

Abstract
The current model of O2 sensing by carotid body chemoreceptor (glomus) cells is that hypoxia inhibits the outward K+ current and causes cell depolarization, Ca2+ influx via voltage-dependent Ca2+ channels and a rise in intracellular [Ca2+] ([Ca2+]i). Here we show that hypoxia (<5% O2), in addition to inhibiting the two-pore domain K+ channels TASK-1/3 (TASK), indirectly activates an ∼20 pS channel in isolated glomus cells. The 20 pS channel was permeable to K+, Na+ and Cs+ but not to Cl− or Ca2+. The 20 pS channel was not sensitive to voltage. Inhibition of TASK by external acid, depolarization of glomus cells with high external KCl (20 mm) or opening of the Ca2+ channel with FPL64176 activated the 20 pS channel when 1 mm Ca2+ was present in the external solution. Ca2+ (10 μm) applied to the cytosolic side of inside-out patches activated the 20 pS channel. The threshold [Ca2+]i for activation of the 20 pS channel in cell-attached patches was ∼200 nm. The reversal potential of the 20 pS channel was estimated to be −28 mV. Our results reveal a sequential mechanism in which hypoxia (<5% O2) first inhibits the K+ conductance and then activates a Na+-permeable, non-selective cation channel via depolarization-induced rise in [Ca2+]i. Our results suggest that inhibition of K+ efflux and stimulation of Na+ influx both contribute to the depolarization of glomus cells during moderate to severe hypoxia.
Introduction
The carotid body glomus cells detect a decrease in arterial O2 tension (hypoxia) and help generate a neural signal that travels to the brainstem cardiorespiratory centre to mediate reflex mechanisms such as arousal from sleep during hypoxia, increased ventilation, and changes in blood pressure and heart rate (Kumar & Prabhakar, 2012). Based on many years of studies, it is now widely accepted that hypoxia produces depolarization of carotid body glomus cells and thereby elicits a cascade of events leading to elevation of transmitter secretion and increased carotid sinus afferent nerve activity (Lopez-Barneo et al. 1988; Delpiano & Hescheler, 1989; Biscoe & Duchen, 1990; Gonzalez et al. 1992; Buckler & Vaughan-Jones, 1994b; Pardal et al. 2000). Critical signalling events that occur within glomus cells involve the initial depolarization, opening of voltage-dependent Ca2+ channels and elevation of intracellular [Ca2+] ([Ca2+]i). The depolarization of glomus cells produced by hypoxia is generally thought to occur as a result of inhibition of the K+ channels that are active near the resting membrane potential (Buckler, 2007; Peers et al. 2010). The types of K+ channels involved in the depolarization process may depend on species. In rat glomus cells, inhibition of large-conductance Ca2+-activated (BK) and TWIK-related acid-sensing K+ (TASK) channels is thought to be the cause of depolarization, but the relative importance of the two channels remains unresolved (Buckler, 2007; Peers & Wyatt, 2007; Wyatt et al. 2007; Evans et al. 2009). The two K+ channels are probably also involved in the chemosensory response in mice, as both channels are well expressed in mice glomus cells (Trapp et al. 2008; Ortega-Saenz et al. 2010; Otsubo et al. 2011).
In addition to K+ channels, a resting Na+ conductance has been demonstrated in rat glomus cells (Buckler & Vaughan-Jones, 1994a; Buckler, 1997; Carpenter & Peers, 2001). The whole-cell holding current was larger with Na+ as the main permeant ion than without Na+ (Buckler, 1997). Substitution of extracellular Na+ with N-methyl-d-glucamine (NMDG) was found to hyperpolarize and reduce the whole-cell holding current to near zero (Carpenter & Peers, 2001). Na+ influx via background Na+ channels presumably keeps the resting membrane potential of glomus cells between −50 and −60 mV, and causes the depolarization when K+ channels are inhibited by hypoxia. In addition to the background Na+ channel, studies suggest that glomus cells also express non-selective cation channels that are permeable to Na+. For example, low glucose in the extracellular medium was found to elicit a Na+-dependent depolarization via activation of a non-selective cation channel (Pardal & Lopez-Barneo, 2002; Bin-Jaliah et al. 2004; Garcia-Fernandez et al. 2007; Zhang et al. 2007). Thus, Na+-permeable channels are expressed in glomus cells, but their biophysical properties are still undefined and it is unknown whether hypoxia affects their activity.
