

The muscular dystrophy protein dysferlin plays a key role in the calcium-activated vesicle fusion of membrane repair. This study establishes calpains as upstream regulators of dysferlin in the membrane repair cascade and further demonstrates that similar C-terminal modules are enzymatically released from other ferlin family members.
Abstract
Dysferlin and calpain are important mediators of the emergency response to repair plasma membrane injury. Our previous research revealed that membrane injury induces cleavage of dysferlin to release a synaptotagmin-like C-terminal module we termed mini-dysferlinC72. Here we show that injury-activated cleavage of dysferlin is mediated by the ubiquitous calpains via a cleavage motif encoded by alternately spliced exon 40a. An exon 40a–specific antibody recognizing cleaved mini-dysferlinC72 intensely labels the circumference of injury sites, supporting a key role for dysferlinExon40a isoforms in membrane repair and consistent with our evidence suggesting that the calpain-cleaved C-terminal module is the form specifically recruited to injury sites. Calpain cleavage of dysferlin is a ubiquitous response to membrane injury in multiple cell lineages and occurs independently of the membrane repair protein MG53. Our study links calpain and dysferlin in the calcium-activated vesicle fusion of membrane repair, placing calpains as upstream mediators of a membrane repair cascade that elicits cleaved dysferlin as an effector. Of importance, we reveal that myoferlin and otoferlin are also cleaved enzymatically to release similar C-terminal modules, bearing two C2 domains and a transmembrane domain. Evolutionary preservation of this feature highlights its functional importance and suggests that this highly conserved C-terminal region of ferlins represents a functionally specialized vesicle fusion module.
INTRODUCTION
In 1998, dysferlin was identified as the genetic cause of recessive limb-girdle muscular dystrophy type 2B (Bashir et al., 1998; Liu et al., 1998). A mouse model of dysferlin deficiency subsequently revealed that dysferlin was required for the calcium-dependent membrane resealing of injured myofibers (Bansal et al., 2003), a process with parallels to synaptic exocytosis and understood to involve calcium-activated vesicle fusion to reseal a breach in the plasma membrane (Steinhardt et al., 1994). Dysferlin contains seven C2 domains (Lek et al., 2010), independently folding protein motifs responsible for calcium sensing, lipid binding, and vesicle fusion in synaptotagmins, the classical mediators of calcium-activated vesicle fusion (Rizo and Südhof, 1998; Shin et al., 2009).
Ferlin family proteins also have identified roles in calcium-regulated vesicle fusion. The Drosophila ferlin, misfire, is required for calcium-dependent vesicular breakdown of the sperm plasma membrane postfertilization (acrosomal exocytosis; Ohsako et al., 2003; Smith and Wakimoto, 2007). Caenorhabditis elegans Fer-1 mutants are infertile due to a defect in calcium-activated fusion of a specialized membranous organelle during spermatogenesis (Achanzar and Ward, 1997; Washington and Ward, 2006). Human otoferlin mutations cause deafness due to a defect in calcium-activated exocytosis of neurotransmitter-containing vesicles at the specialized ribbon synapse of cochlear inner hair cells (Roux et al., 2006). Given the established role of ferlins in calcium-activated vesicle fusion, it is proposed that dysferlin may function as a specialized calcium trigger for the vesicle fusion of membrane repair, a process particularly important for skeletal muscle that is regularly subject to stretch-induced membrane injury (McNeil and Khakee, 1992).
Mitsugumin-53 (MG53, also known as TRIM72) is a TRIM-family (tripartite motif) E3 ubiquitin ligase that also plays a key role in muscle membrane repair (Cai et al., 2009a). Although MG53 has not been implicated in human neuromuscular disease, a mouse knockout model displays a mild muscular dystrophy characterized by defective myofiber membrane resealing (Cai et al., 2009a). Recently we demonstrated that MG53 and dysferlin are rapidly recruited to sites of ballistics injury in cultured human myotubes, forming a lattice-like network that initially specifically labels the exposed edges of the membrane lesion and then expands as the lesion is filled in and resealed (Lek et al., 2013). Intriguingly, we were able to immunodetect only the C-terminus of dysferlin at sites of membrane injury, with three central region or N-terminal antibodies showing negative staining at injury sites (Lek et al., 2013). Western blot analyses subsequently revealed that membrane injury induced cleavage of dysferlin, releasing a C-terminal fragment we termed mini-dysferlinC72 and an N-terminal counterfragment. Cleavage of dysferlin was calcium dependent, occurred most efficiently at neutral pH, and was inhibited by the calpain inhibitor calpeptin, alluding to a role for calpains in dysferlin cleavage with membrane injury (Lek et al., 2013).
Of interest, the C-terminal module released by calpain cleavage of dysferlin, mini-dysferlinC72, bears structural resemblance to a synaptotagmin, with two cytoplasmic C2 domains anchored by a transmembrane domain. Moreover, our previous phylogenetic studies reveal the two most C-terminal C2 domains are the most evolutionarily conserved within the ferlin family tree, with ∼90% amino acid identity between human and mollusk C2E and C2F domains (Lek et al., 2010), suggesting a core and preserved function. Thus our collective results suggest that in the emergency setting of a membrane injury, where intracellular calcium levels rise to acutely high levels within the injury zone, calpain cleavage of dysferlin may provide a means to release a synaptotagmin-like module that plays a specialized role in the vesicle fusion of membrane repair.
The involvement of calpains for membrane repair was first noted in transected giant squid axons that failed to reseal a severed neurite in the presence of the potent calpain inhibitor calpeptin (Xie and Barrett, 1991). Later studies reproduced these findings and further demonstrated that addition of exogenous calpain enhanced resealing of a transected axon and indeed could facilitate resealing even in the absence of calcium (Godell et al., 1997). Studies in various mammalian cell types also highlight a vital role for the ubiquitous calpains-1 and -2 in acute resealing of a plasma membrane injury. Genetic knockdown of calpain-1 and -2 significantly impairs calcium-dependent membrane resealing after injury, as does treatment with calpain inhibitors (Mellgren et al., 2007, 2009; Taneike et al., 2011).
The role of calpain activity in membrane repair might be expected to involve focal adhesion and cytoskeletal remodeling, cutting anchors to allow dynamic microtubule and actomyosin networks to deliver and remove vesicles from the wound site. However, electron microscopy of transected neurites revealed that calpain inhibition did not block vesicle formation or recruitment to the injury site but instead showed a failure of vesicle fusion to restore a dye-impermeable barrier at the severed end (Godell et al., 1997). Thus how calpains regulate the calcium-activated vesicle fusion of membrane repair has remained unclear.
Our study provides a direct link between calpain and dysferlin in the calcium-activated vesicle fusion of membrane repair. We show that dysferlin bears a calpain cleavage site within alternately spliced exon 40a and is specifically cleaved in the setting of a membrane injury in response to high local intracellular calcium. Of importance, we demonstrate that other members of the ferlin family can also be cleaved enzymatically to release similar synaptotagmin-like modules, bearing two C2 domains anchored by their transmembrane domain. Our results suggest a novel functional paradigm for the ferlin family by which calcium signaling activates the enzymatic cleavage of ferlins to release a synaptotagmin-like module. The preservation of this property among different ferlin paralogues suggests that it results in an important modification to their function.
RESULTS
Cleavage of dysferlin to form mini-dysferlin is conferred by exon 40a
In this study, we aimed to identify the mechanisms controlling the injury-activated cleavage of dysferlin and the release of the mini-dysferlinC72 synaptotagmin-like module. Molecular weight calculations of mini-dysferlinC72 (72 kDa) predict that the cleavage site lies between exons 40 and 41, between C2DE and C2E within the dysferlin protein domain structure (Figure 1, A and B). An alternatively spliced exon, exon 40a, occurs between exons 40 and 41. In silico analysis of human dysferlin exon 40a (Liu et al., 2011; ccd.biocuckoo.org), together with a number of dysferlin orthologues, revealed the presence of a putative calpain cleavage site within this alternatively spliced exon, with the highest likelihood of cleavage occurring at the LAPTNTA–SPPSSPH junction in each case (Figure 1C).
