

Abstract
Cystic fibrosis (CF) is caused by dysfunction of the epithelial anion channel cystic fibrosis transmembrane conductance regulator (CFTR). One strategy to restore function to CF mutants is to suppress defects in CFTR processing and function using revertant mutations. Here, we investigate the effects of the revertant mutations G550E and 4RK (the simultaneous disruption of four arginine-framed tripeptides (AFTs): R29K, R516K, R555K and R766K) on the CF mutant G551D, which impairs severely channel gating without altering protein processing and which affects a residue in the same α-helix as G550 and R555. Both G550E and 4RK augmented strongly CFTR-mediated iodide efflux from BHK cells expressing G551D-CFTR. To learn how revertant mutations influence G551D-CFTR function, we studied protein processing and single-channel behaviour. Neither G550E nor 4RK altered the expression and maturation of G551D-CFTR protein. By contrast, both revertants had marked effects on G551D-CFTR channel gating, increasing strongly opening frequency, while 4RK also diminished noticeably the duration of channel openings. Because G551D-CFTR channel gating is ATP independent, we investigated whether revertant mutations restore ATP dependence to G551D-CFTR. Like wild-type CFTR, the activity of 4RK-G551D-CFTR varied with ATP concentration, suggesting that 4RK confers some ATP dependence on the G551D-CFTR channel. Thus, the revertant mutations G550E and 4RK alter the gating pattern and ATP dependence of G551D-CFTR without restoring single-channel activity to wild-type levels. Based on their impact on the CF mutants F508del and G551D, we conclude that G550E and 4RK have direct effects on CFTR structure, but that their action on CFTR processing and channel function is CF mutation specific.
Introduction
The ATP-binding cassette (ABC) transporter cystic fibrosis transmembrane conductance regulator (CFTR; Riordan et al. 1989) is an epithelial anion channel with complex regulation (Gadsby et al. 2006). The importance of CFTR for transepithelial ion transport is dramatically highlighted by its malfunction in the common, life-shortening genetic disease cystic fibrosis (CF; Welsh et al. 2001). By far the most common CF mutation is F508del, the deletion of a phenylalanine residue at position 508 of the CFTR protein sequence (Welsh et al. 2001). The molecular basis for the F508del-CFTR defect is protein misfolding, which causes the retention of the mutant protein by the endoplasmic reticulum (ER) quality control mechanism and its rapid targeting for degradation by the ubiquitin–proteasome pathway (for review, see Lukacs & Verkman, 2012; Farinha et al. 2013b). Any F508del-CFTR that escapes ER quality control to reach the plasma membrane exhibits two further defects: plasma membrane instability (Lukacs et al. 1993) and defective channel gating (Dalemans et al. 1991).
In the search for strategies to rescue the defective processing and trafficking of F508del-CFTR, revertant (second site) mutations in cis with F508del have been identified. These mutations include V510D (Loo et al. 2010), I539T (deCarvalho et al. 2002), G550E (deCarvalho et al. 2002), R553M/Q (Teem et al. 1993) and R555K (Teem et al. 1996) located within nucleotide-binding domain 1 (NBD1) and R1070W (Thibodeau et al. 2010) located within intracellular loop (ICL) 4 of membrane-spanning domain 2 (MSD2). Restoration of the cell surface expression of F508del-CFTR is also achieved by (i) the simultaneous mutation of the four arginine-framed tripeptides (AFTs) in the CFTR gene (R29K, R516K, R555K and R766K termed 4RK; Chang et al. 1999); (ii) the introduction of NBD1 solubilising mutations into the CFTR gene (F494N/Q637R and F429S/F494N/Q637R; Pissarra et al. 2008); (iii) deletion of the regulatory insertion (amino acids F405–L436) from NBD1; Aleksandrov et al. 2010) and (iv) substitution of proline residues at dynamic locations within NBD1 together with the revertant mutation I539T (S422P/S434P/S492P/A534P/I539T; Aleksandrov et al. 2012). Consistent with their effects on F508del-CFTR processing and trafficking, some revertant mutations confer plasma membrane stability on F508del-CFTR (Aleksandrov et al. 2010, 2012; Wang et al. 2011). Moreover, selected revertant mutations attenuate the F508del-CFTR gating defect (Teem et al. 1993; Roxo-Rosa et al. 2006; Pissarra et al. 2008; Aleksandrov et al. 2010, 2012) and have been suggested to act at different sites to repair structural defects caused by the F508del mutation (Farinha et al. 2013a).
With the exception of the CF mutants R560T and A561E (Roxo-Rosa et al. 2006), the effects of revertant mutations have only been investigated on F508del-CFTR. We were therefore interested to learn whether revertant mutations might rescue the cell surface expression and function of other CF mutants. For several reasons, we were particularly interested in G551D, the third commonest CF mutant (Welsh et al. 2001) and first to be treated with a clinically approved CFTR potentiator (ivacaftor (Kalydeco™; VX-770); Van Goor et al. 2009; Ramsey et al. 2011). First, G551D affects the amino acid residue adjacent to G550 in the LSGGQ motif of NBD1, which together with the Walker A and B motifs of NBD2 forms the canonical ATP-binding site (termed site 2) of CFTR located at the NBD1:NBD2 dimer interface (Lewis et al. 2004; Gadsby et al. 2006; Hwang & Sheppard, 2009; Fig.1). Moreover, G551D, G550E and R555K (part of the revertant 4RK) are all situated within the H5 α-helix in the ABC α-subdomain of NBD1 (Lewis et al. 2004; Fig.1). Second, G551D causes a profound defect in channel gating (Li et al. 1996; Cai et al. 2006; Bompadre et al. 2007), without affecting CFTR processing, trafficking and plasma membrane stability (Gregory et al. 1991). Third, both G551D and G550E lead to the insertion of negatively charged amino-acid side-chains into ATP-binding site 2 (Lewis et al. 2004; Gadsby et al. 2006; Hwang & Sheppard, 2009). However, the consequences of these mutations are strikingly different: G550E enhances CFTR activity by prolonging markedly channel openings (Roxo-Rosa et al. 2006), whereas G551D diminishes CFTR activity by abolishing the ATP dependence of channel gating (Bompadre et al. 2007). We were therefore curious to learn how the insertion of two negatively charged residues into ATP-binding site 2 would affect CFTR channel gating.
Figure 1. Amino acid residues in the vicinity of G551 in NBD1 of human CFTR.
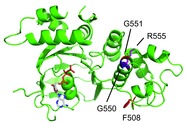
A cartoon view of the crystal structure of NBD1 of human CFTR solved by Lewis et al. (2005) is shown. The positions of F508 (red), G550 (blue), G551 (orange) and R555 (magenta) are indicated. The nucleotide bound by the Walker A and B sequence in NBD1 is also shown. The figure was produced with the pdb file (code: 2BBO) from the Protein Data Bank (http://www.rcsb.org).
To investigate the effects of the revertant mutations G550E and 4RK on G551D-CFTR, we employed a multi-disciplinary approach. Using BHK cells engineered to express high levels of CFTR protein and the iodide efflux technique, we examined the effects of revertant mutations on a large population of G551D-CFTR Cl− channels in intact cells. Then, using biochemical techniques, we explored the effects of the revertants on G551D-CFTR expression, turnover rate and processing efficiency. Next, using high-resolution single-channel recording, we evaluated the impact of the revertants on G551D-CFTR channel gating. Finally, using molecular models of CFTR structure, we investigated how G551D and G550E alter CFTR structure. We discovered that G550E and 4RK are without effect on G551D-CFTR expression and maturation, but alter markedly channel gating. Although neither revertant restored the gating pattern of wild-type CFTR to G551D-CFTR, 4RK conferred it with some ATP dependence. Thus, the revertants G550E and 4RK have direct effects on CFTR structure, but their impact on CFTR processing and function is CF mutation specific.
Methods
Site-directed mutagenesis
For CFTR expression, we used the pNUT expression vector. With the Quikchange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) and complementary pairs of custom-designed HPLC-purified oligonucleotides, we introduced specific mutations into the CFTR coding sequence. The identity of each CFTR construct was verified by sequencing the entire CFTR cDNA.
Cells and CFTR expression
For this study, we used baby hamster kidney (BHK) cells stably expressing CFTR constructs. To generate stable cell lines expressing G551D-, G550E-G551D- and 4RK-G551D-CFTR, we transfected BHK cells with CFTR cDNAs (1.5 μg) using Lipofectin (Life Technologies, Paisley, UK), selecting for stably transfected cells 48 h later using methotrexate (500 μm; AAH Pharmaceuticals Ltd, Coventry, UK), picking surviving clones after 10–15 days treatment with methotrexate. For each CFTR construct, we selected the BHK clone expressing the highest level of CFTR protein, taking care to ensure that the expression of CFTR protein among different CFTR variants was equivalent. BHK cells were cultured and used as described previously (Schmidt et al. 2008) with the exception that for experiments using excised inside-out membrane patches, cells were seeded onto glass coverslips and used within 3–6 days.
Iodide efflux experiments
We measured CFTR-mediated iodide efflux at 23°C as described by Lansdell et al. (1998) using the cAMP agonist forskolin (10 μm) and the CFTR potentiator genistein (50 μm). We used genistein (50 μm) to potentiate CFTR in iodide efflux experiments principally because of the restricted access of extracellular genistein to its binding site on CFTR (Schmidt et al. 2008). BHK cells were first incubated for 1 h in loading buffer containing (mm): 136 NaI, 3 KNO3, 2 Ca(NO3)2, 20 Hepes and 11 glucose, pH 7.4. Next, BHK cells were washed thoroughly with efflux buffer (136 mm NaNO3 replacing 136 mm NaI in the loading buffer) before treatment with agonists. Finally, the amount of iodide in each sample of efflux buffer was determined using an iodide-selective electrode (Orion 9653BNWP; Thermo Scientific, Waltham, MA, USA). Iodide efflux was normalised to the amount of processed CFTR protein as described by Da Paula et al. (2010).
