

Abstract
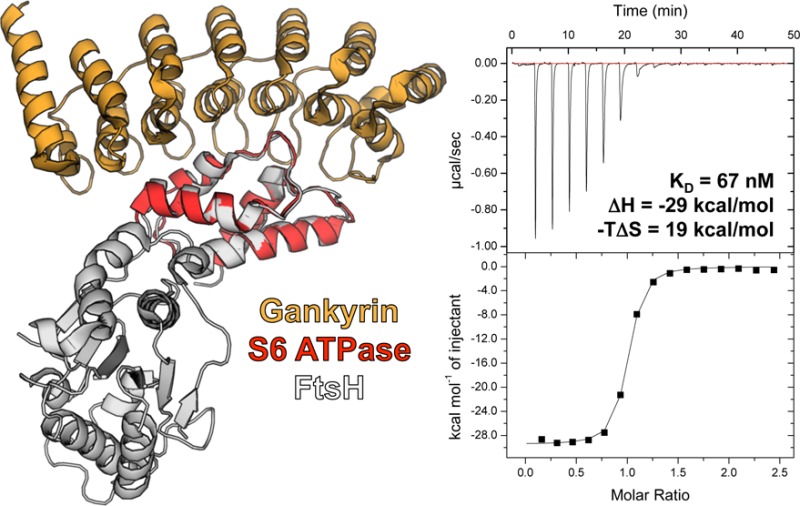
A complex with the C-terminal portion of the proteosomal subunit S6 ATPase is the only available structure of a protein–protein interaction involving the oncoprotein gankyrin. However, difficulties associated with recombinant expression of S6 ATPase alone, or truncations thereof, have limited our understanding of this assembly. We replaced the C-terminal portion of FtsH from Escherichia coli with the structurally homologous C-terminal portion of S6 ATPase and used this grafted protein to characterize the gankyrin–S6 ATPase binding interaction by isothermal titration calorimetry.
Overexpression of gankyrin (Figure 1a, orange) is directly linked to the onset, proliferation, and/or metastasis of breast,1,2 liver,3 oral,4 pancreatic,5 and colorectal cancers.6 In addition, gankyrin plays an essential role in Ras-initiated tumorigenesis, which is operative in ∼30% of all cancers.7 Protein–protein interactions (PPIs) involving gankyrin are of great interest in basic research and as therapeutic targets.
Figure 1.
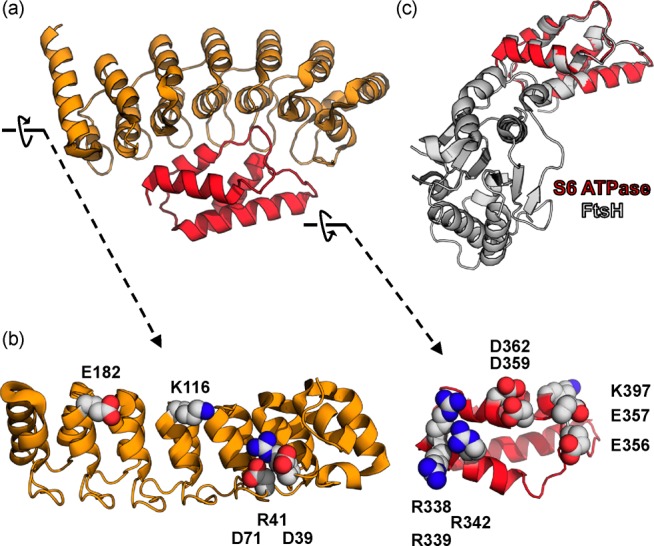
(a) Complex between gankyrin (orange) and the C-terminal portion of the S6 ATPase subunit of the 26S proteosome [red, Protein Data Bank (PDB) entry 2DVW]. The direction of the arrow next to each protein indicates the direction of a 90° rotation, which reveals the binding surfaces, as shown in panel b. (b) Gankyrin and S6 ATPase binding face residues critical to complex stabilization (and mutated in this work). (c) S6 ATPase superimposed on the C-terminal domain of FtsH (PDB entry ILV7).
