

Abstract
To test the hypothesis that different drivers shape the diversity and biogeography of the total and active bacterial community, we examined the bacterial community composition along two transects, one from the inner Pearl River estuary to the open waters of the South China Sea (SCS) and the other from the Luzon Strait to the SCS basin, using 454 pyrosequencing of the 16S rRNA and 16S rRNA gene (V1-3 regions) and thereby characterizing the active and total bacterial community, respectively. The diversity and biogeographic patterns differed substantially between the active and total bacterial communities. Although the composition of both the total and active bacterial community was strongly correlated with environmental factors and weakly correlated with geographic distance, the active bacterial community displayed higher environmental sensitivity than the total community and particularly a greater distance effect largely caused by the active assemblage from deep waters. The 16S rRNA vs. rDNA relationships indicated that the active bacteria were low in relative abundance in the SCS. This might be due to a high competition between active bacterial taxa as indicated by our community network models. Based on these analyses, we speculate that high competition could cause some dispersal limitation of the active bacterial community resulting in a distinct distance-decay relationship. Altogether, our results indicated that the biogeographic distribution of bacteria in the SCS is the result of both environmental control and distance decay.
Keywords: active vs. total bacterial communities, biogeography, diversity, environmental selection, geographic distance, network interaction
Introduction
Marine microbial communities drive many earth system processes, such as the biogeochemical cycles of carbon, nitrogen and sulphur (Falkowski et al. 2008; Fuhrman 2009), in which prokaryotes play a fundamental role (Azam et al. 1983; Karl 2002). For a better understanding of the primary ecological role played by prokaryotes, the biogeographic distribution of prokaryotes represents a fundamental problem. The most classic dictum about microbial biogeography is ‘everything is everywhere; the environment selects’ (Bass-Becking 1934), although this is often challenged by stochastic neutral models (Hubbell 2001; Sloan et al. 2006). Using high-throughput sequencing methods, there is growing evidence of a long-tailed species abundance curve with a large number of rare species present in most ecosystems (Sogin et al. 2006; Fuhrman 2009; Caporaso et al. 2011; Pedrós-Alió 2012), bolstering the likelihood of ‘everything is everywhere’. As for ‘the environment selects’, most studies find a significant correlation between microbial composition and at least one measured environmental or habitat feature, for example the availability of resources such as nutrients and dissolved organic carbon (DOC), and physical parameters such as temperature and salinity (Winter et al. 2010; Agogué et al. 2011; Campbell et al. 2011).
However, ‘the environment selects’ is not the only factor shaping spatial variability in microbial communities across habitats. The current distributions of microorganisms are actually the result of contemporary selection and historical processes. These historical processes include water mass transport and past selection, which overall generate a negative correlation between compositional similarity and geographic distance (the distance-decay relationship) (Green & Bohannan 2006; Hanson et al. 2012). Stochastic neutral models postulate that water mass transport must interact with dispersal limitation to create a distance-decay pattern (Hutchison & Templeton 1999; Hubbell 2001). With dispersal, variation in composition is spatially autocorrelated, whereas, with some dispersal limitation, the distance-decay relationship is strengthened (Hanson et al. 2012). Thus, the geographic distance effect should be relatively weak in habitats where dispersal is high, such as in coastal estuaries. However, at the same time, selective factors such as salinity and nutrients are often organized in a gradient in an estuary, tending to produce a distance effect on spatial variation in microbial composition.
The South China Sea (SCS) is one of the largest semi-enclosed marginal seas, with its deep basin in the tropical-subtropical western North Pacific (Hu et al. 2000). With large amounts of freshwater and nutrient input from the Pearl River, and oceanic water intruding to the continental shelf, the SCS is characterized by sharp physical and chemical gradients over a small spatial scale (Ning et al. 2004). Its meso- and bathypelagic waters are transported into the basin from the Western Pacific through the Luzon Strait (Liu & Liu 1988). Moreover, there is a basin-scale circulation dominated by both the East Asian monsoon and the Kuroshio (the subtropical western boundary current of the North Pacific) in the SCS, seemingly constraining the transverse exchange between coastal and open waters to some extent (Wang et al. 2003). The SCS is thus an ideal environment for examining how microbial community composition and its control mechanisms vary over space; especially, two gradients are particularly relevant to determine environmental control and distance effects, one from the inner Pearl River estuary to the open waters and the other from the Luzon Strait to the basin.
A previous study reported a distinct bacterioplankton community succession at broad phylogenetic levels (Alpha-, Beta- and Gammaproteobacteria and Cytophaga-like) along a salinity gradient from the inner Pearl River estuary to the open waters (Zhang et al. 2006). There was no significant difference in bacterioplankton community composition from the Pearl River estuary to the basin. Finer phylogenetic levels and higher coverage should be taken into account, however, to determine compositional variations, as the likelihood to detect a distance effect might depend on taxonomic resolution (Horner-Devine et al. 2004; Ramette & Tiedje 2007; Logue & Lindstrom 2008; Hanson et al. 2012). In addition, targeting active microbial populations is more likely to detect a distance-decay relationship as the dispersal processes include not only water flow but also successful establishment in the new location (Futuyma 2005; Hanson et al. 2012). Hence, one relevant question is whether total and active microbial communities exhibit a similar or different diversity and biogeography. We hypothesize that different drivers shape diversity and biogeography of total and active bacterial communities.
High-throughput sequencing methods enable efficient deep molecular sampling of the diverse microbial community across different habitats (Sogin et al. 2006; Huber et al. 2007; Huse et al. 2008; Gilbert et al. 2009). High-throughput rRNA and rDNA sequence analyses were used to determine the active and the resident bacterial taxa (Campbell et al. 2011; Campbell & Kirchman 2013; Hunt et al. 2013). In the present study, we examined the bacterial communities collected from both surface and deep waters along two transects, one from the inner Pearl River estuary to the open water of the SCS and the other from the Luzon Strait to the basin using pyrosequencing of the V1-3 hypervariable region of the small subunit rRNA gene from both DNA and RNA [i.e. complementary DNA (cDNA)]. We specifically compared total and active bacterial communities to test whether – and if so, how – environmental selection and dispersal shapes bacterial communities in the SCS.
Materials and methods
Study sites and sampling
A 462-km transect (the P-A transect) from the inner Pearl River estuary to the open waters and a 476-km transect (the S transect) from the Luzon Strait to the SCS basin (Fig.1) were sampled during a winter research cruise (January 2010). In total, 30 samples were collected from the surface and deep waters. The P1 and P2 sites were located in the inner Pearl River estuary with a depth of ∼8.5 m, where the surface water (0.5 m) was sampled. The A3–A8 and S10–S12 sites were sampled from the surface (5 m) and the oxygen minimum layer (OML), which was the near-bottom layer at sites A3–A7 and at ∼800 m for the A8–S12 sites (Fig.1). S9 is the Southeast Asia Time-Series Study station (SEATS) of the SCS central basin with a depth of 3850 m, at which samples were collected from the vertical profile at ten depths from the surface to 3500 m. Two litre samples for DNA or RNA analyses were filtered through 0.2-μm-pore-size polycarbonate filters (Millipore) at a pressure of <0.03 MPa on board. Samples for RNA extraction were collected within 30 min and stored in 2-mL RNase-free tubes with RNAlater RNA stabilization solution (Ambion). All filters were flash-frozen in liquid nitrogen for 10 min and subsequently stored at −80 °C until DNA or RNA extraction.
Fig 1.
