

Abstract
The aim of this study was to fabricate valsartan composite nanoparticles by using the supercritical antisolvent (SAS) process, and to evaluate the correlation between in vitro dissolution and in vivo pharmacokinetic parameters for the poorly water-soluble drug valsartan. Spherical composite nanoparticles with a mean size smaller than 400 nm, which contained valsartan, were successfully fabricated by using the SAS process. X-ray diffraction and thermal analyses indicated that valsartan was present in an amorphous form within the composite nanoparticles. The in vitro dissolution and oral bioavailability of valsartan were dramatically enhanced by the composite nanoparticles. Valsartan–hydroxypropyl methylcellulose–poloxamer 407 nanoparticles exhibited faster drug release (up to 90% within 10 minutes under all dissolution conditions) and higher oral bioavailability than the raw material, with an approximately 7.2-fold higher maximum plasma concentration. In addition, there was a positive linear correlation between the pharmacokinetic parameters and the in vitro dissolution efficiency. Therefore, the preparation of composite nanoparticles with valsartan–hydroxypropyl methylcellulose and poloxamer 407 by using the SAS process could be an effective formulation strategy for the development of a new dosage form of valsartan with high oral bioavailability.
Keywords: supersaturation, bioavailability, solid dispersion, dissolution, supercritical fluid
Introduction
Valsartan, (S)-3-methyl-2-(N-{[2′-(2H-1,2,3,4-tetrazol-5-yl)biphenyl-4-yl]methyl} pentanamido)butanoic acid, is a highly selective antagonist of the angiotensin II receptor.1 Currently, valsartan is widely used to treat hypertension. The oral solid dosage form is considered to be the most suitable for the delivery of active pharmaceutical ingredients (APIs). However, valsartan has poor water solubility (3.08 μg/mL) and high permeability; therefore, it is a class II drug according to the Biopharmaceutics Classification System.2 In addition, valsartan contains a tetrazole derivative-containing acid (pKa =4.73) as well as carboxylic-group acids (pKa =3.9) groups, which may influence its solubility in a pH-dependent manner.3,4 In healthy individuals, valsartan has an absolute bioavailability of 23% after administration of an 80-mg capsule, which might be due to its poor solubility at low pH. In order to improve the oral bioavailability of valsartan, different formulations such as solid dispersion,2,5 self-microemulsifying drug delivery system,6,7 spray-dried emulsion,8 and spray-dried nanosuspension9 have been studied.
Composite formulations, containing polymer and surfactant, are used in the pharmaceutical industry and they can enhance the dissolution and oral bioavailability of poorly water-soluble APIs.10–12 Recently, it was reported that composite nanoparticles could be fabricated by using supercritical fluid technology.13 Carbon dioxide has been widely used as a supercritical fluid because it is nontoxic, nonflammable, and it has relatively low critical parameters (Pc [critical pressure] =7.38 MPa, Tc [critical temperature] =31.1°C). During the last 20 years, supercritical carbon dioxide has been used successfully as a solvent or an antisolvent in the pharmaceutical industry for various purposes such as particle formation and extraction of the active ingredient, and as an alternative green solvent to conventional organic solvents.14–16 Supercritical fluid technologies for particle formation can be classified according to the role of the supercritical carbon dioxide during the process as either a solvent (rapid expansion of supercritical solutions) or an antisolvent (supercritical antisolvent process [SAS]). Recently, the fabrication of composite nanoparticles by using the SAS process has been reviewed.17 In addition, composite nanoparticles prepared by using the SAS process have been demonstrated to enhance the solubility, dissolution, and bioavailability of different poorly water-soluble APIs.18–20
The aim of this study was to fabricate valsartan-containing composite nanoparticles with enhanced dissolution and oral bioavailability by using the SAS process, and to evaluate the correlation between in vitro dissolution and in vivo pharmacokinetic parameters for this poorly water-soluble drug. The effect of the type of polymer and surfactant on the dissolution and oral bioavailability of valsartan was investigated. In addition, physicochemical characterization of composite nanoparticles was performed.
Materials and methods
Valsartan and irbesartan were obtained from Dong-A ST (Yongin-si, Republic of Korea) and Sigma-Aldrich Co. (St Louis, MO, USA), respectively. Hydroxylpropyl cellulose (HPC) (HPC-SSL; Nippon Soda Co., Tokyo, Japan), hydroxypropyl methylcellulose (HPMC) 2910 (HPMC 2910 3cp; Shin-Etsu Chemical Co., Ltd., Tokyo, Japan), polyvinylpyrrolidone K30 (PVP K30; BASF Co. Ltd., Ludwigshafen, Germany), and vinylpyrrolidone-vinyl acetate copolymer (PVP VA64; BASF Co. Ltd.) were used as polymers. D- α-Tocopheryl polyethylene glycol 1000 succinate (TPGS; Eastman Chemical Company, Kingsport, TN, USA), poloxamer 407 (BASF Co. Ltd.), and sucrose laurate (Ryoto sugar ester L1695; Mitsubishi-Kagaku Foods Co., Tokyo, Japan) were used as surfactants. All organic solvents and other chemicals were of high-performance liquid chromatography (HPLC) and analytical grade, respectively.