Gankyrin is reported to bind both cyclin-dependent kinase 4 (CDK4)8 and MDM2,9 resulting in increased efficiency of pRb phosphorylation and p53 polyubiquitination and degradation, respectively. However, the structural basis for these interactions has not yet been reported, and neither the targets nor their putative gankyrin binding domains are expressed as soluble recombinant proteins in Escherichia coli. A more promising venue for studying gankyrin–protein interactions is the co-crystal structure with a C-terminal portion of the S6 ATPase from the 26S proteasome, reported by Yokoyama and co-workers (Figure 1a).10 Preliminary characterization of the recognition interface by Yokoyama and co-workers was achieved by a series of pull-down experiments with gankyrin-His6x and S6 ATPase mutants (concomitantly expressed from a pET-Duet plasmid), in which binding face residues thought to participate in complex stability were mutated to mostly alanine [R342A, R338A/R342A, R338A/R339A/R342A, E356A/E357A, D359A/D362A, and K397E in S6 ATPase and R41A, K116A, D39A/D71A, R41A/K116A, and E182A in gankyrin (highlighted in Figure 1b)].
Efforts to directly probe the gankyrin–S6 ATPase complex are hampered by the tendency of the latter to form inclusion bodies when expressed in the absence of gankyrin. In our hands, such material could not be refolded, and fusion to proteins commonly used to improve stability and solubility was likewise ineffective. An alternative strategy for the display of folded and functional S6 ATPase is protein grafting. In this approach, a protein scaffold is identified that is stable, is expressed well in E. coli, and contains a domain with excellent structural homology to S6 ATPase. If that protein is stable enough to tolerate replacement of the structurally homologous domain with S6 ATPase, it could serve as a generic platform for the display of a folded and functional variant of this otherwise inaccessible protein.
Our initial efforts to identify such a scaffold relied on the recognition by Yokoyama and co-workers that, while the C-terminal portion of FtsH from E. coli has a low level of sequence homology (∼25%) with S6 ATPase, the two proteins have similar tertiary structures [root-mean-square deviation of ∼1.4 Å over 74 main chain residues (Figure 1c)]. Expanding on this finding, we set out to determine if a grafted protein, in which the C-terminal ATPase domain of FtsH is replaced with S6 ATPase, is expressed as a soluble protein in E. coli that mimics the native S6ATPase–gankyrin interaction.
Grafted FtsH-S6 ATPase and wild-type FtSH (wt-FtsH) were expressed as His6x-tagged proteins in E. coli as soluble proteins (Supporting Information, Figure S1). Circular dichroism spectra of the two proteins are virtually identical (Figure 2a), suggesting no appreciable structural change to the FtsH scaffold or grafted S6 ATPase domain. The affinity of this grafted protein for gankyrin was first assessed using a pull-down assay in E. coli. Binding face residues on gankyrin or FtsH-S6 ATPase were mutated to alanine, on the basis of the findings of Yokoyama and co-workers, and their effect on complex stability was qualitatively assessed by measuring the amount of untagged FtsH-S6 ATPase co-purified with gankyrin-His6x.
Figure 2.
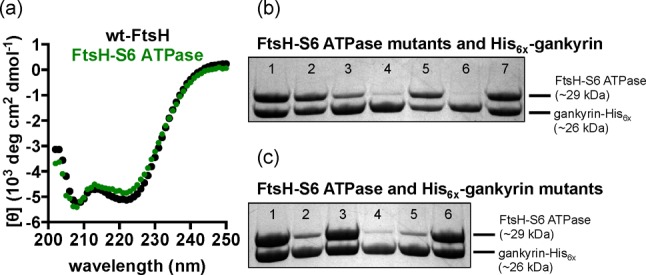
(a) Circular dichroism spectra of wild-type FtsH (wt-FtsH, top) and FtsH-S6 ATPase (bottom). (b) Co-purification of wild-type gankyrin-His6x and FtsH-S6 ATPase mutants: wt-S6 FtsH-S6 ATPase (lane 1), R342A (lane 2), R338A/R342A (lane 3), R338A/R339A/R342A (lane 4), E356A/E357A (lane 5), D359A/D362A (lane 6), and K397E (lane 7). (c) Co-purification of wild-type S6 ATPase and gankyrin-His6x mutants: wt-gankyrin (lane 1), R41A (lane 2), K116A (lane 3), R41A/K116A (lane 4), D39A/D71A (lane 5), and E182A (lane 6).
