

Despite widespread use the extent to which different mammalian transgene methods report on the properties of endogenous proteins has not been systematically compared. This study shows that the choice of fluorescence-tagging method fundamentally influences the ability to image the activity of the mitotic kinase Aurora B.
Abstract
Fluorescence tagging of proteins is a widely used tool to study protein function and dynamics in live cells. However, the extent to which different mammalian transgene methods faithfully report on the properties of endogenous proteins has not been studied comparatively. Here we use quantitative live-cell imaging and single-molecule spectroscopy to analyze how different transgene systems affect imaging of the functional properties of the mitotic kinase Aurora B. We show that the transgene method fundamentally influences level and variability of expression and can severely compromise the ability to report on endogenous binding and localization parameters, providing a guide for quantitative imaging studies in mammalian cells.
INTRODUCTION
Gene tagging with green fluorescent protein (GFP) has revolutionized our understanding of the dynamic properties of the cellular proteome. Powerful fluorescence imaging technologies have made quantitative measurements of protein biophysics and biochemistry feasible in situ (Wu and Pollard, 2005; Maeder et al., 2007). High-throughput imaging and computational data integration make such studies scalable to whole pathways and proteomes (Wu and Pollard, 2005; Maeder et al., 2007). Whereas fusing fluorescent tags to endogenous proteins is routine in yeast, in human cells, the insight gained from quantitative imaging approaches has been limited by the available transgene methodologies. The majority of past studies relied on systems that do not faithfully maintain physiological gene expression, such as cDNA plasmids with heterologous promoters that are randomly integrated into the human cell's genome for stable expression. The resulting overexpression or underexpression is known to affect protein function through numerous mechanisms, such as nonspecific aggregation, alterations in folding, and imbalance with endogenous binding partners (Burgess et al., 2012; Gibson et al., 2013). These considerations have motivated the development of second-generation plasmid-based systems such as bacterial artificial chromosomes (BACs), which retain endogenous cis-acting regulatory sequences and can also be randomly integrated into the genome of the host cell (Poser et al., 2008). However, position-effect variegation in BACs has not been investigated, and all superexpression systems suffer from the continued presence of untagged protein produced from endogenous loci, which cannot be imaged but may carry out all or part of the function of the fusion protein under study (Yue et al., 2008). Although RNA interference (RNAi) knockdown of endogenous protein can be used in combination with RNAi-resistant transgenes, incomplete knockdown and off-target effects make such experiments difficult to control and interpret (Ma et al., 2006). Recently multiple genome-editing technologies, including zinc finger nucleases (ZFNs) and Tale-activated endonucleases (TALENs), have been developed to modify endogenous mammalian loci (Bibikova et al., 2003; Miller et al., 2011). However, whether such knock-in transgenes truly are quantitatively superior to superexpression approaches has not been systematically studied to date.
RESULTS
To address this issue, we created HeLa cell lines stably expressing C-terminal monomeric enhanced GFP (mEGFP) fusions of the key mitotic kinase Aurora kinase B (AURKB-GFP) using four different transgene methods (Figure 1A). We used genome editing by ZFNs (Bibikova et al., 2003) or TALENs (Miller et al., 2011) to target recombination of EGFP from a donor plasmid containing regions of homology to endogenous loci (Figure 1Ai). In addition, we performed random integration of BACs or cDNA plasmids (Figure 1Aii; Poser et al., 2008). For each transgene method (Figure 1A), we created several monoclonal or pooled HeLa lines stably expressing AURKB-GFP, which we validated with a series of assays (Figure 1B; see Materials and Methods for detailed protocols). Briefly, after introduction of the tag, expressing cells were isolated by fluorescence-activated cell sorting (FACS; Figure 1C) and then screened by fluorescence microscopy for specific localization to mitotic organelles as expected for AURKB (Figure 1D; Terada et al., 1998). We distinguished homozygous from heterozygous clones (AURKB has three alleles in HeLa cells; Landry et al., 2013) for the genome-editing methods (Figure 1B; In vitro) by genomic PCR (Figure 1E and Supplemental Figure S1A), Western blot (Figure 1F and Supplemental Figure S1B), and Southern blot (Figure 1G and Supplemental Figure S1, C and D).