Preparation of valsartan-loaded composite nanoparticles
Valsartan–polymer nanoparticles
Valsartan-loaded composite nanoparticles were fabricated using the SAS process (SAS200 model; Thar Technologies, Pittsburgh, PA, USA). To screen polymers, valsartan–polymer nanoparticles were prepared at a 20% drug:80% polymer (w/w) ratio. For HPC, PVP K30, and PVP VA64, valsartan and polymer were dissolved in ethanol at 50 mg/mL (solute concentration). HPMC and valsartan solution (50 mg/mL) was prepared by using a mixture of dichloromethane and ethanol (45:55, w/w). The valsartan–polymer nanoparticles were formed by spraying the drug solution and carbon dioxide into the particle formation vessel at flow rates of 1 mL/min and 11 g/min, respectively. Based on our previous study, the supercritical conditions within the particle formation vessel were set to 40°C and 15 MPa.13 After spraying the drug solution, fresh supercritical carbon dioxide was delivered into the particle formation vessel to remove residual organic solvents, followed by depressurization of the system to atmospheric pressure. The fabricated valsartan-loaded composite nanoparticles were collected from the particle formation vessel basket.
Valsartan–polymer–surfactant nanoparticles
Valsartan was dissolved in ethanol-containing surfactants, including poloxamer 407, Ryoto sugar ester L1695, or TPGS. HPMC was dispersed in this solution, followed by addition of dichloromethane. The components were mixed by gentle stirring until a clear solution was obtained. The solute concentration was 50 mg/mL in a mixture of dichloromethane and ethanol (45:55, w/w). The particle fabrication procedure was performed as described above. The composition of valsartan-loaded composite nanoparticles is described in Table 1.
Table 1.
Formulation, particle size, and specific surface area of the Val composite nanoparticles prepared by using the SAS process
| Formulation (weight/weight) | Drug content (%) | Mean particle size (nm) | Specific surface area (m2/g) |
|---|---|---|---|
| Val:HPC =2:8 | 97.9±1.6 | 302.3±35.2 | 51.22±1.35 |
| Val:HPMC =2:8 | 98.5±2.2 | 277.3±52.3 | 60.23±1.19 |
| Val:PVP K30 =2:8 | 95.8±1.7 | 520.3±50.1 | 31.23±1.23 |
| Val:PVP VA64 =2:8 | 97.7±2.0 | 581.6±62.7 | 28.18±1.11 |
| Val:HPMC:poloxamer 407 =2:7:1 | 98.2±1.6 | 345.3±45.5 | 48.32±0.99 |
| Val:HPMC:Ryoto sugar ester L1695 =2:7:1 | 98.9±1.9 | 759.5±81.2 | 25.21±1.21 |
| Val:HPMC:TPGS =2:7:1 | 97.1±2.3 | 550.2±66.1 | 25.11±0.95 |
Notes: The drug content (%) = weight of the loaded drug/weight of the feeding drug ×100. Data are expressed as the mean ± standard deviation (n=3).
Abbreviations: HPC, hydroxypropyl cellulose; HPMC, hydroxypropyl methyl cellulose; PVP, polyvinylpyrrolidone; PVP VA64, polyvinylpyrrolidone-vinyl acetate; SAS, supercritical antisolvent; TPGS, D-α-Tocopheryl polyethylene glycol 1000 succinate; Val, valsartan.
Characterization of valsartan-loaded composite nanoparticles
The drug amount within composite nanoparticles was measured using a HPLC system (Waters, Milford, MA, USA). Approximately 20 mg of composite nanoparticles was dissolved in 50 mL of dichloromethane and ethanol mixture (45:55, w/w). The solution was filtered through a 0.45-μm syringe filter, and 20 μL of the filtrate was injected onto a C18 analytical column (CAPCELL PAK C18 UG120, 5 μm, 4.6 mm×150 mm; Shiseido Fine Chemicals, Tokyo, Japan). The mobile phase of acetonitrile and water (60:40, v/v), which was adjusted to pH 3.0 with diluted phosphoric acid, was pumped at a flow rate of 1.0 mL/min. The drug was detected spectrophotometrically at 247 nm. The drug content was calculated by using the following equation: loaded drug weight/feeding drug weight ×100. The morphology of valsartan-loaded composite nanoparticles was analyzed by using scanning electron microscopy (JSM-6300; JEOL, Ltd. Tokyo, Japan). The particle size of the valsartan-loaded composite nanoparticles was measured using a laser particle size analyzer (BI-9000; Brookhaven, Upton, NY, USA). Before the measurement, the nanoparticles were dispersed in mineral oil by sonicating for 10 minutes at room temperature. The specific surface area of valsartan-loaded composite nanoparticles was determined with the Brunauer, Emmett, and Teller method by using nitrogen gas adsorption (Autosorb-1; Quantachrome GmbH, Odelzhausen, Germany). The crystallinity of valsartan within composite nanoparticles was determined by using a powder X-ray diffractometer ([PXRD] D/MAX-2200 Ultima/PC; Rigaku Co., Tokyo, Japan) and differential scanning calorimetry ([DSC] S-650; Scinco Co. Ltd., Seoul, Republic of Korea). PXRD measurements were performed from 5° to 50° of 2θ at a scanning speed of 3°/min. DSC measurements were performed by using 2–5 mg samples. The heating rate was 5°C/min from 20°C to 200°C under a nitrogen purge at a flow rate of 20 mL/min. Kinetic solubility tests of valsartan-loaded composite nanoparticles were performed by using a United States Pharmacopeia 37 (USP 37) rotating paddle apparatus (Electrolab, Mumbai, India) at 37°C and 100 rpm. Composite nanoparticles equivalent to 250 mg of valsartan were placed in 250 mL of the dissolution medium (HCl and NaCl, pH 1.2). Three-milliliter samples were collected for analysis at predetermined intervals. The amount of drug dissolved in each medium was determined by using HPLC after filtering through a 0.11-μm syringe filter and diluting with methanol. Dissolution tests of raw material and valsartan-loaded composite nanoparticles were conducted in 900 mL of dissolution medium by using a USP rotating paddle apparatus at 50 rpm. The dissolution media, including HCl and NaCl (pH 1.2), acetate buffer (pH 4.0), and phosphate buffer (pH 6.8), were maintained at 37°C. Samples equivalent to 40 mg of valsartan were placed in the dissolution medium. Three-milliliter samples were collected for analysis at predetermined intervals. After filtering through a 0.11-μm syringe filter and diluting with methanol, the amount of drug dissolved in each medium was determined using HPLC.