In this work, we sought to identify Na+-permeable channels and study their role in the regulation of the excitability of glomus cells under normal and hypoxic conditions. We have identified a 20 pS, non-selective monovalent cation channel that is mostly closed in normoxia but is activated by elevation of cytosolic Ca2+ that occurs in response to depolarizing stimuli, including hypoxia, acid and high external K+. We show evidence for a sequential mechanism in which an initial inhibition of K+ channels such as TASK by hypoxia leads to activation of the 20 pS channel via a depolarization-induced rise in [Ca2+]i. The data suggest that hypoxia (<5% O2)-induced depolarization of glomus cells involves both inhibition of K+ efflux and stimulation of Na+ influx.
Methods
Ethical approval
The Animal Care and Use Committee of Rosalind Franklin University approved the protocol used in this study (protocol #11–49). Rats (Sprague-Dawley; postnatal days 14–18; number of animals: 740) and mice (postnatal days 14–25; number of animals: 32) were anaesthetized by inhalation of isoflurane until cessation of breathing.
TRPM4−/− and TRPM5−/− mice
TRPM4 knockout mice were generated and backcrossed for 10 generations on a C57BL6/N background. Gene targeting in R1 embryonic stem (ES) cells and Cre-loxP-mediated recombination were used to generate a mouse line with a null allele (Trpm4−/−). Additional details are given in the online Supplementary Methods of the original article (Vennekens et al. 2007). Male and female TRPM5 knockout mice (C57BL/6 background) were obtained from Dr Charles Zuker of Columbia University, and bred for two generations. In Dr Zuker's lab, ES clones containing TRPM5 with exons 15 and 19 replaced with PGK-neo'cassette were injected into C57BL/6 blastocysts, and chimeric mice were bred with C57BL/6 mice. Progeny was backcrossed for two generations, and subsequent crossing generated the homozygous knockouts (Zhang et al. 2003).
Cell isolation
Carotid artery bifurcation region was surgically removed from rat and mouse, and carotid bodies were placed in ice-cold low Ca2+, low Mg2+ phosphate buffered saline (low Ca2+/Mg2+ PBS: 137 mm NaCl, 2.8 mm KCl, 2 mm KH2PO4, 0.07 mm CaCl2, 0.05 mm MgCl2, pH 7.4). Each carotid body was cut into 2–3 pieces and placed in a solution containing trypsin (0.4 mg ml−1) and collagenase (0.4 mg ml−1) in low Ca2+/Mg2+ PBS and incubated at 37°C for ∼25 min. Carotid body pieces were gently triturated using a fire polished Pasteur pipette to mechanically dissociate the cells. Carotid body growth medium (Ham's F-12, 10% fetal bovine serum, 23 mm glucose, 4 mm Glutamax-1 (l-alanyl glutamine), 10 kU penicillin/streptomycin and 300 μg ml−1 insulin) was added to stop enzyme activity. After brief trituration, the solution containing the digested carotid bodies was centrifuged for 4 min at ∼2000 g using a microcentrifuge. After removing the supernatant, growth medium was added to gently resuspend the pellet. Suspended cells were placed on glass coverslips coated with poly-l-lysine, and incubated at 37°C for ∼2 h in a humidified atmosphere of 95% air/5% CO2. Cells were used within 6 h after plating.