Western blotting, metabolic labelling and immunoprecipitation
To assess the expression of CFTR protein, we used Western blotting (Farinha et al. 2002). We lysed BHK cells, loaded SDS-polyacrylamide mini-gels (7% (w v−1) polyacrylamide; BioRad, Hercules, CA, USA) with 50 μg total protein and separated protein electrophoretically. After transfer of proteins to Protran nitrocellulose membrane (Schleicher & Schuell BioScience GmbH, Dassel, Germany), we probed membranes with the anti-CFTR monoclonal antibody M3A7, which recognises NBD2 and the C-terminus of CFTR (residues 1197–1480) diluted at 1:2000 (Chemicon, Temecula, CA, USA; Farinha et al. 2004) using a secondary anti-mouse peroxidase-labelled monoclonal antibody at 1:3000 (GE Healthcare Bio-Sciences, Piscataway, CA, USA). Western blots were developed using the SuperSignal West Pico Chemiluminescent Substrate detection system (Pierce, Rockford, IL, USA).
To evaluate the processing efficiency of CFTR, we used metabolic labelling followed by immunoprecipitation (Farinha et al. 2002). We starved cells for 30 min in methionine-free α-modified Eagle's medium (α-MEM; Life Technologies) before radiolabelling for 30 min in the same medium supplemented with 150 μCi ml−1 [35S]-methionine (ICN Biomedicals, Irvine, CA, USA; Farinha et al. 2002). For the chase (0, 0.5, 1, 2 and 3 h), the labelling medium was replaced by α-MEM supplemented with fetal calf serum (5% v v−1) and non-radioactive methionine (1 mm). Cells were then lysed and CFTR protein immunoprecipitated with the anti-CFTR monoclonal antibody M3A7 (Chemicon) and protein G agarose beads (Pierce) before samples were electrophoresed in SDS-PAGE gels and detected by fluorography. Quantification by densitometry was performed with the ImageMaster software (GE Healthcare Bio-Sciences) as described (Farinha et al. 2004).
Electrophysiology
CFTR Cl− channels were recorded in excised inside-out membrane patches using an Axopatch 200B patch-clamp amplifier and pCLAMP data acquisition and analysis software (both from Molecular Devices, Union City, CA, USA; Sheppard & Robinson, 1997; Cai et al. 2006). The pipette (extracellular) solution contained (mm): 140 N-methyl-d-glucamine (NMDG), 140 aspartic acid, 5 CaCl2, 2 MgSO4 and 10 N-tris[hydroxymethyl]-methyl-2-aminoethanesulphonic acid (TES), adjusted to pH 7.3 with Tris ([Cl−], 10 mm). The bath (intracellular) solution contained (mm): 140 NMDG, 3 MgCl2, 1 CsEGTA and 10 TES adjusted to pH 7.3 with HCl, ([Cl−], 147 mm; free [Ca2+], <10−8 m) and was maintained at 37°C. CFTR Cl− channels were activated, channel rundown minimised and the effects of CFTR potentiators tested as described previously (Cai et al. 2006).
To investigate the ATP dependence of CFTR constructs, we used membrane patches containing multiple active channels. For all other studies, we used membrane patches containing ≤ 5 active channels (wild-type CFTR, number of active channels (N) = 1; G551D-CFTR, N ≤ 5; G550E-G551D-CFTR, N ≤ 4; 4RK-G551D-CFTR, N ≤ 5). To determine channel number, we used the maximum number of simultaneous channel openings observed during the course of an experiment. Despite the precautions that we observed (Cai et al. 2006), we cannot exclude the possibility of unobserved mutant Cl− channels in excised membrane patches causing values of open probability (Po) for G551D-, G550E-G551D- and 4RK-G551D-CFTR to be overestimated. In view of this difficulty, Po values for G551D-, G550E-G551D- and 4RK-G551D-CFTR are expressed as apparent Po values (Po(app)).
Single-channel currents were initially recorded on digital audiotape using a digital tape recorder (model DTR-1204, Biologic Scientific Instruments; Intracel Ltd, Royston, UK) at a bandwidth of 10 kHz. On playback, records were filtered with an eight-pole Bessel filter (model F-900C/9L8L, Frequency Devices Inc., Ottawa, IL, USA) at a corner frequency (fc) of 500 Hz and acquired using a DigiData1320A interface (Molecular Devices, CA, USA) and pCLAMP software at a sampling rate of 5 kHz. Unless otherwise specified, for the purpose of illustration, single-channel records were filtered at 500 Hz and digitised at 1 kHz.
To investigate the ATP dependence of CFTR constructs, average current (I) for a specific intervention was determined as the average of all the data points collected during the intervention. NPo was calculated by dividing I by single-channel current amplitude (i) or using lists of open and closed times as described previously (Hughes et al. 2008). To explore the ATP dependence of G551D-, G550E-G551D- and 4RK-G551D-CFTR, NPo values of individual CFTR constructs were expressed as a percentage of the control NPo value measured at ATP (1 mm) in the presence of PKA (75 nm).
To measure i, Gaussian distributions were fitted to current amplitude histograms. For Po (or Po(app)) and burst analyses, lists of open and closed times were created using a half-amplitude crossing criterion for event detection and dwell-time histograms constructed and fitted as described previously (Sheppard & Robinson, 1997); transitions < 1 ms were excluded from the analysis (eight-pole Bessel filter rise time (T10–90) ∼0.73 ms at fc = 500 Hz). Burst analysis was performed as described by Cai et al. (2006) using a tc (the time that separates interburst closures from intraburst closures) determined from analyses of closed time histograms. The mean interburst interval (TIBI) was calculated using the equation (Cai et al. 2006):
| (1) |
where Tb = (mean burst duration) × (open probability within a burst). Mean burst duration (TMBD) and open probability within a burst (Po(burst)) were determined directly from experimental data using pCLAMP software. For wild-type CFTR, only membrane patches that contained a single active channel were used for burst analysis, whereas for G551D-, G550E-G551D- and 4RK-G551D-CFTR, we used membrane patches containing no more than three active channels. We analysed only bursts of single channel openings with no superimposed openings that were separated from one another by a time interval ≥ tc.
Reagents
PKA purified from bovine heart was purchased from Promega Ltd (Southampton, UK) and genistein from LC Laboratories (Woburn, MA, USA). All other chemicals were of reagent grade and supplied by the Sigma-Aldrich Company, Ltd (Gillingham, UK). ATP and 2′-dATP were dissolved in intracellular solution, forskolin in methanol and genistein in DMSO. Stock solutions were stored at −20°C with the exception of those of ATP and 2′-dATP, which were prepared directly before each experiment. Immediately before use, stock solutions were diluted to achieve final concentrations. Precautions against light-sensitive reactions were observed when using genistein. DMSO was without effect on CFTR activity (Sheppard & Robinson, 1997; Schmidt et al. 2008).
Modelling
Models of G551D- and G550E-CFTR NBD1 were generated using available crystal structures of human CFTR NBD1 (pdb codes: 1XMJ and 2BBO). The dimeric Mj0796 NBD structure (pdb code: 1L2T) was used to model the effects of the G550E and G551D equivalent mutations in a tight dimer, ATP-bound NBD state. Modelling was accomplished using VMD and PyMol.
Statistics
Results are expressed as means ± SEM of n observations. To compare sets of data, we used Student's t test. Differences were considered statistically significant when P < 0.05. All tests were performed using SigmaStat (Systat Software Inc., Richmond, CA, USA).
Results
The revertant mutations G550E and 4RK restore CFTR-mediated iodide efflux to the CF mutant G551D-CFTR
The CF mutation G551D disrupts profoundly CFTR channel gating by affecting a key residue in the LSGGQ motif of ATP-binding site 2 (Fig.1; Li et al. 1996; Cai et al. 2006; Bompadre et al. 2007). To investigate whether the revertant mutations G550E and 4RK restore function to G551D-CFTR, we used BHK cells engineered to express high levels of CFTR variants and the iodide efflux technique. With the iodide efflux technique, we evaluate the effects of site-directed mutations and small-molecules on the expression and function of a large population of CFTR Cl− channels in intact cells (Roxo-Rosa et al. 2006; Schmidt et al. 2008).
Figure2A and D demonstrates that the cAMP agonist forskolin (10 μm) and the CFTR potentiator genistein (50 μm) elicited a large transient efflux of iodide from BHK cells expressing wild-type CFTR; untransfected BHK cells fail to respond to these agonists (Schmidt et al. 2008). The magnitude of CFTR-mediated iodide efflux generated by BHK cells expressing G551D-CFTR was attenuated 1.9-fold and delayed in onset by 3 min compared with that of BHK cells expressing wild-type CFTR (Fig.2A and D). Nevertheless, when compared with the response of BHK cells expressing F508del-CFTR studied under identical conditions (Roxo-Rosa et al. 2006; Schmidt et al. 2008), the amount of iodide efflux elicited by G551D-CFTR was sizeable. Interestingly, the magnitude of CFTR-mediated iodide efflux generated by BHK cells expressing the revertant mutations G550E or 4RK in cis with G551D (hereafter termed G550E-G551D-CFTR and 4RK-G551D-CFTR, respectively) was similar to that of wild-type CFTR expressing BHK cells, although efflux was delayed in onset, albeit not as markedly as for G551D-CFTR expressing BHK cells (Fig.2B–D). We interpret these results to suggest that the revertant mutations G550E and 4RK restore to G551D-CFTR substantial amounts of CFTR function.