Pharmacokinetic study
The study protocol was in compliance with the institutional guidelines for the care and use of laboratory animals, and it was approved by the ethics committee of Kyungsung University. Twenty-five male Sprague-Dawley rats weighing 250±10 g (Orient Bio Inc., Seongnam-si, Republic of Korea) were divided into five groups. The rats were fasted for 16 hours prior to the study. The rats were orally administered small gelatin capsules containing raw material, or different composite nanoparticles equivalent to 10 mg/kg of valsartan. One milliliter of water was administered immediately after drug dosing. Blood samples (approximately 0.35 mL) were collected in heparinized tubes from the retro-orbital plexus of rats at specified time intervals. The tubes were centrifuged at 10,000 rpm for 5 minutes at 4°C to obtain the plasma, which was stored at −70°C until analysis. Valsartan and irbesartan (internal standard [IS]) were extracted by protein precipitation as described previously.8 For protein precipitation, 20 μL of the IS (250 ng/mL) and 100 μL of 10 mM ammonium acetate buffer (pH 4.0) were added to 100 μL of heparinized plasma sample followed by addition of 600 μL of methanol. After vortex mixing briefly, the organic phase was separated from the aqueous phase by centrifugation at 13,000 rpm for 5 minutes. A 5-μL aliquot of the organic phase was injected into the liquid chromatography–mass spectrometry (MS)/MS system. The HPLC system included binary pumps, an autosampler, and a column heater (HPLC Agilent 1100 Series; Agilent Technologies, Santa Clara, CA, USA). The HPLC system was operated isocratically at a flow rate of 0.2 mL/min and at 30°C. The mobile phase consisted of methanol:5 mM ammonium acetate (70:30, v/v; pH 4.0). A Phenomenex C18 guard cartridge (3.0×4.0 mm) and Phenomenex Kinetex C18 column (50×2.1 mm, 2.6 μm) were used (Phenomenex; Torrance, CA, USA). The mass spectrometer was a Sciex API 4000 triple quadrupole liquid chromatography–MS/MS system equipped with a TurboIonSpray ionization source operated by using the Analyst 1.4.1 software (Thermo Fisher Scientific, Waltham, MA, USA), as described previously.8 Briefly, the ions monitored by using multiple reaction monitoring were m/z 436.0 (parent) and m/z 291.0 (product) for valsartan, and m/z 429.1 (parent) and m/z 207.2 (product) for the IS. Pharmacokinetic analysis of plasma concentration-time data was carried out by using the WinNonlin Standard Edition software, version 5.3 (Pharsight Corp., St Louis, MO, USA). The area under the curve (AUC0→24 h) was calculated using the trapezoidal method. The maximum concentration of valsartan after oral administration (Cmax) and the time to reach the maximum concentration were determined from the experimentally obtained data.
Statistical analysis
The data were analyzed by a one-way analysis of variance test followed by the Student–Newman–Keuls and least-squares difference tests by using SPSS 21.0 software (IBM Corporation, Armonk, NY, USA).
Results and discussion
In this study, valsartan–polymer–surfactant composite nanoparticles were fabricated using the SAS process to enhance the dissolution and oral bioavailability of valsartan. Valsartan–polymer nanoparticles were prepared to screen HPC, HPMC, PVP K30, and PVP VA64 polymers. As shown in Figure 1 and Table 1, valsartan–HPMC nanoparticles were spherical and had a mean particle size of 277.3 nm, resulting in a specific surface area of 60.23 m2/g. For HPC, the mean particle size was greater than that of valsartan–HPMC nanoparticles, but the difference was not statistically significant (P>0.1). The mean particle size and specific surface area of PVP K30 composite nanoparticles were larger than those of valsartan–HPMC nanoparticles, whereas they were smaller for PVP VA64 particles. The crystallinity of valsartan within composite nanoparticles was determined by using DSC and PXRD. The DSC curve of valsartan exhibited an endothermic peak at approximately 96°C with an enthalpy of 37.06 J/g, corresponding to its melting point. However, the endothermic peak of valsartan was not detected for all composite particles prepared by using the SAS process. In addition, the characteristic crystalline valsartan peaks were not observed in the PXRD patterns of all composite particles (Figure 1). Therefore, the crystallinity of valsartan dramatically decreased, indicating that it was in an amorphous form within the composite nanoparticles (at 20% drug loading within the composite). The kinetic solubility test showed that the maximum valsartan concentration in valsartan–HPMC nanoparticles was 205 μg/mL, and that the concentration at 24 hours was 187 μg/mL. In general, a supersaturated state induced by the amorphous drug is thermodynamically unstable for poorly water-soluble drug-polymer composites, resulting in return to the equilibrium state through drug precipitation.21,22 Drug precipitation has to be inhibited to improve drug absorption from the gastrointestinal tract. Polymers can reduce drug precipitation and maintain the supersaturated state by inhibiting nucleation and crystal growth via surface and steric stabilization, and/or specific interactions with the drug such as hydrogen bonding, hydrophobic interactions, and complex formation.23,24 HPMC has many hydroxyl groups that act as both hydrogen bond acceptors and donors for APIs. This hydrogen bonding is attributed to positive effect for solubilization of poorly water-soluble APIs.25 In fact, valsartan’s solubility was the highest in the 5% HPMC aqueous solution compared to that in hydroxypropyl-β-cyclodextrin (HP-β-CD), PVP K30, and PVP VA64 in our previous study.8 In this study, the HPMC polymer was most effective in preventing valsartan precipitation, followed by HPC. Previously, kinetic solubility tests showed that the oral absorption of poorly water-soluble drugs was strongly correlated to the degree of supersaturation and its maintenance over extended periods. Therefore, HPMC was selected as the polymer for preparation of valsartan composite nanoparticles by using the SAS process.
