

Abstract
Muscle specific tyrosine kinase myasthenia gravis (MuSK MG) is a form of autoimmune MG that predominantly affects women and has unique clinical features, including prominent bulbar weakness, muscle atrophy, and excellent response to therapeutic plasma exchange. Patients with MuSK MG have predominantly IgG4 autoantibodies directed against MuSK on the postsynaptic muscle membrane. Lymphocyte functionality has not been reported in this condition. The goal of this study was to characterize T-cell responses in patients with MuSK MG. Intracellular production of IFN-gamma, TNF-alpha, IL-2, IL-17, and IL-21 by CD4+ and CD8+ T-cells was measured by polychromatic flow cytometry in peripheral blood samples from 11 Musk MG patients and 10 healthy controls. Only one MuSK MG patient was not receiving immunosuppressive therapy. Regulatory T-cells (Treg) were also included in our analysis to determine if changes in T cell function were due to altered Treg frequencies. CD8+ T-cells from MuSK MG patients had higher frequencies of polyfunctional responses than controls, and CD4+ T-cells had higher IL-2, TNF-alpha, and IL-17. MuSK MG patients had a higher percentage of CD4+ T-cells producing combinations of IFN-gamma/IL-2/TNF-gamma, TNF-alpha/IL-2, and IFN-gamma/TNF-alpha. Interestingly, Treg numbers and CD39 expression were not different from control values. MuSK MG patients had increased frequencies of Th1 and Th17 cytokines and were primed for polyfunctional proinflammatory responses that cannot be explained by a defect in Treg function or number.
Keywords: myasthenia gravis, MuSK protein, human, T-lymphocytes, regulatory, autoimmunity
1. Introduction
The most common form of autoimmune myasthenia gravis (MG) is characterized by the presence of circulating acetylcholine receptor (AChR) autoantibodies. Most MG patients with AChR antibodies have prominent weakness of extraocular muscles resulting in drooping of the eyelids (ptosis) and double vision. The weakness usually extends beyond the eyes to the extremities, respiratory muscles, and muscles involved in chewing and swallowing (bulbar muscles). Occasionally the weakness progresses to respiratory failure (MG crisis), which is fatal without treatment. Common treatment strategies include symptomatic therapy with acetylcholinesterase inhibitors, immunosuppression with prednisone or steroid-sparing agents such as azathioprine or mycophenolate mofetil, and mechanical ventilation along with intravenous immunoglobulin or therapeutic plasma exchange when weakness progresses to MG crisis [1].
A less common subset of MG patients who do not have AChR antibodies is characterized by: predominant bulbar, neck and proximal extremity weakness, frequently with muscle atrophy; severe weakness early in the disease often progressing to crisis; poor response or worsening with acetylcholinesterase inhibitors; fewer thymic changes on pathologic examination; and rapid improvement with therapeutic plasma exchange [2–6]. These patients often have autoantibodies directed against muscle specific tyrosine kinase (MuSK) on the postsynaptic membrane of skeletal muscle [7, 8].
MuSK plays important roles in the assembly and stabilization of the AChR and anchoring acetylcholinesterase to the basal lamina at the synapse [9, 10]. The autoantibodies in MuSK MG are typically IgG4, and it has recently been shown that in some patients these autoantibodies bind to the collagen tail subunit (ColQ) of acetylcholinesterase and block the binding of ColQ to MuSK on the postsynaptic muscle membrane [11, 12]. Most immunologic studies in MuSK MG have focused on establishing a pathogenic role for the autoantibodies [13–15]. Other reports have described the beneficial response of MuSK MG to the anti-CD20 monoclonal antibody rituximab [16, 17].
Given that the medical literature is currently devoid of any description of lymphocyte phenotype and functionality in MuSK MG we undertook to determine if T cell abnormalities are present in this condition. We demonstrated that MuSK MG patients have higher frequencies of Th1 and Th17 activity than normal controls, along with an increase in T cell polyfunctionality, and that the increase in T cell functionality cannot be attributed to a breakdown in Treg numbers or CD39 expression.
2. Material and Methods
2.1. Study population and controls
Blood samples were obtained from 11 female MuSK MG patients (mean age: 44.5; range: 19–66 years old) (Table 1) and 10 healthy controls (6 female; mean age: 40.3; range: 25–56 years). MuSK MG patients were recruited during visits to the Duke MG Clinic. All had detectable anti-MuSK antibodies according to commercially available testing (Athena Diagnostics, Worcester, MA) and clinical and electrodiagnostic features consistent with the disease. Clinical data collected from consenting patients included demographics, duration of disease, pharmacologic treatments, antibody results, thymectomy status, and Myasthenia Gravis Foundation of America (MGFA) severity class, MGFA Post-intervention Status (PIS), and MG manual muscle testing (MG-MMT) (Table 1) [18, 19]. The time from onset of symptoms to blood draw was more than 1 year in all MuSK MG patients. Thymectomy had been performed in 6: none had a thymoma or thymic hyperplasia. The maximum MGFA severity class at any point since disease onset was 3 or 4 (moderate to severe generalized weakness) or 5 (crisis) in nearly all patients, while the MGFA PIS at the time of the blood draw was “Minimal Manifestations” or better in 6 and “Improved” in 4. One patient had minimal weakness on MG-MMT and was not on immunosuppressive therapy. The others were on monotherapy with prednisone or mycophenolate mofetil or combination immunosuppressive therapy. Three patients had previously received rituximab.
Table 1.
Clinical characteristics of MuSK MG patients at the time of blood draw (N=11; all were female)
| Age (Yr) | Race | Disease Duration (Mo) | Maximum MGFA Severity Class | MG-MMT | MGFA-PIS | Thymectomy | Immunosuppressives |
|---|---|---|---|---|---|---|---|
| 22 | B | 13 | V | 16 | I | Yes | Prednisone 10mg/d; AZA 100mg/d; monthly TPE |
| 47 | W | 144 | IIIB | 1 | MM | Yes | MMF 750mg/d; RTX 22 months prior to blood draw |
| 59 | B | 180 | IIIB | 2 | MM | No | Prednisone 20mg QOD; MMF 2g/d |
| 22 | B | 144 | V | 7 | I | Yes | MMF 1.5g/d |
| 45 | B | 14 | I | 3 | U | No | Prednisone 17.5mg/d |
| 66 | W | 216 | IIIB | 3 | MM | No | MMF 2g/d |
| 55 | W | 165 | IIIB | 2 | MM | Yes | RTX 13 months prior to blood draw |
| 28 | B | 105 | IIIB | 0 | CSR | Yes | None |
| 64 | W | 139 | V | 3 | MM | Yes | Prednisone 2.5mg QOD; MMF 4g/d |
| 19 | B | 21.5 | IVB | 28 | I | No | MMF 1g/d d/c 11 days prior to blood draw; TPE 11 days prior to blood draw; prednisone 20mg/d |
| 62 | W | 135 | V | 23 | I | No | AZA 150mg/d; prednisone 5mg/d; RTX 3 years prior to blood draw |
Abbreviations: AZA=azathioprine; B=black; BID=twice daily; CSR=complete stable remission; d=day; d/c=discontinued; F=female; g=grams; I=improved; MGFA=Myasthenia Gravis Foundation of America; MM=minimal manifestations; MMF=mycophenolate mofetil; MG-MMT=myasthenia gravis manual muscle testing score at time of blood draw; Mo=months; PIS=post intervention status at time of blood draw; QOD=every other day; RTX=rituximab; TPE=therapeutic plasma exchange; U=unchanged; W=white; Yr=years
Healthy controls weighing more than 110 pounds and not receiving therapy for any chronic disease were recruited and matched as closely as possible for age and gender. This study was approved by the Duke University Institutional Review Board.