Figure 2. Revertant mutations enhance iodide efflux from BHK cells expressing G551D-CFTR.
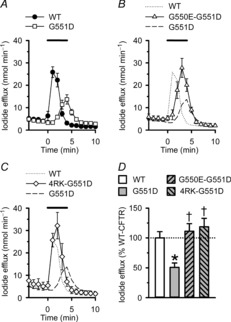
A–C, time courses of iodide efflux from BHK cells expressing the indicated CFTR constructs. During the periods indicated by the bars forskolin (10 μm) and genistein (50 μm) were added to the efflux buffer. Symbols and error bars indicate mean ± SEM (n = 4–6) for each construct. Where not shown, error bars are smaller than symbol size. D, magnitude of peak iodide efflux generated by different CFTR constructs expressed as a percentage of that of wild-type (WT) CFTR. *Significantly different from WT-CFTR (P < 0.05); †significantly different from G551D-CFTR (P < 0.05).
Revertant mutations are without effect on the expression and maturation of G551D-CFTR protein
The CF mutation G551D perturbs channel gating without altering CFTR processing and trafficking (Gregory et al. 1991; Li et al. 1996; Cai et al. 2006; Bompadre et al. 2007). This suggests that revertant mutations might exert their effects on G551D-CFTR by rescuing its gating defect. However, three lines of evidence argue that G550E and 4RK might alter the cell surface expression of G551D-CFTR. First, these revertants restore function to F508del-CFTR predominantly by enhancing its trafficking to the cell surface (Roxo-Rosa et al. 2006). Second, 4RK augments the expression of mature protein for wild-type and V562I-CFTR, a sequence polymorphism in the CFTR gene (Roxo-Rosa et al. 2006). Third, revertant mutations might impose distinct structural changes on F508del- and G551D-CFTR, possibly leading to a decrease in the expression of G551D-CFTR at the cell surface. Thus, we investigated the effects of G550E and 4RK on the expression and maturation of G551D-CFTR protein.
When Western blotting or immunoprecipitation is used to investigate the biosynthesis of CFTR, two different forms of wild-type CFTR protein are visualized: an immature core-glycosylated form that is found in the ER (150 kDa; band B) and a mature fully glycosylated form that has been processed through the Golgi apparatus and delivered to the cell membrane (170–180 kDa; band C; Cheng et al. 1990). Figure3A shows a representative Western blot of wild-type and G551D-CFTR in the absence and presence of G550E and 4RK and Fig.3B quantifies the amount of CFTR protein present in the mature form at steady state for the different CFTR constructs. These data demonstrate that the expression of mature CFTR protein does not differ between wild-type and G551D-CFTR in the absence and presence of revertant mutations. As a result, the magnitude of CFTR-mediated iodide efflux is little changed when it is normalised to the amount of processed CFTR protein (Table1).
Figure 3. Turnover and processing of G551D-CFTR in the absence and presence of revertant mutations.
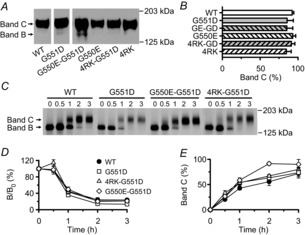
A, Western blot of total protein extracts from BHK cells expressing the indicated CFTR constructs. CFTR was detected with the anti-CFTR monoclonal antibody M3A7. B, quantification of CFTR expression. The amount of CFTR protein present in the mature form (band C) is expressed as a percentage of total CFTR protein (% band C = [band C/(bands B + C)) × 100]. C, autoradiograph of immunoprecipitated CFTR protein from BHK cells expressing the indicated CFTR constructs. Cells were radiolabelled with [35S]-methionine and chased for 0, 0.5, 1, 2 or 3 h before CFTR was immunoprecipitated and separated by SDS-PAGE followed by fluorography. D, turnover of the band B form of CFTR protein. Data are presented as the amount of band B at a given time of the chase (B) relative to the amount at the beginning of the experiment (B0). E, efficiency of processing into band C. Data are presented as the amount of band C at a given time of chase relative to the amount of band B at the beginning of the experiment. In A and C, arrows indicate the positions of the band B and C forms of CFTR protein and the position of molecular weight markers is indicated. In B, D and E, data are means ± SEM (n = 4).
Table 1.
Normalisation of iodide efflux to processed CFTR protein
| CFTR variant | Percentage processeda | Normalised processingb (%) | Normalised iodide effluxc (% peak response) | Iodide efflux to processed proteind (%) |
|---|---|---|---|---|
| Forskolin and genistein | ||||
| WT | 92 ± 2 | 100 | 100 ± 11 | — |
| G550E | 93 ± 4 | 101 | 150 ± 11e | 149 |
| 4RK | 85 ± 6 | 92 | 111 ± 9e | 121 |
| G551D | 86 ± 8 | 93 | 51 ± 7 | 55 |
| G550E-G551D | 85 ± 8 | 92 | 111 ± 13 | 121 |
| 4RK-G551D | 91 ± 5 | 99 | 119 ± 14 | 120 |
| Forskolin and IBMX | ||||
| WT | 92 ± 2 | 100 | 100 ± 7 | — |
| G550E | 93 ± 4 | 101 | ND | ND |
| 4RK | 85 ± 6 | 92 | ND | ND |
| G551D | 86 ± 8 | 93 | 15 ± 2 | 16 |
| G550E-G551D | 85 ± 8 | 92 | 17 ± 2 | 18 |
| 4RK-G551D | 91 ± 5 | 99 | 37 ± 3 | 37 |
CFTR-mediated iodide efflux was evoked by treating BHK cells expressing CFTR variants with forskolin (10 μm) and either genistein (50 μm) or IBMX (100 μm). ND, not determined.
Percentage of CFTR protein processing given by [band C/(band B + band C) × 100].
Ratio of ‘Percentage processed’ value for each CFTR variant to the equivalent value for WT-CFTR.
CFTR-mediated iodide efflux expressed as % of maximum response relative to that of WT-CFTR.
Ratio of ‘Normalised iodide efflux’ value to the corresponding ‘Normalised processing’ value for a given CFTR variant.
Data are from Roxo-Rosa et al. (2006).
To evaluate the effects of the revertant mutations on the turnover rate of band B and the efficiency of its processing into band C, we used pulse-chase analyses followed by CFTR immunoprecipitation. Figure3C again demonstrates that both the immature and mature forms of CFTR were detected in BHK cells expressing wild-type CFTR and the three G551D-CFTR variants. Analysis of these data revealed that the turnover rate of band B for the three G551D-CFTR variants was the same as that of wild-type CFTR (Fig.3D). Moreover, the processing efficiency of G551D- and 4RK-G551D-CFTR was the same as wild-type CFTR, whereas that of G550E-G551D-CFTR was enhanced 1.6-fold at 2 h, but not at other chase times (Fig.3E). We interpret these data to suggest that with the exception of a slight acceleration of protein maturation by G550E, revertant mutations are without effect on the expression and maturation of G551D-CFTR protein.
Revertant mutations alter G551D-CFTR channel gating
The lack of effect of G550E and 4RK on G551D-CFTR protein processing suggests that the augmentation of CFTR-mediated iodide efflux by these mutations is likely to result from a change in channel gating. To test this idea, we used high-resolution single-channel recording to investigate the gating behaviour of CFTR variants in excised inside-out membrane patches. The gating pattern of wild-type CFTR is characterised by bursts of channel openings interrupted by brief flickery closures and separated by longer closures between bursts (Fig.4). By contrast, the gating behaviour of G551D-CFTR is characterised by a marked attenuation of the duration of channel openings and a dramatic prolongation of the closed time interval between openings (Fig.4). Interestingly, both G550E and 4RK increased the frequency, but not the duration of G551D-CFTR channel openings with the effects of 4RK being especially pronounced (Fig.4; for high-resolution recordings of individual openings of the 4RK-G551D-CFTR Cl− channel, see Fig.5). Thus, the gating pattern of G550E-G551D- and 4RK-G551D-CFTR were characterised by frequent, very flickery channel openings.
Figure 4. The single-channel activity of G551D-CFTR in the absence and presence of revertant mutations.
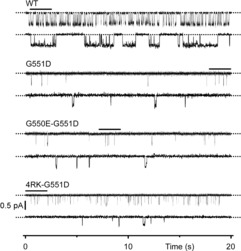
Representative single-channel recordings of WT-, G551D-, G550E-G551D- and 4RK-G551D-CFTR in excised inside-out membrane patches from BHK cells expressing recombinant CFTR. In this and subsequent figures, unless otherwise indicated, ATP (1 mm) and PKA (75 nm) were continuously present in the intracellular solution, voltage was –50 mV, there was a large Cl− concentration gradient across the membrane patch ([Cl−]int = 147 mm; [Cl−]ext = 10 mm) and temperature was 37°C. Dotted lines indicate where channels are closed and downward deflections correspond to channel openings. Beneath each prolonged 20 s recording, 2 s portions indicated by the bars are shown on an expanded time scale. For the purpose of illustration, single-channel records were filtered at 500 Hz and digitised at 1 kHz.
Figure 5. High-resolution openings of the 4RK-G551D-CFTR Cl− channel.
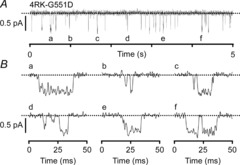
A, representative recording of a single 4RK-G551D-CFTR Cl− channel in an excised inside-out membrane patch. B, six examples of openings of 4RK-G551D-CFTR taken from the indicated positions (a–f) in A. Dotted lines indicate where channels are closed and downward deflections correspond to channel openings. Traces were filtered at 500 Hz and digitised at either 1 kHz (top) or 5 kHz (bottom).
