

Recent studies implicate a role for cell mechanics in cancer progression. Transforming growth factor β–induced epithelial-to-mesenchymal transition results in decreased stiffness and loss of the normal stiffening response to force applied on integrins.
Abstract
Recent studies implicate a role for cell mechanics in cancer progression. The epithelial-to-mesenchymal transition (EMT) regulates the detachment of cancer cells from the epithelium and facilitates their invasion into stromal tissue. Although classic EMT hallmarks include loss of cell–cell adhesions, morphology changes, and increased invasion capacity, little is known about the associated mechanical changes. Previously, force application on integrins has been shown to initiate cytoskeletal rearrangements that result in increased cell stiffness and a stiffening response. Here we demonstrate that transforming growth factor β (TGF-β)–induced EMT results in decreased stiffness and loss of the normal stiffening response to force applied on integrins. We find that suppression of the RhoA guanine nucleotide exchange factors (GEFs) LARG and GEF-H1 through TGF-β/ALK5–enhanced proteasomal degradation mediates these changes in cell mechanics and affects EMT-associated invasion. Taken together, our results reveal a functional connection between attenuated stiffness and stiffening response and the increased invasion capacity acquired after TGF-β–induced EMT.
INTRODUCTION
The reciprocity of mechanical information between cells and their extracellular environment has increased appreciation for the role of physics in cancer metastasis (Butcher et al., 2009). In this complex progression, cancer cells detach from the primary tumor, invade the surrounding stromal space, transmigrate the vascular system, and establish secondary tumors at distal sites. Specific mechanical phenotypes are likely adopted to enable cells to navigate successfully the mechanical environments encountered during metastasis (Wirtz et al., 2011). Epithelial-to-mesenchymal transition (EMT) is an essential physiological process that drives adherent, immotile cells to lose polarity and increase migration. Recently abnormal reactivation of EMT has been implicated in the detachment of cancer cells from epithelial tissue and their subsequent invasion into stromal tissue (Yilmaz and Christofori, 2009; Taylor et al., 2010). The transforming growth factor β (TGF-β) superfamily of growth factors, one of the primary drivers of EMT, initiates this process by altering gene expression (Ranganathan et al., 2007), inducing loss of cell–cell adhesions (Vogelmann et al., 2005), promoting changes to cytoskeletal structure (Moustakas and Stournaras, 1999; Hubchak, 2003), and increasing motility and invasion (Oft et al., 1998; Gordon et al., 2009). These changes in cytoskeletal structure and increased interaction with the extracellular matrix (ECM) implicate a role for altered cell mechanics during EMT (Yilmaz and Christofori, 2009).
Extensive remodeling of the ECM occurs in cancer (Levental et al., 2010), resulting in tumors being stiffer (Paszek et al., 2005) and more heterogeneous than normal tissue (Plodinec et al., 2012); therefore cancer cells moving through tumor ECM experience stiffness gradients not typically experienced by normal epithelial cells. Integrins are transmembrane, mechanosensitive receptors that provide an essential physical connection between the ECM and the actin cytoskeleton (Wang et al., 1993) during cell adhesion and migration. Studies have shown that cells respond to force on integrins by generating a stiffening response to resist the applied force and maintain mechanical reciprocity (Matthews et al., 2006; Lessey et al., 2012). We demonstrated that mechanical response to force is regulated by the activation of the small GTPase RhoA through specific guanine nucleotide exchange factors (GEFs) in fibroblasts (Guilluy et al., 2011). Although RhoA and its effectors have been linked to cancer (Lazer and Katzav, 2011), the molecular mechanisms that regulate its activity and involvement in particular steps of the metastatic cascade, such as during EMT, are not well understood. Here we investigate the mechanistic links between cell stiffness and stiffening response and the increased invasion capacity acquired after TGF-β–initiated EMT cancer progression.
RESULTS
TGF-β–induced EMT alters stiffness and stiffness response to force on integrins
To determine the effect of EMT on cell stiffness and stiffening response, we induced EMT in normal murine mammary gland (NMuMG) epithelial cells, a well-established TGF-β–induced EMT model (Piek et al., 1999; Yu et al., 2002; Xie et al., 2004). A magnetic tweezers system (Fisher et al., 2006; O'Brien et al., 2008) was then used to apply force via integrins (Matthews et al., 2006; Guilluy et al., 2011) to the cytoskeleton through externally attached paramagnetic beads coated with fibronectin (FN). The viscoelastic response of a cell was observed by monitoring the displacement of a bound bead over time during force application (Figure 1A). To quantify the mechanical phenotype in terms of stiffness and stiffening response, we calculated the time-dependent compliance of the cell and fitted it to a Jeffrey model for viscoelastic liquids (Figure 1B; Larson 1999). The spring constant obtained during the first pulse of force provided a measure of stiffness, and by normalizing the spring constants of subsequent force pulses to the first, we obtained the stiffness-response to force, or stiffening response. Two classifications of mechanical response were observed: a stiffening response (Figure 1C) and a softening response (Figure 1D). TGF-β–induced EMT was verified by monitoring reduced E-cadherin levels (see later discussion of Figure 4A) and actin reorganization (insets, Figure 2, A and B).
FIGURE 1:
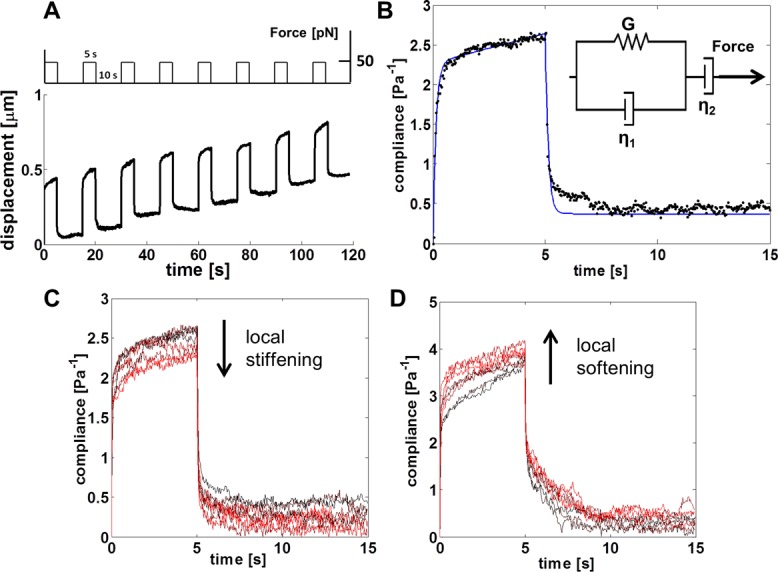
Modeling of cell stiffness and stiffness response. (A) Schematic of the magnetic tweezers experiment: a 50-pN force was applied for 5 s, followed by a 10-s relaxation time, for a total of eight pulls. The time-dependent displacement for a typical bead is shown below the force regimen. (B) The time-dependent compliance (black data points) is calculated from the displacement of a bead and the applied force. The Jeffrey model (inset) is a mechanical circuit that models the elastic (or stiffness, G) and viscous (η1 and η2) responses for a viscoelastic liquid during force application. The Jeffrey model (blue line) was used to quantify the stiffness of the cell as measured during force application. In experiments, the cell stiffness was defined to be the stiffness obtained in modeling the compliance of the cell during the first pulse of force. (C, D) Compliance signatures for representative examples of (C) cell stiffening and (D) cell softening, where time progression is encoded by increasing intensity of red, such that black is the compliance during the first pulse of force. To examine the stiffness of cells in response to force, we used the Jeffrey model to quantify the stiffness for each pulse of force during the 2-min experiment. The obtained stiffness measurements were normalized to the stiffness derived from the first pulse of force (G1) to give relative force response fractions G2/G1 through G8/G1. The full, nonoverlapping compliance signatures are provided in Supplemental Figure S1, C and D.