FIGURE 1:

The calpain cleavage site in dysferlin is predicted to reside in exon 40a. (A) The apparent molecular weight of mini-dysferlinC72 (72 kDa) predicts that cleavage of dysferlin occurs between exons 40 and 41, between C2DE and C2E. (B) Exon 40a bears a consensus site for calpain cleavage (GPS-CCD, ccd.biocuckoo.org; Liu et al., 2011). (C) Alignment of exon 40a between dysferlin paralogues reveals only moderate preservation of amino acid sequence. However, exon 40a sequences in all species possess a putative calpain cleavage site, in each case with maximum likelihood of cleavage LAPTNTA–SPPSSPH.
To examine whether cleavage of dysferlin is conferred by exon 40a, a region flanking exon 40a was PCR amplified from human skeletal muscle and subcloned into our dysferlin expression construct lacking exon 40a. Human embryonic kidney 293 (HEK293) cells, which do not express significant levels of endogenous dysferlin, were transfected with dysferlin expression constructs with and without exon 40a and then subjected to membrane injury via cell scraping in the presence or absence of extracellular calcium. Cell injury induced the calcium-dependent cleavage of dysferlin to release C-terminal mini-dysferlinC72 (Figure 2A, left) and the N-terminal counterfragment (Figure 2A, middle) only in cells transfected with the dysferlin expression construct bearing exon 40a. HEK293 cells expressing the canonical skeletal muscle isoform of dysferlin (without exon 5a, with exon 17, and without exon 40a) did not show injury-activated, calcium-dependent cleavage of dysferlin. We also established that the cleaved mini-dysferlinC72 product bears the extreme luminal/extracellular domain by probing a triplicate membrane with anti-Myc (Figure 2A, right).
FIGURE 2:

Cleavage of dysferlin to form mini-dysferlinC72 is conferred by exon 40a. (A) Untransfected HEK293 cells, as well as HEK293 transfected with dysferlin expression constructs with (+40a) or without exon 40a, were subjected to scrape injury 24 h posttransfection in the presence or absence of calcium. Only dysferlin expression constructs bearing exon 40a demonstrate injury-activated, calcium-dependent formation of the C-terminal mini-dysferlinC72 fragment (lane 6, Hamlet-1 and anti-Myc, black arrows). The N-terminal counterfragment can be detected with Romeo-1 (lane 6, gray arrow). Membranes were reprobed for loading controls GAPDH and β-tubulin. (B) Ubiquitous calpains specifically cleave exon 40a–containing dysferlin. MEFs were transfected by electroporation with dysferlin expression constructs with or without exon 40a and harvested 24 h posttransfection via scrape injury in the presence of calcium. Injury-activated formation of mini-dysferlinC72 requires exon 40a and is observed in wild-type MEFs (WT) but not in MEFs from CAPNS1-knockout mice (−/−) deficient for calpain-1 and -2. Retroviral rescue of CAPNS1 in knockout (−/−R) MEFs restores calpain expression (see CAPN2 immunoblot) to levels exceeding that in WT cells and increases injury-induced dysferlin cleavage. Mini-dysferlinC72 is indicated with asterisks. (C) Dysferlin bearing exon 40a is specifically cleaved by either calpain-1 or -2 in vitro, forming mini-dysferlinC72. Enhanced GFP–dysferlinFLAG was immunoprecipitated with anti-dysferlin (Romeo) and protein G–Sepharose (see Materials and Methods). Sepharose beads were incubated with the indicated dilutions of purified recombinant calpain-1 or -2 at 30°C for 3 min in the presence of 2 mM CaCl2. Digested samples were analyzed by SDS–PAGE and Western blot. Dysferlin bearing exon 40a is specifically cleaved by both calpain-1 and -2 to form mini-dysferlinC72 (black arrowhead), whereas dysferlin without exon 40a remains uncleaved.
Calpains cleave dysferlin within exon 40a to form mini-dysferlinC72
Our previous study established that cleavage of dysferlin is calcium dependent and blocked by treatment with the calpain inhibitor, calpeptin. Calpeptin also exerts inhibitory effects upon the lysosomal cysteine proteases, cathepsins. Thus we used mouse embryonic fibroblasts with CAPNS1 (also known as CAPN4) genetically ablated (Tan et al., 2006) to establish specificity for calpains in the injury-activated cleavage of dysferlin. The ubiquitously expressed conventional calpain-1 and -2 isoforms are heterodimers consisting of the common CAPNS1-encoded regulatory subunit and either a CAPN1- or a CAPN2-encoded catalytic subunit, respectively. CAPNS1 is required for stability and proteolytic activity of the calpain-1 and -2 isoforms. Knockout of CAPNS1 results in complete ablation of calpain-1 and -2 proteolytic activity (Tan et al., 2006).
We transfected mouse embryonic fibroblasts (MEFs) with dysferlin expression constructs with and without exon 40a. Three MEF lines were used: wild-type (WT), CAPNS1 knockout (−/−), and a rescued line stably transduced with a viral vector expressing CAPNS1 (−/−R) that displays elevated levels and activity of calpain-1 and -2 (Tan et al., 2006; Figure 2B, CAPN2 Western blot). Each transfected cell line was subjected to a scrape injury in the presence of calcium. Again, cleavage of dysferlin was observed only in cells transfected with dysferlin containing exon 40a (Figure 2B, middle three lanes). Mini-dysferlinC72 was not detected in CAPNS1−/− MEFs, and mini-dysferlinC72 levels were increased in rescued CAPNS1−/−R cells that express elevated levels of calpain-1 and -2, results consistent with calpains as specific mediators of dysferlin cleavage. Increased levels of immunoreactive degradative bands can be observed in CAPNS1−/− MEFs (although not cleaved mini-dysferlinC72). This is because CAPNS1−/− MEFs have an overt membrane repair defect and suffer cytotoxicity from the AMAXA nucleofection, resulting in elevated proteolysis within the harvested pool of cells.
To provide further evidence supporting calpains as mediators of dysferlin cleavage and establish whether mini-dysferlinC72 is preferentially produced by either calpain-1 or -2, we performed in vitro calpain cleavage experiments using purified calpain and immunopurified dysferlin. Figure 2C shows that immunoprecipitated dysferlin bearing exon 40a is cleaved by both calpain-1 and -2, whereas the canonical dysferlin lacking exon 40a cannot be cleaved.
Exon 40a is widely expressed in human tissues
To determine whether there is one dominant exon 40a-containing dysferlin transcript, we derived primer sets flanking each of the alternately spliced exons 5a, 17, and 40a and analyzed their relative abundance among dysferlin transcripts derived from a panel of human tissues (Clontech Multiple Tissue cDNA panels I and II; Figure 3A). Exon 40a-containing dysferlin transcripts are abundantly expressed in most human tissues (40–60% of transcripts in liver, kidney, lung, placenta, and pancreas; Figure 3A). Skeletal muscle expresses the lowest relative levels of exon 40a–containing transcripts (∼10–15%), with heart and brain expressing intermediate levels (∼15–25%). Exon 5a–containing transcripts dominate in liver, kidney, lung, placenta, and pancreas (80–90% of transcripts). Exon 17 shows variable tissue-specific expression, comprising the dominant mRNA species in liver, heart, brain, and muscle but only a minor mRNA species in kidney, lung, placenta, and pancreas. Our results therefore suggest that in human tissues exon 40a is regularly copresent with exon 5a and sometimes copresent with exon 17; this is consistent with previous results by Pramono et al. (2009), who compared splice isoforms of dysferlin in skeletal muscle and peripheral blood monocytes.
