

Abstract
Background
Brassica rapa is an economically important crop species. During its long breeding history, a large number of morphotypes have been generated, including leafy vegetables such as Chinese cabbage and pakchoi, turnip tuber crops and oil crops.
Results
To investigate the genetic variation underlying this morphological variation, we re-sequenced, assembled and annotated the genomes of two B. rapa subspecies, turnip crops (turnip) and a rapid cycling. We then analysed the two resulting genomes together with the Chinese cabbage Chiifu reference genome to obtain an impression of the B. rapa pan-genome. The number of genes with protein-coding changes between the three genotypes was lower than that among different accessions of Arabidopsis thaliana, which can be explained by the smaller effective population size of B. rapa due to its domestication. Based on orthology to a number of non-brassica species, we estimated the date of divergence among the three B. rapa morphotypes at approximately 250,000 YA, far predating Brassica domestication (5,000-10,000 YA).
Conclusions
By analysing genes unique to turnip we found evidence for copy number differences in peroxidases, pointing to a role for the phenylpropanoid biosynthesis pathway in the generation of morphological variation. The estimated date of divergence among three B. rapa morphotypes implies that prior to domestication there was already considerably divergence among B. rapa genotypes. Our study thus provides two new B. rapa reference genomes, delivers a set of computer tools to analyse the resulting pan-genome and uses these to shed light on genetic drivers behind the rich morphological variation found in B. rapa.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-250) contains supplementary material, which is available to authorized users.
Background
Plants in the Brassica genus display extreme morphological diversity, from cauliflower and broccoli through cabbages and Brussels sprouts to turnips and oil crops. Almost all organs are used for consumption: heads of cabbages and leaves on non-heading vegetable types, inflorescences of cauliflowers, tuberized stems/hypocotyls and/or roots of kohlrabi’s, turnips and swede and enlarged seeds and seedpods of oil types. One of the most important Brassica species, Brassica rapa, also shows this extreme morphological divergence, likely selected for by plant breeders all over the world, with heading and non-heading leafy crops, turnips and both annual and biannual oil crops.
Next to its economic value, B. rapa is also of particular interest in the study of genome evolution, because of its recent genome triplication after divergence from the common ancestor of Arabidopsis and Brassica [1]. The genome sequence of the mesopolyploid crop species B. rapa ssp. pekinesis Chiifu, a Chinese cabbage, was published in 2011 as the first B. rapa reference genome [2]. Interestingly, most retained paralogous genes in this genome still show higher similarity to each other than to their orthologs in A. thaliana. Comparative mapping studies identified a putative ancestral karyotype of the current Arabidopsis and Brassica genomes, with 24 conserved chromosomal blocks, as well as an on-going process of biased gene loss called gene fractionation in three subgenomes of B. rapa[1, 3]. These subgenomes have been reconstructed by grouping the 24 conserved blocks: the least fractionated subgenome (LF), with the highest gene densities; the medium fractionated subgenome (MF1), with moderate gene densities; and the most fractionated subgenome (MF2), with the lowest gene densities [3, 4].
Our main research goal is to understand the genetic drivers underlying the enormous morphological variation between B. rapa subspecies. In this study, we therefore consider two B. rapa genomes – those of a vegetable turnip double haploid line (DH-VT117) and a rapid cycling inbred line (RC-144) – as representatives of the very distinct morphotypes turnip and annual oil (Figure 1). The vegetable turnip has an enlarged hypocotyl/root, whereas the rapid cycling line is developed in Wisconsin by intercrossing mainly annual oils and pakchois/caixins and selecting for earliness in flowering [5]. As recent studies suggested that the genome-wide density of variants is much higher between accessions of one plant species than between lines in one mammalian species, in this study we not only resequenced the turnip and rapid cycling line genomes, but also assembled and re-annotated them, resulting in two new reference genomes [6–11].
Figure 1.

Three Brassica rapa plants. Left: the Chinese cabbage cultivar, Chiifu; middle: an oil-like rapid cycling line (RC-144); right: Japanese vegetable turnip (VT-117).
These two new genomes were combined with the reference Chiifu genome (representative of the heading leafy type) to form an initial B. rapa pan-genome. This concept was raised first in the study of bacterial species, to define the full complement of genes of several closely related strains [12]. In a pan-genome, we can distinguish common genes, present in all accessions of a species; dispensable genes, occurring in more than one genome; and unique genes, specific to a single genome [13]. In the B. rapa pan-genome, we find such genes and explore functional annotations of the unique gene set to find morphotype-specific genes. We also analyze the orthology of genes in the pan-genome to Arabidopsis thaliana and Thellungiella halophila to find lost genes (orthologs missing in one of the three B. rapa genomes) and retained genes (orthologs present in only one of the genomes) (Figure 2). Finally, using orthologous genes we estimate the divergence date of the three B. rapa species and find that it far precedes domestication. The two newly assembled and annotated genomes are available to the community as an online resource at http://www.bioinformatics.nl/brassica/turnip and http://www.bioinformatics.nl/brassica/rapid-cycling, accompanied by the tools developed to explore the pan-genome.
Figure 2.
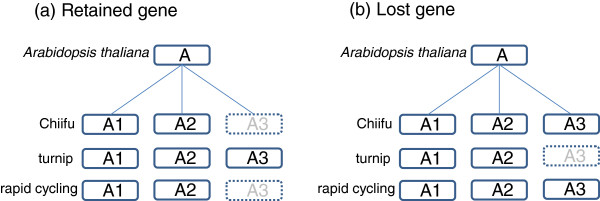
Definition of retained and lost genes. Illustrative examples of a retained and a lost gene in turnip. (a) A. thaliana gene A has three orthologous genes in turnip, but only two in Chiifu and rapid cycling; hence, we call A a retained gene for turnip based on the presence of A3. (b) Gene A is considered a lost gene for turnip based on the absence of A3.
Results
MAKER re-annotation of the reference genome Chiifu
To get a comparable genome annotation for all three B. rapa species, we first re-annotated the Chiifu reference genome using MAKER [14]. This re-annotation covered about 85% of the original 41,019 gene models found in the Brassica database (version 1.2), resulting in 41,052 gene models of which 11,715 were novel predictions (Table 1) [15]. The re-annotation covered about 90% of the exons from the Brassica database (Figure 3). The remaining 6,437 exons, roughly 10%, were mainly (5,615) located in low complexity regions of the genome. As expected, when we decreased the minimum overlap required for matching gene models, the number of recovered gene models increased: only five genes with short lengths (<200 bp) were still missing if the minimum overlap required was 10%. Approximately 75% of re-annotated gene models could be assigned a Gene Ontology term [16].
Table 1.
Comparisons of Chiifu gene models made by MAKER and obtained from BRAD
| BRAD_only | MAKER_only | Overlap_reciprocal | Overlap_split | Overlap_join | Overlap_total | ||
|---|---|---|---|---|---|---|---|
| 100% | Gene | 8,500 | 26,118 | 11,271 | 156 | 2,880 | 32,519 |
| Exons | 30,121 | 42,416 | 164,479 | 26 | 895 | 176,454 | |
| 75% | Gene | 6,437 | 11,715 | 25,848 | 187 | 2,815 | 34,582 |
| Exons | 21,838 | 34,590 | 179,737 | 47 | 964 | 184,743 | |
| 50% | Gene | 5,229 | 8,601 | 29,857 | 203 | 2,762 | 35,790 |
| Exons | 19,385 | 31,639 | 184,909 | 78 | 1,007 | 187,198 | |
| 25% | Gene | 4,239 | 6,544 | 33,617 | 719 | 2,719 | 36,780 |
| Exons | 18,270 | 30,178 | 187,577 | 347 | 1,039 | 188,313 |
Four different minimum overlap requirements (expressed as a fraction of the Chiifu reference gene model) used to compare two gene models at both gene level and exon level. The BRAD_only and MAKER_only columns represent features found only in the reference gene model and MAKER generated gene model respectively. Intersections between two gene models mainly include the fraction overlap reciprocal for both (overlap_reciprocal), overlaps that split one reference feature to many MAKER features (overlap_split) or join many reference features to one MAKER feature (overlap_join).