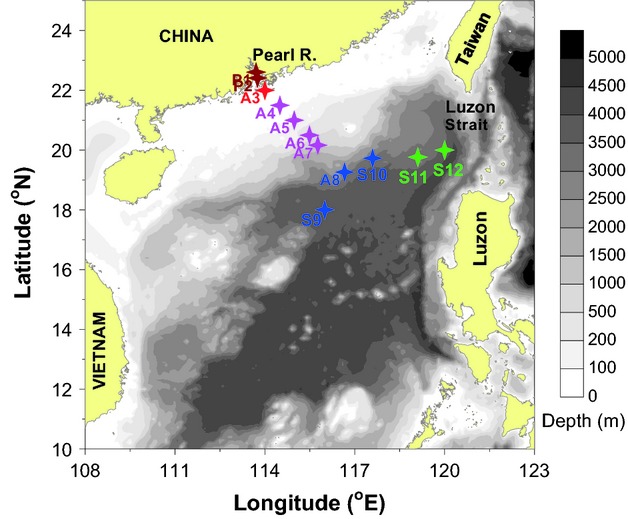
Map of the South China Sea showing sampling stations (coloured stars: consistent colours were used throughout.). Isobaths are used as the background and the grey bar indicates depth.
Biogeochemical analysis
A SeaBird CTD-General Oceanic Rosette sampler with Go-Flo bottles (SBE 9/17 plus, SeaBird Inc., USA) was used to record temperature and salinity and to collect water samples. Samples for inorganic nutrients (nitrate + nitrite, phosphate, silicate) were collected and analysed following Du et al. (2013). Oxygen concentrations were determined on board using the Winkler method (Carpenter 1965). Samples for chlorophyll a (chl a) analysis were collected on 0.7-μm-pore-size GF/F filters (Whatman) and chl a was extracted with 90% acetone in dark. After 24 h of extraction, chl a was determined using a Turner-Designs Trilogy® Laboratory Fluorometer. DOC data were provided by the CHOICE-C project (Dai et al., unpublished).
DNA and RNA extraction, PCR and pyrosequencing
Genomic DNA and RNA were extracted using a MO-BIO UltraClean kit and an RNeasy Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s protocols, respectively. DNA digestion was performed with an RNase-Free DNase Set (Qiagen, Hilden, Germany) during RNA purification. Reverse transcription reactions were performed using the SuperScript RT-PCR system with random hexamers (Invitrogen, Carlsbad, CA, USA) to synthesize first-strand cDNA.
The bacterial hypervariable regions V1–V3 of the 16S rRNA genes were amplified using the primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′), with a unique 10-bp error-correcting Golay barcode at the 5′ end (Zhang et al. 2012). PCRs were carried out in triplicate 25-μL reactions using 20–50 ng of template DNA or cDNA. Negative controls without a template were included for each barcoded primer pair to test for reagent contamination. For PCR with the cDNA template, RNA samples without the RT step were also used as controls to test for residual DNA in the RNA preparations. Thermal cycling consisted of initial denaturation at 94 °C for 3 min followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 45 s, and extension at 72 °C for 45 s, with a final extension at 72 °C for 7 min. Gel-purified amplicon pools were quantified in triplicate with a Quant-iT PicoGreen dsDNA kit (Invitrogen). Equimolar amounts of the PCR amplicons from different samples were mixed and purified via an ethanol precipitation process (Dominguez-Bello et al. 2010). The final concentration of the purified amplicon mixture was determined using a NanoDrop spectrophotometer (ND-2000, Thermo Fisher, Waltham, MA, USA). Pyrosequencing was carried out using a 454 Genome Sequencer GS-FLX Titanium instrument (Roche-454 Life Sciences) at the Chinese National Human Genome Center (Shanghai, China).
Sequence processing
The criteria previously described (Sogin et al. 2006) were used to assess the quality of sequence reads. We eliminated sequences that contained more than one ambiguous nucleotide (N), that did not have a complete barcode and primer at one end, or that were shorter than 150 bp after removal of the barcode and primer sequences. A total of 609 089 reads from the DNA libraries and 232 220 reads from the corresponding RNA libraries passed the pipeline filters. These remaining sequences were assigned to samples by examining the barcode.
Libraries of sequences and operational taxonomic units (OTUs) were analysed in MOTHUR following the standard operating procedure (http://www.mothur.org/wiki/Schloss_SOP) (Schloss et al. 2011). Briefly, sequences were aligned to the silva database, and the resulting alignment was then screened and filtered to ensure that all the sequences overlapped in the same alignment space. To further reduce sequencing errors, the pre.cluster command was used to merge sequence counts that were within 2 bp of a more abundant sequence (Huse et al. 2010). Chimeras were removed using the Chimera Slayer algorithm in MOTHUR (Haas et al. 2011). Finally, classification was carried out using the MOTHUR version of the ‘Bayesian’ classifier with the SILVA reference sequences and taxonomic outline. The confidence cut-off was set to 60%. The sequences that were classified as ‘Cyanobacteria_Chloroplast’ or ‘Mitochondria’ or could not be classified at the kingdom level were removed from the data set. For each sub-data set analysis, all libraries were rarefied to an equal number of sequences prior to downstream analyses using the sub.sample command in MOTHUR. To include all the samples in the analysis, we rarefied the libraries based on the number of sequences of the least deep-sequenced library, which was 4346, 696, 138 and 113 for the heterotrophic bacterial (noncyanobacterial, the same below) DNA- and RNA-based library analysis and cyanobacterial DNA- and RNA-based library analysis, respectively. For the analysis of the latter three sub-data sets, we also tried to rarefy the libraries with 1000 sequences each. Similar results of downstream cluster analysis were obtained although a few samples with a low number of sequences were removed. For the DNA and RNA comparisons of the same sample, all DNA and RNA libraries were analysed as a complete data set in MOTHUR to get a table that indicates the number of times an OTU shows up in each library. The table is called a shared file and can be created using the make.shared command. Then, the DNA and RNA comparisons were based on the frequency of each OTU on the 16S rRNA and rDNA level.
Statistical analysis
Sequences were clustered into OTUs with a cut-off value set at 0.03 for the heterotrophic bacterial assemblage and 0.01 for Cyanobacteria (Melendrez et al. 2011). We chose 0.01 for Cyanobacteria because the 16S rDNA sequence divergence was <3% for Prochlorococcus and Synechococcus strains or genotypes which can be discriminated at a cut-off of 0.01 (Moore et al. 1998). Based on OTU assignment, library richness and diversity estimates (Chao1 and inverse Simpson) were calculated in MOTHUR. Nonmetric multidimensional scaling (NMDS) with two or three dimensions was used to determine the similarity of samples to each other based on thetaYC distance in MOTHUR. Analysis of molecular variance (amova) was applied to test whether the spatial separation of the defined groups visualized in the NMDS plot was statistically significant. Dendrograms relating the similarity in community membership and structure were also generated using MOTHUR with the tree.shared command. The parsimony command was used to test whether the clustering within the tree was statistically significant. Differences were considered significant at P < 0.05. Following the output taxonomy file of classification in MOTHUR, heat maps were created to depict the relative percentage of each classification of bacteria (y axis) within each sample (x axis clustering) with the GENE-E module. Phylogeny-based analyses were also performed using the above similar types of OTU-based analyses in MOTHUR with a phylogenetic tree as input. Unifrac-based metrics were used to assess the similarity between two communities membership (unifrac.unweighted) and structure (unifrac.weighted). After comparing the OTU- and phylogeny-based ordination results, we chose the NMDS based on thetaYC distance due to the stress values below 0.2 and R2 values >0.9.