To quantify single-channel behaviour, we measured i, Po (or Po(app) when there was uncertainty about the number of active channels), mean burst duration (MBD) and interburst interval (IBI). Analysis of single-channel current amplitude histograms revealed that the distribution of open-channel current values for 4RK-G551D-CFTR was shifted slightly towards that of the closed channel with the result that i was statistically smaller than those of wild-type and G551D-CFTR (P < 0.05; Fig.6A and B). By contrast, for G551D- and G550E-G551D-CFTR, i did not differ statistically from that of wild-type CFTR (P > 0.05; Fig.6B). Consistent with previous results (e.g. Cai et al. 2006), Fig.6C–E demonstrates that the Po(app) of G551D-CFTR was reduced 97-fold compared to that of wild-type CFTR because the interburst interval (IBI) of G551D-CFTR was prolonged 39-fold and mean burst duration (MBD) diminished by 79%. In contrast to its effects on F508del-CFTR (Roxo-Rosa et al. 2006), G550E did not restore channel activity to G551D-CFTR. Nevertheless, G550E increased slightly, but significantly (P < 0.05), the Po(app) value of G551D-CFTR by reducing IBI by 61% without altering MBD (P = 0.22); Fig.6C–E). Figure6D and E reveal that 4RK had marked effects on G551D-CFTR channel gating. First, 4RK attenuated the IBI of G551D-CFTR by 92% with the result that the IBI of 4RK-G551D-CFTR was only 3.2-fold longer than that of wild-type CFTR (Fig.6E). Second, 4RK diminished the MBD of G551D-CFTR 3.3-fold so that the MBD of 4RK-G551D-CFTR was 94% shorter than that of wild-type CFTR (Fig.6D). These changes in G551D-CFTR channel gating induced by 4RK caused a small, but statistically significant enhancement of Po(app) (P < 0.05; Fig.6C). However, this enhancement of Po(app) did not restore wild-type function to G551D-CFTR.
Figure 6. Quantification of single-channel data.
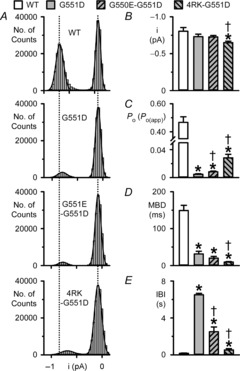
A, current amplitude histograms of single CFTR Cl− channels in excised inside-out membrane patches from BHK cells expressing the indicated CFTR constructs. The continuous lines represent the fit of Gaussian distributions to the data and the vertical dotted lines indicate the positions of the open and closed channel levels of WT-CFTR; the closed channel amplitude is shown on the right. B–E, single-channel current amplitude (i), open probability (Po) or apparent open probability (Po(app)), mean burst duration (MBD) and interburst interval (IBI) of the indicated CFTR constructs. All wild-type CFTR data are from membrane patches containing a single active CFTR Cl− channel, whereas for G551D-, G550E-G551D- and 4RK-G551D-CFTR, Po(app) and burst analysis data are from membrane patches containing ≤ 5 active channels and ≤ 3 active channels, respectively. Data are means + SEM (WT-CFTR, n = 6; G551D-CFTR, i and Po(app), n = 5, MBD and IBI, n = 3; G550E-G551D-CFTR, i and Po(app), n = 12, MBD and IBI, n = 6; 4RK-G551D-CFTR, i and Po(app), n = 12, MBD and IBI, n = 5). *Significantly different from WT-CFTR (P < 0.05); †significantly different from G551D-CFTR (P < 0.05).
The single-channel behaviour of the CFTR constructs G551D-, G550E-G551D- and 4RK-G551D-CFTR necessitates additional controls to minimise analytical error.
Data acquisition
We routinely filter and digitise single-channel records at 500 Hz and 5 kHz, respectively, excluding transitions < 1 ms from analysis. Because G551D-, G550E-G551D- and particularly 4RK-G551D-CFTR were characterised by many very poorly resolved channel openings, we repeated our quantification of single-channel behaviour using single-channel records filtered at a corner frequency (fc) of 1 kHz and digitised using a sampling rate of 10 kHz (Fig.7). Using these conditions, transitions < 0.4 ms were excluded from the analysis (eight-pole Bessel filter rise time (T10–90) ∼0.33 ms at fc = 1 kHz). Figures6 and 7 demonstrate that very similar results were obtained when single-channel records were lightly filtered compared with our standard data acquisition conditions. We interpret these results to suggest that our data acquisition conditions are unlikely to limit the analysis of the brief channel openings observed in this study.
Figure 7. Quantification of single-channel data filtered at 1 kHz and digitised at 10 kHz.
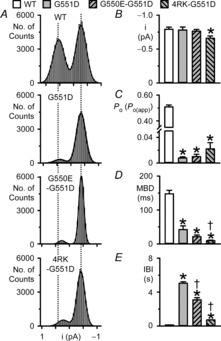
A, current amplitude histograms of single CFTR Cl− channels in excised inside-out membrane patches from BHK cells expressing the indicated CFTR constructs. The continuous lines represent the fit of Gaussian distributions to the data and the vertical dotted lines indicate the positions of the open and closed channel levels of WT-CFTR; the closed channel amplitude is shown on the right. B–E, single-channel current amplitude (i), open probability (Po) or apparent open probability (Po(app)), mean burst duration (MBD) and interburst interval (IBI) of the indicated CFTR constructs. All wild-type CFTR data are from membrane patches containing a single active CFTR Cl− channel, whereas for G551D-, G550E-G551D- and 4RK-G551D-CFTR, Po(app) and burst analysis data are from membrane patches containing ≤ 5 active channels and ≤ 3 active channels, respectively. Data are means + SEM or SD (WT-CFTR, n = 5; G551D-CFTR, n = 2; G550E-G551D-CFTR, i and Po(app), n = 5, MBD and IBI, n = 4; 4RK-G551D-CFTR, i and Po(app), n = 4, MBD and IBI, n = 3). *Significantly different from WT-CFTR (P < 0.05); †significantly different from G551D-CFTR (P < 0.05). To explain the different n values for the data in Figs 6 and 7, not all the membrane patches used in Fig.6 had low enough baseline noise for reanalysis using the conditions of Fig.7.
Number of active channels
To reduce uncertainty about Po(app) values for mutant Cl− channels, we repeated the analysis of Po(app) using membrane patches containing ≤ 3 active channels, the minimum number of active channels observed with G551D-CFTR in the present study. Identical values of Po(app) were obtained for G551D-, G550E-G551D- and 4RK-G551D-CFTR using membrane patches containing ≤ 5 or ≤ 3 active channels (G551D-CFTR: ≤ 5 active channels, Po(app) = 0.0048 ± 0.0008, n = 5; ≤ 3 active channels, Po(app) = 0.0047 ± 0.0011, n = 3; P = 0.91; G550E-G551D-CFTR: ≤ 5 active channels, Po(app) = 0.0084 ± 0.0010, n = 12; ≤ 3 active channels, Po(app) = 0.0086 ± 0.0018, n = 6; P = 0.89; 4RK-G551D-CFTR: ≤ 5 active channels, Po(app) = 0.0283 ± 0.0051, n = 12; ≤ 3 active channels, Po(app) = 0.0280 ± 0.0119, n = 5; P = 0.97). Moreover, the gating behaviour of G551D-CFTR recorded in this study did not differ from our previous data using identical experimental conditions (Fig.6C–E; Cai et al. 2006).
4RK restores some ATP-dependent channel gating to G551D-CFTR
The CF mutation G551D eliminates the ATP dependence of CFTR channel gating (Bompadre et al. 2007). We were therefore interested to learn whether G550E and 4RK might restore ATP dependence to G551D-CFTR. To test this possibility, we used excised inside-out membrane patches containing large numbers of active channels and measured mean current to determine NPo values in the presence of ATP (0.1, 1 and 5 mm) in the intracellular solution. Consistent with the data of Bompadre et al. (2007), Fig.8A–D demonstrates that as the ATP concentration increased, the activity of wild-type CFTR rose, whereas that of G551D-CFTR was largely unaffected except at ATP (5 mm). The revertant mutation G550E had little or no effect at ATP (0.1 mm), but enhanced the magnitude of CFTR current observed at ATP (5 mm) by 2.3-fold, albeit not significantly (P = 0.9; Fig.8E and F). By contrast, the activity of 4RK-G551D-CFTR varied with ATP concentration (Fig.8,G and H). Like wild-type CFTR, at 0.1 mm ATP 4RK-G551D-CFTR Cl− current was reduced by 48%, whereas at 5 mm ATP it was increased by 42% (Fig.8B and H). Consistent with these data, 4RK increased greatly the frequency of G551D-CFTR channel openings (Fig.6E), suggesting that it accelerates the channel opening rate, the ATP-dependent step in CFTR channel gating (Winter et al. 1994; Zeltwanger et al. 1999; Vergani et al. 2003). Taken together, the data suggest that 4RK confers some ATP dependence on G551D-CFTR without rescuing its gating defect.
Figure 8. 4RK, but not G550E, confers some ATP sensitivity on G551D-CFTR.