Figure 3.
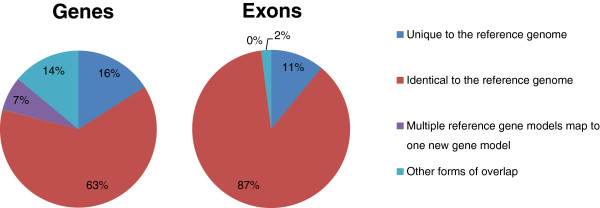
Coverage of published Chiifu reference gene models compared with re-annotated Chiifu gene models. Coverage of published Chiifu reference gene models based on number of genes and exons compared with those re-annotated by MAKER, considering a prediction identical when overlapping the reference gene model by at least 75%.
Genomic variation between the Chinese cabbage, turnip and rapid cycling genotypes
Within a species, the genomic variation between subspecies can vary substantially, as shown in several published intra-species comparative genomic studies [6, 8, 10]. We mapped the turnip and rapid cycling resequenced genomes to the reference Chiifu genome and identified 1,137,171 and 1,308,697 genomic variants respectively (Figure 4). This is less variation than between pairs of A. thaliana accessions [12]. There are 596,323 genomic variations common relative to the reference Chiifu genome (turnip and rapid cycling share the same allele at those sites), 539,747 genomic variations only found between turnip and Chiifu and 711,273 genomic variations only found between rapid cycling and Chiifu. Only 1,101 genomic variations (458,377 bps) are unique, i.e. differ between all three (re) sequenced genomes.
Figure 4.

Genomic variations anchored to chromosomes in resequenced turnip and rapid cycling genomes. Genomic variants including insertions, deletions and SNPs between resequenced turnip, rapid cycling and reference Chiifu genome on each chromosome. On each chromosome (A01-A10), the middle row represents either common or unique variations in the Chiifue genome. Genomic variations between rapid cycling and Chiifu are presented in the top three rows, variations between turnip and Chiifu in the bottom three rows. Common variations have the same sequence composition at the same position in both rapid cycling and turnip; unique variations have different nucleotides between the three genomes at the same position.
Re-annotation of turnip and rapid cycling
The genome sequences of turnip and rapid cycling were reconstructed by applying all genomic variation found to the reference genome. The total lengths of both resulting genomes were almost the same as that of the reference genome (283.84 Mbs): 282.93 Mbs for turnip and 282.69 Mbs for rapid cycling. Before re-annotation of the reference genomes, nearly half of its gene models appeared to be affected by changes in the protein coding region in either turnip (17,052) or rapid cycling (18,734) with moderate to high impact [17] (Additional file 1). Re-annotation of the turnip and rapid cycling genomes resulted in 40,708 and 40,506 predicted gene models respectively, slightly below the number found in the reference genome (41,052). After re-annotation, the number of genes found in the turnip and rapid cycling genomes which changed function or became pseudogenes with respect to the Chiifu genome was only 2,472 resp. 2,270 (Figure 5).
Figure 5.

Number of genes predicted to be functionally affected by genomic variants. Before annotation, genes were considered functionally affected in the rapid cycling line or in turnip when one of the following variants was found w.r.t. the Chiifu genome: SPLICE_SITE_ACCEPTOR, SPLICE_SITE_DONOR, START_LOST, EXON_DELETED, FRAME_SHIFT, STOP_GAINED or STOP_LOST. Genes were considered affected if they had no orthologous gene at the same chromosome/scaffold of Chiifu genome after its re-annotation.
Pan-genome construction and detection of retained and lost genes
Most of the genomic variation maps to intergenic regions, followed by introns, exons and UTRs (Table 2). After re-annotation of the rapid cycling and turnip genomes, 38,186 genes are found to be common, i.e. present in all three genomes (the pan-genome) while 1,090, 1,118 and 1,464 genes are unique to turnip, rapid cycling and Chiifu respectively (Figure 6). Functional annotation of these genes resulted in 172,430 Gene Ontology (GO) assignments to 30,976 genes (Additional file 2).
Table 2.
Number of genomic variants located in exons, introns, UTRs and intergenic regions over three subgenomes
| Turnip | Rapid cycling | |||||
|---|---|---|---|---|---|---|
| Type (alphabetical order) | LF | MF1 | MF2 | LF | MF1 | MF2 |
| EXON-Count | 80,093 | 36,695 | 44,991 | 89,042 | 42,165 | 49,868 |
| EXON-Length | 170,181 | 138,159 | 101,503 | 238,506 | 112,953 | 122,966 |
| INTERGENIC-Count | 243,148 | 129,852 | 179,159 | 280,600 | 154,959 | 192,416 |
| INTERGENIC-Length | 620,502 | 355,733 | 460,022 | 745,887 | 447,983 | 534,174 |
| INTRON-Count | 138,428 | 69,785 | 83,492 | 160,185 | 80,189 | 94,772 |
| INTRON-Length | 312,855 | 160,185 | 179,874 | 357,133 | 166,618 | 216,723 |
| UTR-Count | 8,837 | 4,816 | 5,995 | 10,340 | 5,673 | 6,619 |
| UTR-Length | 20,918 | 9,232 | 11,538 | 34,871 | 11,044 | 12,382 |
Genomic variants mapped on four different types of genome regions grouped by three subgenomes (LF, MF1, MF2) in turnip and rapid cycling. The counts indicate the number of variations in each genomic region; the length is the sum over all genomic variations. SNPs are defined as being 1 bp long.
Figure 6.
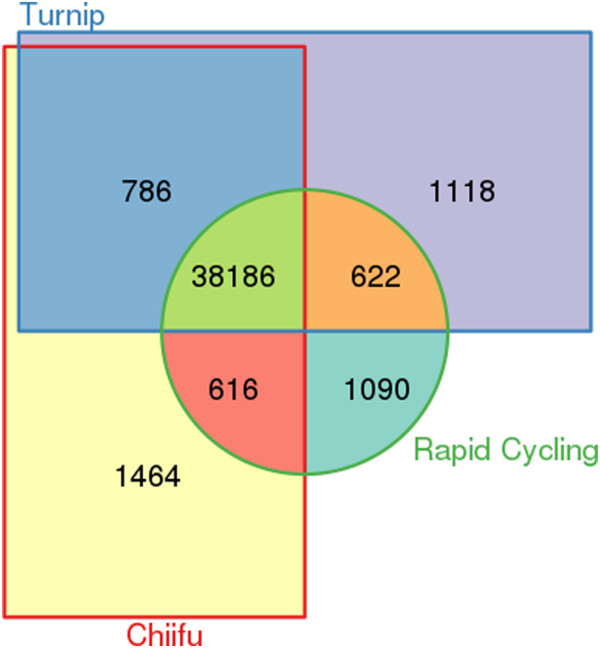
B. rapa pan-genome composition. There are 38,186 genes classified as common in the B. rapa pan genome; the number of unique genes was 1,464 in Chiifu, 1,118 in turnip and 1,090 in rapid cycling.
In total, we thus found 3,672 unique genes, found in only one of the three genomes; all remaining non-unique, non-common genes we called dispensable. About 1,443 out of 2,526 unique and dispensable genes in turnip could be annotated with at least one GO term, as was the case for 1,366 out of 2,328 genes in rapid cycling and 1,649 out of 2,866 in Chiifu. Most of these genes were assigned to only ten biological process GO terms, seven of which were common to the three genomes (Table 3 and Additional file 3). Gene models predicted from contigs that could not be mapped against the Chiifu genome were annotated separately. The number of genes thus found with at least one GO term annotation was 918 for the turnip genome and 548 for the rapid cycling genome (Additional file 4). Most unique and dispensable genes mapped to the LF subgenome, the least mapped to the MF1 subgenome. Corrected for total gene count, the proportion of genes affected by changes to their protein coding region is lowest in the LF subgenome (Figure 7).
Table 3.