FIGURE 1:
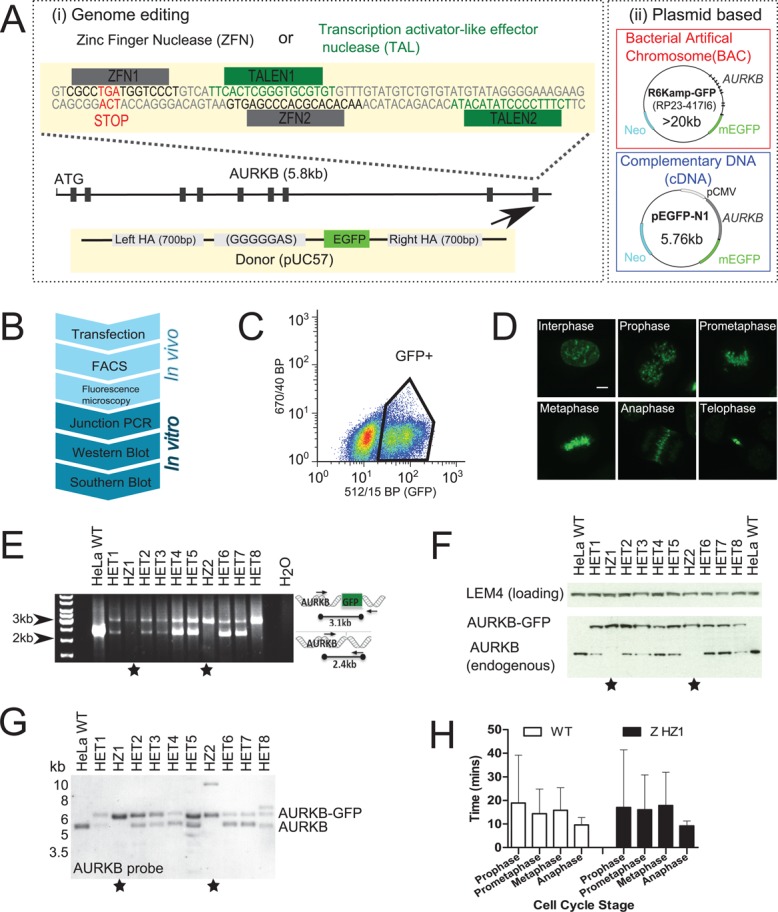
Construction and validation of genome-edited cell lines expressing AURKB-GFP. (A) Schematic of fluorescent gene–tagging systems used to create AURKB-GFP. (i) ZFNs or TALENs cause DNA double-strand breaks at the C-terminus of the AURKB locus, before repair with a donor construct containing EGFP (arrow). (ii) Plasmids containing AURKB-GFP as either the full mouse gene (BAC) or as cDNA are randomly integrated into the genome. (B) Flowchart of assays used to construct genome-edited cell lines. Junction PCR was not used to screen plasmid-based systems since the genomic locations are unknown. (C) Fluorescence-activated cell sorting of GFP-positive cells. Gates were drawn based on comparison to nonfluorescent wild-type parental cells. (D) Fluorescence confocal microscopy maximum-intensity z-projections of genome-edited AURKB-GFP cells (clone HZ2). (E) Junction PCR screening of ZFN genome-edited AURKB-GFP clonal cells. Stars denote clones with all alleles successfully targeted. (F) Western blot screening of nocodazole-arrested ZFN AURKB-GFP cells with anti-AURKB antibody. (G) Southern blot screening of ZFN AURKB-GFP cells using a probe in AURKB intron 2 after Kpnl and Nsil digestion. (H) Mitotic timing from live-cell imaging; cells were automatically classified into cell cycle stages using mCherry-H2B as previously described (Held et al., 2010). n >355 cells from two experiments.
This validation workflow allowed us to select clones with homozygous insertion of EGFP into all three AURKB alleles as well as heterozygous clones for further analysis. Independent of the targeting method used for genome editing, we found some evidence of extra integration sites by Southern blot (Figure 1G and Supplemental Figure S1, C and D). This did not result in unexpected size bands by genomic PCR or Western blot, however (Figure 1, E and F, and Supplemental Figure S1, A and B), and therefore they likely lie in noncoding regions. Homozygous and heterozygous AURKB-GFP cells exhibited indistinguishable cell cycle timing compared with wild-type HeLa cells as measured by automated classification of live-cell imaging data (Held et al., 2010; Figure 1H and Supplemental Figure S1E) or flow cytometry (Supplemental Figure S1F), indicating that the fluorescent fusion protein was fully functional to support cell division. The complete set of cell lines used in this study is listed in Table 1.
TABLE 1:
Cell lines used in this study.
| Expression system | Name | Transgene species |
|---|---|---|
| ZFN | Z HZ1 | Human |
| ZFN | Z HZ2 | Human |
| ZFN | Z HET1 | Human |
| TALEN | T HET1 | Human |
| TALEN | T HET2 | Human |
| mBAC | B P1 | Mouse |
| mBAC | BC1 | Mouse |
| mBAC | B P2 | Mouse |
| mBAC | B C2 | Mouse |
| cDNA | cD P1 | Human |
| cDNA | cD C1 | Human |
| cDNA | cD C2 | Human |
| cDNA | cD C3 | Human |
Having validated this set of AURKB-GFP cells generated from the same parental HeLa line, we systematically analyzed the behavior of the expressed fusion proteins. First, we examined total expression level of AURKB-GFP by Western blotting of whole-cell extracts with anti-AURKB or anti-GFP antibodies (Figure 2A and Supplemental Figure S2A). All genome-edited clones exhibited AURKB-GFP levels similar to the endogenous protein with relatively little variation and independent of the genome-targeting method used to generate them or their zygosity (∼0.6- to 1.7-fold wild type). By contrast, expression of AURKB-GFP from randomly integrated BACs resulted in clearly visible overexpression in the tested clones (∼2.1- to 3.6-fold wild type), suggesting that the presence of the native promoter is not sufficient to ensure physiological expression levels. As expected for a constitutive heterologous promoter (cytomegalovirus; Boshart et al., 1985), different cDNA clones showed the strongest overexpression and the most variable expression between cell lines (∼0.4– to 5.7-fold). Flow cytometry of the intensity distribution of AURKB-GFP from 20,000 cells for each tagging method showed greatest cell-to-cell homogeneity for the two genome-editing methods (CV = 23 [ZFN], 23 [TALEN]), followed by BACs and cDNAs, which exhibited more variability (Figure 2B; CV = 30 and 112, respectively). Similarly, whereas in genome-edited and BAC systems, nearly all cells expressed the fusion protein (ZFN, 99%; BAC, 92–99%), cDNA clones had a variable number of expressing cells (Figure 2B; 28–89%).