Reverse transcriptase PCR
Clusters (∼200) of glomus-like cells isolated from carotid bodies were collected using a polished glass pipette into a centrifuge tube. cDNA was generated using a Single Cell RT-PCR Assay Kit (Signosis, Sunnyvale, CA, USA). Two sets of primer pairs for TRPM4 (set 1: forward 5′-TGGTGGTGTTGCTCCTCATC-3′ and reverse 5′-CTCAGACGCCGGTCATACTC-3′ expected size 240 bp; set 2: forward 5′-ATCTCTCACCTGCGTCTCCT-3′ and reverse 5′-GACGCCGGTCATACTCTCTG-3′ expected size 460 bp) and four sets of primer pairs for TRPM5 (set 1: forward 5′-CATGGTGGCCATCTTCCTGT-3′ and reverse 5′-GGTCACACCATAGGCCACAA-3′ expected size 238 bp; set 2: forward 5′-GGTCTTCAGGAAGGAAGCCC-3′ and reverse 5′-TGGCCTGTGATTCCAGACAC-3′ expected size 251 bp; set 3: forward 5′CCATGTTCAGCTACACATTCCAG3′ and reverse 5′-GAGAACTTGAGTAGGTGCCTCCA-3′ expected size 471 bp set 4: forward 5′-CCATGTTCAGCTACACATTCCAG-3′ and reverse 5′-GTGTGTCAGTCATGGAGGACAAG-3′ expected size 441 bp) were selected using the Primer3Plus software and used in PCR reactions with Taq polymerase. PCR conditions were: initial denaturation at 94°C for 5 min followed by 35 cycles of 95°C for 40 s, 55°C for 50 s and 72°C for 60 s, and a final extension step at 72°C for 10 min. PCR products were run on a 1.2% agarose gel by electrophoresis. When PCR products were not observed, different annealing temperatures (50–60°C) were tested. The PCR products were sequenced at the University of Chicago Sequencing facility. PCR was also performed using TRPM5 cDNA in pcDNA3.1 (obtained from Dr Craig Montell, Johns Hopkins University) as the template for positive control.
Electrophysiology
Electrophysiological recording was performed using a patch clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA, USA). Cell-attached patches were formed with gentle suction with sylgard-coated borosilicate glass pipettes. Channel current was filtered at 2 kHz using an eight-pole Bessel filter (–3 dB; Frequency Devices, Ottawa, IL, USA) and transferred to a computer using the Digidata 1320 interface at a sampling rate of 20 kHz. Single-channel currents were analysed with the pCLAMP program (versions 9/10). Channel openings were analysed to obtain channel activity (NPo, where N is the number of channels in the patch, and Po is the probability of a channel being open). NPo was determined from 5–15 s of current recording. The minimum duration used for analysis of the 20 pS channel was 5 ms, such that TASK channels whose mean open time ranges between 0.5 and 1.0 ms were precluded.
In experiments using cell-attached and inside-out patches, pipette solution contained (mm): 140 KCl (or 140 NaCl, 140 CsCl, 140 NMDG or 100 CaCl2), 1 MgCl2, 5 EGTA, 11 glucose and 10 Hepes (pH 7.3). The [Cl−] in NMDG-Cl solution was ∼100 mm. Extracellular bath (physiological) solution contained (mm): 117 NaCl, 5 mm KCl, 23 NaHCO3, 1 MgCl2, 1 CaCl2, 10 Hepes and 11 glucose (adjusted to pH 7.3 with NaOH). For whole-cell studies, the bath (physiological) solution contained (mm): 117 NaCl, 5 mm KCl, 23 NaHCO3, 1 MgCl2, 1 CaCl2, 10 Hepes and 11 glucose (pH 7.3), and pipette solution contained (mm): 140 KCl, 1 MgCl2, 5 EGTA, 11 glucose, 10 Hepes and 2 MgATP (pH 7.3). For perforated patch experiments, the pipette solution contained (mm): 70 K2SO4, 20 KCl, 1 MgCl2, 1 EGTA, 2 MgATP, 10 Hepes (pH adjusted to 7.3 with KOH) with 240 μg ml−1 amphoteracin-B (from a stock solution of 60 mg ml−1 in DMSO). All solutions containing NaHCO3 were equilibrated with 95% air/5% CO2 and pH adjusted to 7.3 with NaOH, or to pH 6.0 with HCl. The composition of the pH 6.0 solution was identical to that of the extracellular bath solution stated above. The temperature of the perfusion solution at the recording chamber was 34–35°C. [Ca2+] of 10 μm for perfusing inside-out patches was prepared using online software (http://www.stanford.edu/~cpatton/webmaxcS.htm).