Top ten GO biological processes with most genes assigned in Chiifu, turnip and rapid cycling
| Chiifu | Turnip | Rapid cycling | |||
|---|---|---|---|---|---|
| Biological process | #genes | Biological process | #genes | Biological process | #genes |
| Response to stress | 40 | Response to stress | 65 | Response to stress | 31 |
| Response to abiotic stimulus | 31 | Protein modification process | 35 | Protein modification process | 16 |
| Response to endogenous stimulus | 18 | Catabolic process | 32 | Response to abiotic stimulus | 15 |
| Secondary metabolic process | 16 | Transport | 30 | Signal transduction | 13 |
| Signal transduction | 16 | Response to abiotic stimulus | 29 | Cellular component organization | 13 |
| Catabolic process | 14 | Signal transduction | 27 | Response to biotic stimulus | 12 |
| Cellular component organization | 13 | Response to biotic stimulus | 21 | Transport | 12 |
| Anatomical structure morphogenesis | 11 | Carbohydrate metabolic process | 18 | Catabolic process | 11 |
| Response to biotic stimulus | 11 | Cellular component organization | 17 | Response to endogenous stimulus | 9 |
| Protein modification process | 10 | Response to endogenous stimulus | 14 | Lipid metabolic process | 9 |
Only dispensable and unique genes were included in the analysis. The term “response to stress” is the most over-represented and seven out of these ten GO terms are found in Chiifu, turnip as well as rapid cycling.
Figure 7.
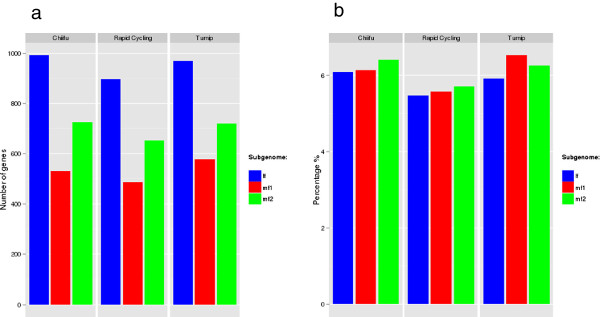
Subgenome composition of dispensable and unique genes in three B. rapa genotypes. The subgenome composition of dispensable and unique genes in three B. rapa genotypes in terms of (a) number of genes; (b) frequency of gene changes, calculated as number of changed genes divided by the total number of total genes in the subgenome. LF: less fractionated subgenome, with the highest gene densities; MF1: more fractionated subgenome 1, with moderate gene densities; MF2: most fractionated subgenome 2, with lowest gene densities.
To study gene copy number variation in the B. rapa pan-genome, orthology between genes in B. rapa and its close relatives Arabidopsis thaliana and Thellungiella halophila was computed. For the common gene set we found 19,301 orthologs in A. thaliana and 20,825 in T. halophile. The numbers of genes with one, two or three orthologous genes in B. rapa were 15,200/3,650/690 in A. thaliana and 17,800/2,950/520 in T. halophila (Table 4). Further analysis of the B. rapa dispensable and unique genes with orthologous genes in either A. thaliana or T. halophila showed that the rapid cycling line and turnip DH line had over 40% resp. 30% less retained genes w.r.t. A. thaliana than Chiifu, yet an approximately equal number of lost genes (Table 5).
Table 4.
Orthologous genes of Arabidopsis thaliana and Thellungiella halophila found in Chiifu, turnip and rapid cycling
| Number of copies in B. rapa | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| Compared genomes | at | th | at | th | at | th |
| Chiifu | 15,237 | 17,880 | 3,706 | 3,041 | 691 | 540 |
| Turnip | 15,190 | 17,812 | 3,676 | 2,907 | 713 | 503 |
| Rapid cycling | 15,225 | 17,774 | 3,595 | 2,960 | 681 | 519 |
The number of A. thaliana (“at”) and T. halophile (“th”) genes having one, two and three copies of orthologous genes in Chiifu, turnip and rapid cycling.
Table 5.
Retained and lost genes in Chiifu, turnip and rapid cycling
| Chiifu | Turnip | Rapid cycling | |
|---|---|---|---|
| Genes with copy number changes | 1,151 | 1,053 | 932 |
| Retained genes | 265 | 180 | 156 |
| Lost genes | 886 | 873 | 906 |
| Retained|lost gene assigned to gene families | 40|23 | 35|19 | 33|20 |
| Genes present in unique and dispensable gene set, without at or th orthologs* | 231 | 280 | 336 |
Dispensable and unique genes having orthologs in A. thaliana or T. halophila were included to determine the retained and lost genes. The latest curated gene family assignment of A. thaliana genes from TAIR was used.
* The number of B. rapa unique and dispensable genes without A. thaliana (“at”) or T. halophile (“th”) orthologous genes.
Functional annotation of dispensable and unique genes in the Chiifu, turnip and rapid cycling genomes
The unique and dispensable genes can be placed in 87 KEGG pathways in turnip, 104 in rapid cycling and 89 in Chiifu, with starch and sucrose metabolism containing the largest proportion of genes (19 in turnip, 20 in rapid cycling and 13 in Chiifu) [18]. These genes are also found in 97, 102 resp. 94 plant-specific pathways hosted by the Plant Metabolic Network in PlantCyc [19]. Genes are much more scattered over different metabolic pathways in PlantCyc than in KEGG [18, 19]. The number of genes found in any pathway in PlantCyc (119 in turnip, 123 in rapid cycling and 112 in Chiifu) is less than half of the number of genes found in KEGG (285 in turnip, 297 in rapid cycling and 254 in Chiifu) since fewer enzymes are associated with each PlantCyc metabolic pathway (Additional file 5).
GO enrichment analysis shows that the dispensable and unique genes in Chiifu have both the most overrepresented (59) and underrepresented (50) GO terms, while the dispensable and unique genes in rapid cycling have the least overrepresented (11) and underrepresented (16) terms and turnip has 35 overrepresented and 13 underrepresented terms. The number of genes assigned to enriched GO terms is higher in turnip (1,095) than in Chiifu (823) and rapid cycling (704).
Genes with association to different morphotypes
Next, we specifically looked for genes potentially related to morphological variation, by considering retained and lost genes with orthologs in both A. thaliana and T. halophila. Only a small percentage of these, 15% of the retained and 10% of the lost genes, could be categorized into known A. thaliana gene families (Table 6). The set of unique and dispensable genes found in turnip is enriched for the GO cellular component term “peroxisome”, and contains Class III peroxidases among both lost (AT5G64120) and retained (AT1G05260 with gene symbol RCI3) genes. To refine our understanding of a possible role of peroxidases in turnip formation, we more closely investigated B. rapa genes orthologous to 155 peroxidase related genes in A. thaliana[20]. We exploited synteny information to support the confidence in orthology predictions and to help distinguishing true orthologs, since A. thaliana and B. rapa are evolutionary very close [21]. B. rapa orthologs of five A. thaliana genes were retained and of four A. thaliana genes were lost in turnip compared to Chiifu and rapid cycling (Figure 8a). We found proteins functionally interacting with these genes using STRING (Figure 8b) [22]. Four of the five retained genes were involved in the phenylpropanoid biosynthesis pathway and the fifth, AT3G63080 (ATGPX5), a glutathione peroxidase, may contribute to glutathione synthesis. Only one of the four A. thaliana orthologs of the lost genes, AT5G64120 (PER71), was predicted to interact with other proteins in STRING, whereas both PER71 and another lost gene AT1G77100 (PER13) are also involved in phenylpropanoid biosynthesis. We then examined all genes known to be involved in the phenylpropanoid biosynthesis pathway in A. thaliana and found that while orthologs of genes encoding a peroxidase (EC number 1.11.1.7) were enriched in turnip, genes encoding a 4-coumarate-CoA ligase (EC 6.2.1.12) or a coniferyl-alcohol glucosyltransferase (EC 2.4.1.111) were underrepresented. The six A. thaliana genes encoding this ligase have ten orthologs in the common gene set of B. rapa, but only two B. rapa genes are orthologous to three A. thaliana genes coding for the glucosyltransferase. This suggests the lower copy number of genes in turnip coding for the glucosyltransferase may cause the reduction of 4-D-glucoside, coniferin, syringin and hence increase the production of different lignins (Figure 8c).
Table 6.