Figure 1.

Scanning electron micrographs (A), kinetic solubility profile (B), differential scanning calorimetry thermograms (C), and powder X-ray diffraction patterns (D) of valsartan–polymer composite nanoparticles prepared by using the SAS process.
Abbreviations: HPC, hydroxypropyl cellulose; HPMC, hydroxypropyl methylcellulose; PVP, polyvinylpyrrolidone; PVP VA64, polyvinylpyrrolidone-vinyl acetate; SAS, supercritical antisolvent.
Ternary compositions, containing polymer in addition to surfactant, may provide effective surface modification and solubilization, resulting in enhanced supersaturation, dissolution rate, and oral absorption of valsartan. However, high amounts of surfactant can induce gastrointestinal tract irritation. Here, the valsartan–HPMC–surfactant composite nanoparticles were fabricated at a 20% drug:70% HPMC:10% surfactant (w/w/w) ratio. As shown in Table 1, the size of valsartan composite nanoparticles increased after the addition of surfactants such as poloxamer 407, Ryoto sugar ester L1695, and TPGS. Valsartan–HPMC–Ryoto sugar ester L1695 particles showed some aggregation and had a specific surface area of 25.2 m2/g (Figure 2). The fusion and aggregation of nanoparticles might be due to the low melting temperature of surfactants, as previously reported.10,18 In fact, the melting points of the used surfactants were below 55°C. However, the mean size of all valsartan–HPMC–surfactant nanoparticles was less than 800 nm. The DSC curves and PXRD patterns of composite nanoparticles did not show the characteristic endothermic melting peak and diffraction peak, respectively, corresponding to crystalline valsartan, indicating that valsartan was present in an amorphous form within the composite nanoparticles (Figure 3). The measured drug content was similar to the theoretical values (Table 1), indicating that valsartan degradation did not occur during the SAS process.
Figure 2.

Scanning electron micrographs of valsartan–HPMC–surfactant composite nanoparticles prepared by using the SAS process.
Notes: (A) Valsartan–HPMC; (B) valsartan–HPMC–poloxamer 407; (C) valsartan–HPMC–Ryoto sugar ester L1695; and (D) valsartan–HPMC–TPGS.
Abbreviations: HPMC, hydroxypropyl methylcellulose; SAS, supercritical antisolvent; TPGS, D-α-Tocopheryl polyethylene glycol 1000 succinate.
Figure 3.
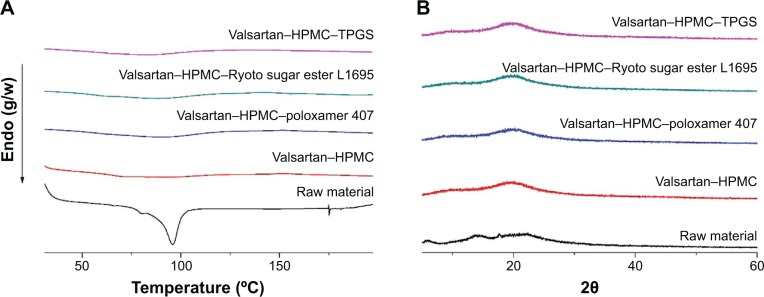
Differential scanning calorimetry thermograms (A), and powder X-ray diffraction patterns (B) of valsartan–HPMC–surfactant composite nanoparticles prepared by using the SAS process.
Abbreviations: HPMC, hydroxypropyl methylcellulose; SAS, supercritical antisolvent; TPGS, D-α-Tocopheryl polyethylene glycol 1000 succinate.
Kinetic solubility tests were conducted in simulated gastric fluids at pH 1.2. The valsartan–HPMC nanoparticles had a maximum solubility of 205 μg/mL. As shown in Figure 4, the maximum solubility of valsartan was dramatically increased by the addition of surfactant to valsartan–HPMC nanoparticles. In particular, the solubility of valsartan–HPMC–poloxamer 407 nanoparticles was 999.1 μg/mL, and their concentration at 24 hours was 992.1 μg/mL. The most effective surfactant tested was poloxamer 407, followed by TPGS and Ryoto sugar ester L1695. Poloxamer 407, a hydrophilic ethylene oxide–hydrophobic propylene oxide block copolymer, is an amphiphilic polymer that acts as a surfactant for solubilization via micelle formation, and as a precipitation inhibitor via steric stabilization and/or hydrogen bonding.26 Poorly water-soluble APIs can be incorporated into the hydrophobic propylene oxide core of micelles formed via self-assembly, which would result in the solubilization of poorly water-soluble APIs. In our previous study, higher solubility of valsartan was observed upon using poloxamer 407 than upon using TPGS.8 In addition, the synergistic effect of poloxamer 407 was observed on the supersaturation of valsartan induced from the HPMC composite. However, this topic needs further study using approaches such as a phase solubility analysis.