Most notably, FtsH-S6 ATPase R338A/R339A/R342A (Figure 2b, lane 4), FtsH-S6 ATPase D359A/D362A (Figure 2b, lane 6), FtsH-S6 ATPase R41A (Figure 2c, lane 2), gankyrin R41A/K116A (Figure 2c, lane 4), and gankyrin D39A/D71A (Figure 2c, lane 5) appear to form complexes that are significantly less stable than the native proteins. This is in contrast to Yokoyama’s original pull-down assay, in which all mutants but R342A S6 ATPase were not appreciably co-purified with gankyrin-His6x. This highlights a potential virtue of our grafting approach. It is unclear if mutations to this unstable form of S6 ATPase appreciably modulate, or abolish, gankyrin–S6 ATPase complex stability or simply further decrease the level of structure and stability of the C-terminal S6 ATPase fragment.
While the FtsH scaffold displays S6 ATPase in a manner that faithfully mimics the native protein (facilitates binding to gankyrin), no information about the exact differences in binding energies can be obtained using the pull-down assay. Moreover, mutational effects that do not dramatically lower, or completely abolish, complex stability cannot be probed using this assay. Only through the described grafting strategy are we able to create a soluble and stable mimic of S6 ATPase, which permits the use of more sensitive biophysical methods for probing this important binding interaction.
We used isothermal titration calorimetry (ITC) to obtain the full thermodynamic signature (ΔH, −TΔS, and ΔG) and stoichiometry (N value) of this interaction, as well as characterize mutational effects on complex stability. Gankyrin binds the grafted FtsH-S6 ATPase with a dissociation constant (KD) of ∼67 nM (Table 1, entry 1). The observed changes in enthalpy (ΔH) and entropy (−TΔS) for this binding interaction were −28.7 and 19.0 kcal/mol, respectively. Gankyrin does not bind wild-type FtsH with any appreciable affinity (Supporting Information, Figure S2), which is unsurprising, given that the S6 and FtsH ATPase subdomains share only ∼24% sequence homology.
Table 1. Analysis of Binding Interactions between Gankyrin and FtsH-S6 ATPase Proteins by ITCa.