Canonical correspondence analysis (CCA) was used to further analyse variations in the bacterial assemblages under the constraint of environmental factors with canoco software (Ter-Braak 1989). Stepwise procedure ‘forward selection’ in canoco was used, which added environmental variables one at a time, until no other variables significantly explain residual variation in species composition, namely when the significance level (P) is no longer <0.05. The null hypothesis that the bacterial assemblage was independent of environmental parameters was tested using constrained ordination with a Monte Carlo permutation test (499 permutations; default setting). Significant explanatory parameters (P < 0.05) without multicolinearity (variance inflation factor <20) (Ter Braak 1986) were obtained for the community structure.
Standard and partial Mantel tests were run in R (VEGAN) to determine correlations between environmental factors or geographic distance between sampling sites and community composition (based on the relative abundance of all OTUs). Correlations with environmental factors indicate environmental filtering, while correlations with geographic distance indicate unequal dispersal among sites (Logares et al. 2013). The standard Mantel test estimates the correlation between two matrices, while the partial Mantel test estimates the correlation between two matrices controlling for the effects of a third matrix. For instance, Mantel test statistics R (distance) and R (environment) examine the correlation between bacterial or cyanobacterial community similarity and geographic distance or environmental factors. Partial Mantel statistic R (distance) estimates the correlation between community similarity and geographic distance, while controlling for the effect of environment. Conversely, R (environment) estimates the correlation between community similarity and environmental factors, while controlling for geographic distance. Dissimilarity matrices of communities were based on Bray–Curtis distances between samples. Environmental parameters were normalized using z-score transformation, and Euclidean distances between samples were calculated. Geographic distance was calculated based on the coordinates of the sampling sites using a spheroidal model of earth. The significance of the Mantel statistic was obtained after 1000 permutations. Mantel tests were performed on the data for all samples, samples from the surface and also samples from the OML. The results of the statistical tests were assumed to be significant at P-values ≤ 0.05.
Network analysis
Beyond the basic inventory descriptions of the composition and diversity of bacterial communities, potential interactions between bacterial taxa were determined through modelling the microbial community in network structure to decipher the structure of complex communities across spatial gradients. Only when a community cluster contains more than eight samples/libraries in cluster analysis, it can be analysed as a complete ecological network, and its constituent samples/libraries can be used as replicates. The data sets were uploaded to the open-accessible network analysis pipeline (Molecular Ecological Network Analysis Pipeline, MENAP, http://ieg2.ou.edu/MENA), and the networks were constructed using random matrix theory-based methods (Zhou et al. 2010, 2011; Deng et al. 2012). In a network graph, each node represents an OTU indicating an individual taxon. The edge between each two nodes represents positive or negative interactions between those two taxa.
Results
Diversity analysis
A total of 609 089 reads from the DNA libraries and 232 220 reads from the corresponding RNA libraries (Table S1 Supporting information) passed the quality control pipeline filters. Rarefaction curves of the number of OTUs vs. sampling effort all indicated that we did not exhaustively sample the communities. For downstream analyses, all libraries were divided into heterotrophic bacterial and cyanobacterial sub-data sets. Diversity analysis showed quite variable diversity indices (Table S2 Supporting information) of the heterotrophic bacterial libraries, ranging from 6.21 of the inverse Simpson index at a depth of 30 m at site A3 to 35.96 at 2000 m at site S9 for the DNA-based libraries and from 6.50 to 79.61 in the surface waters of sites A5 and P1, respectively, for the RNA-based libraries. Overall, cyanobacterial diversity was much lower than heterotrophic bacterial diversity, ranging from 1.92 of the inverse Simpson index to 12.91 for the DNA-based libraries and from 1.20 to 5.62 for the RNA-based libraries. Significant correlations between the inverse Simpson indices of the heterotrophic bacterial communities and temperature were observed when the libraries were separated into the upper 100 m and below 200 m depths. The significantly negative correlation (Pearson correlation = −0.674, P < 0.05) between temperature and the inverse Simpson indices of the heterotrophic bacterial DNA-based libraries at depths below 200 m indicated highest species richness in the bathypelagic water at a temperature of 2.4 °C. In contrast, a significantly positive correlation (Pearson correlation = 0.658, P < 0.05) between temperature and the inverse Simpson indices was observed for the same depth zone for the RNA-based libraries although PCR were not successful with the two samples from 2000 to 3500 m, and a negative relationship (Pearson correlation = −0.771, P < 0.01) for the upper 100 m (except for sites P1 and P2).
Spatial variation in bacterial community structure
Cluster analysis based on 2-D NMDS ordination and the unweighted pair group method with arithmetic mean (UPGMA) trees constructed from thetaYC distances (MOTHUR) was performed for the heterotrophic bacterial and cyanobacterial assemblages. The heterotrophic bacterial DNA-based libraries were separated into one cluster containing communities from all surface samples (except for sites P1 and P2), the 50 m sample of site S9 and the bottom water of the sites with a depth <100 m at 51% similarity, and into one cluster of the 200–1000 m water mass at 77% similarity. In addition, communities from the estuarine sites P1 and P2 and from the bathypelagic water (2000 and 35000 m) of site S9 clustered separately at 44% and 59% similarity, respectively (Fig.2a). The chl a maximum layer (75 m) and the bottom of the euphotic zone (100 m) of site S9 were distinct (at 92% similarity) from similar depths in the DNA-based NMDS, but in the RNA-based NMDS they homogenized with similar depths, whereas the A3 site stood out (at 78% similarity) as having different active communities from other sites (Fig.2b). The estuarine sites (P1-2) were consistently distinct from the others, but there was more dispersion in the RNA-based NMDS ordination space for the mesopelagic samples compared with the DNA-based NMDS. The cluster of 100–1000 m in the DNA-based NMDS separated into two subclusters of 100–500 m (off-shore) and 500 (far-shore) to 1000 m at 51% and 30% similarity, respectively, in the RNA-based NMDS. Furthermore, cluster analysis was also performed for the subsets of the communities from the euphotic surface waters (cluster II in the DNA-based NMDS and cluster III in the RNA-based NMDS) when the estuarine and deep-water samples were excluded. The surface samples from sites S11 and 12 clustered separately from the others at 87% similarity in the DNA-based NMDS (Fig.2c). In the RNA-based NMDS, the samples from the surface of site S10 and 75 m of site S9 were distinct from the others, respectively (Fig.2d). Significant differences between pairwise clusters were obtained using the amova test (P < 0.05).
Fig 2.
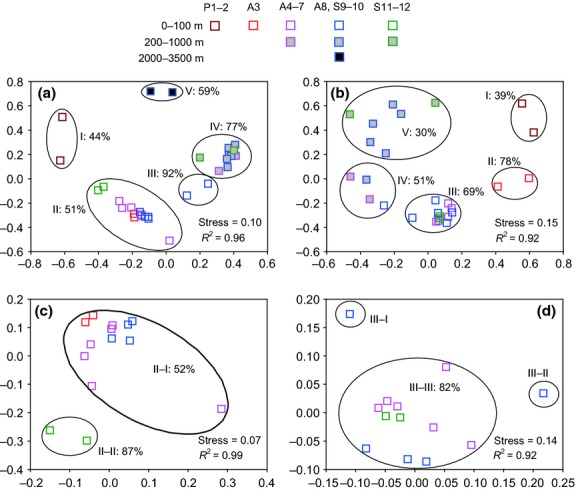
Nonmetric multidimensional scaling (NMDS) ordination with two dimensions based on thetaYC distances between heterotrophic bacterial (noncyanobacterial) DNA- (a: all communities; c: communities form cluster II in a) or RNA-based (b: all communities; d: communities from cluster III in b) communities. Each square represents an individual sample in the NMDS charts. Roman numerals represent cluster serial number. Percentages represent community similarities calculated from thetaYC distance.