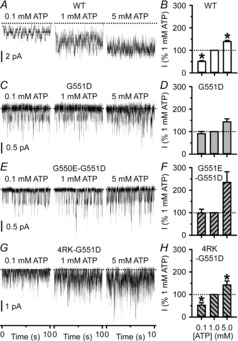
A, C, E and G, representative recordings show the activity of WT-, G551D-, G550E-G551D- and 4RK-G551D-CFTR in excised inside-out membrane patches from BHK cells. The recordings were made in the continuous presence of PKA (75 nm) and the indicated ATP concentrations. Dotted lines indicate where channels are closed and downward deflections correspond to channel openings. B, D, F and H, effects of intracellular ATP concentration on the activity of different CFTR constructs. For each CFTR construct, channel activity, measured as NPo, is expressed as a percentage of that at ATP (1 mm). Data are means ± SEM (WT-CFTR, n = 6–8; G551D-CFTR, n = 4–7; G550E-G551D-CFTR, n = 6–10; 4RK-G551D-CFTR, n = 6–12). *Significantly different from control values at ATP (1 mm) (P < 0.05).
2′-Deoxy-ATP potentiates the single-channel activity of G550E-G551D- and 4RK-G551D-CFTR
Our data demonstrate that in excised inside-out membrane patches, the single-channel activity of G550E-G551D- and 4RK-G551D-CFTR is substantially less than that of wild-type CFTR. However, in the presence of the CFTR potentiator genistein, BHK cells expressing G550E-G551D- and 4RK-G551D-CFTR elicited substantial amounts of iodide efflux like those expressing wild-type CFTR. We were therefore interested to learn whether the single-channel activity of G550E-G551D- and 4RK-G551D-CFTR in excised inside-out membrane patches might be augmented with small molecules. In contrast to its effects on CFTR-mediated iodide efflux (Fig.2), genistein (30 μm), a maximally effective concentration in excised membrane patches (Wang et al. 1998), failed to restore the single-channel activity of G550E-G551D- and 4RK-G551D-CFTR to wild-type levels (n = 12; data not shown). We therefore selected for study the hydrolysable ATP analogue 2′-deoxy-ATP (2′-dATP), which enhances robustly G551D-CFTR channel gating (Cai et al. 2006). To investigate the impact of 2′-dATP on G550E-G551D- and 4RK-G551D-CFTR channel gating, we replaced ATP (1 mm) by 2′-dATP (1 mm) in the intracellular solution and recorded channel activity in the presence of PKA (75 nm). Figure9 demonstrates that 2′-dATP (1 mm) failed to restore wild-type levels of channel activity to G551D-CFTR constructs. However, the Po(app) values of G551D, G550E-G551D- and 4RK-G551D-CFTR were equivalent and 14-, 7.9- and 3.6-fold larger than those recorded in the presence of ATP (1 mm; Fig.9). Thus, in excised inside-out membrane patches G550E and 4RK failed to restore wild-type channel gating to the CF mutation G551D. However, they had pronounced effects on G551D-CFTR channel gating and 4RK restored some ATP dependence to the mutant Cl− channel.
Figure 9. Potentiation of CFTR channel gating by 2′-dATP.
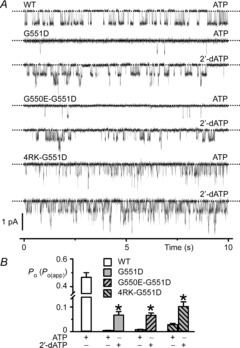
A, representative recordings show the effects of 2′-dATP (1 mm) on CFTR Cl− channels in excised inside-out membrane patches from BHK cells expressing the indicated CFTR constructs. The recordings were made in the continuous presence of PKA (75 nm) and either ATP (1 mm) or 2′-dATP (1 mm) in the intracellular solution. Dotted lines indicate where channels are closed and downward deflections correspond to channel openings. For comparison, the single-channel activity of wild-type CFTR in the presence of ATP (1 mm) and PKA (75 nm) is shown. In the presence of 2′-dATP (1 mm), the Po of wild-type CFTR is 0.70 ± 0.03 (n = 5) (for further information, see Fig.6 of Cai et al. (2006)). B, effects of 2′-dATP (1 mm) on the Po or Po(app) of the indicated CFTR constructs. Data are means + SEM (WT-CFTR, n = 6; G551D-CFTR, n = 5; G550E-G551D-CFTR, n = 9–12; 4RK-G551D-CFTR, n = 7–12). *Significantly different from their control values (P < 0.05).
Structural alterations in the LSGGQ sequence associated with G550E and G551D
The changes in CFTR channel gating associated with alterations in and proximal to the LSGGQ (ABC transporter signature) sequence are consistent with altered interactions between the NBDs. These highly conserved sequences complement the Walker A and Walker B motifs in the ATP-bound state to induce NBD dimerisation and channel opening (Gadsby et al. 2006; Hwang & Sheppard, 2009). To evaluate the impact of the glycine substitutions, molecular models of the G551D mutation were produced using extant NBD crystal structures that include the G550E substitution as a starting template (Lewis et al. 2005). The G550E and G551D substitutions can be accommodated with minimal changes to NBD structure (Fig.10A). The G550E side-chain is highly surface exposed at the N-terminal end of an α-helix. The G551D mutation is also accommodated, but is more constrained by its location in the first turn of the α-helix and by surrounding protein structure. Both substitutions introduce local electrostatic changes to the surface of the NBD that packs tightly against the bound nucleotide from the opposing monomer (Fig.10B). The introduction of steric and electrostatic changes likely results in disrupted NBD–NBD association in the ATP-bound state with the G551D substitution.
Impact of G550E and G551D on CFTR structure.
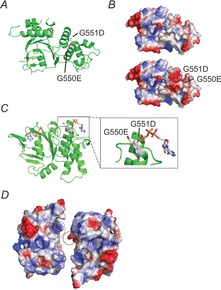
Structural analyses of the impacts of the G550E and G551D mutants were completed to further understand the mechanical basis of CFTR channel gating. A, schematic cartoon of human NBD1 containing the G550E substitution and the modelled G551D mutation (pdb code: 1XMJ). Both sites are shown with side-chain sticks and are labelled. B, surface electrostatic changes associated with the G550E and G551D mutations are shown based on available human NBD1 crystal structures. The native LSGGQ sequence (top) is shown for comparison to the G550E-G551D double mutant (bottom). Regions where surface electrostatics are altered by the glycine substitutions are circled and labelled. C, a cartoon of the Mj0796 NBD is shown with the G550E and G551D equivalent mutations and ATP bound to the complementing Walker A/B half-site. A single NBD is shown for clarity; both nucleotides are indicated for reference. The G551D side-chain is proximal to the γ-phosphate of the bound nucleotide (see the inset). The G550E side-chain is clear of the nucleotide, but may rotate and translate to mimic the γ-phosphate. D, a surface representation of the Mj0796 NBD dimer is shown using calculated electrostatic potentials. The NBDs have been translated apart to display the positions of the complementing electrostatic regions surrounding the nucleotide-binding pocket. The G550E side-chain is circled with a continuous line; the complementing Walker A/B half-site is circled with a dashed line. This interdomain interaction is predicted to stabilise the NBD1:NBD2 dimer and result in an increase in the channel open state, as is observed with G550E and F508del-G550E (Roxo-Rosa et al. 2006).
The changes in CFTR channel gating may in part be explained by interactions of the G551D and G550E side-chains in model NBD dimers. The Mj0796 model NBD dimer was chosen for study as no existing high-resolution structures of CFTR's NBD heterodimer have been solved (Smith et al. 2002). The conservation of the canonical sequences and mechano-chemistry associated with nucleotide binding and hydrolysis suggest that the dimerisation events are likely to be similar at the active CFTR ATPase site. As with CFTR NBD structures, the introduction of the two acidic amino acids required small perturbations to the NBD structure.
In the dimer state, the G550E and G551D substitutions show differences with respect to their proximity to the bound nucleotide. Specifically, the G551D side-chain is positioned proximal to the phosphate groups of the bound nucleotide and would be likely to interfere sterically and electrostatically with ATP-mediated NBD dimerisation (Gadsby et al. 2006; Hwang & Sheppard, 2009; Fig.10C). The G550E mutation is located further from the phosphate groups. The conformational flexibility and increased distance from the γ-phosphate likely tolerates ATP binding and NBD dimerisation. In a post-hydrolytic ADP-bound or nucleotide-free state, the G550E side-chain carboxylate group would be in proximity to interact electrostatically with NBD2, complementing basic surfaces formed within the Walker A/B half-site (Fig.10D). These interactions would require minimal changes in G550E side-chain reorientation or a small translation of the NBDs along the NBD-dimer interface. The enhancement of channel gating by the G550E mutation might be explained by augmented inter-NBD interactions in these models. These models predict that the effect of the G550E substitution would be to enhance Po by decreasing IBI and increasing MBD. This prediction is consistent with our previous studies of the impact of G550E on wild-type and F508del-CFTR (Roxo-Rosa et al. 2006) and in general agreement with the current work, where the IBI of G551D-CFTR was decreased and MBD unaltered by the revertant mutation (Fig.6D and E). One potential mechanism that emerges from the structural model, which is consistent with the functional data, is that the mutations G550E and G551D alter the stability of the NBD1:NBD2 dimer. In general, the G550E mutation increases the apparent NBD–NBD affinity to stabilize the NBD dimer, whereas the G551D mutation probably destabilizes the NBD dimer.
Discussion
In this study, we investigated the impact of the revertant mutations G550E and 4RK on G551D-CFTR, a CF mutant that is correctly delivered to the cell surface, but which disrupts profoundly CFTR function, eliminating ATP-dependent channel gating (Li et al. 1996; Cai et al. 2006; Bompadre et al. 2007). We discovered that G550E and 4RK were without effect on G551D-CFTR protein processing, but altered markedly channel gating. Of note, 4RK restored some ATP-dependent channel gating to G551D-CFTR.