2.2. Isolation and storage of mononuclear peripheral blood cells
Peripheral blood was obtained by venipuncture and collected in acid-citrate-dextrose tubes (BD Vacutainer, Franklin Lake, NJ). Mononuclear cells were separated by Ficoll density gradient centrifugation, washed and counted prior to storage. Cells were resuspended in a 90% FBS (Gemini, West Sacramento, CA) and 10% DMSO (Sigma, St. Louis, MO) solution, and progressively cooled to −80°C in a CoolCell cell freezing container (BioCision, Larkspur, CA). The next day the cells were transferred to liquid nitrogen for long-term storage.
2.3. Intracellular cytokine staining
106 peripheral blood mononuclear cells (PBMCs) were plated in 96-well round bottom plates in RPMI +10% FBS. Cells were left untreated, stimulated with either αCD3 (1μg/mL) and αCD28 (5μg/mL) or phorbol 12-myristate 13-acetate (PMA, 1μg/mL) and ionomycin (IONO, 0.25μg/mL) in the presence of brefeldin A (BD Biosciences, San Jose, CA). Cells were incubated for six hours at 37°C in 6% CO2 in a humidified incubator. After this period, 1–2x106 cells were stained with 50μL of a cocktail mix consisting of titrated volumes of LIVE/DEAD violet dye (Life Technologies, Grand Island, NY), CD14 Pacific Blue, CD3 AmCyan, CD4 Brilliant Violet 605, and CD8 APC-Cy7 conjugates for 30 minutes at 4°C. A combination of LIVE/DEAD dye and CD14 were used as a “dump” channel to eliminate dead cells and monocytes, respectively. CD14, CD3, CD4, and CD8 fluorescent antibodies were obtained from BD Biosciences, San Jose, CA. Following cell surface staining, cells were treated with cytofix/cytoperm (BD Biosciences, San Jose, CA) in accordance with the manufacturer’s recommendations. Intracellular staining was then performed for 30 mins at 4°C using IFN-γ PE-Cy7, TNF-α Alexa Fluor 700, IL-2 APC, IL-17 PcP Cy5.5, and IL-21 PE conjugates. All cytokine fluorescent antibodies were purchased from BD Biosciences, San Jose, CA. Cells were fixed with 1% paraformaldehyde (PFA) and acquired on a LSRII flow cytometer (BD Biosciences, San Jose, CA).
2.4. FOXP3 staining
PBMCs were plated in a 96-well round bottom plate and cells were stained with LIVE/DEAD violet dye (Life Technologies, Grand Island, NY), CD14 Pacific Blue, CD3 AmCyan, CD4 Brilliant Violet 605, CD25 Alexa Fluor 700, and CD39 PE-Cy7 conjugates (eBioscience, San Diego, CA) for 30 minutes in 4°C. CD14, CD3, CD4, and CD25 were obtained from BD Biosciences, San Jose, CA. Following cell surface staining, cells were treated for 1 hour at 4°C with the FOXP3/Transcription Factor Fixation/Permeabilization buffer in accordance with the manufacturer’s recommendations (eBioscience, San Diego, CA). Intra-nuclear staining was then performed for 30 mins at 4°C using FOXP3 Alexa Fluor 647 conjugate. Cells were fixed with 1% PFA and acquired on a LSRII flow cytometer (BD Biosciences, San Jose, CA).
2.5. Data Analysis and Statistics
Data analysis was performed using Flowjo software (Tree Star, Ashland, OR). After the gates for each individual function were created, we used the Boolean gate platform incorporated into the Flowjo software to create an array of possible cytokine combinations. We then created bar graphs and pie charts of the various combinations of intracellular cytokines produced by T cells using “Simplified Presentation of Incredibly Complex Evaluations” (SPICE) software [20]. Student T-tests were used to determine statistical significance between two groups. The p values were calculated using Prism software (Graph Pad, LaJolla, CA).
3. Results
3.1. Cytokine analysis of CD8 T cells in MuSK MG
To generate a comprehensive analysis of cytokine production in MuSK MG patients we developed a nine-color polychromatic flow cytometry panel to test on PBMCs from MUSK MG and healthy controls. Figure 1A depicts our hierarchal gating strategy to identify CD4 and CD8 T cells. Subsequently, cytokine positivity in CD4 and CD8 T cells was determined following stimulation and in unstimulated samples as a control (Figure 1B and C). T cell production of cytokines IFN-γ, TNF-α, and IL-2 was determined following stimulation with αCD3/αCD28 (Fig. 2A) and PMA/IONO (Fig. 2B). Although the mean frequency of cytokine producing cells was higher in the MuSK MG patients than in the controls, none was statistically significant. To further examine the function of CD8 T cells, Boolean gating was performed using Flowjo software to identify CD8 T cells that produced different combinations of cytokines following PMA/IONO stimulation (Fig. 2C). This analysis determines which CD8 T cells are producing one, two, or all three cytokines; cells producing two or more cytokines are deemed polyfunctional. The results are also depicted in pie charts generated using SPICE software that show the color-coded distribution of cytokine producers (Fig. 2D) [21]. The blue slices denote the three-cytokine producers while the red slices represent the cells that produce no cytokines; the color spectrum from red to blue shows the two- and one-cytokine producers. Interestingly, visual analysis of the pie charts shows distinct differences in functionality between MuSK MG and normal donors. CD8 T cells in MuSK MG patients, in comparison with healthy controls, more frequently co-produced IFN-γ; and TNF-α (31% vs 21% ). In contrast, the majority of CD8 T cells in healthy donors produce no cytokines (49% vs 29%). Thus, the increase in cytokine production by CD8 T cells in MuSK MG patients likely represents a pathologic response rather than, for example, the effect of immunotherapy.
Fig 1.

Representative hierarchal gating for T cell analysis. The following gating strategy was used to isolate T cell populations. (A) Initial gating used forward scatter width (FSC-Width) versus height (FSC-Height) plot to remove doublets. Next, events were gated through a forward scatter area (FSC-Area) versus CD14 and LIVE/DEAD dye to remove monocytes and dead cells, respectively. Subsequently, lymphocytes were gated by a FCS-Area versus SSC plot, followed by a gate on CD3+ cells. CD4 and CD8 T cell gates were drawn off of CD3+ cells. (B) Representative detection of cytokines produced by CD4 T cells (IFN-γ, TNF-α, IL-2, IL-17, IL-21) following no stimulation (upper panel) or PMA/IONO stimulation (lower panel). (C) Detection of cytokines (IFN-γ, TNF-α, IL-2) produced by CD8 T cells following no stimulation (lower panel) or PMA/IONO stimulation (upper panel).