FIGURE 4:
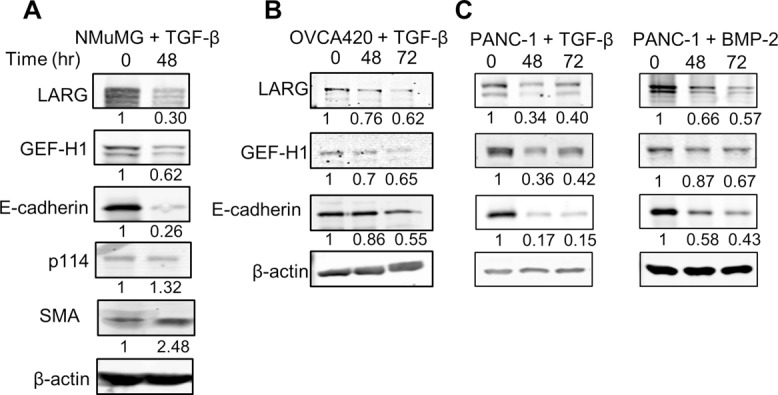
LARG and GEF-H1 expression decreases during EMT. (A, B) Indicated cell lines were treated with 100 pM TGF-β for 48 or 72 h. Cells were then lysed, and protein expression levels were analyzed by Western blot. A representative blot of four independent experiments. (C) PANC-1 cells were treated with 300 ng/ml BMP-2 for 48 and 72 h. Cells were then lysed, and protein expression levels were analyzed by Western blot. A representative blot of four independent experiments.
FIGURE 2:
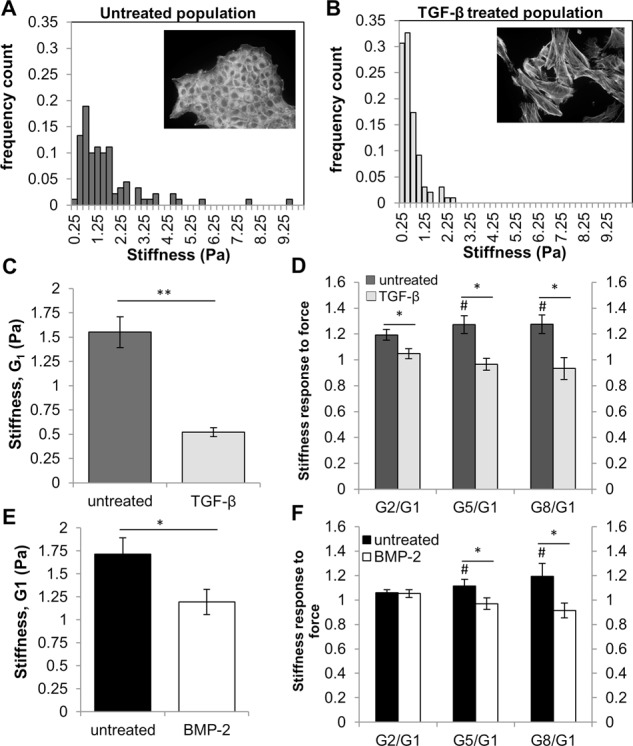
Stiffness and stiffness response to force decrease during TGF-β induced EMT. (A, B) NMuMG cells were treated with 100 pM TGF-β for 48 h to induce EMT. Histogram of NMuMG cell stiffness for (A) epithelial state (untreated) and (B) mesenchymal-state. Insets, fluorescence images are cells stained with phalloidin to show actin structures. (C) Average cell stiffness of NMuMG cells for untreated (n = 90) and TGF-β treated (n = 98) populations. **p < 0.001. (D) Average stiffness response for untreated (n = 30) and TGF-β treated (n = 25) populations. #Stiffness difference of Gx from G1 at the p < 0.05 level. *Stiffness response (Gx/G1) difference between conditions at the p < 0.05 level. (E) PANC-1 cells were treated with 300 ng/mL BMP-2 for 72 h to induce EMT. Average PANC-1 cell stiffness for untreated (n = 86) and BMP-2 treated cells (n = 61). *Stiffness difference relative to untreated cells at the p < 0.05 level. (F) Average PANC-1 stiffness response for untreated (n = 20) and BMP-2 treated (n = 15) cells. #Stiffness difference of Gx from G1 at the p < 0.05 level. *Stiffness response (Gx/G1) difference between conditions at the p < 0.05 level. Error bars represent SEM; data were collected from three independent experiments.
Mechanical characterization demonstrated a population-level shift toward lower stiffness in TGF-β–treated NMuMG cells compared with untreated cells (Figure 2, A and B). In addition, the average stiffness of mesenchymal cells was threefold less than that of epithelial cells (Figure 2C). In response to successive pulses of force, epithelial cells increased their stiffness (Figure 2D) significantly after 1 min or five pulses of force (Figure 2D). After TGF-β–induced EMT, this stiffening response to force was lost, indicating that mesenchymal cells are unable to fully adjust their stiffness in response to external force (Figure 2D). To investigate whether these mechanical changes after EMT are specific to NMuMG cells or to EMT in general, we examined human pancreatic carcinoma (PANC-1) cells, which undergo EMT in response to bone morphogenetic protein-2 (BMP-2; Gordon et al., 2009) and TGF-β. Similar to post-EMT NMuMG cells, PANC-1 cells exhibited decreased stiffness when treated with BMP-2 (Figure 2E) and TGF-β (Supplemental Figure S1C) and a loss of the normal stiffening response to force on integrins after 1 min (Figure 2F and Supplemental Figure S1D, respectively).
To determine whether the decrease in stiffness observed after EMT was a result of the force magnitude applied during the magnetic tweezers (active microrheology) assay, we performed an external passive microrheology assay to measure the basal stiffness of NMuMG cells before and after TGF-β–initiated EMT. Consistent with observations with the magnetic tweezers assay, measurement of the thermal motion of FN-coated beads revealed a threefold decrease in cell stiffness upon TGF-β treatment (Figure 3, A and B).
FIGURE 3:
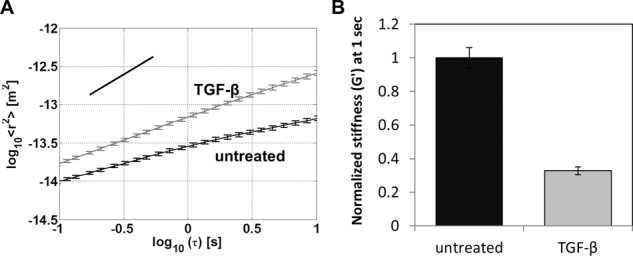
Stiffness of NMuMG cells measured by passive microrheology. (A) Mean-squared displacement (MSD) of 2 μm, FN-coated beads as a function of time scale between 0.1 and 10 s for NMuMG cells with or without TGF-β treatment; each curve is the ensemble average of cell populations: untreated (n = 1050) and TGF-β treated (n = 1010). MSD curves show a slope of <1 (illustrated by the black guide line), indicating subdiffusive viscoelastic response of beads anchored to the cortical actin cytoskeleton through integrin receptors (Wirtz, 2009). Data were taken using a high-throughput microscopy system described previously (Spero et al., 2008). Error bars represent SEM; the MSD of beads attached to untreated and TGF-β treated NMuMG were statistically significant at *p < 0.001 for all time scales. (B) Effective stiffness, G′ (Pa), of NMuMG cells with or without TGF-β treatment at the 1-s time scale. Using the MSD trajectories computed in Supplemental Figure S1B, we calculated the complex, frequency-dependent shear modulus, G*(ω), using the generalized Stokes–Einstein relation, and the elastic contribution is shown at the 1-s time scale.
To dynamically probe areas of increasing and variable stiffness, as seen in cancer ECM (Butcher et al., 2009), cells use integrin-associated focal adhesions as individual and autonomous stiffness sensors (Plotnikov et al., 2012). As such, to examine whether specific binding to integrins was required to elicit a stiffening response during force application, we used poly-d-lysine (PDL)–coated beads, which bind nonspecifically to the cell surface based on charge. In contrast to the response observed with FN-coated beads and consistent with findings in endothelial cells (Collins et al., 2012), force applied to PDL-coated beads did not evoke a stiffening response (Supplemental Figure S1E). Thus the stiffening response to force on FN-coated beads is specific to integrin-mediated attachment to the cytoskeleton.
Cells undergoing EMT undergo changes in the expression of many receptors (Ranganathan et al., 2007). To exclude the possibility that reduction in cell stiffness and stiffening response during EMT was due to loss of integrin expression, we examined expression of α5 and β1 integrins, the primary receptors for FN. We observed no significant reduction in either α5 or β1 level post-EMT in NMuMGs (Supplemental Figure S1F), indicating that the reduction in mechanical properties was not due to reduction of FN receptor expression. Taken together, these data indicate that cells undergo a reduction in stiffness and stiffening response after EMT.