Gene family assignment for retained and lost genes in Chiifu, turnip and rapid cycling
| Chiifu | Turnip | Rapid cycling | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TAIR gene family description | Class | Number of genes in common sets | Number of genes in unique and dispensable sets | TAIR gene family description | Class | Number of genes in common sets | Number of genes in unique and dispensable sets | TAIR gene family description | Class | Number of genes in common sets | Number of genes in unique and dispensable sets |
| C2H2 transcription factor family | LOST | Acyl lipid metabolism family | LOST | C3H transcription factor family | LOST | ||||||
| Core DNA replication machinery | LOST | C2H2 Transcription factor family | LOST | Cytochrome P450 | LOST | ||||||
| Cytochrome P450 | LOST | Class III peroxidase | LOST | Cytoplasmic ribosomal protein gene family | LOST | ||||||
| Cytoplasmic ribosomal protein gene family | LOST | Cytochrome P450 | LOST | Glutathione S-transferase family | LOST | ||||||
| Cytoskeleton | LOST | Cytoplasmic ribosomal protein gene family | LOST | Glycoside hydrolase gene families | LOST | ||||||
| EF-hand containing proteins | LOST | EF-hand containing proteins | LOST | Glycosyltransferase gene families | LOST | ||||||
| Expansins | LOST | FH2 proteins | LOST | Homeobox transcription factor family | LOST | ||||||
| Glutathione S-transferase family | LOST | Glycosyltransferase gene families | LOST | Inorganic solute cotransporters | LOST | ||||||
| Glycosyltransferase gene families | LOST | MIP family | LOST | Lipid metabolism gene families | LOST | ||||||
| Lateral organ boundaries gene family | LOST | Miscellaneous membrane protein families | LOST | MAP kinase kinase kinase kinase (MAPKKKK) family | LOST | ||||||
| Miscellaneous membrane protein families | LOST | Monosaccharide transporter-like gene family | LOST | Miscellaneous membrane protein families | LOST | ||||||
| MYB Transcription factor family | LOST | Trihelix transcription factor family | LOST | Primary pumps (ATPases) gene families | LOST | ||||||
| Primary pumps (ATPases) gene family | LOST | ARF transcription factor family | RETAINED | 15 | 1 | Receptor kinase-like protein family | LOST | ||||
| Acyl Lipid metabolism family | RETAINED | 587 | 3 | Carbohydrate esterase gene families | RETAINED | 75 | 1 | Acyl Lipid metabolism family | RETAINED | 587 | 4 |
| AP2-EREBP transcription factor family | RETAINED | 170 | 1 | Chloroplast and mitochondria gene families | RETAINED | 53 | 1 | BZR transcription factor family | RETAINED | 6 | 1 |
| ARF transcription factor family | RETAINED | 15 | 1 | Class III peroxidase | RETAINED | 71 | 1 | CBL-interacting serione-threonine Protein Kinases | RETAINED | 24 | 1 |
| C2H2 transcription factor family | RETAINED | 215 | 1 | Cytochrome P450 | RETAINED | 144 | 2 | Core cell cycle genes | RETAINED | 65 | 1 |
| C3H transcription factor family | RETAINED | 162 | 1 | Cytoplasmic ribosomal protein gene family | RETAINED | 221 | 1 | Cytochrome P450 | RETAINED | 144 | 3 |
| CCAAT-HAP3 transcription factor family | RETAINED | 13 | 1 | GeBP transcription factor family | RETAINED | 10 | 1 | Glycoside hydrolase gene families | RETAINED | 335 | 1 |
| Cytoplasmic ribosomal protein gene family | RETAINED | 221 | 2 | Glutathione S-transferase family | RETAINED | 37 | 2 | Glycosyltransferase gene families | RETAINED | 280 | 1 |
| EF-hand containing proteins | RETAINED | 175 | 2 | Glycosyltransferase gene families | RETAINED | 280 | 1 | Histidine phosphotransfer proteins | RETAINED | 5 | 1 |
| Eukaryotic initiation factor gene family | RETAINED | 101 | 1 | HSP70s | RETAINED | 4 | 1 | Inorganic solute cotransporters | RETAINED | 93 | 1 |
| Glutathione S-transferase family | RETAINED | 37 | 1 | Lipid metabolism gene families | RETAINED | 106 | 2 | Lipid metabolism gene families | RETAINED | 106 | 1 |
| Glycoside hydrolase gene families | RETAINED | 335 | 2 | MADS-box transcription factor family | RETAINED | 89 | 2 | Miscellaneous membrane protein families | RETAINED | 415 | 3 |
| Glycosyltransferase gene families | RETAINED | 280 | 1 | Myosin | RETAINED | 17 | 1 | NAC transcription factor family | RETAINED | 97 | 1 |
| Lateral organ boundaries gene family | RETAINED | 37 | 2 | Organic solute cotransporters | RETAINED | 272 | 1 | Nodulin-like protein family | RETAINED | 65 | 3 |
| MAP kinase kinase kinase family | RETAINED | 74 | 1 | Receptor kinase-like protein family | RETAINED | 237 | 4 | Organic solute cotransporters | RETAINED | 272 | 2 |
| MIP family | RETAINED | 37 | 1 | REM transcription factor family | RETAINED | 20 | 1 | Pollen coat proteome | RETAINED | 3 | 1 |
| Miscellaneous membrane protein families | RETAINED | 415 | 2 | RCI2 gene family | RETAINED | 6 | 2 | ||||
| MYB transcription factor family | RETAINED | 154 | 1 | SNAREs | RETAINED | 66 | 1 | ||||
| Plant defensins superfamily | RETAINED | 6 | 1 | ||||||||
| Plant U-box protein (PUB) | RETAINED | 66 | 1 | ||||||||
| Primary pumps (ATPases) gene family | RETAINED | 31 | 1 | ||||||||
| Receptor kinase-like protein family | RETAINED | 237 | 2 | ||||||||
Overview of lost and retained genes assigned to A. thaliana gene families. The “LOST” gene family has no value in the dispensable and unique gene set because the inconsistency of counts in the other two genotypes.
Figure 8.
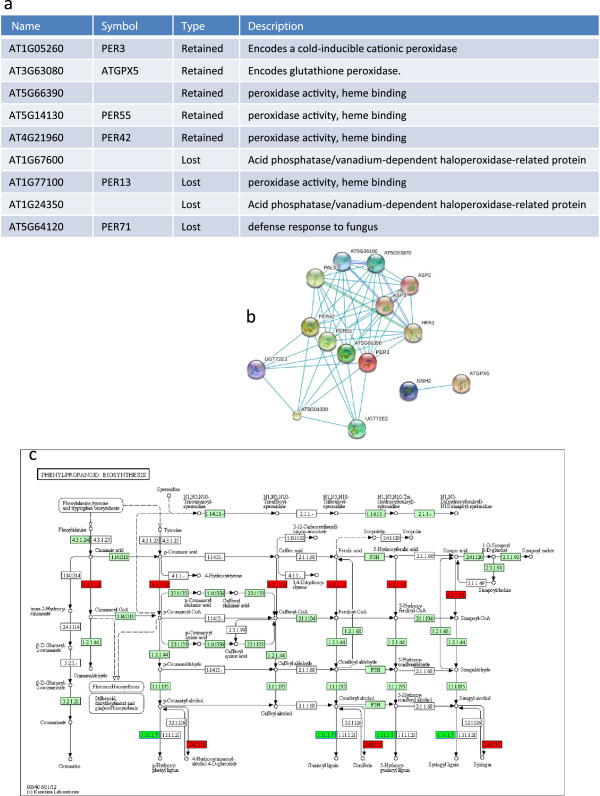
Network analysis of retained and lost genes in turnip. 155 A. thaliana peroxidase-related genes were selected. a) Five retained genes and four lost genes were identified in turnip, five of which were class III peroxidases. b) Summary of the functional protein interaction network found by STRING using five retained genes as input. c) Phenylpropanoid biosynthesis pathway in A. thaliana, including four retained genes and two lost genes. A. thaliana genes that encode enzymes are indicated by light green colored boxes; red resp. dark green boxes indicate genes with less resp. more copies in rapid cycling than in Chiifu and turnip.