FIGURE 2:
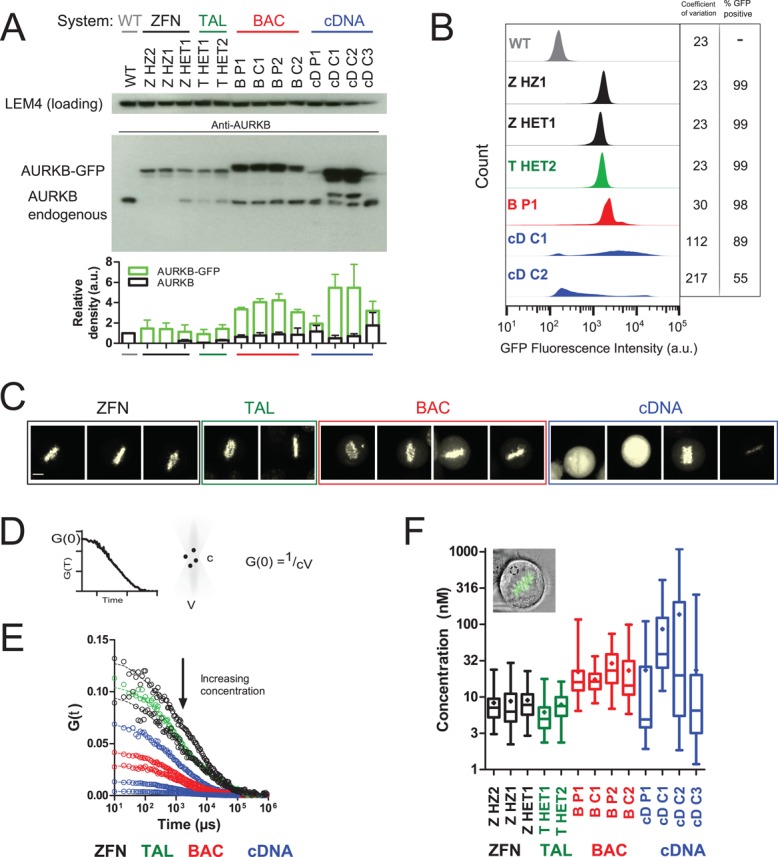
Expression system determines AURKB-GFP levels. (A) Western blot comparison of AURKB levels in nocodazole-arrested cells. BAC-expressed AURKB-GFP runs at a higher molecular weight due to the presence of an S-peptide in addition to the EGFP tag. The graph shows the mean ± SD of three independent experiments. (B) Flow cytometry analysis of AURKB-GFP intensity in nocodazole-arrested cells. Coefficient of variation is from the whole population. GFP+ was defined as 3 SD above autofluorescence from wild-type cells. (C) Sum intensity confocal images of AURKB-GFP using the same imaging conditions throughout. One metaphase cell is shown per panel. Left to right: ZHZ2, ZHZ1, ZHET1, THET1, THET2, BP1, BC1, BP2, BC2, cDP1, cDC1, cDC2, and cDC3. Scale bar, 7 μm. (D) The amplitude of the FCS autocorrelation function (see Materials and Methods), G(0), is inversely proportional to particle number. Absolute protein concentration, c, is calculated using the measured confocal volume, V. (E) AURKB-GFP FCS autocorrelation curves from single representative cells. Top to bottom: ZHZ2, THET1, ZHET1, cDC3, BP2, BC2, cDC2, and cDC1. (F) AURKB-GFP cytoplasmic concentration calculated by fitting FCS autocorrelation curves with a one-component anomalous model of diffusion as described in Materials and Methods. The box and whiskers plot is from >23 cells/sample from two experiments. The mean is depicted as a diamond and the median as a horizontal line, and the whiskers show the minimum and maximum.
Having assessed total expression level and cell-to-cell variation, we next addressed subcellular abundance of the fusion proteins in mitotic metaphase, when AURKB resides mostly on mitotic chromosomes (Carmena et al., 2012). Genome-edited cells exhibited the expected specific localization to metaphase chromosomes, with barely detectable levels of unbound cytoplasmic protein. By contrast, overexpression from the plasmid-based systems led to elevated cytoplasmic signal (Figure 2C and Supplemental Figure S2B, arrowheads). We quantified the absolute cytoplasmic concentration of AURKB-GFP using fluorescence correlation spectroscopy (FCS) in living cells. FCS records fluorescence intensity fluctuations in a small confocal volume as a function of time, allowing determination of physical properties, including the concentration of the fluorescent species from the amplitude of the FCS autocorrelation function (Figure 2D; see Materials and Methods). As such, it is well suited to the detection of mobile cytosolic proteins expressed at nanomolar to micromolar levels. AURKB-GFP genome-edited cells showed a clear autocorrelation signal distinguishable from that of wild-type cells (Supplemental Figure S2C), indicating the presence of a cytoplasmic pool. Consistent with our Western blotting, flow cytometry, and fluorescence microscopy data, FCS analysis showed that AURKB-GFP genome–edited cells had a low cytoplasmic concentration of 8.6 nM ± 5.3 SD (ZFN) or 6.9 nM ± 3.5 (TALEN), whereas overexpression from the plasmid-based systems resulted in significantly higher cytoplasmic concentrations of 23.8 nM ± 17.1 (BAC) or 86.7 nM ± 156.7 (cDNA; Figure 2, E and F, and Supplemental Figure S2D).