Fig 2.

Comparison of CD8 T cell function in normal and MuSK patients. (A) Intracellular cytokine analysis for IFN-γ, TNF-α, and IL-2 production following αCD3 and αCD28 stimulation of PBMCs in normal (black circle) and MuSK patients (square). Unshaded squares are patients who received Rituximab treatment. Columns represent the mean percentages. Percentages were derived from gated CD8 T cells. (B) Intracellular cytokine analysis for IFN-γ, TNF-α, and IL-2 production following PMA/IONO stimulation of PBMCs. (C) The black (normal) and gray (MuSK) bars represents the mean frequency of the CD8 T cell response expressing different combinations of cytokine production in response to PMA/IONO stimulation of PBMCs. “+” sign denotes IFN-γ, TNF-α, or IL-2 positivity. The number on the color-coded pie slice bars represents the number of cytokine positivity. (D) Each pie chart represents the mean response across ten normal individuals and eleven MuSK patients. Responses are grouped by the number of functions and matched to the “pie slice” color bars in panel C. Statistical significance is represented as follows: *p < 0.05. Results are shown from ten normal individuals and eleven MuSK patients.
3.2. Cytokine analysis of CD4 T cells in MuSK MG
For the composite analysis of CD4 T cell function, IL-17 and IL-21 were added to the flow cytometric analysis panel because published evidence suggests a critical role for Th17 cells in autoimmunity [22, 23]. In blood from MuSK MG patients, we observed an increase in the frequency of cells producing IL-17 following stimulation with αCD3/αCD28 (0.53% vs 0.24%) (Fig. 3A) and TNF-α (54% vs 39%), IL-2 (55% vs 42%), and IL-17 (1.8% vs 0.43%) with PMA/IONO stimulation (Fig. 3B). This suggests that the increased frequency of Th1 and Th17 subsets of CD4 T cells may contribute to MuSK MG pathology. To evaluate whether CD4 T cells produce multiple cytokines, we used Boolean gating and SPICE software to generate 32 possible cytokine combinations that could be produced by the CD4 T cells (Fig. 3C and D). As with the CD8 T cells, most CD4 T cells from normal donors produced no cytokine, while a higher frequency of these cells from MuSK MG patients were polyfunctional. However, neither CD8 nor CD4 T cells were capable of producing all three or five cytokines, respectively. On visual inspection, higher percentages of polyfunctional cytokine production in MuSK patients was seen for IFN-γ/IL-2/TNF-α, TNF-α/IL-2, and IFN-γ/TNF-α. Interestingly, MuSK MG patients who had received rituximab had the lowest frequencies of IFN-γ, TNF-α, and IL-2 producing cells following PMA/IONO stimulation (Fig. 3B). Rituximab treatment had no effect on the frequencies of CD4 and CD8 T cells, as these were within the range of normal and rituximab-free MuSK patients (data not shown).
Fig 3.

Comparison of CD4 T cell function in normal and MuSK patients. (A) Intracellular cytokine analysis for IFN-γ, TNF-α, IL-2, IL-17, and IL-21 production following αCD3 and αCD28 stimulation of PBMCs in normal (black circle) and MuSK patients (square). Unshaded squares are patients who received Rituximab treatment. Columns represent the mean percentages. Percentages were derived from gated CD4 T cells. (B) Intracellular cytokine analysis for IFN-γ, TNF-α, IL-2, IL-17, and IL-21 production following PMA/IONO stimulation of PBMCs. (C) The black (normal) and gray (MuSK) bars represents the mean frequency of the CD4 T cells expressing different cytokine combinations in response to PMA/IONO stimulation of PBMCs. “+” sign denotes IFN-γ, TNF-α, IL-2, IL-17, and/or IL-21 positivity. The number on the color-coded pie slice bars represents the number of cytokine positivity. (D) Each pie chart represents the mean response across ten normal individuals and eleven MuSK patients. Responses are grouped by the number of functions and matched to the “pie slice” color bars in panel C. Statistical significance is represented as follows: *p < 0.05; **p < 0.01; ***p < 0.001. Results are shown from ten normal individuals and eleven MuSK patients.
3.3. Increase in T cell function is not due to changes in the Treg population
To determine whether the increase in CD4 T cell function was due to an imbalance of Tregs we examined the frequency and response of Tregs. Tregs are critical regulators of immune tolerance, and autoimmune diseases have been attributed to expansion of autoreactive lymphocytes due to a breakdown of self- tolerance. Tregs were identified by the expression of CD25 and FOXP3 on CD4 T cells (Fig. 4A). Analysis of Treg frequencies revealed no significant changes in MuSK MG patients compared with controls.
Fig 4.
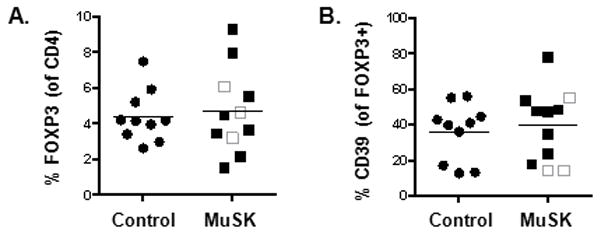
Increase in T cell function is not due to changes in Treg numbers or altered CD39 expression. (A) Tregs were identified as CD25+ and FOXP3+ in the PBMCs of normal (black circle) and MuSK patients (square). The three unshaded squares represent patients who have received rituximab treatment. Frequency of Tregs were derived from total CD4 T cells. (B) CD39 expression in normal and MuSK patients was similar. The frequencies of CD39 were derived from CD25+FOXP3+ Tregs. Results are shown from ten normal individuals and eleven MuSK patients.
To further investigate the role of Tregs in MuSK MG, we examined the expression of CD39, an ectonuclease enzyme responsible for Treg suppressive activity in animal models [24]. CD39+FOXP3+ Tregs have been demonstrated to be critical in suppressing IL-17 production and, these cells are impaired in multiple sclerosis [25]. Since we observed an increase in IL-17 production in MuSK MG patients compared with controls, we examined whether this increase was due to a down-regulation of CD39 expression. In MuSK MG, we observed no differences in the frequency of CD39 expression on FOXP3+ Tregs (Fig. 4B). Interestingly, two out of the three patients who had received rituximab had the lowest frequencies of CD39. Overall, the enhancement of T cell function in MuSK MG patients cannot be atttributed to a defect in Treg numbers nor reduced CD39 expression.
