/mt.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye X, Li Y, Stawicki S, Couto S, Eastham-Anderson J, Kallop D, Weimer R, Wu Y, Pei L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene. 2010;29:5254–5264. doi: 10.1038/onc.2010.268. [DOI] [PubMed] [Google Scholar]
- 35.Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, Carano RA, Kasman I, Mai E, Young J, Zha J, Zhang Z, Ross S, Schwall R, Colbern G, Merchant M. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68:4360–4368. doi: 10.1158/0008-5472.CAN-07-5960. [DOI] [PubMed] [Google Scholar]
- 36.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, Zhang J, Ding P, Apatira A, Chua J, Brandt R, Pine P, Goff D, Singh R, Payan DG, Hitoshi Y. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70:1544–1554. doi: 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]
- 37.Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69:6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, Jang SJ, Park YS, Kim WS, Lee DH, Lee JS, Miller VA, Arcila M, Ladanyi M, Moonsamy P, Sawyers C, Boggon TJ, Ma PC, Costa C, Taron M, Rosell R, Halmos B, Bivona TG. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riely GJ, Kris MG, Zhao B, Akhurst T, Milton DT, Moore E, Tyson L, Pao W, Rizvi NA, Schwartz LH, Miller VA. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin Cancer Res. 2007;13:5150–5155. doi: 10.1158/1078-0432.CCR-07-0560. [DOI] [PubMed] [Google Scholar]
- 40.Pop O, Pirvu A, Toffart AC, Moro-Sibilot D. Disease flare after treatment discontinuation in a patient with EML4-ALK lung cancer and acquired resistance to crizotinib. J Thorac Oncol. 2012;7:e1–2. doi: 10.1097/JTO.0b013e318257fc1d. [DOI] [PubMed] [Google Scholar]