Estimation of divergence date between turnip, rapid cycling and Chiifu
We found 7,768 orthologous gene sets in A. thaliana, Arabidopsis lyrata, Oryza sativa, Vitis vinifera and Zea mays using the latest OMA browser [23] dataset (March 2012); 1,714 of these remained after filtering on a 1:1 orthologous relationship. Combining this set of 1,714 remaining genes with orthologous groups in the B. rapa pan-genome left 104 groups of orthologous genes with a meaningful OMA group description. These were used to infer the divergence date among the three B. rapa genomes, at around 0.259 MYA.
Availability
The two newly-assembled genomes representing the turnip morphotype (turnip) and the oil crop morphotype (rapid cycling), their annotation files, a gene list for the three categories of pan genomes and the Blast2GO project files generated in the study are all provided (Additional files 6, 7, 8 and 9). The genomes can also be browsed at http://www.bioinformatics.nl/brassica/turnip and http://www.bioinformatics.nl/brassica/rapid-cycling. All used software tools in this project can be handled by biologists with some basic bioinformatics skills and the pre-/post-processing scripts are available for download (Table 7, Figure 9 and Additional file 10). These programs were run on an OpenSuSE Linux server with 16 AMD Opteron Processor cores and 128 GB of memory.
Table 7.
Software and scripts used in the project
| Name | Running time (h) | Input format | Output format | Script purpose |
|---|---|---|---|---|
| cortex_var | 24 / genotype | Fastq | vcf | - |
| *cortex_combiner | < 1 | Vcf | fasta | Post-processing of cortex |
| *maker_pre_ws | 24 | Txt | Fasta | Pre-processing for MAKER |
| MAKER | 140/genotype | Fasta | gff, fasta | - |
| *ortholog_assign | 20/genotype | Fasta | csv, fasta | Post-processing of MAKER |
| NCBI BLAST | 200 | Fasta | xml | - |
| *InterProScan_ws | 100 | Fasta | xml | Pre-processing for Blast2GO |
| Blast2GO | < 1 | Xml | csv | - |
| *run_metacyc | < 0.1 | Txt | csv | Post-processing of Blast2GO |
| *beast_pre | < 5 | Txt | nex | Pre-processing of BEAST |
| BEAST | 24 | Nex | png | - |
| *choose_fasta | < 0.1 | Txt | Fasta | Extract sequence from fasta |
The order in the table indicates the flow of the analysis, except for the script “choose_fasta” which can be used anytime when needed. Names starting with an asterisk are scripts generated specifically for this work. The script purpose column indicates when the scripts should be used before or after certain program. All scripts run under Linux and provide a short usage summary when started without arguments. “txt” input format: a list of file names used for the scripts.
Figure 9.
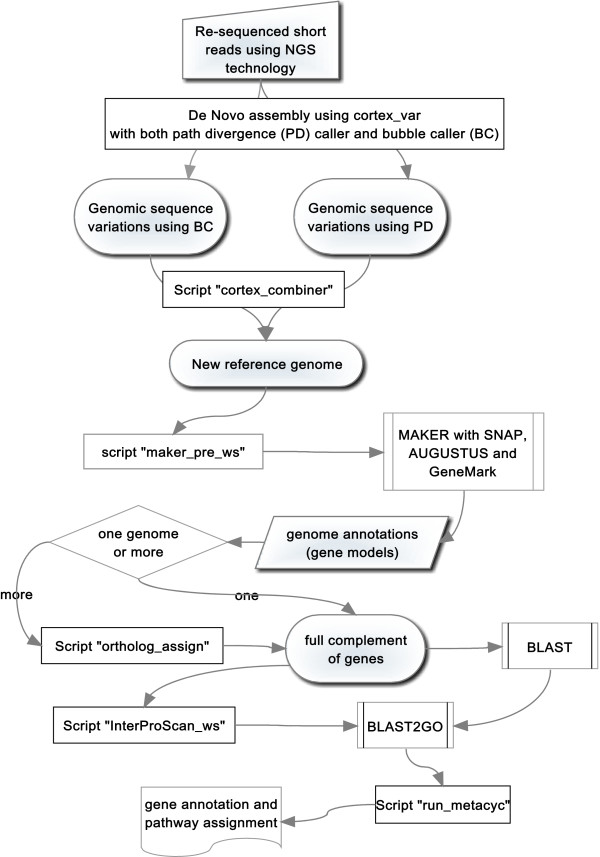
Workflow of the study. The workflow describes the methods and logic used in the study, from raw sequence reads to the annotation of the full complement of genes in a genome. Newly created scripts are marked by “Script”. Any number of genomes can be analyzed using this workflow, provided there is sufficient computational power.
Discussion
Variability in the B. rapa pan-genome
The three B. rapa genomes considered in this work – Chiifu, turnip and rapid cycling – differ by about 0.45 per 100 base pairs, considerably less than the differences between lines of maize (1-2/100 bp) but very close to differences between various accessions of A. thaliana (0.5/100 bp) and of rice (0.4/100 bp) [11, 24, 25]. To further investigate the pan-genomic variation, we focused on the unique genes, on average 1,224 per B. rapa genome. The frequency of functional unique genes over the three subgenomes agrees with the theory that one of the subgenomes (LF) is dominant and hence has the lowest percentage of affected genes (Figure 7).
We expected the number of unique genes in each B. rapa genome to be larger than the average number of unique genes found in different A. thaliana accessions, mainly because the morphological variation between a tuber forming turnip, a heading cabbage and an oilseed rapid cycling is larger than between, say, three A. thaliana accessions. Additionally, the recent genome triplication in B. rapa may have lowered selective pressure on a subset of the genes. However, a recent study analyzing 18 A. thaliana accessions found only 319 unique genes per accession on average. Such a comparison is not completely fair however, as the A. thaliana comparisons used a different definition of unique gene. We thus selected the three A. thaliana accessions (Can, Wil and Sf) with the highest number of genes with predicted major disruptions, and used the protein sequences of their gene models to find unique genes by exactly the same process as in our study. This yielded on average 1,700 unique genes, higher than the 1,224 found in the B. rapa genomes. One explanation may be that the effective population size is much higher for A. thaliana than for B. rapa, which went through several domestication bottlenecks. Additionally, the three B. rapa genotypes are all landraces (or intercrossed genotypes) growing in protected agricultural settings, with varieties selected by breeders and farmers, while A. thaliana is a weed that grows in natural environments under diverse abiotic and biotic stresses (drought, cold, pathogens) with different selection forces.
Our findings are also in line with a previous study, in which the genetic variation in a B. rapa core collection representing all morphotypes and geographical origins was analyzed based on molecular marker profiles [26, 27]. Bayesian clustering implemented in the STRUCTURE software revealed four subpopulations, each representing different morphotypes (I turnip accessions from European origin; II Asian leafy types like Pakchoi plus Asian turnips; III annual oil accessions and IV mainly accessions of Chinese cabbage (CC). AMOVA results indicated that the percentage of variation found within sub-populations/morphotypes is much larger (85%) than the variation among populations (15%), suggesting that only a small percentage of the polymorphisms relate to the sepcific observed morphological differences.
Genomic determinants of morphological variation
Studying the functions of unique and dispensable genes could reveal whether they play a role in the extreme morphological differences between the three plants. Through functional annotation, we found that peroxidases are good candidates for genes involved in the definition of plant morphology. Peroxidases play a role in protection from biotic and abiotic stresses, but also in lignin formation. Four of five turnip specific retained B. rapa genes orthologous to A. thaliana peroxidases are involved in the phenylpropanoid biosynthesis pathway. Phenylpropanoids are a group of plant secondary metabolites and specific compounds differentially accumulate in particular tissues with specialized functions. These results suggest that lignin may be important for turnip tuber formation, which can relate to the increased numbers of xylem vessels in the turnip tuber.