FIGURE 3:

Exon 40a–containing dysferlin is ubiquitously expressed, and mini-dysferlinC72 can be generated in multiple tissues. (A) Exon 40a is widely expressed in human tissues (∼40–60% transcripts), with lower relative levels in skeletal muscle, heart, and brain (∼10–15% transcripts). Dysferlin alternately spliced exons 5a, 17, and 40a were PCR amplified from a human tissue cDNA panel (Clontech) using primers flanking each of the exons. PCR amplification was performed for 30, 35, and 40 cycles to derive a simple standard curve and control for saturation. Ctrl; plasmid control. (B) Endogenous dysferlin from multiple tissues is cleaved by calpains in vitro, releasing mini-dysferlinC72. Mouse tissues were sectioned and lysed in RIPA, and endogenous dysferlin was immunoprecipitated with Romeo and protein G–Sepharose. Dysferlin-bound Sepharose beads were incubated with 0.2 active unit (A.U.) of purified recombinant calpain-1 at 30°C for 10 s in the presence of 2 mM CaCl2. Dysferlin was detected by Western analysis with the C-terminal antibody Hamlet-1. Mini-dysferlinC72 is indicated with a black arrow. (C) An anti–exon 40a antibody (α-40a) is specific to exon 40a-containing dysferlin in transfected HEK293 cells. Membranes were probed with anti–exon 40a and then reprobed with Hamlet-1 to reveal total dysferlin expression. GAPDH indicates even loading. (D) Anti–exon 40a antibody recognizes full-length dysferlin-exon 40a and cleaved mini-dysferlinC72 but not the N-terminal counterfragment. Dysferlin was immunopurified from transfected HEK293 cells and subject to in vitro calpain cleavage. R1 (Romeo) reveals the N-terminal counterfragment, H1 reveals mini-dysferlinC72, and α-40a shows reactivity to full-length dysferlin and mini-dysferlinC72. (E) Dysferlin exon 40a is expressed at similar levels in human muscle and heart. Total dysferlin was immunoprecipitated with Hamlet-1 from three control human muscles (1–3, ages 5, 18, and 37 yr, respectively, from young adults subject to testing for malignant hypothermia and shown to be normal) and two human hearts (1 and 2, donor hearts from young adults). Dysferlin-exon 40a was identified by Western blot with pAb α-40a. Membranes were reprobed with Romeo to reveal total immunoprecipitated dysferlin.
We also performed in vitro calpain digestion of dysferlin immunopurified from murine tissues with Hamlet-1. Results demonstrate that a proportion of expressed dysferlin can be cleaved by calpain to release mini-dysferlinC72, consistent with widespread expression of a dysferlin protein product derived from exon 40a–encoded transcripts (Figure 3B).
We raised a rabbit polyclonal antibody to human exon 40a and demonstrated its specificity by Western analysis of transfected HEK293 cells (Figure 3C). We also performed in vitro calpain cleavage of dysferlin-exon 40a immunoprecipitated from HEK293 cells and found that the pAb-exon 40a recognizes full-length dysferlin bearing exon 40a and cleaved mini-dysferlinC72 but not the N-terminal counterfragment recognized by Romeo-1 (Figure 3D). Despite lower relative levels of exon 40a transcripts in skeletal muscle and heart (compared with other tissues, such as kidney and lung), our anti–human pAb-exon 40a specifically recognized dysferlin-exon 40a protein by Western blot in human skeletal muscle and heart (Figure 3E; unfortunately, the only human tissues available for study). Our results cannot be used to infer the proportion of dysferlin-exon 40a relative to other isoforms, only that dysferlin with exon 40a is expressed at similar levels in heart and skeletal muscle relative to the total dysferlin pool detected by Romeo.
Injury-activated cleavage of dysferlin is a ubiquitous response to membrane injury in multiple cell lineages and occurs independently of MG53 expression
Although skeletal muscle is believed to be particularly prone to membrane injury due to its contractile functions under load, membrane injury is also a feature of ischemic injury to heart and brain and shear stress injury of blood vessel endothelia. Because dysferlin is ubiquitously expressed and readily detected in endothelia and brain, we studied whether the calcium-activated calpain cleavage of dysferlin represents a ubiquitous response to acute membrane injury or is a specific adaptation of skeletal muscle. In primary cultures of human myotubes, human umbilical vein endothelial cells (HUVECs), mouse astrocytes, and microglia, as well as secondary human oligodendrocytes (MO3.13), each displayed calcium-dependent, calpeptin-sensitive formation of mini-dysferlinC72 after scrape damage (Figure 4A). Primary mouse astrocytes and HUVECs do not express detectable levels of MG53 but are still capable of forming mini-dysferlinC72, indicating that MG53 is not required for calpain cleavage of dysferlin (Figure 4B).
FIGURE 4:

Dysferlin is cleaved in multiple cells types independent of MG53. (A, B) Injury-activated formation of mini-dysferlinC72 is calcium dependent and blocked by calpeptin and occurs in multiple cell lineages. (A) Cells were cultured to confluence and damaged by scraping in the presence or absence of Ca2+ or the presence of Ca2+ plus the calpain inhibitor calpeptin (Calp). Cell pellets were lysed in RIPA, and 10 μg of protein was separated by SDS–PAGE and transferred onto PVDF membrane. One PVDF membrane was probed with Hamlet-1, which detects the dysferlin C-terminus and mini-dysferlinC72 (black arrowhead). The duplicate PVDF membrane was probed with Romeo, detecting the dysferlin N-terminus and corresponding cleaved N-terminal fragment (gray arrowhead). Membranes were reprobed with anti-MG53 or anti-GAPDH to show equal loading. (B) Mouse astrocytes and human umbilical vein endothelial cells do not express MG53, and thus formation of mini-dysferlinC72 occurs independently of MG53.
Immunolabeling experiments of differentiated MO3.13 oligodendrocytes (differentiation increases dysferlin expression) subjected to ballistics injury with silica microparticles (4-μm diameter) revealed robust labeling for dysferlin at sites of membrane injury within 10 s of injury (Figure 5, top, left; Hamlet-1, green), concordant with our previous study in cultured human myotubes (Lek et al., 2013). Similarly, injury-induced dysferlin accumulation could be detected only with the C-terminal antibody Hamlet-1 and not the N-terminal antibody Romeo (Figure 5, top, middle; Romeo, red). Staining of primary human myotubes, biasing antibody binding to the dysferlin N-terminus by incubating with the N-terminal Romeo antibody before the C-terminal antibody Hamlet-1, similarly highlights specific recognition of the dysferlin C-terminus but not the dysferlin N-terminus at injury sites (Figure 5, second row).
