

Abstract
Background
Adenosine receptors (ARs) belong to the G protein-coupled receptors (GCPRs) family. The recent release of X-ray structures of the human A2A AR (h A2A AR ) in complex with agonists and antagonists has increased the application of structure-based drug design approaches to this class of receptors. Among them, homology modeling represents the method of choice to gather structural information on the other receptor subtypes, namely A1, A2B, and A3 ARs. With the aim of helping users in the selection of either a template to build its own models or ARs homology models publicly available on our platform, we implemented our web-resource dedicated to ARs, Adenosiland, with the “Best Template Searching” facility. This tool is freely accessible at the following web address: http://mms.dsfarm.unipd.it/Adenosiland/ligand.php.
Findings
The template suggestions and homology models provided by the “Best Template Searching” tool are guided by the similarity of a query structure (putative or known ARs ligand) with all ligands co-crystallized with hA2A AR subtype. The tool computes several similarity indexes and sort the outcoming results according to the index selected by the user.
Conclusions
We have implemented our web-resource dedicated to ARs Adenosiland with the “Best Template Searching” facility, a tool to guide template and models selection for hARs modelling. The underlying idea of our new facility, that is the selection of a template (or models built upon a template) whose co-crystallized ligand shares the highest similarity with the query structure, can be easily extended to other GPCRs.
Keywords: G protein-coupled receptors, Adenosine receptors, Receptor modelling, Bioinformatics platform, Adenosiland
Findings
The template suggestions and homology models provided by the “Best Template Searching”tool are guided by the similarity of a query structure (putative or known ARs ligand) with all ligands co-crystallized with hA2A AR subtype. The tool computes several similarity indexes and sort the outcoming results according to the index selected by the user.
Background
Adenosine receptors (ARs) belong to the G protein-coupled receptors (GCPRs) family. The known four subtypes, termed adenosine A1, A2A, A2B and A3 receptors, are widely distributed in human body and involved in several physio-pathological processes (Fredholm et al. 2001). The release of X-ray structures of the human A2A AR in complex with agonists (Lebon et al. 2011, Xu et al. 2011) and antagonists (Jaakola et al.2008, Doré et al. 2011 Hino et al. 2012, Congreve, et al. 2012, Liu, et al. 2012) has enabled to extend structure-based drug design approaches to this class of receptors. With the use of homology modelling techniques, indeed, structural information on the other subtypes can also be derived. As a key step when building homology models is the selection of a proper template, we have developed a tool to guide the user in this crucial choice by implementing the “Best Template Searching” facility in our web-resource dedicated to ARs, Adenosiland (Floris et al. 2013). This tool is freely accessible at the following web address: http://mms.dsfarm.unipd.it/Adenosiland/ligand.php.
The underlying idea behind this facility is to help the user in selecting the best template or ARs model to get the highest quality receptor for further molecular docking studies. A possible strategy herein presented is to compute the similarity between a known or putative agonist/antagonist and all co-crystallized ARs ligands.
Tool description
The “Best Template Searching” tool works as follows: the user is asked to input a query molecule either by uploading a SMILES string or by directly drawing the 2D structure by using the JME interface; the similarity of the input molecule is then computed against all the ligands co-crystallized with the hA2A AR. The following similarity indexes are calculated: (i) shape similarity (based on the Manhattan distance between USR descriptors), (ii) 2D similarity (based on the Tanimoto and Tversky Similarities of Pubchem Fingerprints), (iii) pharmacophoric similarity (based on the Tanimoto similarity of Pharmacophoric triplets), and (iv) a combined similarity (derived by the following function: 0.6 * pharmacophoric similarity + 0.4 * shape similarity).
The values of the two coefficients composing the latter similarity index have been derived by running a preliminary in-house validation based on all available crystallographic structures: In particular, the two values have been chosen so that by providing as input the structures of the co-crystallized ligand the corresponding receptor structure results the best ranked one according to the combined similarity index. The values obtained for the structures considered for the internal validation are reported in Table 1. For all the structure except one, the suggested template results the corresponding crystal structure. The only exception is represented by NECA for which the structure co-crystallized with adenosine is suggested as best template. Considering the high structural similarity between the two agonist structures, the results is in line with the others.
Table 1.
Values of the in-house validation of the combined similarity index
| Input ligand | Suggested template | Combined similarity value |
|---|---|---|
| Adenosine | 2YDO | 0.83 |
| NECA | 2YDO | 0.72 |
| UK-432,097 | 3QAK | 0.37 |
| ZMA 241385 | 4EIY | 0.69 |
| T4G | 3UZA | 0.84 |
| T4E | 3UZC | 0.92 |
| XAC | 3REY | 0.67 |
| Caffeine | 3RFM | 0.98 |
Simultaneously to the best template searching process, a similarity search screening is also performed against all adenosine agonists and antagonists deposited in ChEMBL, release 14 (Gaulton et al. 2011). In more details, the query is compared to 760 A1, 469 A2A, 559 A2B and 290 A3 AR ligands and the comparison is based on the calculation of the similarity measures previously described. The identified compounds are reported in a table along with the associated binding data available in literature.