Cyanobacterial sequences were retrieved from almost all DNA and RNA samples in the upper 1000 m, but at low abundance in the deep-water samples. A cluster pattern distinctly different from the heterotrophic bacterial assemblage was found in the NMDS and UPGMA tree of the cyanobacterial DNA- and RNA-based libraries. No depth-related distribution pattern of the cyanobacterial assemblages was obtained. Coastal DNA-based communities formed three clusters (Fig.3a), one cluster including communities of the estuarine sites P1 and P2 at 37% similarity, one cluster of site A3 at 81% similarity and one cluster of sites A4 to A7 at 84% similarity, in which open ocean communities from sites S11 and S12 near the Luzon Strait were also included (Fig.3a). Satellite altimetric history indicated that a strong cyclonic eddy was present near the Luzon Strait during our sampling with relatively high chl a concentrations (0.5–0.6 μg L−1 at 5 m). Consequently, episodic input of nutrients from deep water might have shaped the similar cyanobacterial communities at sites S11 and S12, for example highly abundant Synechococcus (Fig.4). Open ocean communities from sites A8, S9 and S10 clustered separately at 47% similarity, together with the bottom community of site A6. The chl a maximum layer (75 m) and the bottom of the euphotic zone (100 m) of site S9 were consistently distinct from adjacent sites in both the DNA- (at 95% similarity) and RNA-based (at 99% similarity) NMDS, as was the A3 site. In addition, the cyanobacterial RNA libraries formed one open ocean cluster including communities from sites A8, S9, S10 and S12 at 81% similarity and one large cluster with coastal and open ocean samples at 40% similarity, in which the estuarine sites P1 and P2 were also included (Fig.3b). Significant differences between pairwise clusters were observed using the amova test (P < 0.05).
Fig 3.
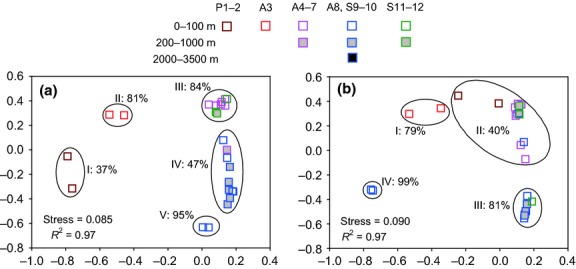
Nonmetric multidimensional scaling (NMDS) ordination with two dimensions based on thetaYC distances between cyanobacterial DNA- (a) or RNA-based (b) communities. Each square represents an individual sample in the NMDS charts. Roman numerals represent cluster serial number. Percentages represent community similarities calculated from thetaYC distance.
Fig 4.

Heat maps showing phylogenetic distribution of bacteria among DNA- (a) or RNA-based (b) samples. Sequences were classified with SILVA reference sequences and taxonomic outline in MOTHUR. The confidence cut-off was set to 60%. The relative percentage of each classification of bacteria (y axis) within each sample (x axis clustering) was shown. Colour bars represent the relative percentage.
Heterogeneity in community composition across spatial gradients
More than 50% of the total OTUs corresponded to Proteobacteria and Cyanobacteria (mainly Synechococcus and Prochlorococcus) in each DNA- or RNA-based library. More than 55% of the total proteobacterial tags were Alphaproteobacteria in the DNA libraries, mostly composed of the SAR11 cluster and Rhodobacteraceae (Fig.4a), while in the RNA libraries 30–89% of the total proteobacterial tags were Alphaproteobacteria, mostly including SAR11, Rhodobacteraceae, SAR116, Methylobacteriaceae, Sphingomonadaceae and Rhodospirillaceae (Fig.4b). Gamma- and Deltaproteobacteria increased with depth in the DNA-based libraries and included a relatively large number of OTUs belonging to the Oceanospirillales and Salinisphaerales, and Nitrospina and the SAR324 cluster. No such trend for the Gammaproteobacteria was found in the corresponding RNA libraries (Fig.4b). Betaproteobacteria were relatively more abundant in the libraries of the inner Pearl River estuary and the deep waters than in other libraries, with most of the OTUs belonging to the Burkholderiaceae. In addition, the Deferribacteres (mainly SAR406), Chloroflexi (mainly SAR202), Planctomycetes, Gemmatimonadetes, Acidobacteria, Firmicutes and Lentisphaerae were also more abundant in the deep water than in the euphotic zone in both DNA- and RNA-based libraries.
Differences in the composition of the total vs. active bacterial community
There was no relationship between the frequency of each OTU on the 16S rRNA and rDNA level (Fig.5). This suggests that the activity of an OTU (frequency on the 16S rRNA level) cannot be estimated based on its relative abundance in the community (frequency on the 16S rDNA). The most abundant taxa (>10%) in the DNA libraries were all below the 1:1 line in Fig.5a, including OTUs in the SAR11 clade, the Actinobacteria Marine group and the Rhodobacteriaceae, whereas the most abundant taxa (>15%) in the RNA libraries were all above the 1:1 line, including OTUs of the Cyanobacteria and Methylobacterium (Fig.5a). In addition, the moderately abundant taxa (5–10%) in the DNA libraries below the 1:1 line included members of the SAR11 and SAR324 clades, the Rhodobacteriaceae, Actinobacteria Marine group and Sporichthyaceae (Fig.5b). The taxa accounting for 5–15% in the RNA libraries above the 1:1 line included members of the Cyanobacteria, Methylobacterium, Erythrobacter, Sphingopyxis, Ralstonia, Pseudomonas, Candidatus Thiobios, Acidimicrobineae, Verrucomicrobia and one SAR11 taxon. In general, low-abundance taxa in the total community had higher 16S rRNA: rDNA ratios, and possibly higher activities, than relatively more abundant taxa.
Fig 5.
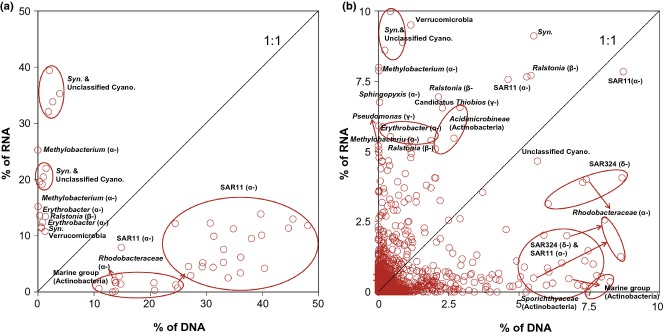
Relationships between 16S rRNA (in RNA libraries) and rDNA (in DNA libraries) frequencies of bacterial OTUs. (a) shows OTUs frequencies more than 10% in either DNA or RNA libraries; (b) shows OTUs frequencies below 10% in both DNA and RNA libraries. Lines are the 1:1 line. Only the taxa of OTUs accounting for more than 5% in either library were shown. Arrows point to the taxa.
Environmental factors explaining spatial variability of the bacterial community
CCA analysis revealed that temperature, salinity, depth, silicate and DOC concentrations were the statistically most significant variables, explaining the pattern of the heterotrophic bacterial community composition based on DNA-based libraries (P < 0.05). Likewise, temperature, salinity, depth, oxygen and chl a concentrations were significant factors determining the composition of the active bacterial community (based on RNA-based libraries; P < 0.05) (Fig.6a and b). However, the CCA models for cyanobacterial assemblages indicated that salinity and chl a concentration, as the statistically significant environmental factors (P < 0.05), were only partly responsible for the community variability over space, yielding low similarity patterns with the NMDS analysis (Fig.6c and d). It appeared that other factors beyond the presently investigated environmental variables might also contribute to the community cluster patterns. In the four CCA models of the heterotrophic bacterial DNA- and RNA-based libraries and the cyanobacterial DNA- and RNA-based libraries, the environmental variables explained approximately 54, 44, 20 and 21% of the total variance in the community composition, respectively. Based on the distance matrix of the significant environmental parameters (without missing values) revealed by the CCA models, Mantel and partial Mantel tests further indicated significant correlations (P < 0.01) between environmental parameters and heterotrophic bacterial community composition (Table1). Moreover, active heterotrophic assemblages displayed tighter correlations to environmental parameters (r = 0.79–0.91) than the total community (r = 0.65–0.82). For cyanobacterial assemblages, environmental factors moderately correlated only with the DNA-based libraries (P < 0.01) but not with the RNA-based libraries (Table1).