FIGURE 5:
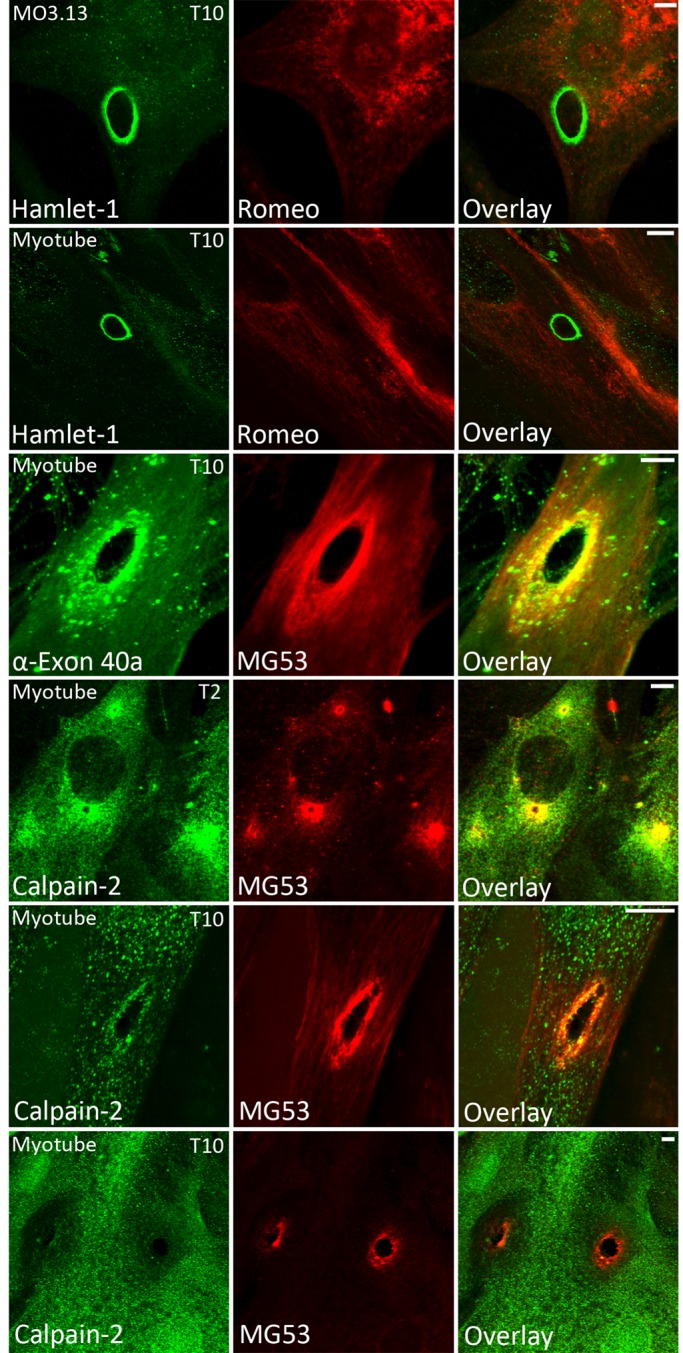
Dysferlin exon 40a and calpain recruit to sites of membrane injury. Cultured MO3.13 secondary oligodendrocytes (row 1) and primary human myotubes (row 2) were shot with 4-μm silica beads using a Bio-Rad Helios Gene Gun, fixed at 10 s postinjury in cold 3% paraformaldehyde, and then permeabilized and immunolabeled (see Materials and Methods). Romeo was applied for 2 h before Hamlet-1 to bias the detection of the N-terminal dysferlin epitope. Dysferlin was detectable only at sites of membrane injury with Hamlet-1 (rows 1 and 2). Staining with an antibody raised to dysferlin exon 40a revealed exon 40a–containing dysferlin recruits to sites of injury within 10 s (row 3). Calpain-2 was detectable at sites of membrane injury at 2 s (T2, row 4) and 10 s postdamage (T10, row 5). Large-injury sites often showed a void of negative labeling for calpain-2 (T10, row 6), suggesting that calpain might be extracted or escape from large injuries. Scale bar, 5 μm.
An antibody specifically recognizing exon 40a intensely labels ballistics injuries in human myotubes
Of interest, an antibody raised specifically to an antigen encoded by exon 40a robustly labels the exposed edges of ballistics lesions in cultured human myotubes 10 s after injury (Figure 5, third row; α-exon 40a, green; MG53, red). These results are consistent with our proposal that the exon 40a isoform of dysferlin plays a specialized role in membrane repair and that the C-terminal module released by calpain cleavage within exon 40a may be the form specifically recruited to injury sites.
Calpain-2 recruits to sites of membrane damage in cultured myotubes
To determine whether injury-activated recruitment of activated calpains to the plasma membrane temporally correlates with that of dysferlin, we damaged primary human myotubes using our ballistics assay and performed immunolabeling experiments with antibodies recognizing calpain. For these studies, we used very light permeabilization (0.01% saponin for 1 min) to minimize extraction of soluble calpain-2. Under these conditions, calpain-2 showed enrichment at injury sites at 2 s (T2) and 10 s (T10) postinjury (Figure 5, rows 4 and 5). Calpain-2 showed bright and consistent enrichment at bruised ballistics lesions and small perforations also labeled by MG53 (Figure 5, T2, row 4), whereas larger lesions often showed a “void” of negative staining around the lesion, suggesting that calpain-2 may escape or be extracted from large injury sites (Figure 5, T10, row 6). Our previous studies showed that MG53 is recruited to the injury zone within 2 s, whereas dysferlin is not detectable until 10 s postinjury (Lek et al., 2013). Thus the temporal recruitment of activated calpains to injury sites is consistent with our proposal that activated calpains are upstream and perhaps primary mediators of a membrane repair cascade, with cleaved dysferlin operating as one effector in this pathway.
Calpains are primary mediators of the calcium-activated repair response triggered by membrane injury
To determine a potential link between calpain activation, cleavage of dysferlin, and cell survival after an acute membrane injury, we developed a flow cytometry assay to quantify membrane resealing across populations of cells injured by scraping. Primary human myotubes (nascently differentiated for 3 d to activate dysferlin expression) were injured by cell scraping, allowed to reseal for 10 min, and then incubated in cell-impermeable propidium iodide, the “cell death” marker. Flow cytometry was then used to analyze the proportion of cells that failed to reseal and were permeable to propidium iodide. Our results confirm a requirement for calcium for membrane resealing, with ∼30% increase in cell survival when calcium is present (Figure 6A). By titrating levels of extracellular calcium, we show that maximal cell survival is achieved with extracellular calcium concentrations >100–200 μM, remarkably consistent with studies published by Steinhardt et al. (1994).
FIGURE 6:

Mini-dysferlinC72 formation requires ∼200 μM extracellular calcium, broadly correlating with the extracellular calcium concentration required for calcium-dependent membrane repair of injured muscle cells. (A) Development of a flow cytometry membrane repair assay reveals 100–200 μM as the activating concentration of extracellular Ca2+ required for calcium-dependent membrane repair pathways in cultured human muscle cells. (B) Treatment of primary human muscle cells with the calpain inhibitor calpeptin shows dose-dependent inhibition of cell survival, with an IC50 of 11.8 ± 5.8 μM (a representative dose–response curve is shown; the calculated IC50 is derived from four independent dose–response curves performed on different days, one with singlet samples at each dose, three in duplicate). C) Representative Western blot of a dose–response curve showing increasing formation of cleaved mini-dysferlinC72 with increasing concentrations of extracellular calcium. (D) Pooled densitometric quantification of levels of cleaved mini-dysferlinC72 from five calcium dose–response curves (EC50 of ∼ 250 μM Ca2+, 95% confidence interval). (E, F) In vitro digestion of dysferlin-exon 40a with 0.2 A.U. of purified calpain-1 (E) and calpain-2 (F). Mini-dysferlinC72 is indicated with black arrows.
Calpeptin potently inhibited cell survival of human myotubes after acute membrane injury, even in the presence of 900 μM extracellular calcium (Figure 6B; calpeptin IC50 = 11.8 ± 5.8 mM). These results support calpains as primary effectors of the calcium-dependent repair response triggered by membrane injury (Godell et al., 1997; Mellgren et al., 2007).
Western blot analysis of cells subjected to the same scrape-injury assay revealed that the calcium dependence of dysferlin cleavage occurs with an EC50 of ∼ 250 μM extracellular calcium (Figure 6C). Our collective results over five independent experiments highlight >200 μM extracellular calcium as a tipping point for dysferlin cleavage in injured cells. Of interest, this mirrors the calcium activation profile of calpain-2 for enzymatic cleavage of immunopurified dysferlin (Figure 6F). Given that calpain-1 shows maximal enzymatic cleavage of dysferlin with <50 μM calcium (Figure 6E), whereas calpain-2 requires >200 μM calcium for enzymatic cleavage of dysferlin (Figure 6F), our results suggest that the extra activity of calpain-2 might have special importance for the injury-activated cleavage of dysferlin in situ.