Hypoxia studies
Cell-attached patches were extracellularly perfused with the physiological solution gassed with 5% CO2/95% air mixture (normoxia) throughout the experimental period. After a steady-state channel current was obtained, the perfusion solution was switched to solution gassed with 5% CO2/95% N2 mixture (hypoxia). The temperature of the perfusion solutions was ∼35°C, and the rate of perfusion was ∼2.4 ml min−1. Stainless steel tubings were used for perfusion lines to prevent gas diffusion. O2 pressure of the solutions was checked using an oxygen meter (ISO2, World Precision Instruments, Sarasota, FL, USA) that was calibrated to 0% with solution gassed with pure nitrogen for 60 min and to 21% with solution gassed with air for 60 min at 35°C. The mean O2 partial pressures as judged by triplicate readings on the meter for the hypoxic solution in the recording chamber used in this study were 12 mmHg (∼1.4%), 29 mmHg (∼2.2%) and 39 mmHg (∼5.1%) for solutions gassed with 0% O2/5% CO2/95% N2, 2% O2/5% CO2/93% N2 and 5% O2/5% CO2/90% N2 mixtures, respectively.
[Ca2+]i measurements
Isolated cells plated on glass coverslips were incubated with 2 μm fura-2 acetoxymethyl ester (fura-2 AM) for 30 min at room temperature (∼24°C) in culture medium, and mounted in a recording chamber positioned on the stage of an inverted microscope (IX71; Olympus America Inc., Center Valley, PA, USA). Fura-2 was alternately excited at 340 and 380 nm and the emitted fluorescence was filtered at 510 nm and recorded using a charged-coupled device (CCD)-based imaging system running SimplePCI software (Hamamatsu Corp.). The cells in the recording chamber were continuously perfused with physiological solution containing (mm): 117 NaCl, 5 KCl, 23 NaHCO3, 1 MgCl2, 1 CaCl2, 10 Hepes and 11 glucose (pH 7.3). The test solution was applied through glass tubing facing the cells. Ratiometric data were calibrated by applying experimentally determined constants to the equation: [Ca2+] = Kd × β × (R–Rmin)/(Rmax–R) (Grynkiewicz et al. 1985). Values for Rmax (12.0), Rmin (0.1) and β (11.6) were determined in vitro as described (Tepikin, 2001), and a Kd value of 300 nm for fura-2 was assumed.
Statistical analysis
Student's t test (for comparison of two sets of data) and one-way analysis of variance (comparison of three or more sets of data) were used. Data were analysed using PRISM software and are given as mean ± standard deviation. Post hoc testing was based on an unpaired t test with Bonferroni correction. Significance level was set at P < 0.05.
Materials
FPL64176, tetraethylammmonium (TEA), iberiotoxin, apamin, 4-aminopyridine (4-AP) and NS-309 were purchased from Tocris (Ellisville, MO, USA). 4,4′ Diisothio-cyanatostilbene-2,2′-disulfonic acid (DIDS), anthracene-9-carboxylic acid (9-AC), amiloride, GSH, GSSH, NAD, 2-aminoethoxydiphenyl borate (2-APB), DMSO, amphoteracin B, NMDG and niflumic acid were purchased from Sigma-Aldrich (St Louis, MO, USA). Fura-2 AM was purchased from Life Technologies (Carlsbad, CA, USA). BAPTA-AM was purchased from Calbiochem (Billerica, MA, USA). For those drugs where DMSO was used to prepare the stock solutions, the final concentration of DMSO used in the perfusion solution were 0.1% or less.
Results
Activation of a 20 pS channel by hypoxia
Cell-attached patches were formed on rat glomus cells with 140 mm K+ in the pipette solution and 5 mm K+/135 mm Na+ in normoxic bath perfusion solution. With pipette potential set at +20 mV, opening of channels with a mean open time duration of ∼0.5 ms and a single-channel conductance of ∼36 pS was present in nearly every patch (Fig.1A: tracing a). The ∼36 pS channel has previously been characterized in detail and shown to represent mostly heteromeric TASK-1/3 with a small contribution from homomeric TASK-3 (Kim et al. 2009a). The ∼16 pS channel, also present at low levels, represents homomeric TASK-1 (Kim et al. 2009a). These K+ channels are referred below as TASK.
Figure 1. Activation of a 20 pS channel by hypoxia.