RhoA GEF expression and recruitment to the adhesion complex during force is lost after TGF-β–induced EMT
Forces applied to integrins increase RhoA activity via Rho GEFs and, in turn, induce a stiffening response through reinforcement and rearrangement of the cytoskeleton and adhesion complexes (Guilluy et al., 2011). On the basis of reduction of stiffness and stiffening response after EMT (Figure 2, C and D), we hypothesized that this could be caused by altered RhoA activity through down-regulation of specific Rho GEFs. Indeed, expression levels of two Rho GEFs, LARG and GEF-H1, were significantly reduced after EMT, whereas expression of p114, another Rho GEF, was unchanged (Figure 4A). Similar results were obtained in TGF-β–induced EMT in OVCA420 and PANC-1 cells and BMP-2–induced EMT in PANC-1 cells, respectively (Figure 4, B and C). These findings suggested specific roles for LARG and GEF-H1 in cell stiffness and stiffening response during EMT.
To test whether EMT affects GEF recruitment to sites of force application, we developed a rotating permanent magnet device (Supplemental Figure S2A) to generate a force regimen consistent in magnitude, duty cycle, and frequency with that produced by the magnetic tweezers (Supplemental Figure S2B) and pulsatile forces to the cells via externally attached FN-coated beads. After force stimulation, we separated the bead fraction containing adhesion complex proteins from the whole lysate (Guilluy et al., 2011) and found that LARG and GEF-H1 were recruited in a time-dependent manner in epithelial-state NMuMG cells. In contrast, TGF-β–induced EMT abrogated this time-dependent recruitment of LARG and GEF-H1 (Figure 5A). Examination of p114, another RhoGEF, showed no recruitment to the adhesion complex and was unchanged after EMT (Figure 5A), suggesting that LARG and GEF-H1 have specific roles in force transduction during EMT. Similar loss of force-dependent recruitment of LARG and GEF-H1 was found in PANC-1 cells treated with BMP-2 (Figure 5B).
FIGURE 5:
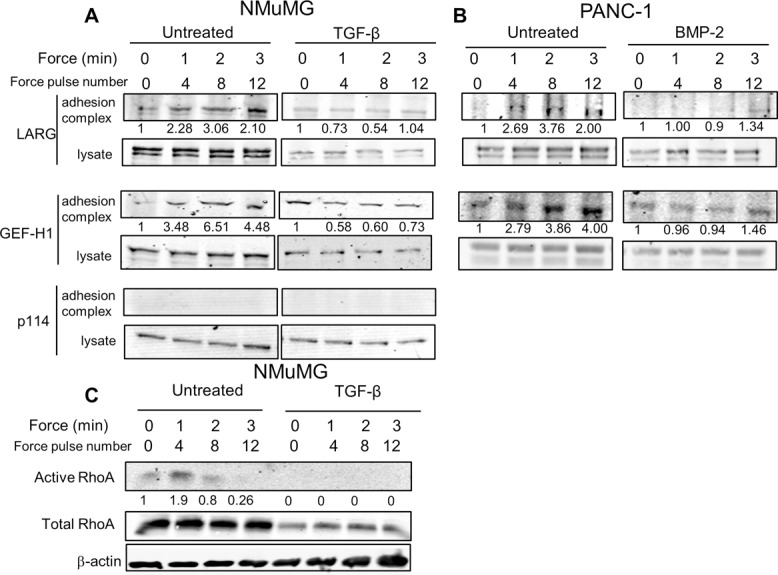
LARG and GEF-H1 recruitment to adhesion complex during force decrease during TGF-β–induced EMT. (A, B) Effect of EMT on RhoA GEF recruitment in either NMuMG (A) or PANC-1 (B) cells. Indicated cells were incubated for 30 min with FN-coated beads (Guilluy et al., 2011) and stimulated with a force regimen (50 pN; 5 s force, 10 s recovery) using a rotating permanent magnet for different amounts of time (Materials and Methods). After magnetic separation of the adhesion complex, both the lysate and adhesion complex fractions were analyzed using Western blots. Representative of four independent experiments. Associated quantification of amount of protein in adhesion complex (bead-to-lysate ratios), relative to untreated cells without force stimulation, is provided. (C) Effect of EMT on RhoA activation in NMuMG cells. Cells were stimulated with a force regimen using a rotating permanent magnet as in A. RhoA activity in lysates was determined as described (Guilluy et al., 2011). Representative of three experiments. Associated quantification of amount of protein in adhesion complex (bead-to-lysate ratio), relative to untreated cells without force stimulation, is provided.
Given our observations of post-EMT reduction in LARG and GEF-H1 expression and force-dependent recruitment, we hypothesized that RhoA activity in response to force would also be reduced after EMT. Using glutathione S-transferase–RBD-coated beads to pull down active RhoA from cell lysates (Guilluy et al., 2011), we found that epithelial-state NMuMG cells activated RhoA within 1 min of force application and that this response was lost after EMT induction (Figure 5C). Of interest, this 1-min time scale was consistent with the point in which the stiffening response in epithelial-state cells becomes significant (Figure 2D). We also found that RhoA expression was significantly reduced after EMT, in line with previous observations (Ozdamar et al., 2005). Taken together, the suppression of force-dependent RhoA activity and the loss of RhoA expression and force-dependent recruitment of LARG and GEF-H1 suggest that disruption of the Rho pathway plays a role in altered stiffness response to force during EMT.
TGF-β regulates Rho GEF expression during EMT by enhancing proteasomal degradation via activin receptor–like kinase 5 signaling
TGF-β can regulate activation of the RhoA pathway via canonical (activin receptor–like kinase 5 [ALK5] dependent) and noncanonical (mitogen-activated protein kinase dependent) TGF-β signaling mechanisms (Bhowmick et al., 2001). To investigate how TGF-β regulates LARG and GEF-H1 expression during EMT, we used the ALK5 inhibitor SB-431542 (Alk et al., 2002) and found that ALK5 inhibition partially rescued TGF-β–mediated LARG and GEF-H1 protein down-regulation in both NMuMG and OVCA420 cells, indicating a requirement for ALK5 in maximal regulation of RhoGEF expression (Figure 6A). In contrast, blocking the mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase pathway with U0126 (Alk et al., 2002), which can also mediate TGF-β responses (Xu et al., 2009), did not ameliorate TGF-β–dependent decreases in LARG and GEF-H1 levels in NMuMG cells (Figure 6A).
FIGURE 6:

TGF-β promotes proteasome degradation of LARG and GEF-H1 during EMT via Alk5. (A) Indicated cells were pretreated for 1 h with 10 μM SB-431542 (ALK5 inhibitor), 10 μM U0126 (MEK inhibitor), or dimethyl sulfoxide (DMSO; negative control), followed by treatment with 100 pM TGF-β for 48 h. (B) NMuMG cells were pretreated for 1 h with 10 μM SB-431542 (ALK5 inhibitor) or DMSO, followed by treatment with 100 pM TGF-β for 48 h or DMSO. Average cell stiffness for DMSO (n = 55), DMSO and TGF-β (n = 84), and SB-431542 and TGF-β (n = 81) populations. *p < 0.01. Error bars represent SEM; data were collected from three independent experiments. (C) NMuMG cells were pretreated for 1 h with 10 μM SB-431542 (ALK5 inhibitor) or DMSO, followed by treatment with 100 pM TGF-β for 48 h or DMSO. Average stiffness response for DMSO (n = 31), DMSO and TGF-β (n = 20), and SB-431542 and TGF-β (n = 26) populations. *p < 0.05. Error bars represent SEM; data were collected from three independent experiments. (D) NMuMG cells transfected with GFP-tagged LARG or GEF-H1cDNA lacking the 3′-UTR were treated with TGF-β for 48 h. Lysates were immunoblotted to determine expression of endogenous and exogenous GFP-tagged LARG/GEFH1. (E) TGF-β treatment does not reduce mRNA levels of LARG or GEF-H1. mRNA levels determined by quantitative PCR were normalized to glyceraldehyde-3-phosphate dehydrogenase. Error bars represent SEM. The dashed line denotes untreated. (F) TGF-β promotes proteasome degradation of LARG and GEF-H1. (D) OVCA420 cells were treated for 72 h with TGF-β and with 10 and 20 μM MG-132 or DMSO as control. (E) NMuMG cells treated with TGF-β for 48 h and 10 μM MG-132 or DMSO as control for the last 16 h of treatment for both D and E. Representative of at least three experiments.