Tool validation
Ligand similarity biased template selection criteria at the basis of the “Best Template Searching” tool has been successfully applied to rationalize the Structure Activity Relationships (SAR) of a series of [5-substituted-4-phenyl-1,3-thiazol-2-yl] furamides as antagonist of the hARs (Inamdar et al. 2013). The most potent derivative of the furamides series, the furan-2-carboxylic acid (4-phenyl-5-pyridin-4-yl-thiazol-2-yl)-amide, has been selected as query molecule: As reported in Table 2, a similarity sorting of the templates based on the combined similarity criteria has been taken into account to select the most suitable models for receptor-based ligand design. The selected workflow is summarized in Figure 1: Starting from the suggested best template, namely the structure with the 3UZA PDB ID, co-crystallized with the 6-(2,6-dimethylpyridin-4-yl)-5-phenyl-1,2,4-triazin-3-amine (T4G), we have constructed A1, A2B and A3 AR models through homology modeling and used the so derived structural information to provide hypotheses of ligand-receptor interaction and ligand-receptor selectivity profile (Inamdar et al. 2013).
Table 2.
Similarity sorting of human A 2A AR templates based on furan-2-carboxylic acid (4-phenyl-5-pyridin-4-yl-thiazol-2-yl)-amide query ligand
| Ligand | PDB ID template | Shape similarity | 2D similarity (Tanimoto) | 2D similarity (Tversky) | Pharmacophore similarity (Tanimoto) | Pharmacophore similarity (Tversky) | Combined similarity (Shape & FP) |
|---|---|---|---|---|---|---|---|
| T4G | 3UZA | 0.33 | 0.86 | 0.89 | 0.46 | 0.65 | 0.52 |
| ZM 241385 | 3PWH | 0.58 | 0.90 | 0.93 | 0.27 | 0.42 | 0.48 |
| T4E | 3UZC | 0.37 | 0.84 | 0.89 | 0.44 | 0.54 | 0.47 |
| ZM 241385 | 4EIY | 0.34 | 0.90 | 0.93 | 0.27 | 0.43 | 0.39 |
| ZM 241385 | 3EML | 0.35 | 0.90 | 0.93 | 0.27 | 0.42 | 0.39 |
| NECA | 2YDV | 0.51 | 0.82 | 0.87 | 0.17 | 0.31 | 0.39 |
| ZM 241385 | 3VG9 | 0.32 | 0.90 | 0.93 | 0.27 | 0.43 | 0.38 |
| XAC | 3REY | 0.21 | 0.89 | 0.94 | 0.25 | 0.48 | 0.37 |
| ZM 241385 | 3VGA | 0.28 | 0.90 | 0.93 | 0.27 | 0.42 | 0.36 |
| Adenosine | 2YDO | 0.33 | 0.82 | 0.86 | 0.18 | 0.31 | 0.31 |
| Caffeine | 3RFM | 0.26 | 0.81 | 0.85 | 0.21 | 0.34 | 0.30 |
| UK-432,097 | 3QAK | 0.16 | 0.87 | 0.93 | 0.14 | 0.35 | 0.27 |
Figure 1.
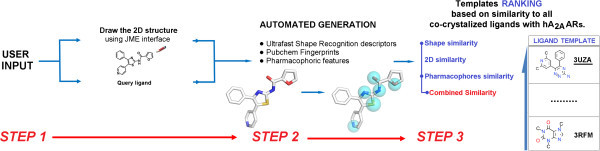
Workflow of the homology modeling template selection based on the structure of furan-2-carboxylic acid (4-phenyl-5-pyridin-4-yl-thiazol-2-yl)-amide.
Methods
The “Best Template Searching” tool is part of the Adenosiland infrastructure, based on Ubuntu 9.10 Linux operating system, which is a patchwork of several informatics tools (for more details see Floris et al. 2013). The similarity indexes are calculated by using different approaches: 2D similarity based on Tanimoto and Tversky indexes (Steinbeck et al. 2003,2006) are calculated from Pubchem Fingerprints (CDK implementation), the shape similarity is calculated by using an in-house implementation of the Ultrafast Shape Recognition method (Floris et al. 2011 Ballester and Richards 2007), and the pharmacophoric features of the pharmacophore-based similarity index are described by Gaussian 3D volumes (Taminau et al. 2008).