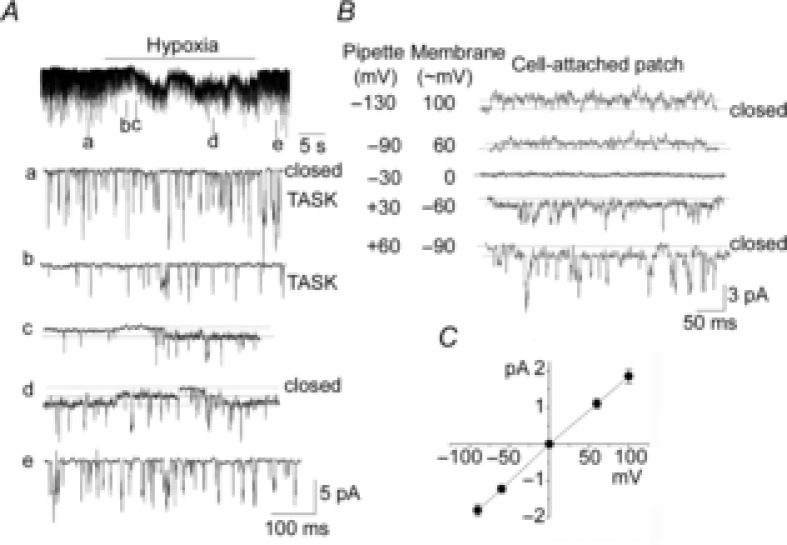
A, TASK channels are active in a cell-attached patch of rat glomus cells in normoxia (tracing a). Pipette solution contained 140 mm K+ and pipette potential was +20 mV. Perfusion with hypoxic solution inhibited TASK activity (tracing b) followed by activation of channels with a long open time duration (tracings c and d). Return to normoxic solution closed the channels with the long open time duration and restored TASK activity to the pre-hypoxia level. B, channel openings from a cell-attached patch are shown at various pipette potentials as indicated during perfusion with hypoxic solution. Opening of both TASK and the 20 pS channel are shown. In this patch, the zero current was observed when the pipette potential is −30 mV. C, I–V relationship. Each point is the mean ± SD (n = 3).
To increase the probability of detecting non-selective cation channels, large cell-attached patches were formed using a pipette with ∼1 MΩ tip resistance. With pipette solution containing 140 mm KCl, TASK was still the main K+ channel present in these cell-attached patches (Fig.1A; tracing a). When the perfusion solution was switched from normoxic to hypoxic (∼1% O2) solution, TASK channel activity decreased quickly to 29 ± 5% (n = 5) of control (Fig.1A; tracings b and c). Hypoxia also reduced the amplitude of TASK by 40 ± 6% (compare tracings a and b in Fig.1A), indicating that hypoxia depolarized the cells from approximately −60 to −36 mV, as estimated from the linear I–V relationship of TASK between 0 and −100 mV (Kim et al. 2009a). Immediately following TASK inhibition, hypoxia activated a channel with a long open time duration, as shown in the expanded current tracings of Fig.1A (tracings c and d). Two open levels can be seen in tracing d, together with opening of TASK channels. Hypoxia (∼1% O2) activated 1–3 channels in ∼75% of cell-attached patches (56 of 74 patches counted). All 74 patches counted showed TASK, and 18 of 74 patches showed only TASK and no 20 pS channel in response to hypoxia. So far, we have not observed activation of the 20 pS channel in cells (presumably Type 2 cells) that did not show TASK (n = 12).
Activation of the channel by hypoxia was reversible and could be repeated in the same patch. The channel remained active throughout the hypoxic period with some fluctuations in channel activity (Fig.1A). Channel openings at various potentials during hypoxia are shown in Fig.1B. In this particular patch, the zero current was observed at a pipette holding potential of approximately −30 mV. The I–V relationship of the hypoxia-activated channel obtained from such patches was linear and the single-channel conductance was 20 ± 1 pS (Fig.1C). Thus, isolated rat glomus cells express an O2-sensitive ion channel whose single channel kinetics are different from those of TASK and BK.