Figure 4.
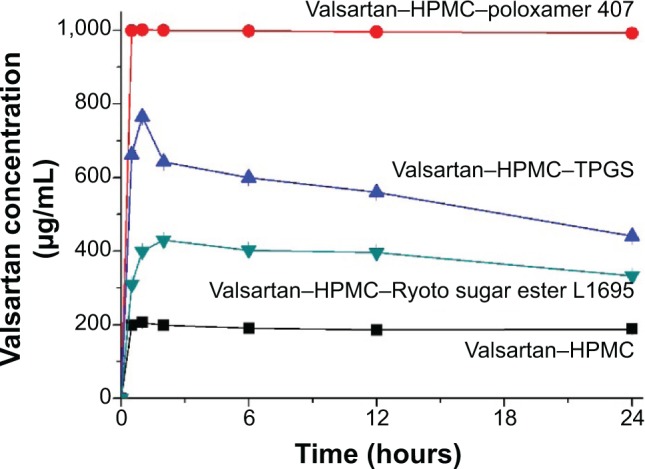
Kinetic solubility profiles of valsartan–HPMC–surfactant composite nanoparticles prepared by using the SAS process.
Note: Data are expressed as the mean ± standard deviation (n=3).
Abbreviations: HPMC, hydroxypropyl methylcellulose; SAS, supercritical antisolvent; TPGS, D-α-Tocopheryl polyethylene glycol 1000 succinate.
The in vitro dissolution profiles of valsartan composite nanoparticles were obtained in four different dissolution media (Figure 5). The raw material had a pH-dependent dissolution profile due to its intrinsic pH-dependent solubility. Approximately 4% of the raw material was detected at pH 1.2 within 2 hours. However, approximately 78% of raw material dissolved at pH 6.8 within the same period. The extent and rate of dissolution of valsartan–HPMC nanoparticles increased according to the pH-dependent solubility of valsartan in the order: pH 1.2< pH 4.0< pH 6.8. Interestingly, pH-independent dissolution profiles of valsartan were obtained after the addition of surfactant into valsartan–HPMC nanoparticles owing to micellar solubilization. In particular, the valsartan–HPMC–poloxamer 407 nanoparticles exhibited faster drug release of up to 90% within 10 minutes under all conditions. Previously, it was reported that higher solubilization of valsartan was observed upon using poloxamer 407 than upon using cremophore EL, sodium lauryl sulfate, solutol HS15, TPGS, Ryoto sugar ester L1695, and Gelucire 44/14.5,8 The significant increase in both the dissolution extent and rate of valsartan might be because of the increased supersaturation caused by the amorphous form, and/or the improved wettability of the particles due to surface modification by the surfactant and HPMC. For the in vitro–in vivo correlation study, dissolution profiles were characterized using the dissolution efficiency (DE%) as defined by Khan and Rhodes.27 DE% for valsartan composite nanoparticles was calculated from the area under the dissolution curves at 120 minutes and expressed as a percentage of the area of the rectangle resulting from 100% dissolution within the same time. As expected, the highest DE% was calculated for the valsartan–HPMC–poloxamer 407 nanoparticles (Table 2).
Figure 5.

Dissolution profiles of valsartan–HPMC–surfactant composite nanoparticles prepared by using the SAS process.
Notes: pH 1.2 (A); pH 4.0 (B); and pH 6.8 (C) dissolution media. Data are expressed as the mean ± standard deviation (n=3).
Abbreviations: HPMC, hydroxypropyl methylcellulose; SAS, supercritical antisolvent; TPGS, D-α-Tocopheryl polyethylene glycol 1000 succinate.
Table 2.
Dissolution efficiency for Val composite nanoparticles prepared by using the SAS process
| Formulation | DE (%)
|
||
|---|---|---|---|
| pH 1.2 | pH 4.0 | pH 6.8 | |
| Raw material | 3.4±0.4 | 5.3±0.4 | 61.1±5.7 |
| Val:HPMC =2:8 | 54.7±2.8 | 61.3±2.2 | 94.2±2.8 |
| Val:HPMC:poloxamer 407 =2:7:1 | 96.1±1.8 | 96.8±1.9 | 97.5±1.5 |
| Val:HPMC:Ryoto sugar ester L1695 =2:7:1 | 87.7±2.5 | 89.9±4.2 | 96.3±2.5 |
| Val:HPMC:TPGS =2:7:1 | 89.7±2.8 | 92.3±3.1 | 96.8±1.9 |
Notes: The dissolution efficiency for Val composite nanoparticles was calculated from the area under the dissolution curves at 120 minutes and expressed as a percentage of the area of the rectangle resulting from 100% dissolution within the same period. Data are expressed as the mean ± standard deviation (n=3).
Abbreviations: DE, dissolution efficiency; HPMC, hydroxypropyl methylcellulose; SAS, supercritical antisolvent; TPGS, D-α-Tocopheryl polyethylene glycol 1000 succinate; Val, valsartan.