4. Discussion
In this study, we evaluated T cell responses in a well characterized cohort of MuSK MG patients. We demonstrated that CD8+ T cells in MuSK MG patients had generally higher frequencies of multiple cytokine producing cells than controls, as well as strong CD4+ T cell activation to stimulation that was characterized by increased TNF-α, IL-2, and IL-17 responses. These CD4+ T cells were primed for polyfunctional proinflammatory responses, with a higher percentage of MuSK patients producing combinations of IFN-γ/IL-2/TNF-α, TNF-α/IL-2, and IFN-γ/TNF-α than controls. Notably, the increased CD4+ T cell responses could not be explained by changes in Treg numbers or function as measured by CD39 expression. Polyfunctionality is often associated with T cell differentiation and the capacity for self-renewal [26, 27]. During T cell maturation, differentiation to memory cells or effector cells occurs at the expense of self-renewal, proliferative capacity and cytokine production. Memory cells have increased self-renewal properties allowing them to persist longer in the host and they immediately respond to antigen upon activation. In contrast, activation of effector cells is slower than memory cells and they are more likely to undergo apoptosis due to their inability to undergo self-renewal and their terminally differentiated state. As such, memory cells tend to exhibit polyfunctional T cell responses compared with effector cells, which are more likely to be monofunctional. These findings suggest that a memory T cell phenotype, which is often associated with polyfunctional responses, may be increased in MuSK MG patients and that their T cells are predisposed to more potent responses.
To date, immunologic studies in patients with MuSK MG have primarily been limited to 1) demonstrating the pathogenic nature of MuSK serum and purified anti-MuSK immunoglobulins in animal transfer studies and in vitro models [8, 13, 28]; 2) establishing that MuSK MG autoantibodies are primarily IgG4 [11, 29–31]; 3) examination of muscle and the thymus gland following thymectomy [4, 5, 32]; and 4) measuring autoantibodies after treatment with rituximab [16, 17].
MG immunology has been most studied in the AChR+ form of the disease (Table 2). In AChR+ MG, the pathogenic role of CD4 T cells has been demonstrated by treatments that diminish CD4 T cell levels, including thymectomy or treatment with anti-CD4 antibodies that deplete CD4 T cells [33, 34]. It has also been observed that the loss of CD4 T cells in AIDS patients correlated with improvement in MG symptoms [35]. Conversely, depletion of CD8 T cells had no effect on the development of myasthenic weakness and anti-AChR antibody synthesis [36]. Based on murine studies, which appear to be supported by studies of MG patients, the dominant subset of CD4 T cells present are Th1 cells. Pro-inflammatory cytokines such as IL-2, IL-12, and IFN-γ are thought to drive the production of complement-fixing immunoglobulins through activation of Th1 cells [37, 38]. More recently, it has been reported that IL-17, which is involved in B cell activation and proliferation, is elevated in MG patient serum [39].
Table 2.
Summary of some known clinical and immunological features of MG
| AChR MG | MuSK MG | LRP4 MG | |
|---|---|---|---|
| Clinical features | |||
| HLA association |
|
||
| Autoantibodies |
|
|
|
| T cells |
|
|
|
| B cells |
|
|
|
| Tregs |
|
|
|
| Bregs |
|
|
|
| Cytokines |
|
|
|
| Complement |
|
||
| Thymic changes |
|
Abbreviations: AChR=acetylcholine receptor; ACheI= acetylcholine esterase inhibitor; AIRE=autoimmune regulator gene; BAFF=B-cell activating facor; HLA=human leukocyte antigen; IVIg=intravenous immunoglobulins; LRP= lipoprotein-related protein 4; MG=myasthenia gravis; MuSK=muscle specific kinase; RTX=rituximab; sIL-2R=soluble IL-2 receptor; TPE=therapeutic plasma exchange
The neuroimmunology of the recently described forms of MG associated with LRP4 and MuSK autoantibodies is less well understood (Table 2). There are significant knowledge gaps in the cellular immunology of these conditions, including regulatory cells and the role of cytokines. The relative rarity of both of these forms of MG makes studying them challenging.
In MuSK MG, the presence of predominantly IgG4 autoantibodies that don’t activate complement has led to the assumption that the disease is mediated by Th2 pathways [16]. Interestingly, we found elevated frequencies of T cells producing cytokines associated with Th1 and Th17 cells. Thus, our future studies will evaluate the role of Th2-associated cytokines to define the predominant pathway of T cell activity in the pathogenesis of MuSK MG.
The potential role of Tregs in the breakdown of self-tolerance in MG has been a focus of recent studies [63–65, 69–71]. The results of these studies have been somewhat mixed, likely due to heterogeneity of the patient populations studied and how Treg populations were defined. Nevertheless, the preponderance of evidence supports a defect in Treg function rather than a decrease in absolute numbers of Tregs. No studies of Tregs in MuSK MG patients have previously been reported, but we have found no changes in Treg numbers in MuSK MG compared with controls. Furthermore, the percentage of these Tregs expressing CD39 was similar to controls. CD39 is an ectonucleotidease that cleaves ATP to form AMP, which can then be cleaved by CD73 to form adenosine [97]. In mice CD39 is expressed on all FOXP3+ T cells and knockdown of CD39 expression reduces the suppressive capacity of Tregs, suggesting that the hydrolysis of ATP by CD39 is a critical mechanism in Treg suppression [24]. In autoimmune diseases such as multiple sclerosis and systemic lupus erythematosus, research suggests a defect in Tregs due to decreased CD39 expression [25, 98]. However, we found no evidence to suggest such a defect in Treg function, by CD39 expression, or numbers, in the pathophysiology of MuSK MG.
There have been many reports of dramatic and often sustained improvement in MuSK MG patients after treatment with the anti-CD20 monoclonal antibody rituximab [17, 42, 99–101]. Several studies reported that this clinical improvement was associated with a significant decline in MuSK autoantibody blood levels [16, 17]. However, the effects of rituximab treatment on lymphocytes have not been reported in detail. In our 3 patients who had previously received rituximab, Treg populations were similar to healthy controls and to rituximab-naïve MuSK MG patients (Fig. 3). Intracellular cytokine production by CD4+ and CD8+ T cells in these patients was in the lower range of the total MuSK cohort with the exception of somewhat higher production of IFN-γ and TNF-α in CD8+ T cells (Fig. 1, 2). The significance of this finding is uncertain and the number of patients who received rituximab is too small to make statistical comparisons. Future studies evaluating the effects of rituximab on B cells in MuSK MG patients should be particularly informative.
A limitation of our study is the use of immunosuppressive medications in nearly all patients and, perhaps, the wide range of disease duration - some of our observations may be affected by these immunosuppressive medications rather than the disease itself [66]. However, the consistency of the responses despite heterogeneous treatment regimens suggests that the altered T cell functionality is not strongly related to immunosuppression and may be a unique feature of MuSK MG.
The one MuSK MG patient not on immunosuppressive therapy also had AChR modulating, but not binding or striated muscle, antibodies. She underwent thymectomy 8 years prior to this study and had achieved pharmacologic remission 3 months after surgery. MuSK autoantibodies were undetectable approximately 3 years after thymectomy and she was in Complete Stable Remission at the time of this study, having taken no immunosuppressives for over 3.5 years. She never received rituximab. Other than an increased percentage of CD4+ T cells, her T cell profile was similar to the other MuSK patients, none of whom had received rituximab. As with the other MuSK MG patients, she had higher percentages of IL-2 and TNF-α producing CD4+ T cells. This patient is somewhat unusual given her remission after thymectomy, but her persistent altered T cell functionality suggests that our overall findings are not due to the use of immunosuppression. Future studies on greater numbers of MuSK MG patients will provide better assessment of factors that may affect immune profiles, such as thymectomy and different immunosuppressive regimens. Due to the rarity of MuSK MG this will likely require a concerted collaborative effort among centers.