O2 sensitivity of the 20 pS channel
Figure2A shows a recording from a cell-attached patch from a rat glomus cell perfused extracellularly with solutions containing two different levels of O2. Activation of the 20 pS channel began at ∼5% O2, albeit to very low levels, and clear channel activation was observed consistently at 1–2% O2 (Fig.2A; tracing b). In most patches, two or three different levels of O2 were tested, and only those patches that showed reversible effects of hypoxia were used for analysis. From such experiments, channel activity was calculated and plotted as a function of O2 content (Fig.2B). Figure2B also shows the inhibitory profile of TASK by hypoxia. At 5% O2, TASK activity was inhibited ∼50%, whereas the 20 pS channel was minimally activated. Thus, TASK is sensitive to a wide range of hypoxia (0–20 % O2), compared to the 20 pS channel that is activated by moderate to severe hypoxia (<5% O2).
Figure 2. Relationship between O2 pressure and channel activity.
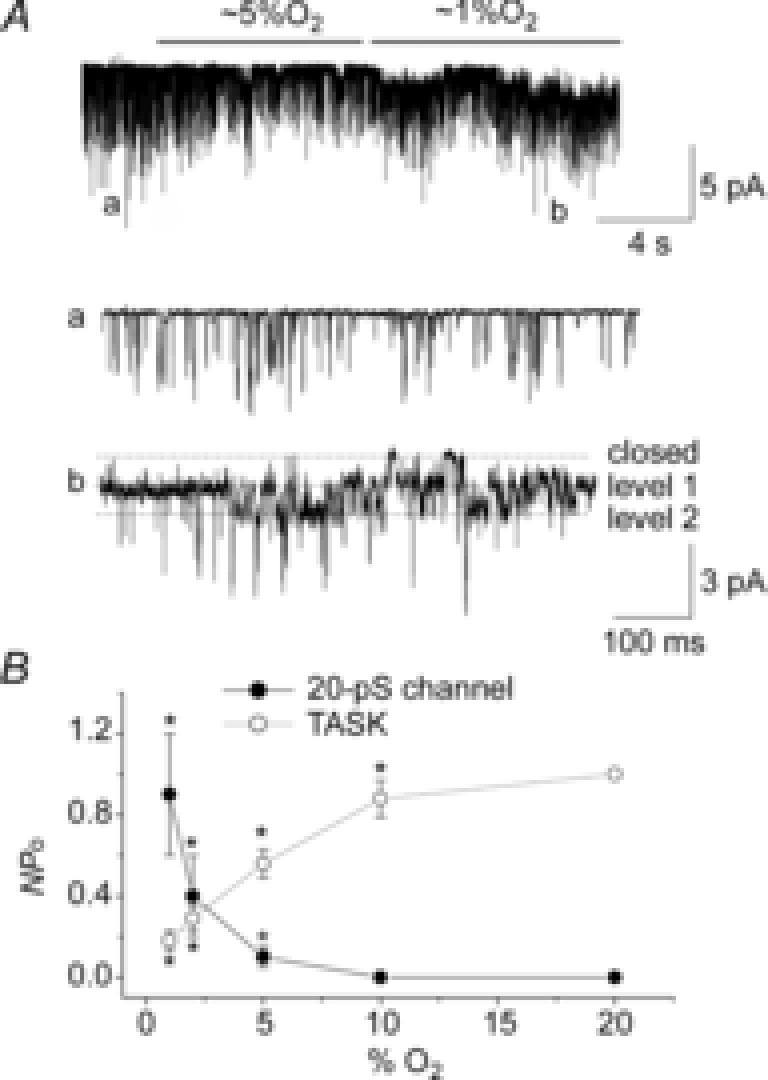
A, example trace of a cell-attached patch recording in response to two different levels of hypoxia. Expanded current tracings are also shown as indicated. Pipette potential was 0 mV and pipette solution contained 140 mm KCl. B, activities for TASK and 20 pS channels are plotted as a function of O2 content. For TASK, activity at 20% O2 was taken as 1.0. For the 20 pS channel, data were not normalized. Each point is mean ± SD (n = 5). *Significantly different from the control values obtained at 20% O2.