AURKB plays several critical roles in genome segregation via the phosphorylation of substrates on chromatin such as core histones and kinetochore proteins and by forming phosphorylation gradients (Tanaka et al., 2002; Wang et al., 2011; Carmena et al., 2012). The balance between soluble cytoplasmic and chromosome-bound kinase is therefore key to interpreting its function. To determine whether the transgene method would perturb this balance, we quantified the amount of AURKB-GFP localized to chromosomes on the metaphase plate using FCS-calibrated confocal imaging in live, dividing cells. Here first we recorded an FCS trace in the cytoplasm to determine the absolute local concentration of GFP and calibrate the corresponding confocal pixel intensity, which thus allows us to transform the entire confocal image into a concentration map (Figure 3A). Despite the different total cell expression levels, BACs showed a similar chromatin-bound AURKB-GFP concentration to genome-edited cells, whereas cDNAs were very variable (Figure 3, B and C). By combining our concentration measurements with three-dimensional reconstructed volumes of mitotic chromosomes and cytoplasm, we obtained the total number of chromatin-bound and free AURKB-GFP proteins. We found that the fraction of AURKB-GFP bound to chromatin is significantly higher in genome-edited cells than in plasmid-based systems (Figure 3D). Whereas in genome-edited cells, ∼75% of total cellular AURKB-GFP resides on chromatin, only 40-50% does so in plasmid-based systems, with the fraction decreasing as the total amount of AURKB increases. This relationship is in agreement with a mathematical model (solid line in Figure 3E; see Materials and Methods) that has a limited number of high-affinity binding sites on chromatin (∼130,000–150,000 molecules), with a binding Kd of 3.4–5.8 nM. Thus, our results suggest that chromatin-binding sites become saturated in overexpression systems, forcing aberrant accumulation of the fusion protein in the cytosol.
FIGURE 3:
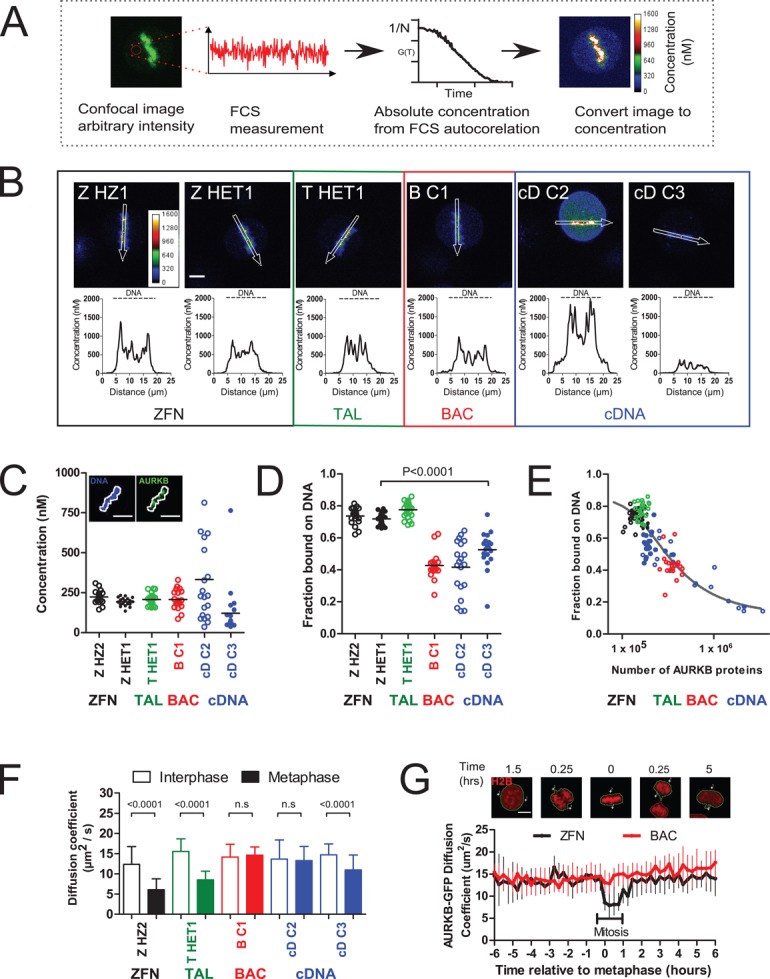
Overexpression alters AURKB-GFP biophysical properties on chromatin and in the cytosol. (A) Schematic of FCS-calibrated imaging. Concentration is determined in one location using FCS, and then an image is taken of the same cell. The image is converted to absolute concentrations using the FCS calibration. (B) Absolute concentration maps of AURKB-GFP in metaphase cells. The line profiles were taken in the areas depicted by the white arrows in the image directly above. Scale bar, 7 μm. (C) AURKB-GFP concentration on the metaphase plate. DNA was automatically identified based on a threshold of Hoechst staining of DNA (white line). Scale bar, 10 μm. (D) Fraction of AURKB-GFP proteins bound on chromatin. Horizontal bar shows the mean. (E) Fraction of chromatin bound AURKB-GFP (dots) as a function of the total number of AURKB proteins. The line is the equilibrium solution of a mass action model of reversible AURKB chromatin binding (see Materials and Methods; total number of chromatin-binding sites CT = 147,000, Kd = 5.21 nM). (F) AURKB-GFP diffusion coefficient from FCS data of >56 cells/sample from four experiments. (G) Single-cell-tracking automated FCS and time-lapse microscopy of AURKB-GFP diffusional mobility through the cell cycle as described in Materials and Methods. Plotted are the median and interquartile range from 9–45 cells. Scale bar, 10 μm. Significance testing by Mann–Whitney test.