Given that LARG and GEF-H1 have been shown to regulate cell mechanics (Guilluy et al., 2011) and expression of LARG and GEFH-1 was regulated via an ALK5-dependent mechanism, we hypothesized that the effect of TGF-β on cell mechanics would be dampened upon ALK5 inhibition. Using the ALK5 inhibitor SB-431542 to partially restore LARG and GEF-H1 levels (Figure 6A), we find that NMuMG cell stiffness (Figure 6B) and stiffness response (Figure 6C) were significantly increased compared with cells treated with only TGF-β. These results directly implicate canonical TGF-β signaling mechanisms downstream of ALK5 in regulating cell mechanics during EMT.
RhoA and the RhoA GEF Net1A have been shown to be down-regulated during TGF-β–induced EMT via a microRNA-based mechanism (Kong et al., 2008; Moustakas and Heldin, 2012) that acts on the 3′-untranslated region (UTR) to induce translational silencing of proteins (He and Hannon, 2004). To test whether microRNAs play a role in TGF-β down-regulation of LARG and GEF-H1, we examined the extent of down-regulation of exogenously expressed LARG or GEF-H1 cDNA containing an N-terminus green fluorescent protein (GFP) tag but lacking a 3′-UTR. We found that exogenous LARG and GEF-H1 were down-regulated to similar extents as their endogenous counterparts (Figure 6D), suggesting that microRNAs may not play a significant role in the TGF-β–induced decreases of LARG and GEF-H1.
Because TGF-β regulates the transcription of many genes during EMT (Xu et al., 2009), we examined whether EMT altered transcription of LARG and GEF-H1. We found that although TGF-β caused down-regulation of E-cadherin, we observed no significant down-regulation of LARG and GEF-H1 at the mRNA level (Figure 6E). Therefore we examined the role of the proteasome in regulating LARG and GEF-H1 levels, which has been shown to regulate TGF-β–dependent levels of RhoA (Wang et al., 2003). We found that the proteasome inhibitor MG-132 was able to rescue down-regulation of LARG and GEF-H1 protein levels after TGF-β–induced EMT in both NMuMG and OVCA420 cells (Figure 6F). These data implicate proteasome-mediated regulation of the RhoGEFs LARG and GEFH-1 during EMT via an ALK5-dependent mechanism.
Epithelial-state cells partially adopt post-EMT mechanical and invasion phenotypes through silencing of LARG and GEF-H1
EMT marks the physical initiation of cancer progression as a cell detaches from the primary tumor and invades the surrounding stromal space. Alterations in the RhoA pathway have been implicated in a variety of cancers (Simpson et al., 2004; Vega et al., 2011). However, whereas RhoA activation in some cancers is associated with increased invasion (Liao et al., 2012), in others, RhoA activation inhibits cell invasion (Bellovin et al., 2006). In addition, RhoA and its associated GEFs LARG and GEF-H1 have been shown to regulate the stiffening response to force applied on integrins (Guilluy et al., 2011). On the basis of these and our findings that LARG and GEF-H1 expression and force-dependent recruitment are reduced in multiple EMT models (Figures 4A and 5A) and that ALK5 inhibition reverses the TGF-β–mediated reduction in cell mechanics, we examined whether LARG and GEF-H1 down-regulation were sufficient for decreased stiffness and stiffening response to force. Using small interfering RNA (siRNA) to reduce GEF expression in NMuMG epithelial-state cells (Figure 7A), we found that silencing LARG, GEF-H1, or both significantly decreased cellular stiffness (Figure 7B) and stiffening response compared with control siRNA–treated cells (Figure 7C). In contrast, siRNA to p114 had no significant effect on stiffness (Figure 7B) or suppression of the stiffening response to force (Figure 7C), suggesting a specific role for LARG and GEF-H1 in determining these mechanical properties.
FIGURE 7:
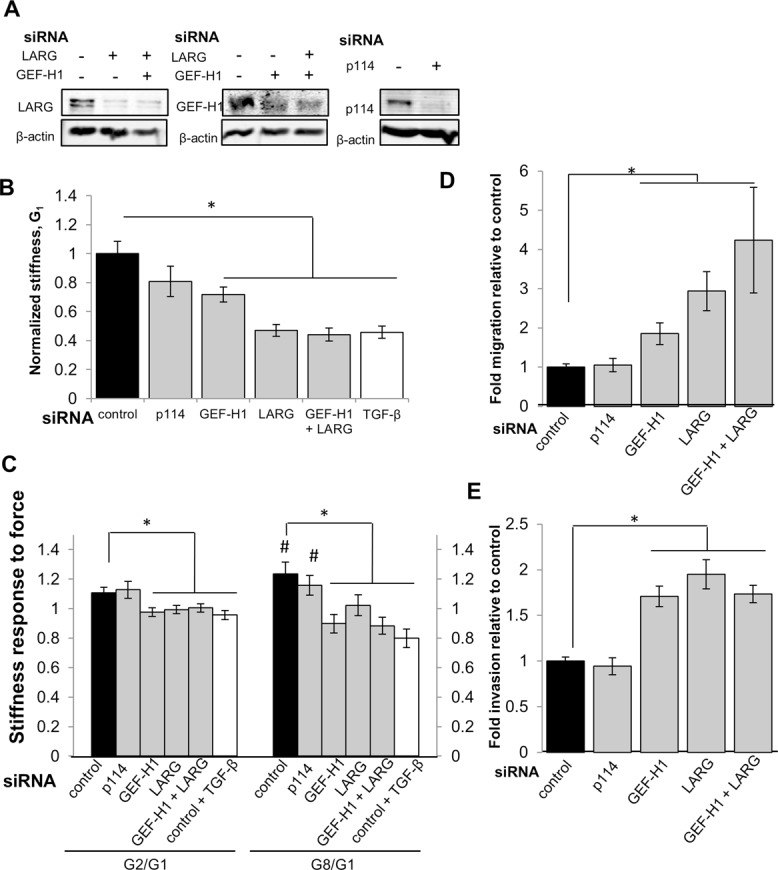
LARG and GEF-H1 knockdowns decrease cell stiffness and stiffness response to force and increase cell migration and invasion. (A) NMuMG cells were transfected with siRNA against LARG, GEF-H1, both, or p114 for 48 h. Quantifications are given normalized to β-actin and relative to control, confirming knockdown of protein expression. (B) Average cell stiffness (G1) for NMuMG cells treated with siRNA control (n = 88), siRNA targeting p114 (n = 70), GEF-H1 (n = 90), LARG (n = 85), GEF-H1 + LARG (n = 100), and NMuMG cells treated with TGF-β and siRNA control (n = 69). *p < 0.01. Error bars represent SEM; data were collected from three independent experiments. (C) Average stiffness response at pulls 2 (G2/G1) and 8 (G8/G1) for NMuMG cells treated with siRNA control (n = 20), siRNA targeting p114 (n = 41), GEF-H1 (n = 19), LARG (n = 21), GEF-H1 + LARG (n = 30), and NMuMG cells treated with TGF-β and siRNA control (n = 19). #p < 0.05 stiffness difference from G1; *p < 0.05 stiffness response difference relative to cells treated with siRNA control. Error bars represent SEM; data were collected from three independent experiments. (D, E) Average migration and invasion for NMuMG cells treated with siRNA as indicated: control, GEF-H1, LARG, both, or p114 for 36 h before plating onto uncoated or Matrigel-coated Transwell filters. *p < 0.05. Error bars represent SEM; data represent mean of three independent experiments.
Given that we and others previously reported an inverse correlation between cell stiffness and invasion (Swaminathan et al., 2011; Xu et al., 2012), we examined the effect of GEF silencing on migration and invasion using siRNA to deplete protein levels in epithelial-state NMuMG cells. We found that silencing LARG, GEF-H1, or both simultaneously significantly increased cell migration and invasion (Figure 7, D and E) compared with control siRNA cells. In contrast, siRNA to p114 did not significantly alter cell migration or invasion (Figure 7, D and E). In line with these results, we found that specific silencing of LARG and GEF-H1 expression in epithelial cells increases invasion capacity toward that of post-EMT mesenchymal cells.
Mesenchymal-state cells recover epithelial-state mechanical and invasion phenotypes through rescue of LARG or GEF-H1 expression
Our results indicate that LARG and GEF-H1 are necessary mediators of stiffness (Figure 7B) and stiffness response (Figure 7C). To examine whether down-regulation of LARG and GEF-H1 was necessary and sufficient to restore post-EMT loss of stiffness and stiffness response phenotypes, we rescued GEF expression after EMT induction and performed mechanical and invasion assays. Post-EMT NMuMG cells were transfected with plasmids containing cDNA to encode GFP-tagged LARG and GEF-H1 to rescue the reduced expression (Figure 8A). Mechanical measurements were performed only on LARG- or GEF-H1–expressing cells, as identified by GFP expression. We found that restoring LARG or GEF-H1 expression rescued the 50% post-EMT reduction in cell stiffness to 70 and 80% of the GFP-control stiffness, respectively (Figure 8B). Similarly, restoring LARG or GEF-H1 expression rescues the 30% post-EMT reduction in stiffening response to 90% of the response observed in GFP-control cells (Figure 8C).