Acknowledgments
This study was supported by a clinician-scientist development award sponsored by the American Academy of Neurology Foundation and the Myasthenia Gravis Foundation of America (Dr. Guptill) and a pilot grant from the Duke Translational Research Institute (CTSA grant UL1RR024128). In addition, this publication was made possible with the help from the Duke University Center for AIDS Research (CFAR), an NIH funded program (P30 AI 64518).
Footnotes
Conflict of interest
The authors have no conflict of interest related to this study.
References
- 1.Meriggioli M, Sanders D. Disorders of neuromuscular transmission. In: Bradley WGDR, Fenichel GM, Jancovic J, editors. Neurology in Clinical Practice. Philadelphia: Butterworths Heinemann Elsevier; 2012. [Google Scholar]
- 2.Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. 2011;44:36–40. doi: 10.1002/mus.22006. [DOI] [PubMed] [Google Scholar]
- 3.Pasnoor M, Wolfe GI, Nations S, Trivedi J, Barohn RJ, Herbelin L, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a U.S. experience. Muscle Nerve. 2010;41:370–4. doi: 10.1002/mus.21533. [DOI] [PubMed] [Google Scholar]
- 4.Leite MI, Strobel P, Jones M, Micklem K, Moritz R, Gold R, et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol. 2005;57:444–8. doi: 10.1002/ana.20386. [DOI] [PubMed] [Google Scholar]
- 5.Lauriola L, Ranelletti F, Maggiano N, Guerriero M, Punzi C, Marsili F, et al. Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology. 2005;64:536–8. doi: 10.1212/01.WNL.0000150587.71497.B6. [DOI] [PubMed] [Google Scholar]
- 6.Stickler DE, Massey JM, Sanders DB. MuSK-antibody positive myasthenia gravis: clinical and electrodiagnostic patterns. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2005;116:2065–8. doi: 10.1016/j.clinph.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Scuderi F, Marino M, Colonna L, Mannella F, Evoli A, Provenzano C, et al. Anti-p110 autoantibodies identify a subtype of “seronegative” myasthenia gravis with prominent oculobulbar involvement. Laboratory investigation; a journal of technical methods and pathology. 2002;82:1139–46. doi: 10.1097/01.lab.0000028144.48023.9b. [DOI] [PubMed] [Google Scholar]
- 8.Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nature medicine. 2001;7:365–8. doi: 10.1038/85520. [DOI] [PubMed] [Google Scholar]
- 9.Cartaud A, Strochlic L, Guerra M, Blanchard B, Lambergeon M, Krejci E, et al. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. The Journal of cell biology. 2004;165:505–15. doi: 10.1083/jcb.200307164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghazanfari N, Fernandez KJ, Murata Y, Morsch M, Ngo ST, Reddel SW, et al. Muscle specific kinase: organiser of synaptic membrane domains. The international journal of biochemistry & cell biology. 2011;43:295–8. doi: 10.1016/j.biocel.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Niks EH, van Leeuwen Y, Leite MI, Dekker FW, Wintzen AR, Wirtz PW, et al. Clinical fluctuations in MuSK myasthenia gravis are related to antigen-specific IgG4 instead of IgG1. J Neuroimmunol. 2008;195:151–6. doi: 10.1016/j.jneuroim.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 12.Kawakami Y, Ito M, Hirayama M, Sahashi K, Ohkawara B, Masuda A, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology. 2011;77:1819–26. doi: 10.1212/WNL.0b013e318237f660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole RN, Reddel SW, Gervasio OL, Phillips WD. Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann Neurol. 2008;63:782–9. doi: 10.1002/ana.21371. [DOI] [PubMed] [Google Scholar]
- 14.Plomp JJ, Huijbers MG, van der Maarel SM, Verschuuren JJ. Pathogenic IgG4 subclass autoantibodies in MuSK myasthenia gravis. Annals of the New York Academy of Sciences. 2012;1275:114–22. doi: 10.1111/j.1749-6632.2012.06808.x. [DOI] [PubMed] [Google Scholar]
- 15.Farrugia ME, Bonifati DM, Clover L, Cossins J, Beeson D, Vincent A. Effect of sera from AChR-antibody negative myasthenia gravis patients on AChR and MuSK in cell cultures. J Neuroimmunol. 2007;185:136–44. doi: 10.1016/j.jneuroim.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Diaz-Manera J, Martinez-Hernandez E, Querol L, Klooster R, Rojas-Garcia R, Suarez-Calvet X, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78:189–93. doi: 10.1212/WNL.0b013e3182407982. [DOI] [PubMed] [Google Scholar]
- 17.Illa I, Diaz-Manera J, Rojas-Garcia R, Pradas J, Rey A, Blesa R, et al. Sustained response to Rituximab in anti-AChR and anti-MuSK positive Myasthenia Gravis patients. J Neuroimmunol. 2008;201–202:90–4. doi: 10.1016/j.jneuroim.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 18.Jaretzki A, 3rd, Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. 2000;55:16–23. doi: 10.1212/wnl.55.1.16. [DOI] [PubMed] [Google Scholar]
- 19.Sanders DB, Tucker-Lipscomb B, Massey JM. A simple manual muscle test for myasthenia gravis: validation and comparison with the QMG score. Annals of the New York Academy of Sciences. 2003;998:440–4. doi: 10.1196/annals.1254.057. [DOI] [PubMed] [Google Scholar]
- 20.Roederer M, Nozzi JL, Nason MC. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2011;79:167–74. doi: 10.1002/cyto.a.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roederer M, Nozzi JL, Nason MX. SPICE: Exploration and analysis of post-cytometric complex multivariate datasets. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2011 doi: 10.1002/cyto.a.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oukka M. Th17 cells in immunity and autoimmunity. Annals of the rheumatic diseases. 2008;67(Suppl 3):iii26–9. doi: 10.1136/ard.2008.098004. [DOI] [PubMed] [Google Scholar]
- 23.Waite JC, Skokos D. Th17 response and inflammatory autoimmune diseases. International journal of inflammation. 2012;2012:819467. doi: 10.1155/2012/819467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O'Farrelly C, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. Journal of immunology. 2009;183:7602–10. doi: 10.4049/jimmunol.0901881. [DOI] [PubMed] [Google Scholar]
- 26.Pantaleo G, Harari A. Functional signatures in antiviral T-cell immunity for monitoring virus-associated diseases. Nature reviews Immunology. 2006;6:417–23. doi: 10.1038/nri1840. [DOI] [PubMed] [Google Scholar]