Other ferlin proteins are enzymatically cleaved by calpain to release similar synaptotagmin-like modules
We next investigated whether calpain cleavage was a unique property of dysferlin or whether other members of the ferlin family share this property. We derived expression constructs of myoferlin and otoferlin and immunopurified each ferlin paralogue from transfected HEK293 cells using a luminal FLAG tag conjugated to the C-terminus. In vitro calpain cleavage revealed that exon 40a–containing dysferlin, myoferlin, and otoferlin were rapidly cleaved at a single, dominant cleavage site, releasing C-terminal (Figure 7A, top) and N-terminal (Figure 7A, bottom) fragments. In each case, the predicted products bear two C2 domains anchored by their transmembrane domain, a structure similar to that of the synaptotagmin family of vesicle fusion proteins (Figure 7C).
FIGURE 7:

Calpain cleaves otoferlin and myoferlin in addition to dysferlin. (A) Calpain rapidly cleaves immunoprecipitated ferlin proteins in vitro. DysferlinMycHis, otoferlinMycFlag, and myoferlinMycFlag were immunoprecipitated with anti-myc and protein G–Sepharose (see Materials and Methods). Dysferlin-bound Sepharose beads were incubated with purified 0.2 A.U. of recombinant calpain-1 at 30°C for 2 or 10 s in the presence of 2 mM CaCl2. Proteolysis was rapidly inhibited by reconstitution of the reaction in SDS lysis buffer and heating to 94°C. Digested samples were analyzed by SDS–PAGE and Western blot. Top, C-terminal fragments detected with anti-myc (dysferlin) or anti-Flag (myoferlin and otoferlin). Bottom, N-terminal fragments detected by N-terminal (Romeo-dysferlin) or internal antibodies (7D2, myoferlin; C12, otoferlin). (B) Dysferlin and otoferlin display damage-dependent cleavage, whereas myoferlin cleavage appears to be constitutive. HEK293 cells were transfected with dysferlinMycHis, otoferlinMycFlag, and myoferlinMycFlag and lysed in calcium-free RIPA (lane 1), RIPA containing 900 μM calcium (permissive for calpain cleavage), or damaged by scraping in the presence of calcium. Scraped cell pellets were lysed in calcium-free RIPA, and 10 μg of protein was separated by SDS–PAGE and transferred onto PVDF membrane. Dysferlin was detected with anti-Myc; otoferlin and myoferlin were detected with anti-Flag. (C) Diagram of the predicted calpain cleavage sites within dysferlin, otoferlin, and myoferlin (schematic produced using DOG 2.0; Ren et al., 2009). Molecular weight calculation of the cleaved C-terminal modules was used to elucidate the most likely calpain cleavage site (ccd.biocuckoo.org). In each case, the C-terminal fragments released by calpain cleavage represent transmembrane-anchored, dual-C2-domain modules.
Although our results show that myoferlin and otoferlin can be cleaved by calpain, we asked whether they are in situ. Transfected HEK293 cells were injured by scraping in the presence of calcium and then harvested in lysis buffer containing EDTA and a protease inhibitor cocktail containing 50 μM calpeptin to inhibit all further calpain activity. Transfected cells were also harvested as uninjured cells either in a “permissive” lysis buffer (plus calcium, without EDTA or calpeptin) to facilitate maximal Ca2+-activated cleavage or a “nonpermissive” buffer (without calcium, plus EDTA and calpeptin; see Materials and Methods).
Exon 40a–containing dysferlin and otoferlin showed injury- and calcium-dependent formation of a C-terminal cleavage module (Figure 7B). Of interest, in multiple experiments, a C-terminal cleavage product of myoferlin was regularly observed in uninjured cells, suggesting that cleavage of myoferlin occurs constitutively within cells (Figure 7B). We have never observed cleavage of exon 40a–containing dysferlin in uninjured cells and believe our results might highlight a key functional difference between dysferlin and myoferlin. Our current research is focused on refining the precise cleavage site within myoferlin and otoferlin and determining the exact role of cellular calpains (or other enzymes) in their cleavage.
DISCUSSION
The ubiquitous calpains (calpain-1 and -2; Mellgren et al., 2007, 2009; Taneike et al., 2011) and dysferlin (Bansal et al., 2003) have been separately identified as key mediators of calcium-dependent plasma membrane repair. In this article, we link roles for calpains and dysferlin in the acute response to a membrane injury, providing an explanation as to how calpains may directly regulate the vesicle fusion of membrane repair (Godell et al., 1997). We demonstrate that the calpain cleavage site within dysferlin in encoded by alternately spliced exon 40a. Of interest, dysferlin exon 40a is comparatively divergent among mammals (52% amino acid identity; Figure 1C) relative to flanking exons 40 and 41 (96 and 88% identity, respectively). Calpain cleavage is determined not by a specific amino acid motif but by both secondary and tertiary structural determinants of the cleavage site (Tompa et al., 2004). Despite the sequence divergence within exon 40a, in silico algorithms strongly predict calpain cleavage at the same position in all higher vertebrates analyzed, suggesting that evolutionary pressure has targeted the maintenance of the calpain cleavage site, regardless of the exact amino acid sequence encoding it.
Calpains are not terminal degradative enzymes but act to selectively modify substrates, often altering protein function as opposed to removing the protein (Goll et al., 2003). Evolutionary preservation of alternate splicing as a means to regulate expression of “calpain-cleavable” dysferlin implies that calpain cleavage of dysferlin in response to high-intensity calcium signaling imparts an important functional modification. We observed the lowest relative expression of exon 40a transcripts in skeletal muscle, heart, and brain—the three major excitable tissues. It is possible that promiscuous production of cleaved mini-dysferlin may not always be desirable, and expression of “cleavable” dysferlin may be more tightly regulated in excitable tissues, where calpain activity may be higher.
Why does dysferlin need to be cleaved by calpains? Our main hypothesis is that separation of the N-terminus from the C-terminus overcomes a regulatory checkpoint in the vesicle fusion activity of dysferlin, perhaps conferring an “overdrive” function by which mini-dysferlinC72 facilitates quicker execution of vesicle fusion activity that is used for emergency membrane repair. Which pool of dysferlin is cleaved? Recent studies of murine muscle fibers transgenically expressing dysferlin-pHluorin (without exon 40a) revealed that membrane injury induced an immediate endocytosis of dysferlin from regions surrounding, and distal to, the injury site (McDade et al., 2014). This raises the intriguing possibility that dysferlin may first be endocytosed and cleaved by calpains and then mini-dysferlinC72–containing vesicles are exocytosed. We do not yet know the fate or function of the N-terminal module released by calpain cleavage. Although this has a clumsy feel for an emergency membrane repair response, perhaps endocytic derivation of “patch vesicles” comprising normal plasma membrane lipid composition presents a significant advantage for cell survival. Collective results suggest that full-length dysferlin may participate in both endocytic and exocytic pathways during the membrane repair response, a paradigm similarly proposed for otoferlin in auditory neurotransmission (Pangrsic et al., 2010), whereas calpain-cleaved mini-dysferlinC72 may represent a specialized exocytic module.
In 2010, a patient bearing a genomic deletion of exons 2–40 within dysferlin was shown to express a truncated dysferlin protein product (Krahn et al., 2010) with striking similarity to naturally occurring calpain-cleaved mini-dysferlinC72. The patient presented with a relatively mild muscle phenotype and expressed a “mini-dysferlin” product via utilization of a cryptic splice site within intron 40, encoding 21 aberrant amino acids before continuing with the normal dysferlin sequence from exon 41. Of importance, transgenic expression of this patient mini-dysferlin in dysferlin-null mouse muscle rescued the membrane repair deficiency but did not correct the dystrophic pathology (Krahn et al., 2010). These data support our evidence that mini-dysferlinC72 plays a critical role in plasma membrane repair and also indicate that the dystrophic pathology in dysferlinopathy patients has a more complicated basis than defective membrane repair alone. Dysferlin is a member of a highly conserved family of secretory proteins, and it is very likely that dysferlin performs a routine day-to-day role in secretory exocytosis, with a role in membrane repair representing an extension of this function. Indeed, calpain cleavage of dysferlin exon 40a may not have evolved specifically for “membrane repair” but instead as a general mechanism to regulate an important and specialized secretory function in settings of intense calcium signaling.