FIGURE 8:
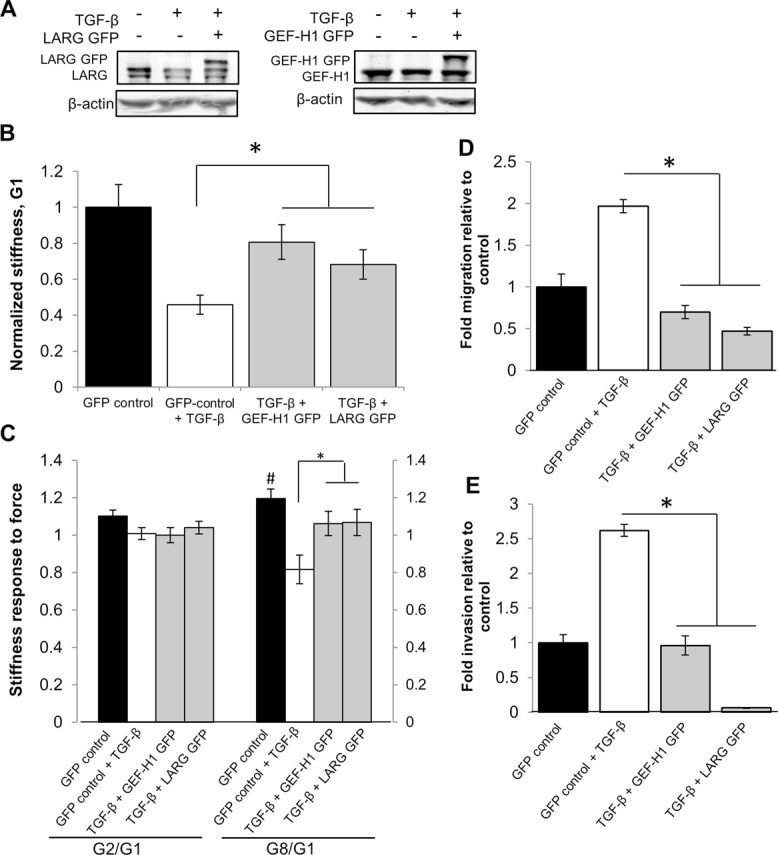
LARG and GEF-H1 overexpression after EMT partially rescues cell stiffness and stiffness response and attenuates migration and invasion. (A) NMuMGs were treated with 100 pM TGF-β for 48 h. Cell were transfected with plasmid containing LARG-GFP or GEF-H1-GFP for the final 16–24 h of treatment. Quantifications are shown normalized to β-actin and relative to control, confirming protein expression. (B) Average cell stiffness (G1) for cells transfected with GFP vector control (n = 65), TGF-β and GFP vector control (n = 55), and GFP-DNA constructs to overexpress GEF-H1 (n = 32) or LARG (n = 54). *p < 0.05 denotes stiffness difference relative to cells treated with TGF-β and GFP vector control. Error bars represent SEM; data were collected from three independent experiments. (C) Average force response at pulls 2 (G2/G1) and 8 (G8/G1) for NMuMG cells transfected with GFP vector control (n = 25), TGF-β and GFP vector control (n = 15), and GFP-DNA constructs to overexpress GEF-H1 (n = 16) or LARG (n = 23). #p < 0.05 denotes stiffness difference from G1; *p < 0.05 denotes stiffness response difference relative to cells treated with TGF-β and GFP vector control. Error bars represent SEM; data were collected from three independent experiments. (D, E) Average migration and invasion for cells treated with TGF-β and transfected with plasmid containing empty vector control (GFP), LARG-GFP, or GEF-H1 GFP for the final 16–24 h of treatment and sorted by flow cytometry for GFP expression. Quantifications given as fold migration or invasion relative to control (n = 3). *p < 0.05.
We previously observed that loss of LARG and GEF-H1 expression in epithelial cells increased migration and invasion (Figure 7, D and E). To determine whether restoring the expression of these GEFs also affects cell migration and invasion, we used fluorescence-activated cell sorting (FACS) to isolate GFP-expressing cells and then performed migration and invasion assays with these cells. We found that overexpression of LARG or GEF-H1 fully suppressed EMT-induced increases in migration and invasion (Figure 8, D and E) compared with control GFP cells, thus establishing LARG and GEF-H1 as being sufficient for the mechanical and invasion phenotypes obtained during TGF-β–initiated EMT.
DISCUSSION
To examine the role of cell mechanics in EMT, we used force-consistent biophysical and biochemical assays to characterize the mechanistic links between stiffness response and cell invasion during EMT. We demonstrate that epithelial-state cells respond to force on integrins by evoking a stiffening response and that after EMT, mesenchymal-state cells have reduced stiffness but also lose the ability to increase their stiffness in response to force. Using loss- and gain-of-function studies, we establish two RhoA activators, LARG and GEF-H1, as both necessary and sufficient mediators of the effect of EMT on stiffness and stiffness response. We determine that TGF-β mediates proteasome degradation of LARG and GEF-H1 via ALK5 and that reduction of these RhoA activators contributes significantly to the increase in migration and invasion behavior during EMT. Here we discuss the potential utility of altered stiffness mechanics in EMT, the significance of GEFs and the RhoA pathway to mesenchymal cell invasion, and the mechanisms by which TGF-β interacts with the RhoA pathway during EMT.
In examining the mechanical phenotype adopted during EMT, we established that post-EMT cells have a decreased stiffness and stiffening response compared with epithelial-state cells. A full understanding of the biological significance of these mechanical changes will require additional investigation, but at present a few hypotheses can be described. First, these results support a model in which epithelial cells resist external deformation by mounting a stiffening response to maintain mechanical equilibrium and prevent potential injury (Glogauer, 1998; Matthews et al., 2006). During EMT and associated epithelium detachment, a stiffening response to force may lose its utility to mesenchymal-state cells, as adaptive stiffness near integrins may increase adhesion size and strength and thus hinder the adhesion turnover required for effective cell migration and invasion. Second, increasing evidence supports the long-standing belief that a certain degree of deformability, or reduced stiffness, is required for metastatic cells to navigate the basement membrane and intravasate and extravasate the vascular system (Suresh et al., 2005; Wirtz et al., 2011). Third, an attenuated cortical stiffness and stiffening response to force after EMT may reduce the internal resistance that cell-generated forces act against to facilitate cancer cell invasion behavior. One consequence of reduced stiffness is that for a given cell-generated force, post-EMT cells could exert higher actomyosin contractile forces to the ECM. Such increased contractile forces have been shown to be correlated with cells of increasing metastatic potential (Kraning-Rush et al., 2012) and are specifically required for mesenchymal invasion in certain cancers (Friedl and Wolf, 2003). Another effect of reduced stiffness to EMT may arise in the generation of contractile forces that are tuned for mechanosensing (de Rooij et al., 2005; Guo et al., 2006). To dynamically probe areas of increasing and variable stiffness, as seen in cancer ECM (Butcher et al., 2009), cells use integrin-associated focal adhesions as individual and autonomous stiffness sensors (Plotnikov et al., 2012). Migration through these regions is believed to involve appropriate temporal regulation of cell-generated, actomyosin-contractile forces (Plotnikov and Waterman, 2013). These forces are transmitted to the ECM via integrin adhesions and must act through an internal stiffness, which is likely the stiffness of the actin cytoskeleton network. Thus our results suggest that reduced stiffness and stiffening response post-EMT to external force application may enable, or increase the efficiency of, mechanosensing mechanisms during cancer cell invasion.