Fig 6.
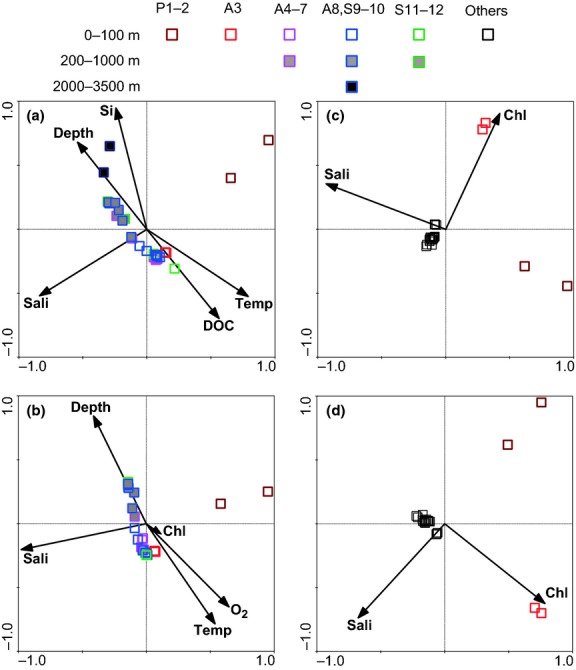
CCA analyses of bacterial communities. (a) Heterotrophic bacterial (noncyanobacterial, the same below) DNA-based libraries; (b) Heterotrophic bacterial RNA-based libraries; (c) Cyanobacterial DNA-based libraries; (d) Cyanobacterial RNA-based libraries. Each square represents an individual sample. Vectors represent statistically significant environmental variables explaining the observed patterns (P < 0.05). Temp: temperature; Sali: salinity; Si: silicate [some of the data are reported in (Du et al. 2013)]; Chl: chlorophyll a; O2: oxygen; DOC: dissolved organic carbon (Dai et al., unpublished).
Table 1.
Mantel and partial Mantel test summary statistics.
| Samples | Mantel | Partial Mantel | ||
|---|---|---|---|---|
| R (distance) | R (environment) | R (distance) | R (environment) | |
| All bacteria | ||||
| DNA | 0.2497** | 0.8228** | 0.217** | 0.8196** |
| RNA | 0.2876** | 0.9136** | 0.2655** | 0.9124** |
| Surface bacteria | ||||
| DNA | 0.3164* | 0.6906** | 0.1548 | 0.6577** |
| RNA | 0.2284 | 0.8313** | −0.0578 | 0.8216** |
| OML bacteria | ||||
| DNA | 0.5072** | 0.7387** | 0.2367 | 0.6501** |
| RNA | 0.6826** | 0.8402** | 0.5595** | 0.7885** |
| All cyanobacteria | ||||
| DNA | 0.4148** | 0.4994** | 0.3191** | 0.4309** |
| RNA | 0.3019** | 0.1556 | 0.2688** | 0.0629 |
The P-values were calculated using the distribution of the Mantel or partial Mantel test statistics estimated from 1000 permutations.
P < 0.05
P < 0.01.
Relation between geographic distance and community composition
Geographic distance between sites correlated with heterotrophic bacterial community composition (standard and partial Mantel tests for all samples; P < 0.01), although the correlation coefficients were much lower than those between environmental properties and community (Table1). However, when samples were divided into surface and OML regions, the changes in community compositions were more strongly related to geographic distance in the OML (r = 0.51–0.68, P < 0.01) than in surface waters (r = 0.32, P < 0.05). Notably, in partial Mantel tests, only the active bacterial community of the OML was related to distance (P < 0.01) (Table1). For cyanobacterial assemblages, geographic distance displayed a moderate correlation with both the DNA- and RNA-based libraries (r = 0.27–0.41, P < 0.01).
Distinct intertaxa relation network for the total vs. active community
Based on the spatial structure of the bacterial community, two large clusters of heterotrophic bacterial communities corresponding to shallow and deep waters (Fig.2a: cluster II and IV; Fig.2b: cluster III and IV + V) were used for ecological network analysis (sample number within each cluster must be ≥8). All curves of network connectivity fitted with the power-law model (R2 > 0.7). Substantial differences were observed in terms of network size and structure between the DNA- and RNA-based libraries (Fig.7; Table S3 and S4 Supporting information). The active bacterial taxa displayed tighter interactions than the total bacterial community as revealed by average path distance and harmonic geodesic distance (Table S3 and S4 Supporting information). Notably, distinctly contrasting interactions between bacterial taxa within the total and active communities were observed. Positive connections dominated the interactions between taxa in the DNA-based networks (Fig. S1a and b Supporting information), whereas negative connections dominated in the RNA-based networks (Fig. S1c and d Supporting information), suggesting more cooperation in the former and more competition in the latter networks.
Fig 7.
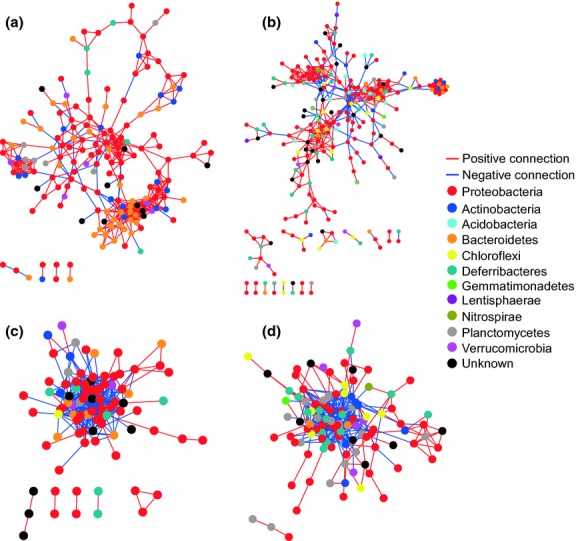
Network interactions of OTUs (force-directed layout). (a) DNA data set of the shallow water; (b) DNA data set of the deep water; (c) RNA data set of the shallow water; (d) RNA data set of the deep water. Each node represents an OTU indicating an individual species. The edge between each two nodes represents positive (red) or negative (blue) interactions between those two species. Colours of the nodes indicate the different major phyla.
Discussion
Total and active bacterial communities displayed different diversity and biogeography
The diversity and biogeographic patterns differed substantially between the total and active bacterial communities. A higher species richness in the bacterial DNA libraries was observed in the deep waters with lower temperature than in the overlying waters, which is consistent with a study in the North Atlantic (Agogué et al. 2011). However, in the RNA libraries, the correlations between species richness and temperature suggested higher richness in the upper mesopelagic waters with a temperature range of 10–15 °C than in the underlying waters. The contrasting trends between the DNA and RNA libraries suggest different mechanisms controlling total and active bacterial community diversity. Active bacterial assemblages are likely to be more dependent on the heterogeneity of available organic substrates potentially introduced by the export of both particulate and dissolved organic matter in combination with the solubilization of sinking particles (Azam & Long 2001; Azam & Malfatti 2007) within the mesopelagic zone. This might result in a greater diversity in the mesopelagic than the euphotic and bathypelagic bacterial communities. However, the DNA libraries in the bathypelagic waters with its high diversity may indicate that these deep waters act as a ‘seed-bank’ of microorganisms. This may be formed by the accumulated genetic material and slow selection under low evolutionary pressure induced by spatially and temporally constant environmental conditions over long timescales in the deep sea (Ghiglione et al. 2012).