Previous work demonstrates that G550E and 4RK are likely to rescue F508del-CFTR expression and function by distinct mechanisms. 4RK allows F508del-CFTR to escape ER quality control and traffic to the cell surface, whereas G550E probably facilitates protein folding by promoting correct domain assembly with the result that the mutant protein is delivered to the cell surface and its gating defect attenuated (Roxo-Rosa et al. 2006; Hoelen et al. 2010). Consistent with these data, rescue of F508del-CFTR by 4RK and G550E is additive (Farinha et al. 2013a). 4RK, but not G550E, enhanced steady-state levels of mature protein (band C) for wild-type CFTR and V562I-CFTR, by increasing, albeit not significantly, protein maturation (Roxo-Rosa et al. 2006). By contrast, the present results demonstrate that revertant mutations have little or no impact on the expression and maturation of G551D-CFTR protein. They also reveal that the impact of G550E and 4RK on F508del- and G551D-CFTR channel gating differs strikingly. First, G550E and 4RK prolong markedly the MBD of F508del-CFTR, but are either without effect or attenuate sharply that of G551D-CFTR (Roxo-Rosa et al. 2006; present study). Second, 4RK is without effect on the IBI of F508del-CFTR, but diminishes strongly that of G551D-CFTR (Roxo-Rosa et al. 2006; present study). As a result, the Po of F508del-CFTR was equivalent to wild-type CFTR, whereas the Po(app) of G551D-CFTR remained profoundly abnormal. We interpret these results to suggest that the effects of G550E and 4RK on CFTR expression and function are CF mutation specific and due to different structural defects. Indeed, rescue of both the NBD1 folding defect and stabilization of domain interfaces is required to overcome the F508del-CFTR folding defect (Mendoza et al. 2012; Rabeh et al. 2012), whereas a single revertant mutation in NBD1 (G550E) endows F508del-CFTR with wild-type levels of channel activity, albeit as a consequence of a different gating pattern (Roxo-Rosa et al. 2006).
In the ATP-driven NBD dimerisation model of CFTR channel gating (Vergani et al. 2003, 2005), ATP binding at site 2 initiates channel opening, while ATP hydrolysis at site 2 leads promptly to channel closure (Gadsby et al. 2006; Hwang & Sheppard, 2009). Evidence from crystal structures of NBD dimers from related ABC transporters argue that G551 forms a critical contact with the γ-phosphate of ATP during its binding to site 2 (for review, see Hwang & Sheppard, 2009). Unsurprisingly, therefore replacement of a hydrogen atom by a large acidic side-chain in the mutation G551D has devastating consequences for CFTR channel gating (Li et al. 1996; Cai et al. 2006; Bompadre et al. 2007). Bompadre et al. (2007) argue that the molecular basis of the G551D mutation is not the prevention of ATP binding at site 2, but the obstruction of conformational changes subsequent to ATP binding that gate the channel pore. In support of this idea, the authors demonstrate that the gating behaviour of G551D represents the spontaneous ATP-independent activity of wild-type CFTR (Bompadre et al. 2007). Interestingly, our structural models of G551D and G550E (Fig.10) are also consistent with this idea. ATP binding at site 2 would probably not be directly altered by either G550E or G551D. Instead, nucleotide-mediated NBD dimerisation events associated with channel regulation are altered by these amino acid substitutions.
The clinically approved CFTR potentiator ivacaftor (Van Goor et al. 2009; Ramsey et al. 2011) enhances strongly ATP-independent channel gating by G551D-CFTR (Eckford et al. 2012; Jih & Hwang, 2013). ATP-independent or constitutive CFTR channel gating is also augmented markedly by site-directed mutations in ICL3 (e.g. K978C; Wang et al. 2010). Like ivacaftor (Eckford et al. 2012; Jih & Hwang, 2013), these mutations restore substantial amounts of function to G551D-CFTR (Wang et al. 2010). Because constitutive mutations enhanced strongly the ATP-sensitivity of CFTR Cl− currents, Wang et al. (2010) interpreted their results to suggest that CFTR channel gating demonstrates aspects of protein allostery exhibited by ligand-gated ion channels such as the nicotinic acetylcholine receptor. These considerations raise the interesting possibility that the restoration of ATP-dependent channel gating to G551D by the revertant mutation 4RK might be an example of allosteric modulation of channel gating. Future studies should test this possibility.
The present work demonstrates that 4RK-G551D-CFTR diminishes current flow through the CFTR pore. This reduction is most likely a consequence of the many very poorly resolved channel openings that characterise the gating behaviour of 4RK-G551D-CFTR. Neither G551D nor 4RK, by themselves, alter current flow through the CFTR pore (Chang et al. 1999; Cai et al. 2006; Roxo-Rosa et al. 2006; Bompadre et al. 2007). Similarly, other mutations in NBD1 and the R domain (RD), the locations of three AFTs, including CF and revertant mutations, are without effect on the i of wild-type CFTR (e.g. Teem et al. 1993; Sheppard et al. 1995; Roxo-Rosa et al. 2006; Pissarra et al. 2008). However, the N-terminus of CFTR, the location of the fourth AFT, is the site of one CF mutation (P5L) that reduces i (Gené et al. 2008). Interestingly, the N-terminus of CFTR contains a cluster of acidic residues (D47, E51, E54 and D58) that bind the RD (Naren et al. 1999), which regulates the interaction of ATP with the NBDs to modulate channel gating (Winter & Welsh, 1997; Mathews et al. 1998). Mutation of a subset of these acidic residues (D47, E54 and D58) shortened MBD, suggesting that the N-terminus might regulate the stability of the open-channel configuration (Fu et al. 2001). However, mutation of the AFT in the N-terminus, by itself, is unlikely to be responsible for the destabilisation of the open-channel configuration in 4RK-G551D-CFTR. Instead, we favour the idea that mutation of multiple AFTs in 4RK specifies the gating phenotype of 4RK-G551D-CFTR.
In intact cells, G550E and 4RK restored substantial amounts of CFTR function to G551D-CFTR, whereas in excised inside-out membrane patches, the single-channel activity of G551D was only marginally enhanced by G550E and 4RK. A likely explanation for this difference is the assays used to investigate CFTR function in intact cells and excised membrane patches. Single-channel recording quantifies the activity of individual channels in excised membrane patches under precisely defined experimental conditions, although CFTR regulation by cytosolic factors is lost using this configuration of the patch-clamp technique. By contrast, the iodide efflux technique measures the activity of a population of CFTR channels in intact cells. Iodide efflux is dependent on a number of factors including anion permeability, membrane potential and pathways for counter-ions (Lansdell et al. 1998). It is feasible that treating BHK cells overexpressing recombinant CFTR with the cAMP agonist forskolin and the CFTR potentiator genistein, which robustly enhances CFTR channel gating (Hwang & Sheppard, 1999), might cause iodide efflux to saturate. In support of this idea, using forskolin (10 μm) and 3-isobutyl-1-methylxanthine (IBMX; 100 μm), which weakly augments CFTR activity by multiple mechanisms (Drumm et al. 1991; Becq et al. 1994; Al-Nakkash & Hwang, 1999), G551D generated a modest efflux of iodide (15% wild-type CFTR), G550E failed to enhance this response and 4RK only augmented it 2.4-fold with the result that the magnitude of iodide efflux produced by 4RK-G551D-CFTR was only 37% wild-type CFTR (Table1). Consistent with these data, work by a number of investigators have demonstrated that the CF mutant G551D-CFTR has little or no Cl− channel function when stimulated by cAMP agonists, but that genistein robustly augments its activity in intact cells (e.g. Illek et al. 1999; Yu et al. 2011).
Alternative explanations for the different effects of revertant mutations in intact cells and excised membrane patches include the phosphorylation status and ATP dependence of G551D-CFTR constructs. Previous studies have demonstrated that genistein augments robustly the activity of weakly phosphorylated CFTR Cl− channels, but has little or no effect on strongly phosphorylated channels (Hwang & Sheppard, 1999). In excised inside-out membrane patches CFTR is highly phosphorylated using PKA, whereas in intact cells its phosphorylation status is less certain. Moreover, our evaluation of the ATP dependence of G551D-CFTR constructs revealed that ATP (5 mm) enhanced the activity of all the constructs tested with the effect on G550E-G551D being particularly marked. These data raise the possibility that differences in ATP concentration between intact cells and excised membrane patches might contribute to the differences observed with G550E and 4RK. However, countering this idea, cells expressing wild-type and 4RK-G551D-CFTR had similar time courses of iodide efflux distinct from those of G551D- and G550E-G551D-CFTR (Fig.2A–C). These data suggest that restoration of some ATP dependence to G551D-CFTR by the revertant mutation 4RK contributes, at least in part, to the observed time course of iodide efflux elicited by 4RK-G551D-CFTR.
To augment the single-channel activity of G551D-CFTR variants in excised membrane patches, we used the hydrolysable ATP analogue 2′-dATP, which enhances strongly Po without exhibiting CF mutation-specific effects, unlike some CFTR potentiators (e.g. phloxine B; Cai et al. 2006; M. Ju, Z. Cai and D. N. Sheppard, unpublished observation). Although 2′-dATP increased the Po(app) of G551D variants, it did not augment those of G550E-G551D- and 4RK-G551D-CFTR above the level achieved by G551D-CFTR. The failure of 2′-dATP to augment further 4RK-G551D-CFTR gating is surprising because 4RK restored some ATP-dependent gating to G551D and because 2′-dATP gates wild-type CFTR more effectively than ATP by interacting with both ATP-binding sites 1 and 2 (Cai et al. 2006). We further proposed that 2′-dATP binds more tightly, but is hydrolysed more slowly than ATP to explain the increased frequency and duration of CFTR channel openings in its presence (Cai et al. 2006). Consistent with these ideas, structural studies of the interaction of ATP and 2′-dATP with ribonuclease reductase show minor reorientation of the nucleotide in the active site (Fairman et al. 2011). The loss of the 2-hydroxyl moves the ribose moiety deeper into the binding pocket and alters the position of the phosphate groups in the active site. These changes are associated with increased binding affinity for 2′-dATP and changes in domain–domain packing in the oligomeric holo-enzyme (Fairman et al. 2011).