The pharmacokinetic study of valsartan composite nanoparticles and raw material was conducted in rats. The oral absorption of valsartan from the composite nanoparticles was dramatically higher than that of the raw material, as shown in the plasma concentration-time curves of valsartan after oral administration (Figure 6). In particular, the valsartan–HPMC–poloxamer 407 nanoparticles exhibited higher oral bioavailability than that of the raw material, with approximately 7.2- and 4.6-fold higher Cmax and AUC0→24 h, respectively (Table 3). In addition, the AUC0→24 h of valsartan increased in the order: raw material < HPMC nanoparticles < HPMC–Ryoto sugar ester L1695 nanoparticles = HPMC–TPGS nanoparticles < HPMC–poloxamer 407 nanoparticles.
Figure 6.
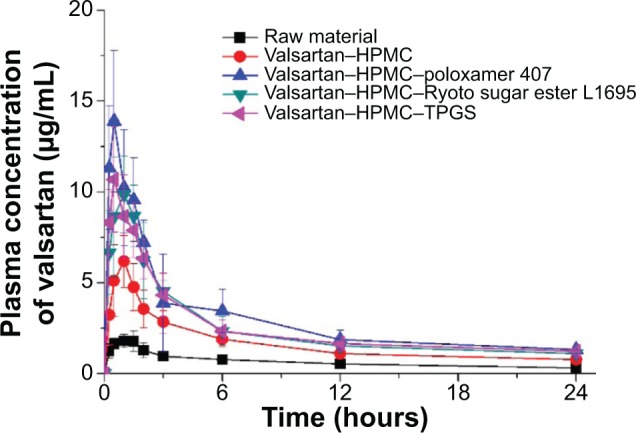
Plasma concentration-time profile of valsartan in rats after oral administration of the raw material and valsartan composite nanoparticles prepared by using the SAS process.
Note: Data are expressed as the mean ± standard deviation (n=5).
Abbreviations: HPMC, hydroxypropyl methylcellulose; SAS, supercritical antisolvent; TPGS, D-α-Tocopheryl polyethylene glycol 1000 succinate.
Table 3.
Pharmacokinetic parameters for Val composite nanoparticles prepared by using the SAS process
| Formulation | AUC0–24 h (μg·hour/mL) | Cmax (μg/mL) | Tmax (hours) |
|---|---|---|---|
| Raw material | 16.0±3.57 | 1.9±0.4 | 1.9±0.4 |
| Val:HPMC =2:8 | 40.4±6.23a | 6.2±0.9a | 0.9±0.4 |
| Val:HPMC:poloxamer 407 =2:7:1 | 73.8±14.0a–d | 13.6±3.5a–d | 0.5±0.3 |
| Val:HPMC:Ryoto sugar ester L1695 =2:7:1 | 58.0±9.11a,b | 9.9±1.6a,b | 0.8±0.3 |
| Val:HPMC:TPGS =2:7:1 | 61.2±10.7a,b | 10.4±1.9a,b | 0.6±0.3 |
Notes:
Indicates P<0.05 versus raw material
Indicates P<0.05 versus Val:HPMC =2:8
Indicates P<0.05 versus Val:HPMC:Ryoto sugar ester L1695 =2:7:1
Indicates P<0.05 versus Val:HPMC:TPGS =2:7:1. Data are expressed as the mean ± standard deviation (n=5).
Abbreviations: AUC, area under the concentration-time curve; Cmax, peak plasma concentration; HPMC, hydroxypropyl methylcellulose; SAS, supercritical antisolvent; Tmax, time to peak concentration; TPGS, D-α-Tocopheryl polyethylene glycol 1000 succinate; Val, valsartan.
To determine whether there was a correlation between in vitro dissolution and in vivo parameters, a regression analysis was conducted by using DE%, Cmax, and AUC0→24 h. As shown in Figure 7, there was a positive linear correlation between the pharmacokinetic parameters (Cmax and AUC0→24 h) and the DE% calculated in pH 1.2 and pH 4.0 dissolution media (R2>0.90). However, there was a poor correlation with DE% calculated in pH 6.8 dissolution media compared to that calculated in the other dissolution medium. Valsartan contains a tetrazole derivative-containing acid (pKa =4.73) as well as carboxylic-group acids (pKa =3.9) groups, which may influence its solubility in a pH-dependent manner, resulting in an extremely low dissolution rate at acidic pH conditions. However, valsartan showed a high dissolution rate for raw material and composite nanoparticles with pH 6.8 dissolution medium compared to that observed with an acidic pH dissolution medium. Thus, a poor correlation between dissolution data and pharmacokinetic data was observed for the pH 6.8 dissolution medium compared to that observed for the other dissolution medium. In fact, the oral bioavailability of valsartan was increased by enhancing the in vitro dissolution in pH 1.2 and pH 4.0 dissolution media, in agreement with our previous in vitro–in vivo correlation results using a spray-dried emulsion containing valsartan. The dissolution of valsartan was very slow due to poor water solubility at low pH because of its carboxylic acid-containing molecular structure. Generally, the oral absorption of a drug is proportional to the dissolution process within the gastrointestinal tract.28 Therefore, the dissolution and solubilization steps could be rate-limiting for the absorption of a poorly water-soluble drug. In this study, the supersaturated solution of valsartan induced by composite nanoparticles resulted in increased absorption through the gastrointestinal epithelial membrane, thereby enhancing the oral bioavailability of valsartan. Considering the intrinsic solubility of valsartan and the physiological pH conditions of the gastrointestinal tract, it is important to increase the extent and rate of dissolution at conditions below pH 4.0 to develop a new dosage form of valsartan with high oral bioavailability.