Although the SCS is characterized by sharp physical and chemical horizontal gradients over a small spatial scale, space difference in the DNA-based libraries between the coastal and open ocean was much weaker than that in depth gradient. Prominent depth-related clustering of heterotrophic bacterial communities was observed throughout the SCS. The distribution patterns of bacterial phylotypes across spatial gradients reflect the cluster patterns of communities. Pronounced stratification among specific bacterial groups was observed, which is consistent with recent phylogenetic surveys (DeLong 2005; Hewson et al. 2006; Treusch et al. 2009; Galand et al. 2010; Kirchman et al. 2010; Agogué et al. 2011). For instance, the SAR324 (Deltaproteobacteria), SAR406 (Deferribacteres) and SAR202 (Chloroflexi) clusters were relatively abundant in the meso- and bathypelagic waters of the SCS in both DNA and RNA libraries. These groups are reported as typical deep-water clades also in the deep Atlantic and Pacific (Wright et al. 1997; Morris et al. 2004; DeLong et al. 2006; Pham et al. 2008; Agogué et al. 2011). Notably, active heterotrophic bacterial assemblages clustered more pronouncedly than the total communities in both vertically and horizontally according to the CCA analysis. The different biogeographic patterns suggest that the composition of the total heterotrophic bacterial community was shaped by water mass stratification, while active heterotrophic bacterial assemblages are relatively more sensitive to environmental gradients. There was no clear depth-related distribution pattern in the cyanobacterial assemblages. Communities from the same sites basically clustered together, suggesting that the sequences retrieved from the deep waters are largely originating from the upper ocean through sinking or vertical movement of the water mass.
Comparison of the community structure based on OTU percentages between the total and active communities indicates a divergent distribution pattern of individual bacterial taxa. Low-abundant OTUs in the DNA libraries were highly abundant in the RNA libraries, indicating that low-abundant bacteria might be active, such as some Cyanobacteria and Methylobacterium. In contrast, high-abundant bacteria might have low activity, such as most of the SAR11 clades and Rhodobacteriaceae. A similar conclusion was reached in a survey of two lakes and the surface waters off Delaware (Jones & Lennon 2010; Campbell et al. 2011). Overall, the active bacterial assemblages consisting of low-abundance members in the total communities displayed a distinctly different diversity and biogeography than the total communities.
Environmental filtering in the biogeography of total and active bacterial communities
Environmental filters appear to act strongly in shaping diversity and biogeography of total and active heterotrophic bacterial communities in the SCS, but with greater selection pressure on active assemblages as environmental conditions pronouncedly vary in the SCS. As indicated by the cluster pattern of the active heterotrophic bacterial assemblages, the most distinct community difference was present in the estuarine area influenced by freshwater input of nutrients and particles. Another region of rapid changes in bacterial community composition is the upper mesopelagic zone influenced by the input of organic matter from the surface waters and by laterally advected suspended particles from the continental shelf.
The environmental variables, temperature, salinity and depth explained the cluster patterns of total and active heterotrophic bacterial communities (Fig.6a and b). As the main factors discriminating different water masses, this observation supports the notion that water masses may act as dispersal barriers for bacterioplankton (Aristegui et al. 2009; Agogué et al. 2011) contributing to the formation of the spatial structure of communities. Silicate is reported to be one of the most common indicators of river water in the ocean (Moore et al. 1986) and thus significantly contributed to the DNA-based cluster pattern in our study region. In addition, specific environmental properties, such as the concentrations of DOC for the total heterotrophic bacterial community, and the concentrations of O2 and chl a for the active assemblage, also contribute to the presence of distinct bacterial communities in the SCS. It appears that the factors tightly associated with heterotrophic activity, such as chl a as a tracer of labile organic carbon available for those heterotrophic organisms, might be more important for the composition of the active heterotrophic bacterial community than other factors (Fig.6b). Mantel and partial Mantel tests also support the notion that environmental conditions strongly structure the heterotrophic bacterial communities in the SCS, in particular the active assemblages. Compared with the heterotrophic bacterial community, environmental factors play a much weaker role in shaping the structure of cyanobacterial assemblages in the SCS.
Distinct distance effect on the biogeography of total and active bacterial communities
Overall, geographic distance between sites displayed weak (low r-value), but significant (P < 0.05) correlations with total and active bacterial community composition. However, the relation between geographic distance and heterotrophic bacterial community composition substantially differed between the surface and deep (OML) layers. A tighter relation was found between geographic distance and the OML communities than in surface waters (Table1), however, only for the RNA-based OML communities. This suggests that the active heterotrophic bacterial community in deep waters might be more constrained by water mass transport and dispersal processes than the surface community. The active deep-water bacterial community tends to produce a geographic distance effect by spatial variation in microbial composition (Ghiglione et al. 2012). In contrast, the surface community composition is driven by modern environmental selection as partial Mantel tests revealed statistically significant correlations (P < 0.01) with environmental traits rather than with geographic distance. Compared with the heterotrophic bacterial community, geographic distance played a slightly stronger role in shaping the cyanobacterial assemblage structure in the SCS. Based on the above analysis, we found that the biogeography of the total bacterial community based on DNA analysis was driven by strong environmental selection and weak geographic distance, in which the active heterotrophic bacterial assemblage based on RNA analysis displayed higher environmental sensitivity and particularly a greater geographic distance effect than the total community. We speculate that a high dispersal induces a relatively weak distance effect on spatial variation in the composition of the DNA-based community in the hydrologically dynamic SCS. However, some species might fail to establish in the new location to which they were introduced through dispersal, resulting in a relatively distinct distance-decay relationship in the RNA-based active heterotrophic bacterial community.
High competition between active bacterial taxa causing dispersal limitation
What could induce dispersal limitations and thereby not allowing some species to successfully establish themselves in a new location moved there through water mass transport? Functional traits of species, such as survival strategy, limited adaptive ability towards the environment and interaction with other species could be the main causes. Community network models were constructed to compare intertaxa interactions within the total and active community. These interactions could be competition or cooperation based on nutrients, material, information and space (Deng et al. 2012). We found that community network size and structure differed substantially between the total and active communities and distinct interactions between bacterial taxa dominated total and active communities. Positive interactions within the total community may be the result of mutualism between species in long-term co-evolution processes, whereas negative interactions within the active community hint to an intertaxa competition for the same resource (e.g. food or living space) or feed on the same prey (Montoya et al. 2006; Bascompte 2007). This competitive interaction could explain why active phylotypes are low in abundance in the SCS as indicated in Fig.5. The competition for resources is also consistent with the above CCA analysis (Fig.6), indicating that the factors tightly associated with heterotrophic activity, such as chl a (here used as a tracer of labile organic carbon available for heterotrophic organisms), might be more crucial for the composition of the active community than other factors. Based on this network analysis, the active phylotypes of the SCS are assumed to be impaired by high competition with each other and thus be less abundant and even could not successfully inhabit a new location to which they are introduced through water mass transport.