Interestingly, the Po(app) of G551D-CFTR achieved with 2′-dATP is equivalent to that attained with the clinically approved CFTR potentiator ivacaftor (Van Goor et al. 2009, but see Jih & Hwang, 2013), which partially restores airway surface liquid volume and ciliary beat frequency to improve noticeably lung function in CF patients with the G551D mutation (Van Goor et al. 2009; Ramsey et al. 2011). To restore the gating behaviour of wild-type CFTR to G551D-CFTR, it might be necessary to use combinations of CFTR potentiators with distinct mechanisms of action (Yu et al. 2011). However, the observation that the hydrolysable ATP analogue N6-(2-phenylethyl)-2′-deoxy-ATP (P-dATP) enhanced G551D-CFTR activity 36-fold by binding to ATP-binding site 1 (Miki et al. 2010) argues that small molecule CFTR potentiators might be designed, which dock specifically with the ATP-binding sites of CFTR. Because P-dATP restored wild-type channel gating to F508del-CFTR (Miki et al. 2010), this approach should be a priority for CF drug development efforts.
In conclusion, this study demonstrated that the revertant mutations G550E and 4RK alter markedly the gating behaviour of the CF mutant G551D-CFTR and 4RK conferred the mutant channel with some ATP dependence. Although neither revertant mutation returned to G551D-CFTR the single-channel behaviour of wild-type CFTR, the cAMP agonist forskolin and the CFTR potentiator genistein restored substantial amounts of CFTR-mediated iodide efflux to cells expressing G550E-G551D- and 4RK-G551D-CFTR. Based on their impact on the CF mutants F508del and G551D (Roxo-Rosa et al. 2006; present study), we conclude that the revertant mutations G550E and 4RK have direct effects on CFTR structure, but that their actions on CFTR processing and single-channel function is CF mutation-specific due to different structural defects. These and other data (Hoelen et al. 2010; Dong et al. 2012) caution that revertant mutations might have opposing effects on CFTR processing and single-channel function that might diminish the restoration of function to CF mutants.
Acknowledgments
We thank Professor Y. Sohma and our departmental colleagues for valuable discussions and assistance.
Glossary
- ABC (transporter)
ATP-binding cassette (transporter)
- AFT
arginine-framed tripeptide
- BHK (cells)
baby hamster kidney (cells)
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- ER
endoplasmic reticulum
- i
single-channel current amplitude
- IBI
interburst interval
- ICL
intracellular loop
- MBD
mean burst duration
- MSD
membrane-spanning domain
- N
number of active channels
- NBD
nucleotide-binding domain
- Po
open probability
- Po(app)
apparent open probability
- PKA
protein kinase A
- RD
regulatory domain
- TES
N-tris[hydroxymethyl]methyl-2-aminoethanesulphonic acid
Key points
Malfunction of the cystic fibrosis transmembrane conductance regulator (CFTR), a gated pathway for chloride movement, causes the common life-shortening genetic disease cystic fibrosis (CF).
Gene changes (called second-site mutations or revertants) that restore function to F508del, the most common CF mutation, also alter the behaviour of the CF mutant G551D.
Revertants have direct impact on the structure of CFTR, but they exert their effects in a mutation-specific way.
Information about the action of revertants assists the development of new therapies that target the root cause of CF.
Additional information
Competing interests
None declared.
Author contributions
Conception and design of the experiments: C.M.F., Z.C., P.H.T., M.D.A. and D.N.S.; collection, analysis and interpretation of data: Z.X., L.S.P., C.M.F., J.L., Z.C., P.H.T., M.D.A. and D.N.S.; drafting the article or revising it critically for important intellectual content: C.M.F., Z.C., P.H.T., M.D.A. and D.N.S. All authors approved the final version of the manuscript. Molecular biology, cell line generation, protein biochemistry and iodide efflux experiments were performed at the University of Lisboa. Electrophysiological experiments were undertaken at the University of Bristol. Molecular modelling was carried out at University of Pittsburgh School of Medicine.
Funding
This work was supported by the Biotechnology and Biological Sciences Research Council (grant no. BB/C517517/1), the Cystic Fibrosis Trust, the Fundação para a Ciência e a Tecnologia (FCT; research grant: PTDC/SAU-GMG/122299/2010 to M.D.A. and strategic grant PEst-OE/BIA/UI4046/2011 to BioFIG), the National Institutes of Health (NIH/NIDDK DK083284) and the Cystic Fibrosis Foundation (CFFT; THIBOD09XX0). L.S.P. was supported by an FCT doctoral fellowship (SFRH/BD/9095/2002) and Z.X. and J.L. by scholarships from the University of Bristol and the Overseas Research Student Awards Scheme of Universities UK.
Authors' present addresses
Z. Xu: Hamilton Robotics China, 50 Boxia Road, Shanghai Pudong Software Park, Shanghai 201203, China; L. S. Pissarra: Abbott GmbH & Co. KG, Max-Planck-Ring 2, 65205 Wiesbaden, Germany.
References
- Aleksandrov AA, Kota P, Aleksandrov LA, He L, Jensen T, Cui L, Gentzsch M, Dokholyan NV. Riordan JR. Regulatory insertion removal restores maturation, stability and function of ΔF508 CFTR. J Mol Biol. 2010;401:194–210. doi: 10.1016/j.jmb.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov AA, Kota P, Cui L, Jensen T, Alekseev AE, Reyes S, He L, Gentzsch M, Aleksandrov LA, Dokholyan NV. Riordan JR. Allosteric modulation balances thermodynamic stability and restores function of ΔF508 CFTR. J Mol Biol. 2012;419:41–60. doi: 10.1016/j.jmb.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Nakkash L, Hwang T-C. Activation of wild-type and ΔF508-CFTR by phosphodiesterase inhibitors through cAMP-dependent and -independent mechanisms. Pflügers Arch. 1999;437:553–561. doi: 10.1007/s004240050817. [DOI] [PubMed] [Google Scholar]
- Becq F, Jensen TJ, Chang X-B, Savoia A, Rommens JM, Tsui L-C, Buchwald M, Riordan JR. Hanrahan JW. Phosphatase inhibitors activate normal and defective CFTR chloride channels. Proc Natl Acad Sci U S A. 1994;91:9160–9164. doi: 10.1073/pnas.91.19.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompadre SG, Sohma Y, Li M. Hwang T-C. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J Gen Physiol. 2007;129:285–298. doi: 10.1085/jgp.200609667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Taddei A. Sheppard DN. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. J Biol Chem. 2006;281:1970–1977. doi: 10.1074/jbc.M510576200. [DOI] [PubMed] [Google Scholar]
- Chang X-B, Cui L, Hou Y-X, Jensen TJ, Aleksandrov AA, Mengos A. Riordan JR. Removal of multiple arginine-framed trafficking signals overcomes misprocessing of ΔF508 CFTR present in most patients with cystic fibrosis. Mol Cell. 1999;4:137–142. doi: 10.1016/s1097-2765(00)80196-3. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR. Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq J-P. Lazdunski M. Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature. 1991;354:526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- Da Paula AC, Sousa M, Xu Z, Dawson ES, Boyd AC, Sheppard DN, Amaral MD. Folding and rescue of a cystic fibrosis transmembrane conductance regulator trafficking mutant identified using human-murine chimeric proteins. J Biol Chem. 2010;285:27033–27044. doi: 10.1074/jbc.M110.120352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deCarvalho ACV, Gansheroff LJ. Teem JL. Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator ΔF508. J Biol Chem. 2002;277:35896–35905. doi: 10.1074/jbc.M205644200. [DOI] [PubMed] [Google Scholar]
- Dong Q, Ostedgaard LS, Rogers C, Vermeer DW, Zhang Y. Welsh MJ. Human-mouse cystic fibrosis transmembrane conductance regulator (CFTR) chimeras identify regions that partially rescue CFTR-ΔF508 processing and alter its gating defect. Proc Natl Acad Sci U S A. 2012;109:917–922. doi: 10.1073/pnas.1120065109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, Dawson DC. Collins FS. Chloride conductance expressed by ΔF508 and other mutant CFTRs in Xenopus oocytes. Science. 1991;254:1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- Eckford PDW, Li C, Ramjeesingh M. Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem. 2012;287:36639–36649. doi: 10.1074/jbc.M112.393637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, Prendergast J, Welin RM, Flodin S, Roos A, Nordlund P, Li Z, Walz T. Dealwis CG. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat Struct Mol Biol. 2011;18:316–323. doi: 10.1038/nsmb.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, King-Underwood J, Sousa M, Correia AR, Henriques BJ, Roxo-Rosa M, Da Paula AC, Williams J, Hirst S, Gomes CM. Amaral MD. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem Biol. 2013a;20:943–955. doi: 10.1016/j.chembiol.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Farinha CM, Matos P. Amaral MD. Control of cystic fibrosis transmembrane conductance regulator membrane trafficking: not just from the endoplasmic reticulum to the Golgi. FEBS J. 2013b;280:4396–4406. doi: 10.1111/febs.12392. [DOI] [PubMed] [Google Scholar]