- 27.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nature reviews Cancer. 2012;12:671–84. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boneva N, Frenkian-Cuvelier M, Bidault J, Brenner T, Berrih-Aknin S. Major pathogenic effects of anti-MuSK antibodies in myasthenia gravis. J Neuroimmunol. 2006;177:119–31. doi: 10.1016/j.jneuroim.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 29.McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55:580–4. doi: 10.1002/ana.20061. [DOI] [PubMed] [Google Scholar]
- 30.Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, et al. IgG1 antibodies to acetylcholine receptors in 'seronegative' myasthenia gravis. Brain : a journal of neurology. 2008;131:1940–52. doi: 10.1093/brain/awn092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohta K, Shigemoto K, Fujinami A, Maruyama N, Konishi T, Ohta M. Clinical and experimental features of MuSK antibody positive MG in Japan. Eur J Neurol. 2007;14:1029–34. doi: 10.1111/j.1468-1331.2007.01870.x. [DOI] [PubMed] [Google Scholar]
- 32.Shiraishi H, Motomura M, Yoshimura T, Fukudome T, Fukuda T, Nakao Y, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol. 2005;57:289–93. doi: 10.1002/ana.20341. [DOI] [PubMed] [Google Scholar]
- 33.Ahlberg R, Yi Q, Pirskanen R, Matell G, Swerup C, Rieber EP, et al. Treatment of myasthenia gravis with anti-CD4 antibody: improvement correlates to decreased T-cell autoreactivity. Neurology. 1994;44:1732–7. doi: 10.1212/wnl.44.9.1732. [DOI] [PubMed] [Google Scholar]
- 34.Morgutti M, Conti-Tronconi BM, Sghirlanzoni A, Clementi F. Cellular immune response to acetylcholine receptor in myasthenia gravis: II. Thymectomy and corticosteroids Neurology. 1979;29:734–8. doi: 10.1212/wnl.29.5.734. [DOI] [PubMed] [Google Scholar]
- 35.Nath A, Kerman RH, Novak IS, Wolinsky JS. Immune studies in human immunodeficiency virus infection with myasthenia gravis: a case report. Neurology. 1990;40:581–3. doi: 10.1212/wnl.40.4.581. [DOI] [PubMed] [Google Scholar]
- 36.Wang ZY, Karachunski PI, Howard JF, Jr, Conti-Fine BM. Myasthenia in SCID mice grafted with myasthenic patient lymphocytes: role of CD4+ and CD8+ cells. Neurology. 1999;52:484–97. doi: 10.1212/wnl.52.3.484. [DOI] [PubMed] [Google Scholar]
- 37.Conti-Fine BM, Milani M, Wang W. CD4+ T cells and cytokines in the pathogenesis of acquired myasthenia gravis. Annals of the New York Academy of Sciences. 2008;1132:193–209. doi: 10.1196/annals.1405.042. [DOI] [PubMed] [Google Scholar]
- 38.Utsugisawa K, Nagane Y, Obara D, Kondoh R, Yonezawa H, Tohgi H. Interleukin-2 production by peripheral blood mononuclear cells from patients with myasthenia gravis. Eur Neurol. 2003;49:160–3. doi: 10.1159/000069079. [DOI] [PubMed] [Google Scholar]
- 39.Roche JC, Capablo JL, Larrad L, Gervas-Arruga J, Ara JR, Sanchez A, et al. Increased serum interleukin-17 levels in patients with myasthenia gravis. Muscle Nerve. 2011;44:278–80. doi: 10.1002/mus.22070. [DOI] [PubMed] [Google Scholar]
- 40.Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve. 2008;37:141–9. doi: 10.1002/mus.20950. [DOI] [PubMed] [Google Scholar]
- 41.Juel VC, Massey JM. Myasthenia gravis. Orphanet J Rare Dis. 2007;2:44. doi: 10.1186/1750-1172-2-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diaz-Manera J, Rojas Garcia R, Illa I. Treatment strategies for myasthenia gravis: an update. Expert Opin Pharmacother. 13:1873–83. doi: 10.1517/14656566.2012.705831. [DOI] [PubMed] [Google Scholar]
- 43.Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, Schalke B, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol. 2012;259:427–35. doi: 10.1007/s00415-011-6194-7. [DOI] [PubMed] [Google Scholar]
- 44.Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. 2011;69:418–22. doi: 10.1002/ana.22312. [DOI] [PubMed] [Google Scholar]
- 45.Janer M, Cowland A, Picard J, Campbell D, Pontarotti P, Newsom-Davis J, et al. A susceptibility region for myasthenia gravis extending into the HLA-class I sector telomeric to HLA-C. Human immunology. 1999;60:909–17. doi: 10.1016/s0198-8859(99)00062-2. [DOI] [PubMed] [Google Scholar]
- 46.Giraud M, Beaurain G, Yamamoto AM, Eymard B, Tranchant C, Gajdos P, et al. Linkage of HLA to myasthenia gravis and genetic heterogeneity depending on anti-titin antibodies. Neurology. 2001;57:1555–60. doi: 10.1212/wnl.57.9.1555. [DOI] [PubMed] [Google Scholar]
- 47.Chen WH, Chiu HC, Hseih RP. Association of HLA-Bw46DR9 combination with juvenile myasthenia gravis in Chinese. Journal of neurology, neurosurgery, and psychiatry. 1993;56:382–5. doi: 10.1136/jnnp.56.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsuki K, Juji T, Tokunaga K, Takamizawa M, Maeda H, Soda M, et al. HLA antigens in Japanese patients with myasthenia gravis. J Clin Invest. 1990;86:392–9. doi: 10.1172/JCI114724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giraud M, Vandiedonck C, Garchon HJ. Genetic factors in autoimmune myasthenia gravis. Annals of the New York Academy of Sciences. 2008;1132:180–92. doi: 10.1196/annals.1405.027. [DOI] [PubMed] [Google Scholar]
- 50.Niks EH, Kuks JB, Roep BO, Haasnoot GW, Verduijn W, Ballieux BE, et al. Strong association of MuSK antibody-positive myasthenia gravis and HLA-DR14-DQ5. Neurology. 2006;66:1772–4. doi: 10.1212/01.wnl.0000218159.79769.5c. [DOI] [PubMed] [Google Scholar]
- 51.Bartoccioni E, Scuderi F, Augugliaro A, Chiatamone Ranieri S, Sauchelli D, Alboino P, et al. HLA class II allele analysis in MuSK-positive myasthenia gravis suggests a role for DQ5. Neurology. 2009;72:195–7. doi: 10.1212/01.wnl.0000339103.08830.86. [DOI] [PubMed] [Google Scholar]
- 52.Wang ZY, Diethelm-Okita B, Okita DK, Kaminski HJ, Howard JF, Conti-Fine BM. T cell recognition of muscle acetylcholine receptor in ocular myasthenia gravis. J Neuroimmunol. 2000;108:29–39. doi: 10.1016/s0165-5728(00)00250-2. [DOI] [PubMed] [Google Scholar]
- 53.Wang ZY, Okita DK, Howard JF, Jr, Conti-Fine BM. CD4+ T cell repertoire on the epsilon subunit of muscle acetylcholine receptor in myasthenia gravis. J Neuroimmunol. 1998;91:33–42. doi: 10.1016/s0165-5728(98)00146-5. [DOI] [PubMed] [Google Scholar]