| entry | gankyrin | FtsH-S6 ATPase | KD (nM) | ΔG (kcal/mol) | ΔH (kcal/mol) | –TΔS (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | wild-type | wild-type | 67.3 ± 5.7 | –9.8 ± 0.1 | –28.7 ± 0.5 | 19.0 ± 0.6 |
| 2 | wild-type | R342A | 216.6 ± 25.8 | –9.1 ± 0.1 | –22.0 ± 0.8 | 12.9 ± 0.8 |
| 3 | wild-type | R338A/R342A | 2549 ± 353 | –7.6 ± 0.1 | –6.1 ± 0.7 | –1.5 ± 0.8 |
| 4 | wild-type | R338A/R339A/R342A | 7471 ± 301 | –7.0 ± 0.1 | –2.2 ± 0.1 | –4.7 ± 0.1 |
| 5 | wild-type | E356A/E357A | 71.8 ± 5.9 | –9.8 ± 0.1 | –27.3 ± 0.9 | 17.5 ± 0.8 |
| 6 | wild-type | D359A/D362A | no binding | – | – | – |
| 7 | wild-type | K397E | 95.2 ± 12.2 | –9.7 ± 0.2 | –25.6 ± 2.5 | 15.9 ± 2.7 |
| 8 | R41A | wild-type | 313.3 ± 17.6 | –8.1 ± 1.2 | –17.1 ± 1.7 | 9.0 ± 2.8 |
| 9 | K116A | wild-type | 71.3 ± 15.5 | –9.7 ± 0.2 | –24.0 ± 0.9 | 14.3 ± 1.1 |
| 10 | D39A/D71A | wild-type | 93.0 ± 5.6 | –9.7 ± 0.2 | –25.0 ± 0.7 | 15.4 ± 0.8 |
| 11 | R41A/K116A | wild-type | 3633 ± 404 | –7.4 ± 0.1 | –4.9 ± 0.6 | –2.5 ± 0.7 |
| 12 | E182A | wild-type | 140.6 ± 9.7 | –9.4 ± 0.1 | –28.2 ± 2.1 | 18.8 ± 2.0 |
All errors represent the standard deviation of three separate experiments. ITC conditions were as follows: 20 mM sodium phosphate, 150 mM NaCl, and 2.5 mM 2-mercaptoethanol (pH 7.4) at 25 °C.
Alanine mutation of S6 ATPase R342, which engages gankyrin through a salt bridge with gankyrin E182, modestly lowers complex stability [KD = 216.6 ± 25.8 nM (Table 1, entry 2)]. Double (R338A/R342A) and triple (R338A/R339A/R342A) mutation of a positively charged patch on the S6 ATPase face, which disrupts a salt bridge between S6ATPase R342 and gankyrin E182, dramatically lowers complex stability [KD values of 2.5 ± 0.4 and 7.5 ± 0.2 μM (Table 1, entries 3 and 4, respectively)]. Interestingly, both of these mutations result in favorable binding entropies (−TΔS values of −1.5 ± 0.8 and −4.7 ± 0.1 kcal/mol, repectively, compared to a −TΔS of 19.0 ± 0.6 for the native interaction). While the molecular mechanism for this dramatic change is unclear, one possible rationale is a lower energy of desolvation for the alanine mutants, compared to that of the native protein. While Yokoyama’s original pull-down data suggest a significant role for S6 ATPase E356/E357 in complex stability, double alanine mutation did not appreciably lower binding affinity (Table 1, entry 5). Conversely, removal of a negatively charged patch on the S6 ATPase binding face [D359A/D362A (Table 1, entry 6)] abolished binding. An E182A mutation in gankyrin, which further probes a salt bridge with S6 ATPase residue 342, was found to modestly lower bindng affinity [KD = 140.6 ± 9.7 nM (Table 1, entry 12)], further suggesting a relatively minor role of this interaction in complex stability. Residue K397 in S6 ATPase makes a salt bridge with gankyrin D39/D71. However, a mutant that reverses the ionic nature of this residue (K397E) binds gankyrin with an affinity similar to that of the native protein [KD = 95.2 ± 12.2 nM (Table 1, entry 7)], suggesting a relatively minor role of this particular salt bridge in complex stability. While gankyrin mutation K116A and double mutation D39A/D71A had minimal effects on binding affinity (Table 1, entries 9 and 10, respectively), an R41A mutation significantly decreased affinity [KD = 313.3 ± 17.6 nM (Table 1, entry 8)]. While the single K116A mutation had a minimal effect on binding, an R41A/K116A double mutation, which is designed to test the role of a larger hydrogen bond/salt bridge network, dramatically lowers affinity [KD = 3.6 ± 0.4 μM (Table 1, entry 11)]. The R41A/K116A mutant, however, binds gankyrin with a favorable binding entropy (−TΔS = −2.5 ± 0.7 kcal/mol), possibly due to the lower energy of desolvation for the alanine mutants, compared to that of the native protein. The binding stoichiometry (n) for each interaction was found to be ∼1 [n = 0.91–1.02 (Supporting Information)]. Reversing the titration did not appreciably alter the binding data (data not shown).