- Farinha CM, Mendes F, Roxo-Rosa M, Penque D. Amaral MD. A comparison of 14 antibodies for the biochemical detection of the cystic fibrosis transmembrane conductance regulator protein. Mol Cell Probes. 2004;18:235–242. doi: 10.1016/j.mcp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Farinha CM, Nogueira P, Mendes F, Penque D. Amaral MD. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem J. 2002;366:797–806. doi: 10.1042/BJ20011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Ji H-L, Naren AP. Kirk KL. A cluster of negative charges at the amino terminal tail of CFTR regulates ATP-dependent channel gating. J Physiol. 2001;536:459–470. doi: 10.1111/j.1469-7793.2001.0459c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P. Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gené GG, Llobet A, Larriba S, de Semir D, Martínez I, Escalada A, Solsona C, Casals T. Aran JM. N-terminal CFTR missense variants severely affect the behavior of the CFTR chloride channel. Hum Mutat. 2008;29:738–749. doi: 10.1002/humu.20721. [DOI] [PubMed] [Google Scholar]
- Gregory RJ, Rich DP, Cheng SH, Souza DW, Paul S, Manavalan P, Anderson MP, Welsh MJ. Smith AE. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol Cell Biol. 1991;11:3886–3893. doi: 10.1128/mcb.11.8.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelen H, Kleizen B, Schmidt A, Richardson J, Charitou P, Thomas PJ. Braakman I. The primary folding defect and rescue of ΔF508 CFTR emerge during translation of the mutant domain. PLoS One. 2010;5:e15458. doi: 10.1371/journal.pone.0015458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes LK, Ju M. Sheppard DN. Potentiation of cystic fibrosis transmembrane conductance regulator (CFTR) Cl− currents by the chemical solvent tetrahydrofuran. Mol Membr Biol. 2008;25:528–538. doi: 10.1080/09687680802487967. [DOI] [PubMed] [Google Scholar]
- Hwang T-C. Sheppard DN. Molecular pharmacology of the CFTR Cl− channel. Trends Pharmacol Sci. 1999;20:448–453. doi: 10.1016/s0165-6147(99)01386-3. [DOI] [PubMed] [Google Scholar]
- Hwang T-C. Sheppard DN. Gating of the CFTR Cl− channel by ATP-driven nucleotide-binding domain dimerisation. J Physiol. 2009;587:2151–2161. doi: 10.1113/jphysiol.2009.171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illek B, Zhang L, Lewis NC, Moss RB, Dong J-Y. Fischer H. Defective function of the cystic fibrosis-causing missense mutation G551D is recovered by genistein. Am J Physiol Cell Physiol. 1999;277:C833–C839. doi: 10.1152/ajpcell.1999.277.4.C833. [DOI] [PubMed] [Google Scholar]
- Jih K-Y. Hwang T-C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci U S A. 2013;110:4404–4409. doi: 10.1073/pnas.1215982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell KA, Kidd JF, Delaney SJ, Wainwright BJ. Sheppard DN. Regulation of murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in Chinese hamster ovary cells. J Physiol. 1998;512:751–764. doi: 10.1111/j.1469-7793.1998.751bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X. Emtage S. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004;23:282–293. doi: 10.1038/sj.emboj.7600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, Maloney PC, Guggino WB, Hunt JF. Emtage S. Impact of the ΔF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem. 2005;280:1346–1353. doi: 10.1074/jbc.M410968200. [DOI] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Wang W, Garami E, Hewryk M, Lee D, Rommens JM, Galley K. Bear CE. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271:28463–28468. doi: 10.1074/jbc.271.45.28463. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC. Clarke DM. The V510D suppressor mutation stabilizes ΔF508-CFTR at the cell surface. Biochemistry. 2010;49:6352–6357. doi: 10.1021/bi100807h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Chang X-B, Bear C, Kartner N, Mohamed A, Riordan JR. Grinstein S. The ΔF508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane: determination of functional half-lives on transfected cells. J Biol Chem. 1993;268:21592–21598. [PubMed] [Google Scholar]
- Lukacs GL. Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med. 2012;18:81–91. doi: 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews CJ, Tabcharani JA, Chang X-B, Jensen TJ, Riordan JR. Hanrahan JW. Dibasic protein kinase A sites regulate bursting rate and nucleotide sensitivity of the cystic fibrosis transmembrane conductance regulator chloride channel. J Physiol. 1998;508:365–377. doi: 10.1111/j.1469-7793.1998.365bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, Feranchak AP, Brautigam CA. Thomas PJ. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148:164–174. doi: 10.1016/j.cell.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H, Zhou Z, Li M, Hwang T-C. Bompadre SG. Potentiation of disease-associated cystic fibrosis transmembrane conductance regulator mutants by hydrolyzable ATP analogs. J Biol Chem. 2010;285:19967–19975. doi: 10.1074/jbc.M109.092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naren AP, Cormet-Boyaka E, Fu J, Villain M, Blalock JE, Quick MW. Kirk KL. CFTR chloride channel regulation by an interdomain interaction. Science. 1999;286:544–548. doi: 10.1126/science.286.5439.544. [DOI] [PubMed] [Google Scholar]
- Pissarra LS, Farinha CM, Xu Z, Schmidt A, Thibodeau PH, Cai Z, Thomas PJ, Sheppard DN. Amaral MD. Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem Biol. 2008;15:62–69. doi: 10.1016/j.chembiol.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A. Lukacs GL. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148:150–163. doi: 10.1016/j.cell.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordoñez C. Elborn JS for the VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1671. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, Drumm ML, Iannuzzi MC, Collins FS. Tsui L-C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Roxo-Rosa M, Xu Z, Schmidt A, Neto M, Cai Z, Soares CM, Sheppard DN. Amaral MD. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc Natl Acad Sci U S A. 2006;103:17891–17896. doi: 10.1073/pnas.0608312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Hughes LK, Cai Z, Mendes F, Li H, Sheppard DN. Amaral MD. Prolonged treatment of cells with genistein modulates the expression and function of the cystic fibrosis transmembrane conductance regulator. Br J Pharmacol. 2008;153:1311–1323. doi: 10.1038/sj.bjp.0707663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Ostedgaard LS, Winter MC. Welsh MJ. Mechanism of dysfunction of two nucleotide binding domain mutations in cystic fibrosis transmembrane conductance regulator that are associated with pancreatic sufficiency. EMBO J. 1995;14:876–883. doi: 10.1002/j.1460-2075.1995.tb07069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN. Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J Physiol. 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ. Hunt JF. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teem JL, Berger HA, Ostedgaard LS, Rich DP, Tsui L-C. Welsh MJ. Identification of revertants for the cystic fibrosis ΔF508 mutation using STE6-CFTR chimeras in yeast. Cell. 1993;73:335–346. doi: 10.1016/0092-8674(93)90233-g. [DOI] [PubMed] [Google Scholar]
- Teem JL, Carson MR. Welsh MJ. Mutation of R555 in CFTR-ΔF508 enhances function and partially corrects defective processing. Recept Channels. 1996;4:63–72. [PubMed] [Google Scholar]
- Thibodeau PH, Richardson JM, III, Wang W, Millen L, Watson J, Mendoza JL, Du K, Fischman S, Senderowitz H, Lukacs GL, Kirk K. Thomas PJ. The cystic fibrosis-causing mutation ΔF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem. 2010;285:35825–35835. doi: 10.1074/jbc.M110.131623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M. Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC. Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433:876–880. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Nairn AC. Gadsby DC. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J Gen Physiol. 2003;121:17–36. doi: 10.1085/jgp.20028673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zeltwanger S, Yang ICH, Nairn AC. Hwang T-C. Actions of genistein on cystic fibrosis transmembrane conductance regulator channel gating: evidence for two binding sites with opposite effects. J Gen Physiol. 1998;111:477–490. doi: 10.1085/jgp.111.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Okeyo GO, Tao B, Hong JS. Kirk KL. Thermally unstable gating of the most common cystic fibrosis mutant channel (ΔF508): “rescue” by suppressor mutations in nucleotide binding domain 1 and by constitutive mutations in the cytosolic loops. J Biol Chem. 2011;286:41937–41948. doi: 10.1074/jbc.M111.296061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wu J, Bernard K, Li G, Wang G, Bevensee MO. Kirk KL. ATP-independent CFTR channel gating and allosteric modulation by phosphorylation. Proc Natl Acad Sci U S A. 2010;107:3888–3893. doi: 10.1073/pnas.0913001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh MJ, Ramsey BW, Accurso F. Cutting GR. Cystic fibrosis. In: Valle D, editor; Scriver CR, Beaudet AL, Sly WS, editors. The Metabolic and Molecular Basis of Inherited Disease. 8th edn. New York: McGraw-Hill Inc; 2001. pp. 5121–5188. [Google Scholar]
- Winter MC, Sheppard DN, Carson MR. Welsh MJ. Effect of ATP concentration on CFTR Cl− channels: a kinetic analysis of channel regulation. Biophys J. 1994;66:1398–1403. doi: 10.1016/S0006-3495(94)80930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter MC. Welsh MJ. Stimulation of CFTR activity by its phosphorylated R domain. Nature. 1997;389:294–296. doi: 10.1038/38514. [DOI] [PubMed] [Google Scholar]
- Yu Y-C, Miki H, Nakamura Y, Hanyuda A, Matsuzaki Y, Abe Y, Yasui M, Tanaka K, Hwang T-C, Bompadre SG. Sohma Y. Curcumin and genistein additively potentiate G551D-CFTR. J Cyst Fibros. 2011;10:243–252. doi: 10.1016/j.jcf.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltwanger S, Wang F, Wang GT, Gillis KD. Hwang TC. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J Gen Physiol. 1999;113:541–554. doi: 10.1085/jgp.113.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]