- 54.Protti MP, Manfredi AA, Straub C, Howard JF, Jr, Conti-Tronconi BM. Immunodominant regions for T helper-cell sensitization on the human nicotinic receptor alpha subunit in myasthenia gravis. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:7792–6. doi: 10.1073/pnas.87.19.7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moiola L, Karachunski P, Protti MP, Howard JF, Jr, Conti-Tronconi BM. Epitopes on the beta subunit of human muscle acetylcholine receptor recognized by CD4+ cells of myasthenia gravis patients and healthy subjects. J Clin Invest. 1994;93:1020–8. doi: 10.1172/JCI117050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yi Q, Ahlberg R, Pirskanen R, Lefvert AK. Acetylcholine receptor-reactive T cells in myasthenia gravis: evidence for the involvement of different subpopulations of T helper cells. J Neuroimmunol. 1994;50:177–86. doi: 10.1016/0165-5728(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 57.Levinson AI, Zweiman B, Lisak RP. Immunopathogenesis and treatment of myasthenia gravis. Journal of clinical immunology. 1987;7:187–97. doi: 10.1007/BF00915723. [DOI] [PubMed] [Google Scholar]
- 58.Lisak RP, Laramore C, Levinson AI, Zweiman B, Moskovitz AR. Suppressor T cells in myasthenia gravis and antibodies to acetylcholine receptor. Ann Neurol. 1986;19:87–9. doi: 10.1002/ana.410190119. [DOI] [PubMed] [Google Scholar]
- 59.Levinson AI, Dziarski A, Lisak RP, Zweiman B, Moskovitz AR, Brenner T, et al. Polyclonal B-cell activity in myasthenia gravis. Neurology. 1981;31:1198–201. doi: 10.1212/wnl.31.9.1198. [DOI] [PubMed] [Google Scholar]
- 60.Lisak RP, Laramore C, Levinson AI, Zweiman B, Moskovitz AR, Witte A. In vitro synthesis of antibodies to acetylcholine receptor by peripheral blood cells: role of suppressor T cells in normal subjects. Neurology. 1984;34:802–5. doi: 10.1212/wnl.34.6.802. [DOI] [PubMed] [Google Scholar]
- 61.Kohler S, Keil TO, Swierzy M, Hoffmann S, Schaffert H, Ismail M, et al. Disturbed B cell subpopulations and increased plasma cells in myasthenia gravis patients. J Neuroimmunol. 2013;264:114–9. doi: 10.1016/j.jneuroim.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 62.Skeie GO, Apostolski S, Evoli A, Gilhus NE, Illa I, Harms L, et al. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 17:893–902. doi: 10.1111/j.1468-1331.2010.03019.x. [DOI] [PubMed] [Google Scholar]
- 63.Matsui N, Nakane S, Saito F, Ohigashi I, Nakagawa Y, Kurobe H, et al. Undiminished regulatory T cells in the thymus of patients with myasthenia gravis. Neurology. 2010;74:816–20. doi: 10.1212/WNL.0b013e3181d31e47. [DOI] [PubMed] [Google Scholar]
- 64.Xu WH, Zhang AM, Ren MS, Zhang XD, Wang F, Xu XC, et al. Changes of Treg-associated molecules on CD4+CD25 +Treg cells in myasthenia gravis and effects of immunosuppressants. Journal of clinical immunology. 2012;32:975–83. doi: 10.1007/s10875-012-9685-0. [DOI] [PubMed] [Google Scholar]
- 65.Thiruppathi M, Rowin J, Ganesh B, Sheng JR, Prabhakar BS, Meriggioli MN. Impaired regulatory function in circulating CD4(+)CD25(high)CD127(low/-) T cells in patients with myasthenia gravis. Clinical immunology. 2012;145:209–23. doi: 10.1016/j.clim.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fattorossi A, Battaglia A, Buzzonetti A, Ciaraffa F, Scambia G, Evoli A. Circulating and thymic CD4 CD25 T regulatory cells in myasthenia gravis: effect of immunosuppressive treatment. Immunology. 2005;116:134–41. doi: 10.1111/j.1365-2567.2005.02220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Battaglia A, Di Schino C, Fattorossi A, Scambia G, Evoli A. Circulating CD4+CD25+ T regulatory and natural killer T cells in patients with myasthenia gravis: a flow cytometry study. Journal of biological regulators and homeostatic agents. 2005;19:54–62. [PubMed] [Google Scholar]
- 68.Li X, Xiao BG, Xi JY, Lu CZ, Lu JH. Decrease of CD4(+)CD25(high)Foxp3(+) regulatory T cells and elevation of CD19(+)BAFF-R(+) B cells and soluble ICAM-1 in myasthenia gravis. Clinical immunology. 2008;126:180–8. doi: 10.1016/j.clim.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 69.Masuda M, Matsumoto M, Tanaka S, Nakajima K, Yamada N, Ido N, et al. Clinical implication of peripheral CD4+CD25+ regulatory T cells and Th17 cells in myasthenia gravis patients. J Neuroimmunol. 2010;225:123–31. doi: 10.1016/j.jneuroim.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Wang HB, Chi LJ, Wang WZ. The role of FoxP3+CD4+CD25hi Tregs in the pathogenesis of myasthenia gravis. Immunology letters. 2009;122:52–7. doi: 10.1016/j.imlet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 71.Balandina A, Lecart S, Dartevelle P, Saoudi A, Berrih-Aknin S. Functional defect of regulatory CD4(+)CD25+ T cells in the thymus of patients with autoimmune myasthenia gravis. Blood. 2005;105:735–41. doi: 10.1182/blood-2003-11-3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scarpino S, Di Napoli A, Stoppacciaro A, Antonelli M, Pilozzi E, Chiarle R, et al. Expression of autoimmune regulator gene (AIRE) and T regulatory cells in human thymomas. Clinical and experimental immunology. 2007;149:504–12. doi: 10.1111/j.1365-2249.2007.03442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Strobel P, Rosenwald A, Beyersdorf N, Kerkau T, Elert O, Murumagi A, et al. Selective loss of regulatory T cells in thymomas. Ann Neurol. 2004;56:901–4. doi: 10.1002/ana.20340. [DOI] [PubMed] [Google Scholar]
- 74.Link J, Navikas V, Yu M, Fredrikson S, Osterman PO, Link H. Augmented interferon-gamma, interleukin-4 and transforming growth factor-beta mRNA expression in blood mononuclear cells in myasthenia gravis. J Neuroimmunol. 1994;51:185–92. doi: 10.1016/0165-5728(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 75.Confalonieri P, Antozzi C, Cornelio F, Simoncini O, Mantegazza R. Immune activation in myasthenia gravis: soluble interleukin-2 receptor, interferon-gamma and tumor necrosis factor-alpha levels in patients' serum. J Neuroimmunol. 1993;48:33–6. doi: 10.1016/0165-5728(93)90055-4. [DOI] [PubMed] [Google Scholar]
- 76.Hartung HP, Reiners K, Schmidt B, Stoll G, Toyka KV. Serum interleukin-2 concentrations in Guillain-Barre syndrome and chronic idiopathic demyelinating polyradiculoneuropathy: comparison with other neurological diseases of presumed immunopathogenesis. Ann Neurol. 1991;30:48–53. doi: 10.1002/ana.410300110. [DOI] [PubMed] [Google Scholar]
- 77.Utsugisawa K, Sano M. Interleukin-2 production by peripheral blood mononuclear cell from patients with myasthenia gravis--correlation with clinical severity. Rinsho shinkeigaku = Clinical neurology. 1992;32:474–8. [PubMed] [Google Scholar]