We previously observed that dysferlin is only identifiable at sites of ballistics injury in cultured cells using a C-terminal antibody, Hamlet-1, and not by three central region or N-terminal antibodies (Lek et al., 2013). In this study, we derived an antibody raised to human dysferlin exon 40a, recognizing the full-length exon 40a–containing dysferlin and cleaved mini-dysferlinC72. This exon 40a antibody robustly labels dysferlin at sites of membrane injury, providing another line of evidence suggesting that the calpain-cleaved C-terminal module is the form specifically recruited to injury sites (Figure 5). Although we cannot exclude the possibility that the full-length exon 40a–containing dysferlin (and other dysferlin isoforms) may be present at injury sites, immunolabeling using three separate N-terminal or central regions antibodies yielded negative results, despite intensive efforts with antigen retrieval and fixation methods. Therefore, if full-length dysferlin is present at injury sites, our results indicate that it adopts a conformation that structurally precludes the accessibility of antibodies.
When calpain activity is inhibited by calpeptin, cell survival is greatly reduced after membrane scrape injury, even in the presence of calcium (Figure 6A; Mellgren and Huang, 2007; Mellgren et al., 2007, 2009; Taneike et al., 2011). Ubiquitous calpains are abundantly expressed and show enrichment at sites of membrane injury before dysferlin is detected (Figure 5), temporally consistent with calpains as upstream regulators of a downstream membrane repair cascade that involves the cleavage of dysferlin. Our results lead to a paradigm in which calpain cleavage of dysferlin releases mini-dysferlinC72, which subsequently recruits to, and aids repair of, damaged membranes.
A major finding of this study is that other ferlin family members can be enzymatically cleaved to release N- and C-terminal modules. Myoferlin and otoferlin are cleaved to form tail-anchored, dual-C2-domain C-terminal fragments similar to mini-dysferlinC72 (Figure 7A). The C2 domains of the mini-ferlin modules, C2E and C2F, are the most highly conserved C2 domains of the ferlin family (Lek et al., 2010). Moreover, the dual-C2-domain C-terminal modules have structural similarity to the synaptotagmin family of vesicle fusion proteins (Figure 7B). Our results imply that cleavage of ferlin proteins is an evolutionarily conserved event with origins predating gene expansion of the ferlin family.
We hypothesize the synaptotagmin-like modules released through enzymatic cleavage of ferlin proteins have specialized vesicle-fusion modality. For dysferlin and otoferlin, we show that the unregulated calcium influx after membrane disruption activates calpains to release these C-terminal vesicle-fusion modules. In contrast, our evidence suggests that myoferlin is cleaved constitutively, highlighting a potential difference in the functional modulation between the highly homologous dysferlin and myoferlin paralogues.
MATERIALS AND METHODS
Cell culture
Human myoblast cultures were established as described in Lek et al. (2013). Myoblasts were cultured in 1:1 DMEM:F12 (Life Technologies, Carlsbad, CA) containing 20% fetal bovine serum (FBS; Life Technologies), 10% Amniomax (Life Technologies), and 1:200 gentamicin (Life Technologies). Myoblasts were differentiated in 1:1 DMEM:F12 containing 3% horse serum (HS; Life Technologies) and 1:200 gentamicin for up to 6 d before use. Astrocytes and microglia were isolated from neonatal C57BL/6J mice as described by Mecha et al. (2011) and cultured in DMEM (Life Technologies) containing 10% FBS and 10% HS with 1% penicillin/streptomycin (Pen/Strep; Life Technologies). Cell identity was verified by CD-11c expression (microglia) and glial-fibrillary acidic protein expression (astrocytes). Neonatal cardiomyocytes were isolated from 2- to 4-d-old C57BL/6 mice using the Neonatal Cardiomyocyte Isolation System (Worthington). Cardiomyocytes were plated at 0.5 × 105 cells/cm2 in 1:1 DMEM:F12 containing 20% FBS, 5% HS, 5% Amniomax, 2 mM l-glutamine (Life Technologies), 1% nonessential amino acids (Life Technologies), 3 mM sodium pyruvate (Life Technologies), 1:200 gentamicin, and 1% Pen/Strep. Cultures were left to dedifferentiate and grow to confluence for ∼5 d before use.
HUVECs were purchased from Life Technologies and cultured in M-200 medium (Life Technologies) containing low-serum growth supplement and 1:200 gentamicin. Human secondary oligodendroglial MO3.13 cells were cultured in DMEM containing 10% FBS and 1:200 gentamicin, then differentiated for 7 d in serum-free DMEM containing 100 nM phorbol-12-myristate-13-acetate (Sigma-Aldrich, St. Louis, MO) and 1:200 gentamicin before use. HEK293 cells were cultured in DMEM containing 10% FBS and 1:200 gentamicin. Wild-type, CAPNS1−/−, and CAPNS1−/− lentiviral rescued MEF lines are described in Tan et al., 2006. MEFs were cultured in DMEM containing 10% FBS and 1:200 gentamicin.
Cell scrape injury
Cells were grown to confluence in six-well plates (BD Biosciences, San Jose, CA), washed twice with phosphate-buffered saline (PBS [Life Technologies]; either with calcium or without calcium plus 10 mM EDTA [Sigma-Aldrich]), and then scraped with a rubber policeman (BD Bioscience). Cells were transferred from the well 30 s postscraping and quenched with 50 mM EDTA and 1:500 protease inhibitor cocktail (Sigma-Aldrich) on ice to prevent all further proteolysis. Cells were pelleted at 300 × g for 3 min, the supernatant removed, and the cell pellet solubilized in 150 μl of radioimmunoprecipitation buffer (RIPA; 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 1 mM EDTA, 1:500 protease inhibitor cocktail [Sigma-Aldrich]). Protein content was determined by bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA).
SDS–PAGE and Western blotting
Samples were separated by SDS–PAGE on 4-12% Bis-Tris polyacrylamide gels (Life Technologies) using PAGE Ruler (Thermo Fisher Scientific) as a size marker and transferred onto polyvinylidene fluoride (PVDF) membrane (Merck Millipore, Billerica, MA). Membranes were blocked using 5% milk (or 5% BSA [Sigma-Aldrich]) in PBS containing 0.1% Tween-20 (Amresco, Solon, OH). Primary antibodies were incubated overnight, followed by washing and incubation with horseradish peroxidase–conjugated secondary antibodies (Thermo Fisher Scientific). The membrane was washed and developed using ECL reagent Hyperfilm (GE Healthcare, Buckinghamshire, UK). Western blot quantification was performed using ImageJ (National Institutes of Health, Bethesda, MD).
Ballistics-induced membrane injury, immunocytochemistry, and microscopy
Ballistics-induced injury was performed using a Helios Gene Gun (Bio-Rad, Hercules, CA) with bullet cartridges prepared according to the manufacturer's instructions using silica microparticles (4-μm diameter; Sigma-Aldrich) substituted for gold microparticles. Differentiated myotubes cultured on Thermanox coverslips (Thermo Fisher Scientific) were transferred into a 24-well plate containing 200 μl of PBS (Life Technologies) and shot at 300 psi. Immediately after discharge of the gun, a further 200 μl of PBS was immediately added to the well to refresh the fluid expelled by the helium blast. All solutions used were equalized to room temperature (23–26°C).
Cells were fixed in cold 3% paraformaldehyde (Sigma-Aldrich) at 10 s postinjury and then permeabilized with 0.1% Triton (Sigma-Aldrich; calpain-2 antibody), 0.1% saponin (Sigma-Aldrich; anti–exon 40a antibody), or 1:1 methanol: acetone (Sigma-Aldrich; Hamlet-1 and Romeo antibodies). Cells were blocked in 2% BSA in PBS for 30 min and incubated in primary antibodies overnight. After washing, cells were reblocked for 15 min and incubated in secondary antibody for 2 h. Cells were then washed and mounted using Fluorsave (Merck Millipore). Images were captured using a Leica SP5 scanning confocal microscope with a 63× oil objective.