Conclusions
Diversity and biogeographic patterns differed substantially between the total and active bacterial communities in the SCS. Our results suggest that environmental filters act strongly on the heterotrophic bacterial community in the SCS, particularly with greater selection for active assemblages, whereas they relatively weakly affect cyanobacterial community structure. Geographic distance between sites weakly affected total bacterial community composition (including Cyanobacteria), but greatly impacted the active heterotrophic bacterial community of deep waters and the active cyanobacterial community. Thus, our hypothesis that different drivers shape diversity and biogeography of total and active bacterial communities was verified. Taken together, the community structure, its correlations with environmental traits and geographic distance and community network models further indicate that in the SCS, the active bacteria experience high competition among each other and thus are low in abundance. This might cause dispersal limitation in that some species might be unsuccessful in becoming established in the new location to which they were introduced through water mass transport. Therefore, the active assemblages exhibited a relatively distinct distance-decay relationship. This study highlights the importance of examining both bacterial 16S rRNA and 16S rDNA to understand community biogeography and diversity and its ecological driving forces. Although the results supported our hypothesis, further experiments are necessary, for example in other ecosystems, to determine the relationships between environmental conditions, geographic distance and community composition for a better understanding of global microbial biogeography.
Acknowledgments
We thank Professor J. Gan, the chief scientist of the PRE cruise, for providing the sampling opportunity and the temperature and salinity data, Professor J. Hu for the temperature and salinity data from the SCS cruise, and Professor J. Sun for the chl a data. Dr. Y. Deng is thanked for his assistance with network analysis. This research was funded by 973 program 2013CB955700, the NSFC projects 41176095, 91028001, 41121091 and 41023007, the NSFF project 2012J01182 and the projects GASI-03-01-02-03 and GASI-03-01-02-05. The cruises were supported by 973 CHOICE-C 2009CB421200.
Data accessibility
DNA and cDNA sequences: NCBI SRA: SRP026056.
Analysis input files for programs, the OTU tables and environmental data: Dryad doi:10.5061/dryad.s8c2c.
Y.Z. provided funding for research, performed research, analysed data and wrote the manuscript. Z.Z. performed research and data analyses. M.D. provided research cruises and background data, interpreted data and edited the manuscript. N.J. provided funding for research, laboratory equipment and space for research to be conducted, interpreted data and edited the manuscript. G.J.H. interpreted data and edited the manuscript.
Supporting Information
Additional supporting information may be found in the online version of this article.
Table S1 Numbers of clean sequences passing the quality control pipeline filters for each DNA and corresponding RNA pool.
Table S2 The inverse Simpson index of heterotrophic bacterial (HeteroBac) and cyanobacterial libraries (Cyano) for each DNA and corresponding RNA pool.
Table S3 Network global properties of DNA-based heterotrophic bacterial communities.
Table S4 Network global properties of the RNA-based heterotrophic bacterial communities.
Fig. S1 Network interactions of OTUs (circular layout). (a) DNA data set of the shallow water; (b) DNA data set of the deep water; (c) RNA data set of the shallow water; (d) RNA data set of the deep water. Each node represents an OTU indicating an individual species. The edge between each two nodes represents positive (red) or negative (blue) interactions between those two species. Colours of the nodes indicate different major phyla.
References
- Agogué H, Lamy D, Neal PR, Sogin ML, Herndl GJ. Water mass-specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Molecular Ecology. 2011;20:258–274. doi: 10.1111/j.1365-294X.2010.04932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aristegui J, Gasol JM, Duarte CM, Herndl G. Microbial oceanography of the dark ocean’s pelagic realm. Limnology and Oceanography. 2009;54:1501–1529. [Google Scholar]
- Azam F, Long RA. Oceanography: Sea snow microcosms. Nature. 2001;414:495–498. doi: 10.1038/35107174. [DOI] [PubMed] [Google Scholar]
- Azam F, Malfatti F. Microbial structuring of marine ecosystems. Nature Reviews Microbiology. 2007;5:782–791. doi: 10.1038/nrmicro1747. [DOI] [PubMed] [Google Scholar]
- Azam F, Fenchel T, Field J, Gray J, Meyer-Reil L, Thingstad F. The ecological role of water-column microbes in the sea. Marine Ecology Progress Series. 1983;10:257–263. [Google Scholar]
- Bascompte J. Networks in ecology. Basic and Applied Ecology. 2007;8:485–490. [Google Scholar]
- Bass-Becking LGM. Geobiologie of Inleiding tot de Milieukunde. The Hague, the Netherlands: WP Van Stockum & Zoon; 1934. [Google Scholar]
- Campbell BJ, Kirchman DL. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME Journal. 2013;7:210–220. doi: 10.1038/ismej.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BJ, Yu L, Heidelberg JF, Kirchman DL. Activity of abundant and rare bacteria in a coastal ocean. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12776–12781. doi: 10.1073/pnas.1101405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter JH. The Chesapeake Bay Institute technique for the Winkler dissolved oxygen method. Limnology and Oceanography. 1965;10:141–143. [Google Scholar]
- DeLong EF. Microbial community genomics in the ocean. Nature Reviews Microbiology. 2005;3:459–469. doi: 10.1038/nrmicro1158. [DOI] [PubMed] [Google Scholar]
- DeLong EF, Preston CM, Mincer T, et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science. 2006;311:496–503. doi: 10.1126/science.1120250. [DOI] [PubMed] [Google Scholar]
- Deng Y, Jiang Y-H, Yang Y, He Z, Luo F, Zhou J. Molecular ecological network analyses. BMC Bioinformatics. 2012;13:113. doi: 10.1186/1471-2105-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Liu Z, Dai M, et al. Impact of the Kuroshio intrusion on the nutrient inventory in the upper northern South China Sea: insights from an isopycnal mixing model. Biogeosciences Discussions. 2013;10:6939–6972. [Google Scholar]
- Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth’s biogeochemical cycles. Science. 2008;320:1034–1039. doi: 10.1126/science.1153213. [DOI] [PubMed] [Google Scholar]
- Fuhrman JA. Microbial community structure and its functional implications. Nature. 2009;459:193–199. doi: 10.1038/nature08058. [DOI] [PubMed] [Google Scholar]
- Futuyma DJ. Evolution. Sunderland, MA: Sinauer Associates Inc; 2005. [Google Scholar]
- Galand PE, Potvin M, Casamayor EO, Lovejoy C. Hydrography shapes bacterial biogeography of the deep Arctic Ocean. ISME Journal. 2010;4:564–576. doi: 10.1038/ismej.2009.134. [DOI] [PubMed] [Google Scholar]
- Ghiglione J-F, Galand PE, Pommier T, et al. Pole-to-pole biogeography of surface and deep marine bacterial communities. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17633–17638. doi: 10.1073/pnas.1208160109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JA, Field D, Swift P, et al. The seasonal structure of microbial communities in the Western English Channel. Environmental Microbiology. 2009;11:3132–3139. doi: 10.1111/j.1462-2920.2009.02017.x. [DOI] [PubMed] [Google Scholar]
- Green J, Bohannan BJ. Spatial scaling of microbial biodiversity. Trends in Ecology and Evolution. 2006;21:501–507. doi: 10.1016/j.tree.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Gevers D, Earl AM, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JB. Beyond biogeographic patterns: processes shaping the microbial landscape. Nature Reviews Microbiology. 2012;10:497–506. doi: 10.1038/nrmicro2795. [DOI] [PubMed] [Google Scholar]