Conclusions
We have implemented a novel tool, called “Best Template Searching” to provide template suggestions and homology models of all four hARs based on the similarity between a query structure provided by the user and all co-crystallized ARs ligands. It is well known that ligand-driven induced fit of the receptor is a key feature to facilitate the identification or the optimization of novel potent and selective agonists and antagonists, in particular through molecular docking studies. We therefore believe that choosing as template the structure co-crystallized with the ligand that shares the highest structural similarity with the scaffold of interest may represent an effective strategy. This is in facts the underlying idea of our platform implementation: By using the “Best Template Searching” option, users can upload a SMILES string or directly draw the 2D structure by using the JME interface of the scaffold of interest and search the most similar ligand co-crystallized so far with the hA2A AR. Several similarity indexes are calculated by using different approaches such as a 2D similarity, shape similarity, pharmacophore-based similarity, and simple consensus shape- and pharmacophore-based similarity index.
We are also confident that the proposed strategy can be easily and effectively extended to other GPCRs.
Acknowledgements
The molecular modeling work coordinated by S.M. has been carried out with financial support of the University of Padova, Italy and the Italian Ministry for University and Research (MIUR), Rome, Italy. S.M. is also very grateful to Chemical Computing Group, YASARA Biosciences GmbH and Acellera for the scientific and technical partnership. Finally, we desire to give our appreciations to Peter Ertl for his courtesy in using its the Java Molecule Editor (JME).
Abbreviations
- ARs
Adenosine receptors
- GPCRs
G protein-coupled receptors
- NECA
N-ethyl-5′-carboxamido adenosine
- T4E
4-(3-amino-5-phenyl-1,2,4-triazin-6-yl)-2-chlorophenol
- T4G
6-(2,6-dimethylpyridin-4-yl)-5-phenyl-1,2,4-triazin-3-amine
- ZM 241385
4-(2-(7-amino-2-(2-furyl)(1,2,4)triazolo(2,3-a)(1,3,5)triazin-5-yl-amino)ethyl)phenol
- XAC
N-(2-aminoethyl)-2-[4-(2,6-dioxo-1,3-dipropyl- 2,3,6,7-tetrahydro-1H-purin-8-yl)phenoxy]acetamide.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MF, DS and RM developed and engineered the web tool. DS, ACi and ACu carried out the experiments, analyzed the data, and interpreted the results. ACi and SM designed the research protocol and wrote the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Matteo Floris, Email: floris@crs4.it.
Davide Sabbadin, Email: davide.sabbadin@studenti.unipd.it.
Antonella Ciancetta, Email: antonella.ciancetta@unipd.it.
Ricardo Medda, Email: medda@crs4.it.
Alberto Cuzzolin, Email: alberto.cuzzolin@studenti.unipd.it.
Stefano Moro, Email: stefano.moro@unipd.it.
References
- Ballester PJ, Richards WG. Ultrafast shape recognition to search compound databases for similar molecular shapes. J Comput Chem. 2007;28:1711–1723. doi: 10.1002/jcc.20681. [DOI] [PubMed] [Google Scholar]
- Congreve M, Andrews SP, Doré AS, Hollenstein K, Hurrell E, Langmead CJ, Mason JS, Ng IW, Tehan B, Zhukov A, Weir M, Marshall FH. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J Med Chem. 2012;55:1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris M, Masciocchi J, Fanton M, Moro S. Swimming into peptidomimetic chemical space using pepMMsMIMIC. Nucleic Acids Res. 2011;39:W261–W269. doi: 10.1093/nar/gkr287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris M, Sabbadin D, Medda R, Bulfone A, Moro S. Adenosiland: walking through adenosine receptors landscape. Eur J Med Chem. 2013;58:248–257. doi: 10.1016/j.ejmech.2012.10.022. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino T, Arakawa T, Iwanari H, Yurugi-Kobayashi T, Ikeda-Suno C, Nakada-Nakura Y, Kusano-Arai O, Weyand S, Shimamura T, Nomura N, Cameron AD, Kobayashi T, Hamakubo T, Iwata S, Murata T. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012;482:237–240. doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamdar GS, Pandya AN, Thakar HM, Sudarsanam V, Kachler S, Sabbadin D, Moro S, Klotz K-N, Vasu KK. New insight into adenosine receptors selectivity derived from a novel series of [5-substituted-4-phenyl-1,3-thiazol-2-yl] benzamides and furamides. Eur J Med Chem. 2013;63:924–934. doi: 10.1016/j.ejmech.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, Cherezov V, Stevens RC. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E. The chemistry development kit (CDK): an open-source Java library for chemo- and bioinformatics. J Chem Inf Comput Sci. 2003;43:493–500. doi: 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck C, Hoppe C, Kuhn S, Floris M, Guha R, Willighagen EL. Recent developments of the chemistry development kit (CDK) an open-source java library for chemo- and bioinformatics. Curr Pharm Des. 2006;12:2111–2120. doi: 10.2174/138161206777585274. [DOI] [PubMed] [Google Scholar]
- Taminau J, Thijs G, De Winter H. Pharao: pharmacophore alignment and Optimization. J Mol Graph Model. 2008;27:161–169. doi: 10.1016/j.jmgm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao Z-G, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]