In this paper we focus on the DNA level, but it is entirely possible that turnip formation is (additionally) regulated at the transcriptional or even post-translational level. Gene loss occurred more in rapid cycling than in turnip and Chiifu (906 in rapid cycling, 873 in turnip and 886 in Chiifu). Rapid cycling may have a different composition of flowering time genes because it was generated by crossing early flowering B. rapa genotypes to create a morphotype with a short life cycle for educational purposes [5]. To verify this hypothesis, we looked for genes in the three B. rapa genomes orthologous to 367 known flowering related A. thaliana genes. These flowering genes were classified into five different categories, including flower development, gibberellin-, photoperiod/circadian rhythm- and vernalization pathway and metabolic processes (Additional file 11). In rapid cycling, there are five lost genes related to flowering time (covering all five categories), compared to only three in Chiifu and turnip (from a single category, photoperiod in turnip and vernalization in Chiifu) (Table 8).
Table 8.
Flowering time related lost genes in three B. rapa genotypes
| Genotype | Ara ID | Gene name | Pathway | Gene full name | Protein function |
|---|---|---|---|---|---|
| Rapid cycling | AT2G32950 | COP1 | Photoperiod | CONSTITUTIVE PHOTOMORPHOGENIC 1 | E3 ubiquitin ligase |
| Rapid cycling | AT3G11440 | MYB65 | Gibberellin | MYB65 | MYB transcription factor |
| Rapid cycling | AT3G20740 | FIS3 | Vernalization | FERTILIZATION-INDEPENDENT ENDOSPERM | Encodes a protein similar to the transcriptional regular of the animal Polycomb group |
| Rapid cycling | AT5G03790 | LMI1 | Flower development | LATE MERISTEM IDENTITY 1 | HD-Zip transcription factor |
| Rapid cycling | AT5G47010 | LBA1 | Metabolic process | LOW-LEVEL BETA-AMYLASE 1 | Required for nonsense-mediated mRNA decay |
| Chiifu | AT1G04440 | CKL13 | Vernalization | CASEIN KINASE LIKE 13 | Protein serine/threonine kinase activity |
| Chiifu | AT4G25470 | CBF2 | Vernalization | C-REPEAT/DRE BINDING FACTOR 2 | Encodes a member of the DREB subfamily A-1 of ERF/AP2 transcription factor family |
| Chiifu | AT5G59710 | VIP2 | Vernalization | VIRE2 INTERACTING PROTEIN 2 | Encodes a nuclear-localized NOT (negative on TATA-less) domain-containing |
| Turnip | AT1G53090 | SPA4 | Photoperiod | SPA1-RELATED 4 | WD-40 and protein kinase-like domain |
| Turnip | AT4G27430 | CIP7 | Photoperiod | COP1-INTERACTING PROTEIN 7 | - |
| Turnip | AT5G64813 | LIP1 | Photoperiod | LIGHT INSENSITIVE PERIOD 1 | GTPase |
Five lost genes are related to flowering time in Rapid cycling, covering all five categories of lowering time genes. In the other genomes just three genes are found, related only to photoperiod in turnip and vernalization in Chiifu.
Evolutionary divergence
Chiifu, rapid cycling and turnip are estimated to have diverged 259,000 years ago, far preceding domestication (around 10,000 years ago). This may seem to imply that prior to domestication there was already considerably divergence among B. rapa genomes; however, domestication can accelerate selection and hence influence divergence time estimates. We do not know whether there was already variation in appearance, such as enlarged hypocotyls, leaves that form heads, multi tillering types etc. prior to domestication, or whether (more likely) there was a common wild type, and that breeders merely combined mutations and allelic variation by crossing which gave rise to diverse morphotypes. It is also unknown whether early plant breeders could breed for all different morphotypes starting from the same genetic materials, or that specific B. rapa materials (in geographical niches) resulted in certain morphotypes. Resequencing more B. rapa genotypes belonging to turnip, leafy and oil types, especially from diverse geographical regions (e.g. European and Asian turnips) may shed light on these questions.The percentage of B.rapa genes that have orthologs in T. halophila is higher than the percentage with orthologs in A. thaliana. The divergence date of A. thaliana and B. rapa is estimated at 17 MYA, earlier than that of T. halophila and B. rapa at 12 MYA and earlier than the whole genome triplication event dated 5–9 MYA, after speciation of A. thaliana/B. rapa and T. halophila/B. rapa[28]. In other words, B. rapa genes are expected to be more similar to T. halophila genes than to A. thaliana genes.
Computational analysis
As practical considerations make it hard to obtain the sequencing depth required for de novo genome assembly, in this work we took a hybrid approach in which we first mapped reads to a reference genome and then created new genomes by applying all variation found. The trade-off between the number of detected variants and mapping accuracy is important. A low mapping quality threshold setting leads to many candidate genes for further experimental validation, but can also introduce false positive discoveries.
For the purposes of this study, we developed a number of scripts for variant calling, re-annotation and functional annotation that can help biologists to answer similar questions on genotype-phenotype relations. The re-annotation is a particulary time-consuming step, which may be extended by considering RNA-seq data or available gene model GFF and FASTA files.
Conclusions
Here we present two novel reference genomes and their annotations representing the morphotypes turnip and rapid cycling to the B. rapa community, which provides reliable templates for studying genetic variation between these two morphotypes and the reference, Chiifu. In addition, this paper offers a complete workflow for those having limited computational resources and bioinformatics expertise studying similar biological questions. We investigated the resulting B. rapa pan-genome, paying specific attention to potential drivers of morphological variation. The number of genes with protein-coding changes among the three B. rapa genomes was lower than that among three diverse accessions of Arabidopsis thaliana. We found peroxidases, mainly involved in phenylpropanoid biosynthesis, enriched in the genes retained in turnip. Analysis of the gene content of the B. rapa pan-genome revealed that the divergence date between the three morphotypes was dated long before domestication (250,000 versus 5,000-10,000 years ago).
Methods
DNA material and sequencing
The genomes of a Japanese turnip doubled haploid line DH-VT117 and a rapid cycling oil-like inbred line RC-144 were resequenced. DH-VT117 is a purple red peeled round vegetable turnip derived through microspore culture from a donor plant of B. rapa ssp. campestris cv. Toya, CGN15201. RC-144 (Osborn FIL501) is self-compatible and has a rapid life cycle of about 24 days from sowing to flowering. Libraries with insert sizes of 300 bp, 500 bp and 2kbp were sequenced on Illumina Hiseq 2000 (Illumina Inc., San Diego CA), yielding ~100 million 71 bp paired reads for the turnip genome and ~100 million for the rapid cycling genome. Read data of the three libraries of both genotypes were independently converted to single-color binary files for fast reloading with removal of PCR duplicates, using the cortex_var assembly software tool [29]. All seven resulting binary files (including the reference genome) were then used to call variants using the Bubble Caller algorithm. Separately, the two genotypes were compared to the reference Chiifu genome using the Path Divergence Caller algorithm.
Genome annotation
Gene level differences between turnip, rapid cycling and the reference genome (B. rapa ssp pekinensis, Chiifu) can only be meaningfully compared when gene models are comparable, i.e. predicted using the same method. Gene models for the reference genomecan be downloaded from the BRAD website (http://brassicadb.org/brad/), but the methods used to determine these unfortunately are not described in sufficient detail to allow reproduction of the annotation. We therefore used the MAKER genome annotation pipeline to re-annotate all three genomes [14]. Next to MAKER’s default reference gene models, all available unigenes of B. rapa, Brassica napus, Brassica oleracea were imported as closely related EST evidences and unigenes from other Brassica species as alternate evidence. Protein homology evidence was based on protein sequences of Brassicaceae in the NCBI RefSeq database [30]. SNAP, AUGUSTUS and GENEMARK [31–33] were used to predict genes. All other MAKER parameters were set to default values.
Genomic variation detection
Genomic variation was detected using the cortex_var software suite with k-mer size set to 31, by both the Bubble Caller and Path Divergence Caller algorithms [29]. Variants found by these two algorithms are defined as overlapping when they either shared the same position on the chromosome or their mapped positions were close enough that one of the contigs could cover another. Such overlapping variants were merged by choosing the one yielding the longest assembled sequence. After filtering out genomic variants where the 5′ flank of the contig maps to the reference with a mapping quality score Q < 30, genomes of rapid cycling and turnip were reconstructed by applying all remaining variants to the reference genome. Both unmapped contigs and contigs with Q < 30 were annotated by MAKER, using the same settings as used for the whole genome re-annotation. The NCBI non-redundant protein database was searched for predicted gene models (BLAST, default settings) to exclude bacterial sequences. Genes on unmapped contigs with no bacterial hits were subjected to functional annotation and pathway assignment, but not included in pan genome composition because they cannot be used to detect positional orthologs.