- Hewson I, Steele JA, Capone DG, Fuhrman JA. Remarkable heterogeneity in meso- and bathypelagic bacterioplankton assemblage composition. Limnology and Oceanography. 2006;51:1274–1283. [Google Scholar]
- Horner-Devine MC, Lage M, Hughes JB, Bohannan BJ. A taxa–area relationship for bacteria. Nature. 2004;432:750–753. doi: 10.1038/nature03073. [DOI] [PubMed] [Google Scholar]
- Hu J, Kawamura H, Hong H, Qi Y. A review on the currents in the South China Sea: seasonal circulation, South China Sea warm current and Kuroshio intrusion. Journal of Oceanography. 2000;56:607–624. [Google Scholar]
- Hubbell SP. The Theoretical Derivation of Neutral Theory as Applied to Multispecies Communities. Princeton, NJ: Princeton University Press; 2001. [Google Scholar]
- Huber JA, Welch DBM, Morrison HG, et al. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- Hunt DE, Lin Y, Church MJ, et al. Relationship between abundance and specific activity of bacterioplankton in open ocean surface waters. Applied And Environmental Microbiology. 2013;79:177–184. doi: 10.1128/AEM.02155-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genetics. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environmental Microbiology. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison DW, Templeton AR. Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution, the International Journal of Organic Evolution. 1999;53:1898–1914. doi: 10.1111/j.1558-5646.1999.tb04571.x. [DOI] [PubMed] [Google Scholar]
- Jones SE, Lennon JT. Dormancy contributes to the maintenance of microbial diversity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5881–5886. doi: 10.1073/pnas.0912765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl DM. Nutrient dynamics in the deep blue sea. Trends In Microbiology. 2002;10:410–418. doi: 10.1016/s0966-842x(02)02430-7. [DOI] [PubMed] [Google Scholar]
- Kirchman DL, Cottrell MT, Lovejoy C. The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environmental Microbiology. 2010;12:1132–1143. doi: 10.1111/j.1462-2920.2010.02154.x. [DOI] [PubMed] [Google Scholar]
- Liu C-T, Liu R-J. The deep current in the Bashi Channel. Acta Oceanographica Taiwanica. 1988;20:107–116. [Google Scholar]
- Logares R, Lindström ES, Langenheder S, et al. Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME Journal. 2013;7:937–948. doi: 10.1038/ismej.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue JB, Lindstrom ES. Biogeography of bacterioplankton in inland waters. Freshwater Reviews. 2008;1:99–114. [Google Scholar]
- Melendrez MC, Lange RK, Cohan FM, Ward DM. Influence of molecular resolution on sequence-based discovery of ecological diversity among Synechococcus populations in an alkaline siliceous hot spring microbial mat. Applied and Environmental Microbiology. 2011;77:1359–1367. doi: 10.1128/AEM.02032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya JM, Pimm SL, Solé RV. Ecological networks and their fragility. Nature. 2006;442:259–264. doi: 10.1038/nature04927. [DOI] [PubMed] [Google Scholar]
- Moore WS, Sarmiento JL, Key RM. Tracing the Amazon component of surface Atlantic water using 228Ra, salinity and silica. Journal of Geophysical Research. 1986;91:2574–2580. [Google Scholar]
- Moore LR, Rocap G, Chisholm SW. Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature. 1998;393:464–467. doi: 10.1038/30965. [DOI] [PubMed] [Google Scholar]
- Morris R, Rappe M, Urbach E, Connon S, Giovannoni S. Prevalence of the Chloroflexi-related SAR202 bacterioplankton cluster throughout the mesopelagic zone and deep ocean. Applied and Environmental Microbiology. 2004;70:2836–2842. doi: 10.1128/AEM.70.5.2836-2842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning X-r, Chai F, Xue H, Cai Y, Liu C, Shi J. Physical-biological oceanographic coupling influencing phytoplankton and primary production in the South China Sea. Journal of Geophysical Research. 2004;109:C10005. [Google Scholar]
- Pedrós-Alió C. The rare bacterial biosphere. The Annual Review of Marine Science. 2012;4:449–466. doi: 10.1146/annurev-marine-120710-100948. [DOI] [PubMed] [Google Scholar]
- Pham VD, Konstantinidis KT, Palden T, DeLong EF. Phylogenetic analyses of ribosomal DNA-containing bacterioplankton genome fragments from a 4000 m vertical profile in the North Pacific Subtropical Gyre. Environmental Microbiology. 2008;10:2313–2330. doi: 10.1111/j.1462-2920.2008.01657.x. [DOI] [PubMed] [Google Scholar]
- Ramette A, Tiedje JM. Biogeography: an emerging cornerstone for understanding prokaryotic diversity, ecology, and evolution. Microbial Ecology. 2007;53:197–207. doi: 10.1007/s00248-005-5010-2. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE. 2011;6:e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, Curtis TP. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environmental Microbiology. 2006;8:732–740. doi: 10.1111/j.1462-2920.2005.00956.x. [DOI] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Braak CJF. Canonical correspondence analysis: a new eigenvector technique for multivariate direct gradient analysis. Ecology. 1986;67:1167–1179. [Google Scholar]
- Ter-Braak CJF. CANOCO - an extension of DECORANA to analyze species-environment relationships. Hydrobiologia. 1989;184:169–170. [Google Scholar]
- Treusch AH, Vergin KL, Finlay LA, et al. Seasonality and vertical structure of microbial communities in an ocean gyre. ISME Journal. 2009;3:1148–1163. doi: 10.1038/ismej.2009.60. [DOI] [PubMed] [Google Scholar]
- Wang D, Wang W, Shi P, Guo P, Gan Z. Establishment and adjustment of monsoon-driven circulation in the South China Sea. Science in China Series D: Earth Sciences. 2003;46:173–181. [Google Scholar]
- Winter C, Bouvier T, Weinbauer MG, Thingstad TF. Trade-offs between competition and defense specialists among unicellular planktonic organisms: the “killing the winner” hypothesis revisited. Microbiology and Molecular Biology Reviews. 2010;74:42–57. doi: 10.1128/MMBR.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright TD, Vergin KL, Boyd PW, Giovannoni SJ. A novel delta-subdivision proteobacterial lineage from the lower ocean surface layer. Applied and Environmental Microbiology. 1997;63:1441–1448. doi: 10.1128/aem.63.4.1441-1448.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jiao N, Cottrell MT, Kirchman DL. Contribution of major bacterial groups to bacterial biomass production along a salinity gradient in the South China Sea. Aquatic Microbial Ecology. 2006;43:233–241. [Google Scholar]
- Zhang Y, Zhao Z, Chen C-TA, Tang K, Su J, Jiao N. Sulfur metabolizing microbes dominate microbial communities in andesite-hosted shallow-Sea hydrothermal systems. PLoS ONE. 2012;7:e44593. doi: 10.1371/journal.pone.0044593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Deng Y, Luo F, He Z, Tu Q, Zhi X. Functional molecular ecological networks. MBio. 2010;1:e00169–00110. doi: 10.1128/mBio.00169-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Deng Y, Luo F, He Z, Yang Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. MBio. 2011;2:e00122–00111. doi: 10.1128/mBio.00122-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Numbers of clean sequences passing the quality control pipeline filters for each DNA and corresponding RNA pool.
Table S2 The inverse Simpson index of heterotrophic bacterial (HeteroBac) and cyanobacterial libraries (Cyano) for each DNA and corresponding RNA pool.
Table S3 Network global properties of DNA-based heterotrophic bacterial communities.
Table S4 Network global properties of the RNA-based heterotrophic bacterial communities.
Fig. S1 Network interactions of OTUs (circular layout). (a) DNA data set of the shallow water; (b) DNA data set of the deep water; (c) RNA data set of the shallow water; (d) RNA data set of the deep water. Each node represents an OTU indicating an individual species. The edge between each two nodes represents positive (red) or negative (blue) interactions between those two species. Colours of the nodes indicate different major phyla.
Data Availability Statement
DNA and cDNA sequences: NCBI SRA: SRP026056.
Analysis input files for programs, the OTU tables and environmental data: Dryad doi:10.5061/dryad.s8c2c.