A key prerequisite for systems microscopy is to detect complexes incorporating the tagged protein inside the cell. It is known that AURKB engages in numerous complexes in mitotic cells, including binding to members of the so-called chromosomal passenger complex (Wang et al., 2011; Carmena et al., 2012), which should cause a decrease in its mobility in the cytoplasm. To assess whether the cytoplasmic accumulation in overexpressing clones would interfere with our ability to detect AURKB-containing complexes of low mobility, we determined the diffusion coefficient of AURKB-GFP by FCS. During interphase, AURKB-GFP cytoplasmic diffusion coefficients were indistinguishable between different transgene methods (ZFN, 13.2 μm2/s ± 6.4 SD; BAC, 14.0 μm2/s ± 4.6; Figure 3F, unfilled bars). However, metaphase cells exhibited significant differences in diffusion coefficient between expression systems (Figure 3F, filled bars). Genome-edited cells had reduced AURKB-GFP mobility compared with interphase (ZFN, 6.5 μm2/s ± 3.9 SD), indicative of being bound to complexes of a larger Stokes radius, whereas in BAC cells, no significant change was detected (BAC, 14.7 μm2/s ± 3.9). Of interest, cDNA cells showed variable behavior, with a decrease in diffusional mobility detectable only in cells with low AURKB-GFP expression level (cD C3; ∼10 nM cytoplasmic) but not in higher-expressing clones (cD C2). Using automated time-lapse FCS to track single living nuclei through cell division, we acquired images and spectroscopy data every 15 min in dividing cells. Again, the cell cycle–dependent reversible change in diffusion coefficient from G2 to mitosis to G1 indicative of complex formation could readily be detected in single genome-edited cells but was not detectable in BAC-derived cells (Figure 3G).
DISCUSSION
Overall our findings show that the choice of transgene system strongly affects expression level and variability of fluorescently tagged genes in human cells. These expression changes have important consequences for the accurate quantitation of endogenous binding parameters using microscopy. Our data indicate a clear anticorrelation between overexpression—with free ligand dominating the overall pool—and the ability to detect both specifically localized protein and protein engaged in large complexes. In terms of achieving a close to physiological expression level, the genome editing methods clearly outperformed plasmid-based approaches that suffer from heterologous promoters (cDNA) and position-effect variegation (cDNA and BAC). Furthermore, genome-edited cell lines showed far greater homogeneity, with almost any clone obtained by thorough screening (Figure 1) closely matching endogenous levels. Consistent with previous work (Doyon et al., 2011), this has implications for understanding the biology of cellular systems tagged with fluorescent proteins in human cells. However, it is also an important practical consideration when making cell lines stably expressing fluorescent fusion proteins, since it is often impossible to screen for true endogenous levels of expression in all subcellular pools. In addition, for essential genes, homozygous genome-edited cells ensure biological functionality of the fusion protein and exclude the possibility that complex formation and specific binding are obscured by the presence of unlabeled endogenous protein.
A frequent argument for randomly integrated plasmid-based transgenes has been that for functionally important tagged genes, surviving clones would automatically select for physiological expression level (Rabut et al., 2004). Our case study with Aurora B provides reason for caution. We could generate stable plasmid-based cell clones with up to 100-fold overexpression that did not reveal gross phenotypic differences in cell division timing or histone H3 phosphorylation (Supplemental Figure S3, A–C). Nevertheless, the relevant biophysical properties of Aurora B were completely obscured in these cells due to a massive unbound pool in the cytoplasm. Thus, phenotypically normal cells may still be unsuitable for quantitative fluorescence microscopy. If quantitative imaging of in vivo proteomic parameters is the objective, genome editing appears to be the method of choice to fluorescently tag genes in human cells.
MATERIALS AND METHODS
Constructs, cell line construction, and growth
Zinc finger nucleases were purchased from Sigma-Aldrich (St. Louis, MO) with DNA-binding sequences in the 5′–3′ direction of CGCCTGATGGTCCCT and CACTCGGGTGCGTGTGTT. TALENs were from Cellectis Bioresearch (Romainville, France) with DNA-binding domains in the 5′–3′ direction of TTCACTCGGGTGCGTGT and TATGTATAGGGGAAAGA. The donor plasmid consisted of mEGFP cDNA sequence flanked by a left homology arm (ENSEMBL release 75, ENST00000316199, chromosome 17:8108992-8108191) and a right homology arm (ENSEMBL release 75, ENST00000316199, chromosome 17:8107923-8108440), consisting of AURKB genomic sequence. ZFN/TALEN and donor plasmid transfection was with jetPRIME reagent (Polyplus, Illkirch-Graffenstaden, France). We incubated 0.5 μg of each ZFN/TALEN and 1 μg of donor plasmid for 15 min with 200 μl of jetPRIME buffer and 4 μl of jetPRIME before addition to cells grown to 80% confluency in a six-well dish with 2 ml of growth medium. After 4 h, the transfection mix was changed to normal medium.
HeLa Kyoto cells were grown in DMEM supplemented with 10% fetal bovine serum at 37°C. Cells expressing AURKB-GFP mouse BAC as pool were previously described (Neumann et al., 2010), and further pools were also obtained as a kind gift from T. Hyman and I. Poser (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany). cDNA AURKB-GFP was cloned by inserting GFP into the Sal1 and BamH1 sites of the vector AURKB-mCherry pEGFP-N1 (a kind gift of M. Isokane, European Molecular Biology Laboratory, Heidelberg, Germany). Cells expressing cDNA AURKB-GFP were selected in 500 μg/ml neomycin and cells expressing cDNA H2B-mCherry with 0.3 μg/ml puromycin.