Membrane repair assay
Primary human myotubes nascently differentiated for 3 d to induce dysferlin expression were trypsinized and replated 3 h before damage. Cells were washed twice with PBS ± Ca2+ ± calpeptin (Tocris Bioscience, Bristol, UK), depending on scrape conditions. Cells were scraped in PBS containing 1:2500 TOPRO (Invitrogen) for 15 s and then transferred into a fluorescence-activated cell sorting tube (BD Bioscience) and incubated at 23–26°C for 10 min. After recovery, cells were pelleted at 300 × g for 1 min at 23–26°C, the supernatant discarded, and the cell pellet resuspended in PBS containing 1:200 propidium iodide (Invitrogen). Cells were analyzed on a BD FACSDiva flow cytometer to assess damaged and repaired cells.
Transfection
HEK293 cells were transfected in 10-cm dishes using CaPO4. Briefly, 350 μl of 0.25 M CaCl2 (Sigma-Aldrich) was added dropwise to 14 μg of supercoiled plasmid DNA. Then 350 μl of BES solution (50 mM BES, 280 mM NaCl, 1.5 mM NaHPO4⋅7H2O [Sigma-Aldrich]) was added to the CaCl2/DNA solution and incubated at room temperature for 15 min. The transfection solution was added to cells, and cells were harvested 16 h posttransfection. Mouse embryonic fibroblasts were transfected by standard protocol using an AMAXA Nucleofector 2b with MEF1 solution (Lonza AG, Basel, Switzerland).
Immunoprecipitation
Epitope-tagged ferlin constructs were immunoprecipitated from transfected HEK293 cells. Cells were treated with 50 μM calpeptin 1 h before lysis. Cells were washed with PBS without calcium and lysed on ice in 1 ml of RIPA. After lysis, cell lysate was precleared with 25 μl of protein G–Sepharose (GE Healthcare), and insoluble material and protein G–Sepharose were pelleted at 20,000 × g for 15 min. A 250-ng amount of either anti–epitope tag antibody (rabbit anti-DDDDK or rabbit anti-Myc; Abcam, Cambridge, UK) or 250 ng of Romeo antibody was added per plate of cell lysate and incubated for 16 h at 4°C. Antibody-bound dysferlin was then immunopurified with 25 μl of protein G–Sepharose and incubated for 2 h, and beads were washed three times with RIPA.
In vitro calpain cleavage
Purified calpain-1 (porcine) and -2 (human) was purchased from Millipore. In vitro cleavage of dysferlin was performed using a modified protocol from Mandic et al. (2002). Protein G–Sepharose–bound dysferlin was washed three times in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Sigma-Aldrich; pH 7.5), 50 mM NaCl, and 1 mM MgCl2 containing 2 mM CaCl2. Diluted recombinant calpain was added directly to the protein G–Sepharose–bound dysferlin and incubated at 30°C for 2 s to 3 min, as indicated. Digestion was quenched by reconstitution into SDS lysis buffer (2% SDS, 10% glycerol, 50 mM Tris, pH 7.4, and 10 mM dithiothreitol [Sigma-Aldrich]), and samples were heated to 94°C for 3 min.
Antibodies
Exon 40a antibody.
A rabbit polyclonal antibody to the peptide SSLAPTNTASPPSSPH encoded by dysferlin exon 40a was derived by EuroGentec (Liege, Belgium). Affinity-purified immunoglobulin G was used at 1:100 for Western blot and 1:25 for immunocytochemistry.
Western blot.
We used Hamlet 1 (1:500; Leica Microsystems, Wetzlar, Germany; Mueller et al., 2014), Romeo (1:1000; Abcam; Mueller et al., 2014), mitsugumin-53/TRIM72 (1:5000; a generous gift from Jianjie Ma, University of Medicine and Dentistry of New Jersey, Piscataway, NJ; Cai et al., 2009b), c-Myc (1:500; Santa Cruz Biotechnology, Dallas, TX), β-tubulin (1:1000; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH: 1:5000; Merck, Millipore), OTOF-C12 (Santa Cruz Biotechnology), and anti-myoferlin 7D2 (Abcam; Leung et al., 2012).
Immunocytochemistry.
We used Hamlet 1 (1:100), Romeo (1:1000), mitsugumin-53/TRIM72 (1:5000; a gift from J. Ma), and calpain-2 (1:25; Cell Signaling Systems; Pilop et al., 2009).
PCR and primers
PCR was carried out on human cDNA panels purchased from Clontech (Mountain View, CA; Human MTC Panel I and Human Immune MTC Panel). Primers to dysferlin exon 5a were as follows: forward, 5′ CTAGTCTGTCCGCCAGCTTC 3′, and reverse, 5′ CAGGGTCCAGGTGATCGA 3′. Primers to exon 17 were as follows: forward, 5′ GGAGGACATTGAAAGCAACC 3′, and reverse, 5′ CCTCTATGGCAGTCCCAGAG 3′. Primers to exon 40a were as follows: forward, 5′ ATCACCGTCAAGGTCATCG 3′, and reverse, 5′ GTAACACCTTCAAGCTGTACCGG 3′. GAPDH primers were supplied as part of the Clontech cDNA panel kit.
Constructs
Our dysferlin construct (EGFP-FL-DYSF pcDNA3.1, National Center for Biotechnology Information [NCBI] reference sequence NP_003485.1) was a generous gift from Kate Bushby (Institute of Human Genetics, International Centre for Life, Newcastle upon Tyne, UK) and subcloned into pIRES2 EGFP (OriGene). Our exon 40a–containing construct (40a-DYSF, NCBI reference sequence NM_001130978.1) was generated by PCR amplifying exon 40a from human skeletal muscle cDNA. Exon 40a–containing products were isolated and digested with ClaI (New England Bioscience, Ipswich, MA) and inserted into our FL-DYSF-pIRES2 construct. Otoferlin (NCBI reference sequence NM_194248.2) and myoferlin-pCMV6 (NCBI reference sequence NM_013451.3) were purchased from OriGene (Rockville, MD) and subcloned into pIRES2-EGFP.
Protein alignment and calpain cleavage prediction
Protein alignment was performed using ClustalW. Calpain cleavage prediction was performed using GPS-CCD (ccd.biocuckoo.org; Liu et al., 2011).
Statistics
Statistics was performed using Prism 6 (GraphPad).
Animal studies
Ethical approval for this project was provided by the Children's Hospital at Westmead/Children's Medical Research Institute Animal Care and Ethics Committee (K289).
Acknowledgments
We gratefully acknowledge Jianjie Ma and Noah Weisleder (University of Medicine and Dentistry of New Jersey, Piscataway, NJ) for the polyclonal anti-MG53 antibody. We thank the Children's Medical Research Institute Flow Cytometry Facility for their technical assistance and Laurence Cantrill for technical assistance with confocal microscopy. We thank the funding bodies supporting our work: the Australian National Health and Medical Research Council (Project Grant APP1048814 to S.T.C. and K.M.; Career Development Fellowship APP1048816 to S.T.C.; and Ph.D. Scholarship to F.L.), the Jain Foundation (S.T.C.), the University of Sydney for postgraduate awards (G.R., N.W., and A.K.P.), the Brain Foundation (S.T.C. and K.N.), and the Canadian Institutes of Health Research (P.A.G.). The Leica SP5 in the Correlative Light and Electron Microscope Suite at Kids Research Institute, Children's Hospital at Westmead, was supported by the Cancer Institute New South Wales (Research Equipment 10/REG/1-23), the Australian National Health and Medical Research Council (2009-02759), the Ian Potter Foundation (20100508), the Perpetual Foundation (730), the Ramaciotti Foundation (3037/2010), and the Sydney Medical School Research Infrastructure Major Equipment Scheme.