EMT marks the physical initiation of cancer progression as a cell detaches from the primary tumor and invades the surrounding stromal space. Invasion is a complex process, however, and it is known that cancer cells can adjust between amoeboid and mesenchymal motility modes depending on the particular ECM environment (Wolf et al., 2003; Sanz-Moreno et al., 2008; Sanz-Moreno and Marshall, 2010). During EMT, generally immobile epithelial cells acquire mesenchymal motility, characterized by protease-dependent degradation of the ECM, Rac1 GTPase-regulated lamellipodial protrusions at the leading edge, and tightly controlled and appropriate RhoA- and RhoC-dependent actomyosin contractility at the cell rear that results in disassembly of FAs and retraction of the trailing edge (Yilmaz and Christofori, 2009). Although evidence indicates that the GEF/RhoA pathway is altered in numerous cancers (Simpson et al., 2004; Vega et al., 2011), its role is complicated, with often conflicting reported results. RhoA activation in some cancers is associated with increased invasion (Liao et al., 2012), whereas in other models, its activation inhibits cell invasion (Bellovin et al., 2006). Studies using HeLa cells (Nalbant et al., 2009) and retinal pigment epithelia cells (Tsapara et al., 2010) showed that GEF-H1 mediates cell migration in wound-healing assays and standard invasion assays. Another group used NMuMG cells and found that GEF-H1 is required for invasion across compliant collagen gels (Heck et al., 2012). Of interest, observations in keratinocytes have shown that the RhoA GEF Net1A is specifically down-regulated during TGF-β–induced EMT (Papadimitriou et al., 2011). Furthermore, work with human breast cancer cells suggests that regulation of RhoA by GEFs alone may initiate EMT invasion, as mesenchymal invasion was promoted over amoeboid by knockdown of either Net1A or RhoA (Carr et al., 2013). We find that LARG and GEF-H1 are down-regulated during EMT (TGF-β and BMP-2 induced) in multiple epithelial models. In addition, silencing expression of LARG and GEF-H1 increases invasive capacity toward that of post-EMT mesenchymal cells. Our data are consistent with a model in which mesenchymal invasion occurs concurrently with decreased stiffness and is mediated via TGF-β regulation of RhoA activity to enable passage through the basement membrane and ECM required during EMT.
TGF-β mediates cellular functions during EMT via both canonical Smad-dependent pathways, as well as by noncanonical Smad-independent pathways, with canonical pathways typically being downstream of the type I TGF-β receptor (ALK5). Mechanisms of TGF-β regulation include alterations of gene transcription (Massagué, 2012), microRNA-mediated translational silencing (Winter et al., 2009), and enhanced proteasome degradation via increased polyubiquitination (Ozdamar et al., 2005). We established that TGF-β enhances proteasomal degradation of LARG and GEF-H1 via an ALK5-dependent pathway and that microRNAs do not seem to play a prominent role in the regulation of these GEFs. TGF-β was previously reported to target Net1A, another RhoA GEF, for proteasome degradation, although translational silencing by miR-24 also contributes to its down-regulation (Papadimitriou et al., 2011). Of interest, TGF-β has also been shown to target RhoA for proteasome degradation by activating the ubiquitin ligase Smurf1 via polarity protein Par6 (Ozdamar et al., 2005). Tight spatial and temporal regulation of RhoA and associated GEFs by TGF-β is crucial during the EMT program. Early in EMT, loss of RhoA activation and destabilization of microtubules at the basal surface of epithelial cells causes loss of cell–basement membrane interactions (Nakaya et al., 2008). Of interest, transient up-regulation of Net1A has been shown to be required for EMT initiation, but by 24 h, Net1A levels are subsequently depleted as the cell progresses through EMT (Papadimitriou et al., 2011), perhaps to allow cells to acquire mesenchymal motility as discussed earlier.
In this work, we established that LARG and GEF-H1 down-regulation is critical for the mechanical alterations that occur during EMT, and that the down-regulation of these proteins facilitates cell invasion. Our results connect for the first time the origin of the stiffness and invasion relationship to EMT and highlight the importance of mechanics and physical forces in cancer cell invasion.
MATERIALS AND METHODS
Cell culture and transfection
NMuMG cells were maintained in high-glucose DMEM (Life Technologies) containing 10% fetal bovine serum (FBS), 10 μg/ml insulin, penicillin/streptomycin (Life Technologies, Carlsbad, CA), and 15 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer. PANC-1 cells were maintained in high-glucose DMEM (Life Technologies) containing 10% FBS, penicillin/streptomycin, and 15 mM HEPES buffer. OVCA420 cells were maintained in RPMI containing 10% FBS and penicillin/streptomycin. The NMuMG, PANC-1 and OVCA420 cell lines were maintained as described previously (Elbendary et al., 1994; Kowanetz et al., 2004; Gordon et al., 2009).
Immunoblotting
Cell lysates were prepared in Laemmli sample buffer. Before loading, they were heated at 95°C for 5–10 min and centrifuged at 13,000 rpm for 1 min. Samples were resolved by 12% SDS–PAGE for RhoA or 7.5% SDS–PAGE for other proteins. Proteins were transferred to nitrocellulose or Immobilon-FL membranes, blocked with Odyssey blocking buffer (LI-COR), and then immunoblotted with primary antibodies overnight. Fluorophore-conjugated secondary antibodies were used to bind primary antibodies, and fluorescence was detected and quantified using the Odyssey system (LI-COR).
The following antibodies were used: GEF-H1 (1:1000; 4076S; Cell Signaling Technology, Danvers, MA), LARG (1:1000; ab86095; Abcam, Cambridge, UK), p114 (1:1000; a gift from Keith Burridge, University of North Carolina at Chapel Hill), RhoA (1:1000; 2117; Cell Signaling Technology), E-cadherin (1:1000; 610182; BD Transduction: BD Biosciences, San Jose, CA), smooth muscle actin (1:2500; A5228; Sigma-Aldrich), and β-actin (1:2500; A5441; Sigma-Aldrich).
Real-time PCR
Cells were plated and treated in six-well plates. Total RNA was harvested using the RNeasy Mini Kit (Qiagen, Venlo, Netherlands), and 600 ng of RNA per sample was reverse transcribed to cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). Real-time (RT) PCR was then performed by mixing iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) with the respective forward and reverse primers. The following primers were used for RT-PCR:
GEF-H1 forward: 5′-CAG TTT GAG AGG AAC CCG GC-3′
GEF-H1 reverse: 5′-ACT CCT CCC AGA TCA AGG CA-3′
LARG forward: 5′-GGG ACC GAA GCC GAC G-3′
LARG reverse: 5′-TTC GTC ACA GAG GGA AGT CG-3′
P114 forward: 5′-CTA GAG GAG GGC AGT GAT CG-3′
P114 reverse: 5′-CCT TCA TTC TCT CCC CGG TT-3′
E-cadherin forward: 5′-CCT TCC CCC AAC ACG TCC CCC C-3′
E-cadherin reverse: 5′-TCT CCA CCT CCT TCT TCA TC-3′
GAPDH forward: 5′-TTG ACC TCA ACT ACA TGG TCT A-3′
GAPDH reverse: 5′-ACC AGT AGA CTC CAC GAC ATA C-3′
RNA interference and vector overexpression
siRNAs were purchased from Thermo Scientific. Cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). siRNA was mixed with Lipofectamine 2000 reagent in Opti-MEM media (Life Technologies) at manufacturer-recommended concentrations. The transfection media was removed after 12 h, and RNA interference was allowed to proceed for a total of 48 h. The following siRNAs were used.
Scramble nontargeting control (Thermo Scientific ON-TARGET plus D-001810-01-05):
Anti–GEF-H1: 5′-GGG CGA CGC UUU AUA CUU G-3′
Anti-LARG: 5′-GAU UAC AAC CGA ACG GCU A-3′
DNA plasmid transfection for overexpression assays was also carried out using Lipofectamine 2000. DNA was mixed with Lipofectamine 2000 reagent in Opti-MEM at the manufacturer-recommended concentrations. The transfection medium was removed after 12 h, and overexpression was allowed to continue for 48–72 h. Overexpressed proteins were GFP-tagged at the N-terminus. To select for cells overexpressing the protein of interest, we performed FACS for GFP-expressing cells. GEF-H1 cDNA was cloned into a PEGFP-n1 vector, and LARG cDNA was cloned into a pENTR 2B vector (Invitrogen).