Figure 7.
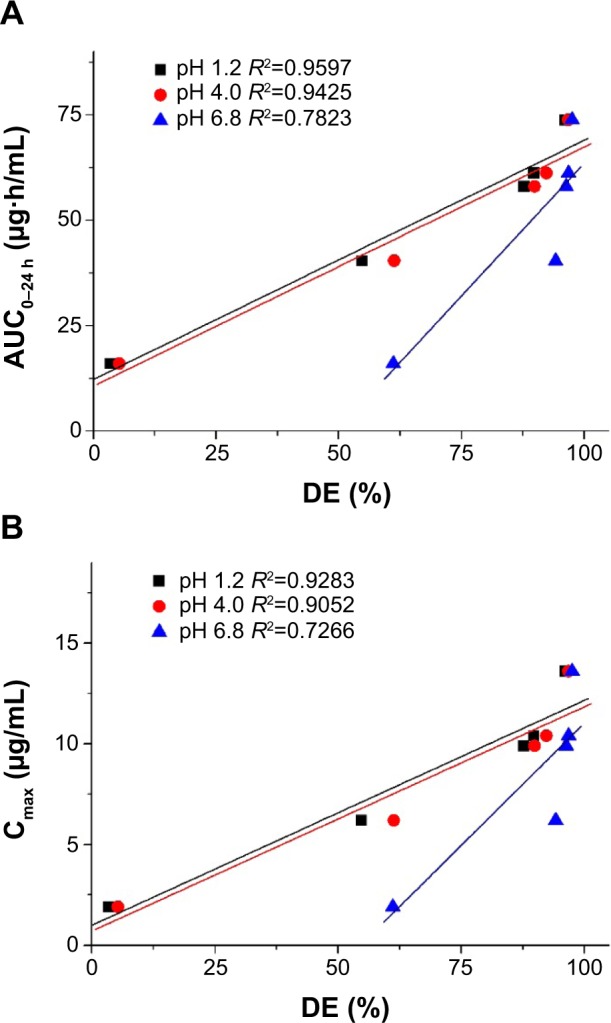
Correlation between the in vitro dissolution efficiency and in vivo pharmacokinetic parameters.
Notes: (A) AUC0–24h; (B) Cmax.
Abbreviations: AUC0–24 h, area under the concentration-time curve; Cmax, peak concentration; DE, dissolution efficiency; R2, coefficient of determination.
Conclusion
In this study, spherical composite nanoparticles smaller than 400 nm, which contained valsartan, were fabricated by using the SAS process. The in vitro dissolution and oral bioavailability of valsartan were dramatically enhanced by the composite nanoparticles. Valsartan–HPMC–poloxamer 407 nanoparticles exhibited faster drug release of up to 90% within 10 minutes under all dissolution conditions, and higher oral bioavailability than that of the raw material, with approximately 7.2- and 4.6-fold higher Cmax and AUC0→24 h, respectively. Therefore, the preparation of composite nanoparticles with HPMC and poloxamer 407 by using the SAS process could be an effective formulation strategy for the development of a new dosage form of valsartan with high oral bioavailability.
Acknowledgments
This research was supported by Kyungsung University Research Grants in 2013.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Markham A, Goa KL. Valsartan. A review of its pharmacology and therapeutic use in essential hypertension. Drugs. 1997;54(2):299–311. doi: 10.2165/00003495-199754020-00009. [DOI] [PubMed] [Google Scholar]
- 2.Yan YD, Sung JH, Kim KK, et al. Novel valsartan-loaded solid dispersion with enhanced bioavailability and no crystalline changes. Int J Pharm. 2012;422(1–2):202–210. doi: 10.1016/j.ijpharm.2011.10.053. [DOI] [PubMed] [Google Scholar]
- 3.Flesch G, Müller P, Lloyd P. Absolute bioavailability and pharmacokinetics of valsartan, an angiotensin II receptor antagonist, in man. Eur J Clin Pharmacol. 1997;52(2):115–120. doi: 10.1007/s002280050259. [DOI] [PubMed] [Google Scholar]
- 4.Mbah CJ. Physicochemical properties of valsartan and the effect of ethyl alcohol, propylene glycol and pH on its solubility. Pharmazie. 2005;60(11):849–850. [PubMed] [Google Scholar]
- 5.Park YJ, Lee HK, Im YB, Lee W, Han HK. Improved pH-independent dissolution and oral absorption of valsartan via the preparation of solid dispersion. Arch Pharm Res. 2010;33(8):1235–1240. doi: 10.1007/s12272-010-0814-0. [DOI] [PubMed] [Google Scholar]
- 6.Poudel BK, Marasini N, Tran TH, Choi HG, Yong CS, Kim JO. Formulation, characterization and optimization of valsartan self-microemulsifying drug delivery system using statistical design of experiment. Chem Pharm Bull (Tokyo) 2012;60(11):1409–1418. doi: 10.1248/cpb.c12-00502. [DOI] [PubMed] [Google Scholar]
- 7.Beg S, Swain S, Singh HP, Patra ChN, Rao ME. Development, optimization, and characterization of solid self-nanoemulsifying drug delivery systems of valsartan using porous carriers. AAPS Pharm Sci Tech. 2012;13(4):1416–1427. doi: 10.1208/s12249-012-9865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baek IH, Kim JS, Ha ES, et al. Oral absorption of a valsartan-loaded spray-dried emulsion based on hydroxypropylmethyl cellulose. Int J Biol Macromol. 2014;69:222–228. doi: 10.1016/j.ijbiomac.2014.05.059. [DOI] [PubMed] [Google Scholar]
- 9.Ma Q, Sun H, Che E, et al. Uniform nano-sized valsartan for dissolution and bioavailability enhancement: influence of particle size and crystalline state. Int J Pharm. 2013;441(1–2):75–81. doi: 10.1016/j.ijpharm.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 10.Kim MS, Kim JS, Cho W, et al. Supersaturatable formulations for the enhanced oral absorption of sirolimus. Int J Pharm. 2013;445(1–2):108–116. doi: 10.1016/j.ijpharm.2013.01.067. [DOI] [PubMed] [Google Scholar]
- 11.Baek IH, Kim MS. Improved supersaturation and oral absorption of dutasteride by amorphous solid dispersions. Chem Pharm Bull (Tokyo) 2012;60:1468–1473. doi: 10.1248/cpb.c12-00563. [DOI] [PubMed] [Google Scholar]