- 78.Link J, He B, Navikas V, Palasik W, Fredrikson S, Soderstrom M, et al. Transforming growth factor-beta 1 suppresses autoantigen-induced expression of pro-inflammatory cytokines but not of interleukin-10 in multiple sclerosis and myasthenia gravis. J Neuroimmunol. 1995;58:21–35. doi: 10.1016/0165-5728(94)00183-o. [DOI] [PubMed] [Google Scholar]
- 79.Huang YM, Kivisakk P, Ozenci V, Pirskanen R, Link H. Increased levels of circulating acetylcholine receptor (AChR)-reactive IL-10-secreting cells are characteristic for myasthenia gravis (MG) Clinical and experimental immunology. 1999;118:304–8. doi: 10.1046/j.1365-2249.1999.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yi Q, Lefvert AK. Idiotype- and anti-idiotype-reactive T lymphocytes in myasthenia gravis. Evidence for the involvement of different subpopulations of T helper lymphocytes. Journal of immunology. 1994;153:3353–9. [PubMed] [Google Scholar]
- 81.Shimada K, Koh CS, Yanagisawa N. Detection of interleukin-6 in serum and cerebrospinal fluid of patients with neuroimmunological diseases. Arerugi = [Allergy] 1993;42:934–40. [PubMed] [Google Scholar]
- 82.Link J, Fredrikson S, Soderstrom M, Olsson T, Hojeberg B, Ljungdahl A, et al. Organ-specific autoantigens induce transforming growth factor-beta mRNA expression in mononuclear cells in multiple sclerosis and myasthenia gravis. Ann Neurol. 1994;35:197–203. doi: 10.1002/ana.410350211. [DOI] [PubMed] [Google Scholar]
- 83.Kim JY, Yang Y, Moon JS, Lee EY, So SH, Lee HS, et al. Serum BAFF expression in patients with myasthenia gravis. J Neuroimmunol. 2008;199:151–4. doi: 10.1016/j.jneuroim.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 84.Ragheb S, Lisak R, Lewis R, Van Stavern G, Gonzales F, Simon K. A potential role for B-cell activating factor in the pathogenesis of autoimmune myasthenia gravis. Archives of neurology. 2008;65:1358–62. doi: 10.1001/archneur.65.10.1358. [DOI] [PubMed] [Google Scholar]
- 85.Nakano S, Engel AG. Myasthenia gravis: quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology. 1993;43:1167–72. doi: 10.1212/wnl.43.6.1167. [DOI] [PubMed] [Google Scholar]
- 86.Yin W, Allman W, Ouyang S, Li Y, Li J, Christadoss P, et al. The increased expression of CD21 on AchR specified B cells in patients with myasthenia gravis. J Neuroimmunol. 2013;256:49–54. doi: 10.1016/j.jneuroim.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 87.Selcen D, Fukuda T, Shen XM, Engel AG. Are MuSK antibodies the primary cause of myasthenic symptoms? Neurology. 2004;62:1945–50. doi: 10.1212/01.wnl.0000128048.23930.1d. [DOI] [PubMed] [Google Scholar]
- 88.Schluep M, Willcox N, Vincent A, Dhoot GK, Newsom-Davis J. Acetylcholine receptors in human thymic myoid cells in situ: an immunohistological study. Ann Neurol. 1987;22:212–22. doi: 10.1002/ana.410220205. [DOI] [PubMed] [Google Scholar]
- 89.Leite MI, Jones M, Strobel P, Marx A, Gold R, Niks E, et al. Myasthenia gravis thymus: complement vulnerability of epithelial and myoid cells, complement attack on them, and correlations with autoantibody status. The American journal of pathology. 2007;171:893–905. doi: 10.2353/ajpath.2007.070240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scadding GK, Vincent A, Newsom-Davis J, Henry K. Acetylcholine receptor antibody synthesis by thymic lymphocytes: correlation with thymic histology. Neurology. 1981;31:935–43. doi: 10.1212/wnl.31.8.935. [DOI] [PubMed] [Google Scholar]
- 91.Guigou V, Emilie D, Berrih-Aknin S, Fumoux F, Fougereau M, Schiff C. Individual germinal centres of myasthenia gravis human thymuses contain polyclonal activated B cells that express all the Vh and Vk families. Clinical and experimental immunology. 1991;83:262–6. doi: 10.1111/j.1365-2249.1991.tb05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet neurology. 2009;8:475–90. doi: 10.1016/S1474-4422(09)70063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Emilie D, Crevon MC, Cohen-Kaminsky S, Peuchmaur M, Devergne O, Berrih-Aknin S, et al. In situ production of interleukins in hyperplastic thymus from myasthenia gravis patients. Human pathology. 1991;22:461–8. doi: 10.1016/0046-8177(91)90132-9. [DOI] [PubMed] [Google Scholar]
- 94.Cohen-Kaminsky S, Delattre RM, Devergne O, Klingel-Schmitt I, Emilie D, Galanaud P, et al. High IL-6 gene expression and production by cultured human thymic epithelial cells from patients with myasthenia gravis. Annals of the New York Academy of Sciences. 1993;681:97–9. doi: 10.1111/j.1749-6632.1993.tb22873.x. [DOI] [PubMed] [Google Scholar]
- 95.Cohen-Kaminsky S, Devergne O, Delattre RM, Klingel-Schmitt I, Emilie D, Galanaud P, et al. Interleukin-6 overproduction by cultured thymic epithelial cells from patients with myasthenia gravis is potentially involved in thymic hyperplasia. European cytokine network. 1993;4:121–32. [PubMed] [Google Scholar]
- 96.Aime C, Cohen-Kaminsky S, Berrih-Aknin S. In vitro interleukin-1 (IL-1) production in thymic hyperplasia and thymoma from patients with myasthenia gravis. Journal of clinical immunology. 1991;11:268–78. doi: 10.1007/BF00918185. [DOI] [PubMed] [Google Scholar]
- 97.Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem. 2010;285:7176–86. doi: 10.1074/jbc.M109.047423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Loza MJ, Anderson AS, O'Rourke KS, Wood J, Khan IU. T-cell specific defect in expression of the NTPDase CD39 as a biomarker for lupus. Cell Immunol. 2011;271:110–7. doi: 10.1016/j.cellimm.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hain B, Jordan K, Deschauer M, Zierz S. Successful treatment of MuSK antibody-positive myasthenia gravis with rituximab. Muscle Nerve. 2006;33:575–80. doi: 10.1002/mus.20479. [DOI] [PubMed] [Google Scholar]
- 100.Thakre M, Inshasi J, Marashi M. Rituximab in refractory MuSK antibody myasthenia gravis. J Neurol. 2007;254:968–9. doi: 10.1007/s00415-006-0442-2. [DOI] [PubMed] [Google Scholar]
- 101.Nowak RJ, Dicapua DB, Zebardast N, Goldstein JM. Response of patients with refractory myasthenia gravis to rituximab: a retrospective study. Therapeutic advances in neurological disorders. 2011;4:259–66. doi: 10.1177/1756285611411503. [DOI] [PMC free article] [PubMed] [Google Scholar]