RhoA activity assay
Cells were plated onto 10-cm dishes and treated with 100 pM TGF-β or not for 48 h. They were then incubated with fibronectin-coated magnetic beads and subject to force as described. Immediately after force application, cells were lysed in ice-cold lysis buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 500 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 200 μM sodium orthovanadate, 1% Triton X, inhibitors). The lysis reaction was allowed to proceed for 30 min in a rotator at 4°C. Beads and cell debris were then separated via centrifugation at 13,000 rpm for 15 min at 4°C. The clarified lysate was incubated with 30 μg of Rhotekin-RBD beads (Cytoskeleton, Denver, CO) for 1 h in a rotator at 4°C. The bead and lysate fractions were then separated via centrifugation at 5000 rpm for 2 min. The bead fractions were washed three times with wash buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton-X, inhibitors), and samples were prepared for SDS–PAGE with Laemmli sample buffer. The lysate fractions were also prepared for SDS–PAGE with Laemmli sample buffer.
Preparation of paramagnetic beads
Tosyl-activated magnetic Dynabeads (4.5 μm; Invitrogen) were incubated with FN for 1 h at room temperature on a spinning rotator. Beads were centrifuged and resuspended in a 10% Tris solution (0.86 M, 7.4 pH) to quench unoccupied tosyl groups on the bead surface. After 45 min of rotation at room temperature or overnight at 4°C, bead were concentrated again and resuspended in phosphate-buffered saline (PBS). Beads were vortexed or sonicated before use to prevent aggregation.
Magnetic tweezers mechanical assay
The University of North Carolina three-dimensional force microscope was used as a magnetic tweezers system to apply a 50-pN force pulse for 5 s, followed by a 10-s relaxation, to fibronectin-coated magnetic beads (4.5-μm diameter). The time-dependent displacement of bound beads was recorded using a high-speed video camera at 30 frames/s (Pulnix; JAI) and was tracked using CISMM Video Spot Tracker software (Center for Computer Integrated Systems for Microscopy and Manipulation [http://cismm.cs.unc.edu]). Before experiments began, we calibrated the magnetic tweezers system by applying a force ramp to magnetic beads (4.5-μm diameter) in a Newtonian fluid of known viscosity. By recording bead trajectories and computing bead velocities, we used Stokes' law, F = 6πaηv, to determine the force, where a is the bead radius, η is the fluid viscosity, and v is the bead velocity. Knowledge of the bead displacement r(t) and the applied force F(t) allowed us to compute the compliance signature, J(t) = 6πar(t)/F(t), which we then fitted to a Jeffrey (modified Kelvin–Voigt) mechanical circuit model for viscoelastic liquids. The spring constant is reported as the stiffness in pascals. The ability of a cell to locally respond to applied force, termed the stiffness response, was computed by normalizing stiffness values of subsequent force pulses to the stiffness. Two types of force response were observed: a stiffening response, for which Gn1/G1 > 1 (Supplemental Figure S1A), and a softening response, for which Gn1/G1 < 1 (Supplemental Figure S1B). Mechanical measurements were taken for 1 h after 20 min of bead incubation. To minimize unintentional force application, measurements were taken only for cells spaced several hundred micrometers apart. In addition, bead displacements below system resolution (10 nm) and greater than the diameter of the bead (4.5 μm) were not included in the analysis. Mechanical measurements are reported as the mean ± SEM.
Rotating permanent magnet and adhesion complex recruitment assay
To closely link force-dependent biology results from mechanical (magnetic tweezers) and biochemical approaches (protein expression and recruitment), we engineered a rotating permanent magnet system designed to be lowered into a standard 10–cm cell culture dish (Supplemental Figure S2A). A DC motor (ServoCIty, Winfield, KS) was used to rotate two custom-made, axially magnetized, 120° arc magnets (N52 grade; K&J Magnetics, Pipersville, PA) at 4 rpm at a height of 16 mm above the cells to generate a time-varying force of a magnitude (50 pN), duty cycle (5 s of force every 15 s, or 33%), and frequency (6.7 mHz) consistent with the force regimen used with the magnetic tweezers (Supplemental Figure S2B). To validate the device, we used engineering software (COMSOL Multiphysics) to simulate the magnetic field gradient and MATLAB to calculate the resulting force, given the material properties of the magnetic beads (Supplemental Figure S2B).
For the adhesion complex recruitment assay, cells were plated onto 10-cm dishes and treated with 100 pM TGF-β or not for 48 h. They were then incubated for 30 min at 37°C in serum-free medium with FN-coated magnetic beads. Force was applied using a rotating magnet described later. Immediately after force application, cells were lysed in ice-cold lysis buffer (20 mM Tris, 150 mM NaCl, 2 mM MgCl2, 1% NP-40, inhibitors). Beads were isolated from the lysate using a magnetic separation stand. The lysate was clarified with centrifugation at 13,000 rpm for 15 min at 4°C. Proteins in both fractions were denatured and reduced in Laemmli buffer.
Passive microrheology mechanical assay
Passive microrheology measurements used nonmagnetic beads (2 μm; Invitrogen) coated with fibronectin. Video data were acquired at 54 frames/s using an automated microscope system described previously (Spero et al., 2008). Brownian motion of the beads was tracked using CISMM Video Spot Tracker software, and the mean squared displacement (MSD) of the trajectories was computed using
where t is the elapsed time and τ is the time lag, or time scale. The stiffness of the cell, or the elastic modulus, was computed from the MSD using the generalized Stokes–Einstein relation (GSER). In short, the frequency-dependent elastic modulus was calculated using
where the magnitude of the complex viscoelastic modulus is given by
Γ is the gamma function, and α is the local logarithmic slope of <r2(τ)>. Values for G′ at a time lag of 1 s, the maximum time scale where thermal fluctuations are predominately Brownian, are reported as stiffness in pascals. Owing to computational challenges in evaluating statistical significance in the GSER formulism, statistical analysis was performed on the value of the MSD at the 1-s time scale to evaluate differences between cell populations.
Matrigel invasion and Transwell migration assays
Cells were trypsinized and seeded at a density of 50,000 cells/well on either Matrigel-coated (BD Biosciences) or uncoated (Corning) Transwell filters in a 24-well plate and allowed to invade for 12 h toward 10% FBS in the lower chamber. Cells invading and migrating through the Matrigel-coated and uncoated filters, respectively, were stained with Three Step stain (Richard-Allan Scientific). Each filter was counted in its entirety with four 10× fields, and invasion or migration was quantified as fold relative to control.
Immunofluorescence staining
Cells were plated onto coverslips at a sparse density. They were fixed with 4% paraformaldehyde for 5 min with 0.1% Triton-X in PBS and blocked with 1% BSA in PBS. Alexa Fluor 594 Phalloidin (Invitrogen) was then used to stain for actin. Stained cells on coverslips were then mounted onto slides using ProLong Gold Anti-Fade Reagent (Invitrogen) and imaged on a fluorescence microscope with a 40× air objective.
Statistical analysis
Measurements are reported as the mean ± SEM, and differences between conditions were analyzed with either a two-tailed, unpaired Student's t test or a two-tailed, paired Student's t test for difference measurements between corresponding samples.
Supplementary Material
Acknowledgments
We thank Keith Burridge for generous use of reagents; Christophe Guilluy, Caitlin Collins, Jeremy Cribb, and Vinay Swaminathan for helpful discussions; and Lisa Sharek for technical support. This work was supported by Ovarian Cancer Research Fund Grant 25875 (K.M.), Susan G. Komen for the Cure Grant SAC100002 (G.C.B.), and National Institutes of Health/National Cancer Institute Grants R33-CA155618 (R.S.) and R01-CA135006 (G.C.B.). G.L. is a Howard Hughes Medical Institute Research Fellow.
Abbreviations used:
- ALK5
activin receptor–like kinase 5
- BMP
bone morphogenetic protein
- EMT
epithelial-to-mesenchymal transition
- GEF
guanine nucleotide exchange factor
- NMuMG
normal murine mammary gland
- PANC-1
human pancreas carcinoma
- TGF-β
transforming growth factor β.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-05-1015) on August 20, 2014.