Collectively, our findings represent the first quantitative assessment of the binding interaction, and binding thermodynamics, of a physiologically relevant complex involving the oncoprotein gankyrin. These data also potentially establish a target affinity for therapeutic reagents designed to inhibit gankyrin-dependent protein–protein interactions.11,12
Acknowledgments
We thank R. Tennyson for assistance with circular dichroism experiments.
Supporting Information Available
Experimental details, polyacrylamide gel electrophoresis gel analysis, sequences of all proteins used in this work, and representative isothermal titration calorimetry binding isotherms for each entry in Table 1. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding was provided by National Institutes of Health Grant R01GM107520. A.M.C. was supported in part by a Lou Hegedus fellowship.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zhen C.; Chen L.; Zhao Q.; Liang B.; Gu Y. X.; Bai Z. F.; Wang K.; Xu X.; Han Q. Y.; Fang D. F.; Wang S. X.; Zhou T.; Xia Q.; Gong W. L.; Wang N.; Li H. Y.; Jin B. F.; Man J. H. (2013) Oncogene 32, 3452–3460. [DOI] [PubMed] [Google Scholar]
- Kim Y. H.; Kim J. H.; Choi Y. W.; Lim S. K.; Yim H.; Kang S. Y.; Chung Y. S.; Lee G. Y.; Park T. J. (2013) Exp. Mol. Pathol. 94, 360–365. [DOI] [PubMed] [Google Scholar]
- Fu X. Y.; Wang H. Y.; Tan L.; Liu S. Q.; Cao H. F.; Wu M. C. (2002) World J. Gastroenterol. 8, 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Knobloch T. J.; Kresty L. A.; Zhang Z.; Lang J. C.; Schuller D. E.; Weghorst C. M. (2011) Anticancer Res. 31, 2683–2692. [PubMed] [Google Scholar]
- Meng Y.; He L.; Guo X.; Tang S.; Zhao X.; Du R.; Jin J.; Bi Q.; Li H.; Nie Y.; Liu J.; Fan D. (2010) Cancer Lett. 297, 9–17. [DOI] [PubMed] [Google Scholar]
- Tang S.; Yang G.; Meng Y.; Du R.; Li X.; Fan R.; Zhao L.; Bi Q.; Jin J.; Gao L.; Zhang L.; Li H.; Fan M.; Wang Y.; Wu K.; Liu J.; Fan D. (2010) Cancer Biol. Ther. 9, 88–95. [DOI] [PubMed] [Google Scholar]
- Man J. H.; Liang B.; Gu Y. X.; Zhou T.; Li A. L.; Li T.; Jin B. F.; Bai B.; Zhang H. Y.; Zhang W. N.; Li W. H.; Gong W. L.; Li H. Y.; Zhang X. M. (2010) J. Clin. Invest. 120, 2829–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. N.; Tsai M. D. (2002) Biochemistry 41, 3977–3983. [DOI] [PubMed] [Google Scholar]
- Higashitsuji H.; Higashitsuji H.; Itoh K.; Sakurai T.; Nagao T.; Sumitomo H.; Masuda T.; Dawson S.; Shimada Y.; Mayer R. J.; Fujita J. (2005) Cancer Cell 8, 75–87. [DOI] [PubMed] [Google Scholar]
- Nakamura Y.; Nakano K.; Umehara T.; Kimura M.; Hayashizaki Y.; Tanaka A.; Horikoshi M.; Padmanabhan B.; Yokoyama S. (2007) Structure 15, 179–189. [DOI] [PubMed] [Google Scholar]
- Nanaware P. P.; Ramteke M. P.; Somavarapu A. K.; Venkatraman P. (2014) Proteins 82, 1283–1300. [DOI] [PubMed] [Google Scholar]
- Chapman A. M.; McNaughton B. R. (2014) ACS Chem. Biol. 9, 2223–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.