Fluorescence-activated cell sorting and flow cytometry
At 7–10 d posttransfection of genome-editing constructs, cells were synchronized with 2 mM thymidine for 18 h and subsequently released into fresh medium for 7–8 h. Mitotic cells were collected via mitotic shakeoff before sorting. GFP+ HeLa Kyoto cells were sorted with a MoFlo Legacy cell sorter (Beckman Coulter) equipped with a 100-μm nozzle. BD FACSFlow sheath (BD Biosciences, San Jose, CA) was used as sheath fluid, filtered in-line through a PALL Fluorodyne II filter 0.2 μm (Pall). A 488 nm–tuned Coherent Sabre laser (TEM00 mode, 200 mW) was used for the excitation of GFP. Fluorescence was collected and detected after filtering through 488-nm StopLine single-notch and 512/15 BP filters. Laser illumination, Moflo's L-configuration optical layout, and sorting were optimized using Flow-Check Fluorospheres (Beckman Coulter, Pasadena, CA). Sort decision was based on a combination of regions drawn around FSC-area versus SSC-area and FSC-area versus pulse width together with 488 nm–derived fluorescence 512/15 BP (GFP) versus 670/40 BP (autofluorescence) plots. Parental HeLa Kyoto cells transfected only with donor plasmid were used as negative control. No correlation was seen between the level of AURKB-GFP expression and the number of alleles integrated by PCR and Western analysis.
Cell cycle analysis was performed on an LSR-Fortessa SORP cytometer (BD Biosciences) after staining with propidium iodide (Invitrogen, Carlsbad, CA); cells were washed in phosphate-buffered saline (PBS), fixed in cold 70% ethanol for >2 h, washed in PBS, and then stained with 40 μg/ml propidium iodide containing 0.1% Triton-100 and RNase A overnight at room temperature to ensure saturation of DNA staining. Cell cycle–stage analysis was performed by fitting with the cell cycle Watson (pragmatic) algorithm in FlowJo v7.6.4 software (Tree Star, Ashland, OR).
Genomic PCR
Junction genomic PCR was performed at endogenous AURKB loci to detect the insertion of EGFP at the C-terminus using two separate sets of primers, one of which annealed inside GFP and one outside, as follows: AURKB forward (AGCTGGAGGTCCATCCTTGT), AURKB reverse (ATCACTGCTGCTTCTATTGGCT), and mEGFP reverse (gatgttgccgtcctccttga). PCR was performed using HotStar HiFidelity (Qiagen, Venlo, Netherlands) according to the supplier's manual and with a primer annealing temperature of 53ºC.
Southern blotting
Genomic DNA was prepared with the MasterPure DNA Purification Kit (Epicentre Biotechnologies, Madison, WI) according to the supplier's manual. Twenty micrograms of genomic DNA was digested with either XbaI or KpnI/NsiI (NEB) at 37°C overnight. We performed 0.8% agarose gel electrophoresis, and the gel was denatured with 2× 20-min washes in 0.5 M NaOH/1.5 M NaCl. After neutralization by 2× 20-min incubation in 1.5 M NaCl/0.5 M Tris-HCl, pH 7.0, the DNA fragments were transferred via capillary forces from the gel to a Genescreen Plus R Nylon membrane (PerkinElmer, Boston, MA) in 10× SSC (1.5 M NaCl/0.15 M Na3 citrate⋅H2O). After cross-linking of the DNA via ultraviolet light (UVStratalinker 1800; Stratagene, La Jolla, CA), hybridization was performed using DIG Easy Hyb (Roche, Basel, Switzerland) accordingly to the manufacturer's guidelines. Digoxigenin-dUTP (DIG-dUTP)–labeled AURKB or mEGFP probes for detection were generated using the PCR DIG Probe Synthesis Kit (Roche).
The sequence of the AURKB 3′ probe was TAAACAGTTCTGTACCCTAGAAGCTGTCTTTGATTATTTCACTGCTCTGGTCAGGATCTAGTACAGGAGCCAATAGAAGCAGCAGTGATTCTGTCTTGTTCCTGACATGAGGTTACACTGTTATGCATGATGCTTGCTGTAGGTTTTTGATAGATACCCTTTATCAGGTTAAACAGGGCCTATTCTACTCATAGTTTACTCAGAGTCTT.
The sequence of AURKB intron 2 probe was ATGAAGAAGTTGAAAGTGAGAAGTGGTCCAGGGCCATCAGGGTAGCTCTTAGTGTCCTTAAAGCCTGTTCTTGCAAAAGTGTAAATCCTGGTTATTGCTATAACATTCAAGGCCTCAAGCATCTTATCTAAGCCTTAATAAACTAAGCTTTTTAGGAGACTTTGAGAATACCTCTGTGCTAAACCCCTTGAATTGTGGGTGTTCATTGACGCAGCTCTCTACTGTGGTGTAAGT.
Live-cell time-lapse imaging
Live-cell microscopy was at 37°C in CO2-independent medium without phenol red (Invitrogen). A Zeiss 780 laser scanning confocal microscope with a 63× PlanApochromat, numerical aperture (NA) 1.4 oil objective was used to acquire 512 × 512 pixel images with a pixel size of 0.439 μm at 5-min intervals. The z-stacks of eight slices spaced equally through 22.4 μm were acquired and formed into maximum-intensity z-projections. Automatic detection of nuclei in the H2B-mCherry channel and subsequent analysis of movies for mitotic timing were with CellCognition software as described previously (Held et al., 2010). Briefly, six morphological classes were defined: interphase, prophase, prometaphase, metaphase, anaphase, and telophase. The training set contained >1000 manually labeled nuclei, which were annotated with an overall accuracy of >95% in the cross-validation. To reduce the effect of classification errors on phase length measurements, classification results were corrected with hidden Markov models afterward.
Western blotting
Cells were lysed for 20 min on ice in RIPA buffer (50 mM Tris HCl, pH 8, 150 mM NaCl, 1% NP40, 0.5 M sodium deoxycholate, 0.1% SDS, complete protease inhibitor cocktail, PhosSTOP [Roche]). Protein concentration was quantitated using the bicinchoninic acid method (Sigma-Aldrich). Antibodies used in overnight incubations were anti-AIM-1 1:400 (611082; BD Bioscience), anti-LEM4 (1:2000), and anti-GFP (Yavuz et al., 2010). Bands were quantified in Fiji using the Gel Analyzer tool and normalized relative to a standard on each gel consisting of either wild-type HeLa cells or ZFN clone HZ2.