- 12.Choi JH, Lee K, Hong S, et al. Effect of biocompatible polymers on the physicochemical and dissolution properties of fenofibrate in nanoparticle system. Journal of Pharmaceutical Investigation. 2013;43:507–512. [Google Scholar]
- 13.Kim MS. Influence of hydrophilic additives on the supersaturation and bioavailability of dutasteride-loaded hydroxypropyl-β-cyclodextrin nanostructures. Int J Nanomedicine. 2013;8:2029–2039. doi: 10.2147/IJN.S44795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nalawade SP, Picchioni F, Janssen L. Supercritical carbon dioxide as a green solvent for processing polymer melts: Processing aspects and applications. Prog Polym Sci. 2006;31:19–43. [Google Scholar]
- 15.Kim MS, Lee S, Park JS, Woo JS, Hwang SJ. Micronization of cilostazol using supercritical antisolvent (SAS) process: effect of process parameters. Powder Technol. 2007;177:64–70. [Google Scholar]
- 16.Karn PR, Cho W, Park HJ, Park JS, Hwang SJ. Characterization and stability studies of a novel liposomal cyclosporin A prepared using the supercritical fluid method: comparison with the modified conventional Bangham method. Int J Nanomedicine. 2013;8:365–377. doi: 10.2147/IJN.S39025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cocero MJ, Martín Á, Mattea F, Varona S. Encapsulation and co-precipitation processes with supercritical fluids: fundamentals and applications. J Supercrit Fluids. 2009;47:546–555. [Google Scholar]
- 18.Kim MS, Kim JS, Park HJ, Cho WK, Cha KH, Hwang SJ. Enhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int J Nanomedicine. 2011;6:2997–3009. doi: 10.2147/IJN.S26546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim MS, Kim JS, Cho W, Park HJ, Hwang SJ. Oral absorption of atorvastatin solid dispersion based on cellulose or pyrrolidone derivative polymers. Int J Biol Macromol. 2013;59:138–142. doi: 10.1016/j.ijbiomac.2013.03.068. [DOI] [PubMed] [Google Scholar]
- 20.Ha ES, Choo GH, Baek IH, et al. Dissolution and bioavailability of lercanidipine-hydroxypropylmethyl cellulose nanoparticles with surfactant. Int J Biol Macromol. 2014;72C:218–222. doi: 10.1016/j.ijbiomac.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 21.Cha KH, Cho KJ, Kim MS, et al. Enhancement of the dissolution rate and bioavailability of fenofibrate by a melt-adsorption method using supercritical carbon dioxide. Int J Nanomedicine. 2012;7:5565–5575. doi: 10.2147/IJN.S36939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim MS, Jin SJ, Kim JS, et al. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2008;69(2):454–465. doi: 10.1016/j.ejpb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Wen H, Morris KR, Park K. Hydrogen bonding interactions between adsorbed polymer molecules and crystal surface of acetaminophen. J Colloid Interface Sci. 2005;290:325–335. doi: 10.1016/j.jcis.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 24.Kojima T, Higashi K, Suzuki T, Tomono K, Moribe K, Yamamoto K. Stabilization of a supersaturated solution of mefenamic acid from a solid dispersion with EUDRAGIT(®) EPO. Pharm Res. 2012;29:2777–2791. doi: 10.1007/s11095-011-0655-7. [DOI] [PubMed] [Google Scholar]
- 25.Eedara BB, Kankane M, Jukanti R, Nagabandi VK, Bandari S. Enhanced solubility and permeability of exemestane solid dispersion powders for improved oral delivery. Journal of Pharmaceutical Investigation. 2013;43:229–242. [Google Scholar]
- 26.Adeli E, Mortazavi SA. Design, formulation and evaluation of Azithromycin binary solid dispersion using Kolliphor series for the solubility and in vitro dissolution rate enhancement. Journal of Pharmaceutical Investigation. 2014;44:119–131. [Google Scholar]
- 27.Khan KA, Rhodes CT. Effect of compaction pressure on the dissolution efficiency of some direct compression systems. Pharm Acta Helv. 1972;47(10):594–607. [PubMed] [Google Scholar]
- 28.Palanisamy M, Khanam J. Effect of physiochemical variables on phase solubility and dissolution behavior of indomethacin solid dispersion system. Journal of Pharmaceutical Investigation. 2014;44:147–162. [Google Scholar]