REFERENCES
- Alk K, Alk R, Inman GJ, As FJN, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Bellovin DI, Simpson KJ, Danilov T, Maynard E, Rimm DL, Oettgen P, Mercurio AM. Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene. 2006;25:6959–6967. doi: 10.1038/sj.onc.1209682. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9:108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr HS, Zuo Y, Oh W, Frost JA. Regulation of FAK activation, breast cancer cell motility and amoeboid invasion by the RhoA GEF Net1. Mol Cell Biol. 2013;33:2773–2786. doi: 10.1128/MCB.00175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C, Guilluy C, Welch C, O'Brien ET, Hahn K, Superfine R, Burridge K, Tzima E. Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr Biol. 2012;22:2087–2094. doi: 10.1016/j.cub.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol. 2005;171:153–164. doi: 10.1083/jcb.200506152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbendary A, Berchuck A, Davis P, Havrilesky L, Bast RC, Jr, Iglehart JD, Marks JR. Transforming growth factor beta 1 can induce CIP1/WAF1 expression independent of the p53 pathway in ovarian cancer cells. Cell Growth Differ. 1994;5:1301–1307. [PubMed] [Google Scholar]
- Fisher JK, Cribb J, Desai KV, Vicci L, Wilde B, Keller K, Taylor RM, II, Haase J, Bloom K, O'Brien ET, Superfine R. Thin-foil magnetic force system for high-numerical-aperture microscopy. Rev Sci Instrum. 2006;77:023702. doi: 10.1063/1.2166509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- Glogauer M. The role of actin-binding protein 280 in integrin-dependent mechanoprotection. J Biol Chem. 1998;273:1689–1698. doi: 10.1074/jbc.273.3.1689. [DOI] [PubMed] [Google Scholar]
- Gordon KJ, Kirkbride KC, How T, Blobe GC. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis. 2009;30:238–248. doi: 10.1093/carcin/bgn274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilluy C, Swaminathan V, Garcia-Mata R, O'Brien ET, Superfine R, Burridge K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol. 2011;13:722–727. doi: 10.1038/ncb2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Frey MT, Burnham NA, Wang Y. Substrate rigidity regulates the formation and maintenance of tissues. Biophys J. 2006;90:2213–2220. doi: 10.1529/biophysj.105.070144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Heck JN, Ponik SM, Garcia-Mendoza MG, Pehlke CA, Inman DR, Eliceiri KW, Keely PJ. Microtubules regulate GEF-H1 in response to extracellular matrix stiffness. Mol Biol Cell. 2012;23:2583–2592. doi: 10.1091/mbc.E11-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubchak SC. Cytoskeletal rearrangement and signal transduction in TGF-1-stimulated mesangial cell collagen accumulation. J Am Soc Nephrol. 2003;14:1969–1980. doi: 10.1097/01.asn.0000076079.02452.92. [DOI] [PubMed] [Google Scholar]
- Kong W, Yang H, He L, Zhao J, Coppola D, Dalton WS, Cheng JQ. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784. doi: 10.1128/MCB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowanetz M, Valcourt U, Bergström R, Heldin C-H, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol. 2004;24:4241–4254. doi: 10.1128/MCB.24.10.4241-4254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraning-Rush CM, Califano JP, Reinhart-King Ca. Cellular traction stresses increase with increasing metastatic potential. PloS One. 2012;7:e32572. doi: 10.1371/journal.pone.0032572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson R. The Structure and Rheology of Complex Fluids. New York: Oxford University Press; 1999. [Google Scholar]
- Lazer G, Katzav S. Guanine nucleotide exchange factors for RhoGTPases: good therapeutic targets for cancer therapy. Cell Signal. 2011;23:969–979. doi: 10.1016/j.cellsig.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry. 2012;51:7420–7432. doi: 10.1021/bi300758e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2010;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YC, Ruan JW, Lua I, Li MH, Chen WL, Wang JRY, Kao RH, Chen JH. Overexpressed hPTTG1 promotes breast cancer cell invasion and metastasis by regulating GEF-H1/RhoA signalling. Oncogene. 2012;31:3086–3097. doi: 10.1038/onc.2011.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BD, Overby DR, Mannix R, Ingber DE. Cellular adaptation to mechanical stress: role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J Cell Sci. 2006;119:508–518. doi: 10.1242/jcs.02760. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin C-H. Induction of epithelial-mesenchymal transition by transforming growth factor β. Semin Cancer Biol. 2012;22:446–454. doi: 10.1016/j.semcancer.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Stournaras C. Regulation of actin organisation by TGF-beta in H-ras-transformed fibroblasts. J Cell Sci. 1999;112:1169–1179. doi: 10.1242/jcs.112.8.1169. [DOI] [PubMed] [Google Scholar]
- Nakaya Y, Sukowati EW, Wu Y, Sheng G. RhoA and microtubule dynamics control cell-basement membrane interaction in EMT during gastrulation. Nat Cell Biol. 2008;10:765–775. doi: 10.1038/ncb1739. [DOI] [PubMed] [Google Scholar]
- Nalbant P, Chang Y, Chang Z, Bokoch GM. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol Biol Cell. 2009;20:4070–4082. doi: 10.1091/mbc.E09-01-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien TE, Cribb J, Marshburn D, Taylor RM, 2nd, Superfine R. Magnetic manipulation for force measurements in cell biology. In: Correia JJ, Detrich HW, editors. Biophysical Tools for Biologists, Volume 2: In Vivo Techniques. Burlington, MA: Academic Press; 2008. pp. 433–450. [Google Scholar]
- Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang H-R, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- Papadimitriou E, Vasilaki E, Vorvis C, Iliopoulos D, Moustakas A, Kardassis D, Stournaras C. Differential regulation of the two RhoA-specific GEF isoforms Net1/Net1A by TGF-β and miR-24: role in epithelial-to-mesenchymal transition. Oncogene. 2011;31:2862–2875. doi: 10.1038/onc.2011.457. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P. TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci. 1999;112:4557–4568. doi: 10.1242/jcs.112.24.4557. [DOI] [PubMed] [Google Scholar]
- Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti-Dallenbach R, Oertle P, Hyotyla JT, Aebi U, Bentires-Alj M, Lim RYH, Schoenenberger C. The nanomechanical signature of breast cancer. Nat Nanotechnol. 2012;7:757–765. doi: 10.1038/nnano.2012.167. [DOI] [PubMed] [Google Scholar]
- Plotnikov SV, Pasapera AM, Sabass B, Waterman CM. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell. 2012;151:1513–1527. doi: 10.1016/j.cell.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov SV, Waterman CM. Guiding cell migration by tugging. Curr Opin Cell Biol. 2013;25:619–626. doi: 10.1016/j.ceb.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan P, Agrawal A, Bhushan R, Chavalmane AK, Kalathur RKR, Takahashi T, Kondaiah P. Expression profiling of genes regulated by TGF-beta: differential regulation in normal and tumour cells. BMC Genomics. 2007;8:98. doi: 10.1186/1471-2164-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V, Marshall CJ. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr Opin Cell Biol. 2010;22:690–696. doi: 10.1016/j.ceb.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Simpson KJ, Dugan AS, Mercurio AM. Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Res. 2004;64:8694–8701. doi: 10.1158/0008-5472.CAN-04-2247. [DOI] [PubMed] [Google Scholar]
- Spero RC, Vicci L, Cribb J, Bober D, Swaminathan V, O'Brien ET, Rogers SL, Superfine R. High throughput system for magnetic manipulation of cells, polymers, and biomaterials. Rev Sci Instrum. 2008;79:083707. doi: 10.1063/1.2976156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh S, Spatz J, Mills JP, Micoulet A, Dao M, Lim CT, Beil M, Seufferlein T. Connections between single-cell biomechanics and human disease states: gastrointestinal cancer and malaria. Acta Biomater. 2005;1:15–30. doi: 10.1016/j.actbio.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Swaminathan V, Mythreye K, O'Brien ET, Berchuck A, Blobe GC, Superfine R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. 2011;71:5075–5080. doi: 10.1158/0008-5472.CAN-11-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor Ma, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15:169–190. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsapara A, Luthert P, Greenwood J, Hill CS, Matter K, Balda MS. The RhoA activator GEF-H1/Lfc is a transforming growth factor-β target gene and effector that regulates smooth muscle actin expression and cell migration. Mol Biol Cell. 2010;21:860–870. doi: 10.1091/mbc.E09-07-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega FM, Fruhwirth G, Ng T, Ridley AJ. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J Cell Biol. 2011;193:655–665. doi: 10.1083/jcb.201011038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelmann R, Nguyen-Tat M-D, Giehl K, Adler G, Wedlich D, Menke A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J Cell Sci. 2005;118:4901–4912. doi: 10.1242/jcs.02594. [DOI] [PubMed] [Google Scholar]
- Wang H-R, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science. 2003;302:1775–1779. doi: 10.1126/science.1090772. [DOI] [PubMed] [Google Scholar]
- Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Bröcker E-B, Friedl P. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia. 2004;6:603–610. doi: 10.1593/neo.04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Mezencev R, Kim B, Wang L, McDonald J, Sulchek T. Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PloS One. 2012;7:e46609. doi: 10.1371/journal.pone.0046609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- Yu L, Hébert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.