Common, unique and dispensable genes
Two genes found in two genomes are considered positionally orthologous when they are reciprocal best BLAST hits and located on the same chromosome or scaffold. We first detected such positional orthologs between A. thaliana and T. halophila on the one hand and Chiifu, turnip and rapid cycling on the other. A gene is defined as lost when a positional ortholog is missing in only one of the three genomes and as retained when it is present in only one of the three genomes (Figure 2).
Next, we detected positional orthologs between each two of the three B. rapa genomes. We define common genes as genes found in two out of three comparisons (i.e. present in all three genomes) and unique genes as genes not occurring in any comparison. Genes that are in neither the common gene set nor the unique gene set are called dispensable.
Subgenome assignments
Genes in the Chiifu genome were assigned to the same subgenome as the one published if the new gene model mapped to the same coordinates [3]. For the other two genomes, genes were assigned to subgenomes by transferring assignment from the reciprocal best BLAST hit of Chiifu when available, or from the closest flanking genes (one gene upstream and one gene downstream) if these are in the same subgenome. Genes were not assigned to a subgenome if these rules did not apply.
Functional annotation
To annotate genes in the B. rapa pan-genome, first common, dispensable and unique genes were searched in the NCBI non-redundant protein database (2012/06/07) using BLAST with default settings [34]. For dispensable and unique genes an additional InterProScan analysis was performed [35]. Gene function was predicted using the Blast2GO pipeline version B2G4Pipe version 2.5.0, integrating the BLAST and InterProScan results and KEGG pathways (based on gene EC numbers) if applicable [36]. Plant specific metabolic pathways were added as supplementary resource using the latest PlantCyc database files (release 6.0) [19].
Candidate genes for morphological differences
Orthology between genes in A. thaliana and T. halophila on the one hand and B. rapa common, dispensable and unique genes on the other hand, was assessed using Inparanoid 4.1 [37]. A. thaliana and T. halophila protein sequences were downloaded from ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v8.0. For each gene in A. thaliana and T. halophila the numbers of orthologous genes in Chiffu, turnip and rapid cycling were counted.
Enriched Gene Ontology terms for dispensable and unique genes without orthologs in either A. thaliana or T. halophila were determined for each genome using Fisher’s exact test [16]. Gene family information of the Brassica genes was inferred from A. thaliana orthologs using the latest curated gene family assignment, downloaded from TAIR (ftp://ftp.arabidopsis.org/Genes/Gene_families/).
Inferring divergence time
In order to estimate the time of divergence of Chiifu, turnip and rapid cycling, first orthologous genes were found between these three subspecies of B. rapa and A. thaliana, Arabidopsis lyrata, Oryza sativa, Vitis vinifera and Zea mays. Orthologous relationships among the five non-B. rapa species were retrieved from the OMA browser [23] and only genes with a one-to-one relationship between two species (i.e. genes with only one orthologous gene in another species) were taken into account, to prevent inclusion of in-paralogs. Orthologous pairs between A. thaliana and B. rapa Chiifu were used to connect orthologous relationships between the non-B. rapa species and the three B. rapa subspecies. Multiple sequence alignments generated by EMMA (EMBOSS, [38]) using orthologous gene groups were independently analysed (i.e. with unlinked trees) under a codon-position specific estimated generalized time reversible (GTR) substitution model with lognormal relaxed clock in BEAST [39].
Electronic supplementary material
Additional file 1: Change rate on each chromosome and different effect types. Excel file containing details of the change rate in turnip and rapid cycling. (XLSX 15 KB)
Additional file 2: Functional annotation of common genes in Chiifu, turnip and rapid cycling. Excel file containing details of the functional annotation of common genes in Chiifu, turnip and rapid cycling. (XLSX 3 MB)
Additional file 3: The GO term enrichment analysis results on unique and dispensable genome of Chiifu, turnip and rapid cycling. Excel file of results obtained from GO term enrichment analysis using unique and dispensable genome of Chiifu, turnip and rapid cycling. (XLSX 345 KB)
Additional file 4: Functional annotation of dispensable, unique and unmapped genes in Chiifu, turnip and rapid cycling. Excel file containing details of the functional annotation of dispensable, unique and unmapped genes on Chiifu, turnip and rapid cycling. The unmapped genes only apply to the turnip and rapid cycling genomes. (XLSX 1 MB)
Additional file 5: KEGG and PlantCyc pathway assignment of dispensable and unique genes in Chiifu, turnip and rapid cycling. Excel file containing details of KEGG and PlantCyc pathway assignments of dispensable and unique genes in Chiifu, turnip and rapid cycling. (XLSX 83 KB)
Additional file 6: The reference genomes of turnip and rapid cycling. Fasta file containing details of the chromosomes and scaffolds on turnip and rapid cycling. The two newly assembled genomes, their annotation files, a gene list for the three categories of pan genomes and the Blast2GO project files. (ZIP 73 MB)
Additional file 7: The annotation files of Chiifu, turnip and rapid cycling genome. Archived file containing three GFF files, including details of the gene features in Chiifu, turnip and rapid cycling. (ZIP 73 MB)
Additional file 8: Common, dispensable and unique genes in Chiifu, turnip and rapid cycling. Excel file containing details of the full complement of genes on Chiifu, turnip and rapid cycling. (ZIP 16 MB)
Additional file 9: Blast2GO project files of dispensable and unique genes in Chiifu, turnip and rapid cycling. Archived file containing three DAT files, each including a project file which can be imported in the Blast2GO program for further analysis. (ZIP 752 KB)
Additional file 10: All new created scripts in the study. Archived file containing all new created scripts used in the study. (ZIP 15 MB)
Additional file 11: The flowering time candidate gene list in A. thaliana. Excel file listing candidate genes related to flowering time in A. thaliana, categorized into flower development, gibberellin, photoperiod, vernalization and metabolic process. (XLSX 253 KB)
Acknowledgements
First, I would like to send my deepest thanks and appreciation to my former supervisor and promoter Prof. Jack A.M. Leunissen, who passed away after a long illness on May 14 2012. We thank Sander Peters, Ram Kumar Basnet, Aalt-Jan van Dijk and Gabino Sanchez-Perez for useful discussions on this project. We received financial support from the Programme Strategic Scientific Alliance (project number: 08-PSA-BD-02) of the Royal Netherlands Academy of Arts and Sciences (KNAW).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KL carried out the variant calling, genome annotation, participated in the comparative genomics study and drafted the manuscript. NZ prepared DNA samples. NZ, EIS and HN participated in the comparative genomics study. FC performed the quality check on the sequencing data. XW participated in the experimental design. DdR and RGFV were involved in the writing of the manuscript. GB conceived the study, participated in its design and coordination and helped draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ke Lin, Email: ke.lin@wur.nl.
Ningwen Zhang, Email: ningwen.zhang@wur.nl.
Edouard I Severing, Email: edouard.severing@wur.nl.
Harm Nijveen, Email: harm.nijveen@wur.nl.
Feng Cheng, Email: biojoiner@gmail.com.
Richard GF Visser, Email: richard.visser@wur.nl.
Xiaowu Wang, Email: wangxw@mail.caas.net.cn.
Dick de Ridder, Email: dick.deridder@wur.nl.
Guusje Bonnema, Email: guusje.bonnema@wur.nl.