Fluorescence correlation spectroscopy
Fluorescence correlation spectroscopy was performed with a Zeiss LSM 780 ConfoCor3 system as previously described (Maeder et al., 2007; Schmidt et al., 2009). Samples were incubated at 37°C and excited with a 488-nm laser at minimal power (<1 kW/cm2) to reduce bleaching, photophysical effects, and cellular toxicity. Light was focused with a water immersion 40×/1.2 NA objective lens and collected by two avalanche photodiodes in the spectrally distinct regions 505–540 nm (Fg) and 600–650 nm (Fr) after passing through a pinhole set to 1 Airy unit. The instrument was calibrated before each experiment using Alexa 488 and Alexa 568 to align the pinholes and perform coverglass corrections. Each measurement was taken for 30 s in total.
From the fluorescence time traces Fg and Fr, temporal autocorrelation functions were calculated according to
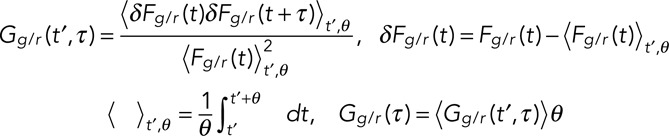 (1) (1)
|
Here local averaging with a typical window size of θ ∼ 1 s allows for the correction of slow processes such as cellular movement and photobleaching. Resulting autocorrelation functions were then fitted with a model function for anomalous diffusion including molecular blinking as sources of fluctuations (Schmidt et al., 2009):
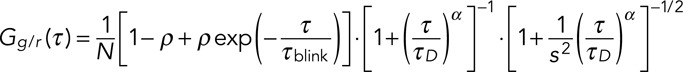 (2) (2)
|
where N = cVeff is the number of fluorescent molecules in the focal volume Veff and c their concentration, ρ is the fraction of molecules in a nonfluorescent state with a lifetime τblink, and α is the anomaly parameter of diffusion resulting in a mean dwell time τdiff of the molecules in the focus. The diffusion coefficient D = w02/4τdiff and the concentration c were extracted after calibrating the focal volume Veff = π3/2w03s and the lateral and axial focal radii w0 and sw0 using FCS measurements of Alexa 488 with a known diffusion coefficient. For FCS data processing we used the Fluctuation Analyzer software (written by Malte Wachsmuth, European Molecular Biology Laboratory, Heidelberg, Germany; available upon request).
Automated FCS with single-cell tracking was performed using an in-house-adapted Zeiss 780 ConfoCor3 system. Image and FCS acquisition were coordinated using a custom-written VBA macro (AutofocusScreen, available on request), which coordinated autofocusing, image acquisition, image analysis, and FCS. mCherry-H2B images of nuclei were thresholded on the fly in Fiji (Schindelin et al., 2012) such that two FCS measurement positions per cell in the cytoplasm could be specified. FCS measurements were acquired for 10 s per position, with imaging and FCS repeated at 15 min intervals. Cells were aligned to metaphase after data acquisition.
FCS-calibrated imaging
One or a few FCS measurements were performed in the cytoplasm as described in Fluorescence correlation spectroscopy to extract the concentration at the respective locations. A confocal slice was then taken in the same cell with the same microscope configuration, using, however, the photomultiplier detectors such that intensities were not clipped off, nor was the detector saturated. The image intensities were averaged locally at the FCS measurement spots and subsequently related to the measured concentration. This was performed for all cells, yielding a linear relationship between intensity and concentration. This linear relationship between concentration and intensity was also confirmed in a serial dilution of Alexa 488 dye in water. Images were transformed to absolute concentrations after background intensity subtraction, where background was defined as an area of the image containing no cells.
The number of moles, n, in a cellular compartment was estimated using
|
|
where c is the molar concentration, obtained either directly from an FCS measurement in the cytoplasm or from FCS-calibrated imaging of the chromatin. V is the volume, using estimates of 750 μm3 for DNA and 5750 μm3 for the cytoplasm (provided by Julius Hossain, European Molecular Biology Laboratory, Heidelberg, Germany) in metaphase cells. The number of proteins N is related to n by Avogadro's number, NA,
|
|
Mathematical modeling
For AURKB binding to chromatin, C, we assume mass-action kinetics and reversible binding according to
AURKB + C ↔ AURKB-C
This leads to an equation for the concentration of AURKB bound to chromatin, AC,
|
|
where C and A are the concentration of chromatin-binding sites and AURKB in the cytoplasm, respectively. In equilibrium and assuming conservation of chromatin, CT = AC + C, and AURKB, AT = A + AC, one obtains for the fraction of molecules bound to chromatin
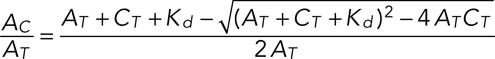 (6) (6)
|
with the dissociation constant Kd = k−/k+. The measured fraction (AC/AT)m also includes cytoplasmic proteins diffusing in the DNA region but not bound to DNA:
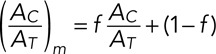 (7) (7)
|
where
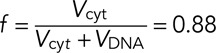 (8) (8)
|
The unknown parameters Kd and CT have been fitted to the data shown in Figure 3E. The number of untagged nonfluorescent AURKB proteins is assumed to be constant in all systems and equal to one-third of the amount from homozygous knock-in cells. Therefore for cDNA and BAC cells, we assume that the total AURKB number (tagged plus untagged) is the sum of the measured protein number from the homozygous ZFN clone Z HZ2 plus the measured fluorescent protein number.