Abbreviations used:
- HUVEC
human umbilical vein endothelial cell
- MEF
mouse embryonic fibroblast
- RIPA
radioimmunoprecipitation buffer
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-04-0947) on August 20, 2014.
REFERENCES
- Achanzar WE, Ward S. A nematode gene required for sperm vesicle fusion. J Cell Sci. 1997;110:1073–1081. doi: 10.1242/jcs.110.9.1073. [DOI] [PubMed] [Google Scholar]
- Bansal D, Miyake K, Vogel SS, Groh S, Chen C-C, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, Ko J-K, Lin P, Thornton A, Zhao X, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009a;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, Weisleder N, Ko J-K, Komazaki S, Sunada Y, Nishi M, Takeshima H, Ma J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J Biol Chem. 2009b;284:15894–15902. doi: 10.1074/jbc.M109.009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godell CM, Smyers ME, Eddleman CS, Ballinger ML, Fishman HM, Bittner GD. Calpain activity promotes the sealing of severed giant axons. Proc Natl Acad Sci USA. 1997;94:4751–4756. doi: 10.1073/pnas.94.9.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Krahn M, Wein N, Bartoli M, Lostal W, Courrier S, Bourg-Alibert N, Nguyen K, Vial C, Streichenberger N, Labelle V, et al. A naturally occurring human minidysferlin protein repairs sarcolemmal lesions in a mouse model of dysferlinopathy. Sci Transl Med. 2010;2:50ra69. doi: 10.1126/scitranslmed.3000951. [DOI] [PubMed] [Google Scholar]
- Lek A, Evesson FJ, Lemckert FA, Redpath GMI, Lueders A-K, Turnbull L, Whitchurch CB, North KN, Cooper ST. Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. J Neurosci. 2013;33:5085–5094. doi: 10.1523/JNEUROSCI.3560-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek A, Lek M, North K, Cooper S. Phylogenetic analysis of ferlin genes reveals ancient eukaryotic origins. BMC Evol Biol. 2010;10:231. doi: 10.1186/1471-2148-10-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C, Shaheen F, Bernatchez P, Hackett T-L. Expression of myoferlin in human airway epithelium and its role in cell adhesion and zonula occludens-1 expression. PLoS One. 2012;7:e40478. doi: 10.1371/journal.pone.0040478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- Liu Z, Cao J, Gao X, Ma Q, Ren J, Xue Y. GPS-CCD: a novel computational program for the prediction of calpain cleavage sites. PLoS One. 2011;6:e19001. doi: 10.1371/journal.pone.0019001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandic A, Viktorsson K, Strandberg L, Heiden T, Hansson J, Linder S, Shoshan MC. Calpain-mediated bid cleavage and calpain-independent Bak modulation: two separate pathways in cisplatin-induced apoptosis. Mol Cell Biol. 2002;22:3003–3013. doi: 10.1128/MCB.22.9.3003-3013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade JR, Archambeau A, Michele DE. Rapid actin-cytoskeleton–dependent recruitment of plasma membrane–derived dysferlin at wounds is critical for muscle membrane repair. FASEB J 28, 3660–3670. 2014 doi: 10.1096/fj.14-250191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Khakee R. Disruptions of muscle fibre plasma membranes. Role in exercise-induced damage. Am J Pathol. 1992;140:1097–1109. [PMC free article] [PubMed] [Google Scholar]
- Mecha M, Iñigo PM, Mestre L, Hernangómez M, Borrell J, Guaza C. An easy and fast way to obtain a high number of glial cells from rat cerebral tissue: a beginners approach. Protocol Exchange, DOI:10.1038/protex.2011.218. 2011 [Google Scholar]
- Mellgren RL, Huang X. Fetuin A stabilizes m-calpain and facilitates plasma membrane repair. J Biol Chem. 2007;282:35868–35877. doi: 10.1074/jbc.M706929200. [DOI] [PubMed] [Google Scholar]
- Mellgren RL, Miyake K, Kramerova I, Spencer MJ, Bourg N, Bartoli M, Richard I, Greer PA, McNeil PL. Calcium-dependent plasma membrane repair requires m- or μ-calpain, but not calpain-3, the proteasome, or caspases. Biochim Biophys Acta. 2009;1793:1886–1893. doi: 10.1016/j.bbamcr.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellgren RL, Zhang W, Miyake K, McNeil PL. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem. 2007;282:2567–2575. doi: 10.1074/jbc.M604560200. [DOI] [PubMed] [Google Scholar]
- Mueller AL, Desmond PF, Hsia R-C, Roche JA. Improved immunoblotting methods provide critical insights into phenotypic differences between two murine dysferlinopathy models. Muscle Nerve. 2014;50:286–289. doi: 10.1002/mus.24220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsako T, Hirai K, Yamamoto MT. The Drosophila misfire gene has an essential role in sperm activation during fertilization. Genes Genet Syst. 2003;78:253–266. doi: 10.1266/ggs.78.253. [DOI] [PubMed] [Google Scholar]
- Pangrsic T, Lasarow L, Reuter K, Takago H, Schwander M, Riedel D, Frank T, Tarantino LM, Bailey JS, Strenzke N, et al. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010;13:869–876. doi: 10.1038/nn.2578. [DOI] [PubMed] [Google Scholar]
- Pilop C, Aregger F, Gorman RC, Brunisholz R, Gerrits B, Schaffner T, Gorman JH, Matyas G, Carrel T, Frey BM. Proteomic analysis in aortic media of patients with Marfan syndrome reveals increased activity of calpain 2 in aortic aneurysms. Circulation. 2009;120:983–991. doi: 10.1161/CIRCULATIONAHA.108.843516. [DOI] [PubMed] [Google Scholar]
- Pramono Z, Tan C, Seah I, See J, Kam S, Lai P, Yee W. Identification and characterisation of human dysferlin transcript variants: implications for dysferlin mutational screening and isoforms. Hum Genet. 2009;125:413–420. doi: 10.1007/s00439-009-0632-y. [DOI] [PubMed] [Google Scholar]
- Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19:271–273. doi: 10.1038/cr.2009.6. [DOI] [PubMed] [Google Scholar]
- Rizo J, Südhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879–15882. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Shin O-H, Xu J, Rizo J, Südhof TC. Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc Natl Acad Sci USA. 2009;106:16469–16474. doi: 10.1073/pnas.0908798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MK, Wakimoto BT. Complex regulation and multiple developmental functions of misfire, the Drosophila melanogaster ferlin gene. BMC Dev Biol. 2007;7:21. doi: 10.1186/1471-213X-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhardt R, Bi G, Alderton J. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA. Conditional disruption of ubiquitous calpains in the mouse. Genesis. 2006;44:297–303. doi: 10.1002/dvg.20216. [DOI] [PubMed] [Google Scholar]
- Taneike M, Mizote I, Morita T, Watanabe T, Hikoso S, Yamaguchi O, Takeda T, Oka T, Tamai T, Oyabu J, et al. Calpain protects the heart from hemodynamic stress. J Biol Chem. 2011;286:32170–32177. doi: 10.1074/jbc.M111.248088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P, Buzder-Lantos P, Tantos A, Farkas A, Szilágyi A, Bánóczi Z, Hudecz F, Friedrich P. On the sequential determinants of calpain cleavage. J Biol Chem. 2004;279:20775–20785. doi: 10.1074/jbc.M313873200. [DOI] [PubMed] [Google Scholar]
- Washington NL, Ward S. FER-1 regulates Ca2+-mediated membrane fusion during C. elegans spermatogenesis. J Cell Sci. 2006;119:2552–2562. doi: 10.1242/jcs.02980. [DOI] [PubMed] [Google Scholar]
- Xie XY, Barrett JN. Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca2+-triggered protease activity and cytoskeletal disassembly. J Neurosci. 1991;11:3257–3267. doi: 10.1523/JNEUROSCI.11-10-03257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]