References
- 1.Mun JH, Kwon SJ, Yang TJ, Seol YJ, Jin M, Kim JA, Lim MH, Kim JS, Baek S, Choi BS, Yu HJ, Kim DS, Kim N, Lim KB, Lee SI, Hahn JH, Lim YP, Bancroft I, Park BS. Genome-wide comparative analysis of the Brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome Biol. 2009;10(10):R111. doi: 10.1186/gb-2009-10-10-r111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun JH, Bancroft I, Cheng F, Huang S, Li X, Hua W, Wang J, Wang X, Freeling M, Pires JC, Paterson AH, Chalhoub B, Wang B, Hayward A, Sharpe AG, Park BS, Weisshaar B, Liu B, Li B, Liu B, Tong C, Song C, Duran C, et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43(10):1035–1039. doi: 10.1038/ng.919. [DOI] [PubMed] [Google Scholar]
- 3.Cheng F, Wu J, Fang L, Sun S, Liu B, Lin K, Bonnema G, Wang X. Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS One. 2012;7(5):e36442. doi: 10.1371/journal.pone.0036442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang H, Woodhouse MR, Cheng F, Schnable JC, Pedersen BS, Conant G, Wang X, Freeling M, Pires JC. Altered patterns of fractionation and exon deletions in Brassica rapa support a two-step model of paleohexaploidy. Genetics. 2012;190(4):1563–1574. doi: 10.1534/genetics.111.137349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams PH, Hill CB. Rapid-cycling populations of brassica. Science. 1986;232(4756):1385–1389. doi: 10.1126/science.232.4756.1385. [DOI] [PubMed] [Google Scholar]
- 6.Ding J, Araki H, Wang Q, Zhang P, Yang S, Chen JQ, Tian D. Highly asymmetric rice genomes. BMC Genomics. 2007;8:154. doi: 10.1186/1471-2164-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dopman EB, Hartl DL. A portrait of copy-number polymorphism in Drosophila melanogaster. Proc Natl Acad Sci USA. 2007;104(50):19920–19925. doi: 10.1073/pnas.0709888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santuari L, Hardtke CS. The case for resequencing studies of Arabidopsis thaliana accessions: mining the dark matter of natural genetic variation. Biol Reprod. 2010;2:85. doi: 10.3410/B2-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. Diversity of human copy number variation and multicopy genes. Science. 2010;330(6004):641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swanson-Wagner RA, Eichten SR, Kumari S, Tiffin P, Stein JC, Ware D, Springer NM. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res. 2010;20(12):1689–1699. doi: 10.1101/gr.109165.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, Lyngsoe R, Schultheiss SJ, Osborne EJ, Sreedharan VT, Kahles A, Bohnert R, Jean G, Derwent P, Kersey P, Belfield EJ, Harberd NP, Kemen E, Toomajian C, Kover PX, Clark RM, Ratsch G, Mott R. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature. 2011;477(7365):419–423. doi: 10.1038/nature10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, MargarityRos I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci USA. 2005;102(39):13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15(6):589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sanchez Alvarado A, Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng F, Liu S, Wu J, Fang L, Sun S, Liu B, Li P, Hua W, Wang X. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 2011;11:136. doi: 10.1186/1471-2229-11-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Pujar A, Shearer AG, Travers M, Weerasinghe D, Zhang P, Karp PD. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40(Database issue):D742–D753. doi: 10.1093/nar/gkr1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Passardi F, Cosio C, Penel C, Dunand C. Peroxidases have more functions than a Swiss army knife. Plant Cell Rep. 2005;24(5):255–265. doi: 10.1007/s00299-005-0972-6. [DOI] [PubMed] [Google Scholar]
- 21.Kristensen DM, Wolf YI, Mushegian AR, Koonin EV. Computational methods for Gene Orthology inference. Brief Bioinform. 2011;12(5):379–391. doi: 10.1093/bib/bbr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider A, Dessimoz C, Gonnet GH. OMA Browser–exploring orthologous relations across 352 complete genomes. Bioinformatics. 2007;23(16):2180–2182. doi: 10.1093/bioinformatics/btm295. [DOI] [PubMed] [Google Scholar]
- 24.Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J, Ware DH, Buckler ES. A first-generation haplotype map of maize. Science. 2009;326(5956):1115–1117. doi: 10.1126/science.1177837. [DOI] [PubMed] [Google Scholar]
- 25.Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Lin Z, Buckler ES, Qian Q, Zhang QF, Li J, Han B. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42(11):961–967. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 26.Pino Del Carpio D, Basnet RK, De Vos RC, Maliepaard C, Visser R, Bonnema G. The patterns of population differentiation in a Brassica rapa core collection. Theor Appl Genet. 2011;122(6):1105–1118. doi: 10.1007/s00122-010-1516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Wang X, Deng B, Lou P, Wu J, Sun R, Xu Z, Vromans J, Koornneef M, Bonnema G. Genetic relationships within Brassica rapa as inferred from AFLP fingerprints. Theor Appl Genet. 2005;110(7):1301–1314. doi: 10.1007/s00122-005-1967-y. [DOI] [PubMed] [Google Scholar]
- 28.Lysak MA, Koch MA, Pecinka A, Schubert I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005;15(4):516–525. doi: 10.1101/gr.3531105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat Genet. 2012;44(2):226–232. doi: 10.1038/ng.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40(Database issue):D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33(20):6494–6506. doi: 10.1093/nar/gki937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanke M, Tzvetkova A, Morgenstern B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 2006;7(Suppl 1):S11–S18. doi: 10.1186/gb-2006-7-s1-s11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mulder N, Apweiler R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol. 2007;396:59–70. doi: 10.1007/978-1-59745-515-2_5. [DOI] [PubMed] [Google Scholar]
- 36.Conesa A, Gotz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berglund AC, Sjolund E, Ostlund G, Sonnhammer EL. InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 2008;36(Database issue):D263–D266. doi: 10.1093/nar/gkm1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olson SA. EMBOSS opens up sequence analysis. European Molecular Biology Open Software Suite. Brief Bioinform. 2002;3(1):87–91. doi: 10.1093/bib/3.1.87. [DOI] [PubMed] [Google Scholar]
- 39.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Change rate on each chromosome and different effect types. Excel file containing details of the change rate in turnip and rapid cycling. (XLSX 15 KB)
Additional file 2: Functional annotation of common genes in Chiifu, turnip and rapid cycling. Excel file containing details of the functional annotation of common genes in Chiifu, turnip and rapid cycling. (XLSX 3 MB)
Additional file 3: The GO term enrichment analysis results on unique and dispensable genome of Chiifu, turnip and rapid cycling. Excel file of results obtained from GO term enrichment analysis using unique and dispensable genome of Chiifu, turnip and rapid cycling. (XLSX 345 KB)
Additional file 4: Functional annotation of dispensable, unique and unmapped genes in Chiifu, turnip and rapid cycling. Excel file containing details of the functional annotation of dispensable, unique and unmapped genes on Chiifu, turnip and rapid cycling. The unmapped genes only apply to the turnip and rapid cycling genomes. (XLSX 1 MB)
Additional file 5: KEGG and PlantCyc pathway assignment of dispensable and unique genes in Chiifu, turnip and rapid cycling. Excel file containing details of KEGG and PlantCyc pathway assignments of dispensable and unique genes in Chiifu, turnip and rapid cycling. (XLSX 83 KB)
Additional file 6: The reference genomes of turnip and rapid cycling. Fasta file containing details of the chromosomes and scaffolds on turnip and rapid cycling. The two newly assembled genomes, their annotation files, a gene list for the three categories of pan genomes and the Blast2GO project files. (ZIP 73 MB)
Additional file 7: The annotation files of Chiifu, turnip and rapid cycling genome. Archived file containing three GFF files, including details of the gene features in Chiifu, turnip and rapid cycling. (ZIP 73 MB)
Additional file 8: Common, dispensable and unique genes in Chiifu, turnip and rapid cycling. Excel file containing details of the full complement of genes on Chiifu, turnip and rapid cycling. (ZIP 16 MB)
Additional file 9: Blast2GO project files of dispensable and unique genes in Chiifu, turnip and rapid cycling. Archived file containing three DAT files, each including a project file which can be imported in the Blast2GO program for further analysis. (ZIP 752 KB)
Additional file 10: All new created scripts in the study. Archived file containing all new created scripts used in the study. (ZIP 15 MB)
Additional file 11: The flowering time candidate gene list in A. thaliana. Excel file listing candidate genes related to flowering time in A. thaliana, categorized into flower development, gibberellin, photoperiod, vernalization and metabolic process. (XLSX 253 KB)