Immunofluorescence
Cells grown on coverslips were fixed in 4% paraformaldehyde/PBS for 5 min and permeabilized in 0.1% Triton X-100/0.1% Tween-20 for 5 min. Cells were blocked in 3% bovine serum albumin for 1 h before primary antibody incubation for 2 h at room temperature. Antibodies used were anti–Aurora B pT232 (1:2000; Rockland Immunochemicals, Pottstown, PA), anti–histone H3 pS28 (1:500; 10543; Abcam), and anti–histone H3 (1:500; 46765; Abcam).
Supplementary Material
Acknowledgments
We thank Tony Hyman and Ina Poser for donating the mouse BAC AURKB-GFP and thank all members of the Ellenberg laboratory for support. We thank Kota Miura for help in setting up the automated Fiji analysis and Bianca Nijmeijer for help with Southern blotting. This study was supported by funding from the European Commission within the MitoSys and SystemsMicroscopy consortia (FP7/2007-2013-241548 and FP7/2007-2013-258068). R.M. was supported by an EMBO fellowship (ALTF 416-2012, GA-2010-267146) and a Wellcome Trust Henry Wellcome Fellowship (100090/Z/12/Z).
Abbreviation used:
- AURKB
Aurora kinase B
- BAC
bacterial artificial chromosome
- FCS
fluorescence correlation spectroscopy
- GFP
green fluorescent protein
- TALEN
Tale-activated endonuclease
- ZFN
zinc finger nuclease.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-06-1091) on September 17, 2014.
B.K., R.M., A.P.G., and J.M. constructed cell lines. R.M., M.W., and A.Z.P. performed and analyzed FCS measurements. All authors analyzed data. R.M conceived the project. R.M and J.E. wrote the manuscript.
The authors declare no competing financial interests.
REFERENCES
- Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- Boshart M, Weber F, Jahn G, Dorsch-Häsler K, Fleckenstein B, Schaffner W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell. 1985;41:521–530. doi: 10.1016/s0092-8674(85)80025-8. [DOI] [PubMed] [Google Scholar]
- Burgess A, Lorca T, Castro A. Quantitative live imaging of endogenous DNA replication in mammalian cells. PLoS One. 2012;7:e45726. doi: 10.1371/journal.pone.0045726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol. 2012;13:789–803. doi: 10.1038/nrm3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon JB, Zeitler B, Cheng J, Cheng AT, Cherone JM, Santiago Y, Lee AH, Vo TD, Doyon Y, Miller JC, et al. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat Cell Biol. 2011;13:331–337. doi: 10.1038/ncb2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson TJ, Seiler M, Veitia RA. The transience of transient overexpression. Nat Methods. 2013;10:715–721. doi: 10.1038/nmeth.2534. [DOI] [PubMed] [Google Scholar]
- Held M, Schmitz MHA, Fischer B, Walter T, Neumann B, Olma MH, Peter M, Ellenberg J, Gerlich DW. CellCognition: time-resolved phenotype annotation in high-throughput live cell imaging. Nat Methods. 2010;7:747–754. doi: 10.1038/nmeth.1486. [DOI] [PubMed] [Google Scholar]
- Landry JJ, Pyl PT, Rausch T, Zichner T, Tekkedil MM, Stütz AM, Jauch A, Aiyar RS, Pau G, Delhomme N, et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda) 2013;3:1213–1224. doi: 10.1534/g3.113.005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Creanga A, Lum L, Beachy PA. Prevalence of off-target effects in Drosophila RNA interference screens. Nature. 2006;443:359–363. doi: 10.1038/nature05179. [DOI] [PubMed] [Google Scholar]
- Maeder CI, Hink MA, Kinkhabwala A, Mayr R, Bastiaens PIH, Knop M. Spatial regulation of Fus3 MAP kinase activity through a reaction-diffusion mechanism in yeast pheromone signalling. Nat Cell Biol. 2007;9:1319–1326. doi: 10.1038/ncb1652. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Neumann B, Walter T, Hériché JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser I, Sarov M, Hutchins JR, Hériché JK, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW, et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabut G, Doye V, Ellenberg J. Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat Cell Biol. 2004;6:1114–1121. doi: 10.1038/ncb1184. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U, Im K-B, Benzing C, Janjetovic S, Rippe K, Lichter P, Wachsmuth M. Assembly and mobility of exon-exon junction complexes in living cells. RNA. 2009;15:862–876. doi: 10.1261/rna.1387009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJR, Nasmyth K. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 2002;108:317–329. doi: 10.1016/s0092-8674(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J. 1998;17:667–676. doi: 10.1093/emboj/17.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Ballister ER, Lampson MA. Aurora B dynamics at centromeres create a diffusion-based phosphorylation gradient. J Cell Biol. 2011;194:539–549. doi: 10.1083/jcb.201103044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J-Q, Pollard TD. Counting cytokinesis proteins globally and locally in fission yeast. Science. 2005;310:310–314. doi: 10.1126/science.1113230. [DOI] [PubMed] [Google Scholar]
- Yavuz S, Santarella-Mellwig R, Koch B, Jaedicke A, Mattaj IW, Antonin W. NLS-mediated NPC functions of the nucleoporin Pom121. FEBS Lett. 2010;584:3292–3298. doi: 10.1016/j.febslet.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Yue Z, Carvalho A, Xu Z, Yuan X, Cardinale S, Ribeiro S, Lai F, Ogawa H, Gudmundsdottir E, Gassmann R, et al. Deconstructing Survivin: comprehensive genetic analysis of Survivin function by conditional knockout in a vertebrate cell line. J Cell Biol. 2008;183:279–296. doi: 10.1083/jcb.200806118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.