Activation of the 20 pS channel by low external pH and high external KCl
A careful inspection of current recordings obtained after perfusion with normoxic and hypoxic solutions showed that hypoxia inhibited TASK first and then activated the 20 pS channel (see Figs 1A and 2A). As this sequential effect was observed consistently, we hypothesized that an initial inhibition of TASK may be the trigger for the activation of the 20 pS channel. Consistent with this mechanism, bath perfusion of cell-attached patches with an acidic solution (pH 6.0) that inhibits TASK activated the 20 pS channel in 7 of 9 patches (Fig.3A). Because the pH of the pipette solution was 7.3, these results indicated that the effect of acid was not directly on the channel itself. Commonly used K+ channel blockers that do not affect TASK such as TEA (5 mm, n = 18) and 4-AP (2 mm, n = 7) failed to activate the 20 pS channel when applied to the bath perfusion solution.
Figure 3. Activation of the 20 pS channel by acid and 20 mm KCl.
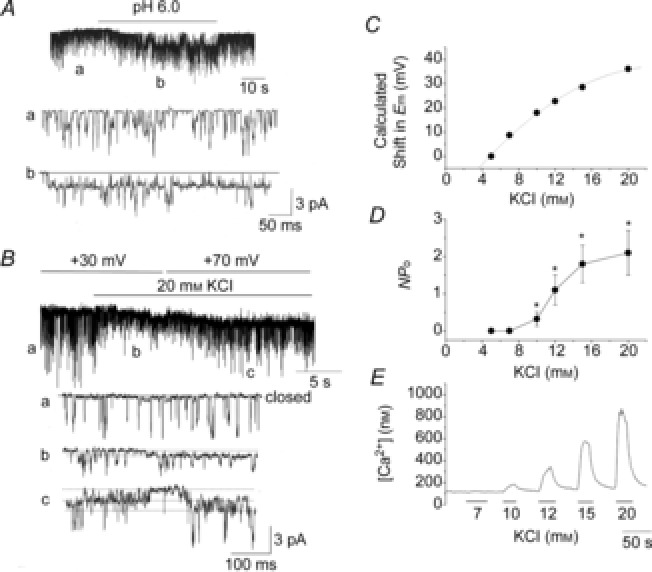
A, external acid solution activated the 20 pS channel in a cell-attached patch of rat glomus cells. Pipette potential was 0 mV and pipette solution contained 140 mm K+. B, application of 20 mm KCl to the external solution perfusing the cell-attached patch activated the 20 pS channel (tracing b). Channel openings at pipette potential of +30 mV (tracings a and b) and 70 mV (tracing c) are shown. C, calculated shift in Em plotted as a function of external [KCl] based on the Nernst equation. D, activity of the 20 pS channel activity plotted as a function external [KCl]. Each point is the mean ± SD of 9–12 patches. E, tracing of typical changes in [Ca2+]i (from fura-2 signal) at different external [KCl] from a glomus cell.
As acid- and hypoxia-mediated inhibition of the K+ conductance cause depolarization of glomus cells, could it be the depolarization that activates the 20 pS channel? Therefore, high KCl was used to further test the effect of depolarization without causing TASK inhibition (Fig.3B). When the bath [KCl] perfusing cell-attached patches was increased from 5 to 20 mm, TASK activity (NPo) was not significantly affected (0.18 ± 0.03 to 0.16 ± 0.02; n = 6, P > 0.05), but the amplitude of TASK quickly decreased from 3.0 ± 0.1 to 1.2 ± 0.1 pA (n = 6, P < 0.05), as predicted from the reduced transmembrane potential across the patch membrane. The decrease in TASK amplitude was followed by opening of a channel with kinetics similar to those of the 20 pS channel (Fig.3B; tracing b). When the pipette potential was changed from +30 to +70 mV, the amplitude levels of both TASK and the channel activated by high KCl increased proportionally (Fig.3B; tracings b and c). The single-channel conductance calculated from the amplitudes at two potentials was 20 ± 1 pS for the channel with long-lasting openings (n = 3). These results show that KCl-induced depolarization of glomus cells activates the 20 pS channel.
To determine the degree of depolarization necessary to activate the 20 pS channel, we tested the effect of different extracellular [KCl] on 20 pS channel activation. Figure3C shows the calculated shift in membrane potential as a function of [KCl]. At 5 and 7 mm KCl, 20 pS channels were not activated (n = 12 each; Fig.3D). At 10 mm KCl, the 20 pS channel was present in 4 of 12 patches with a low open probability. At 12 mm (8 of 10 patches), 15 mm (8 of 9 patches) and 20 mm KCl (8 of 9 patches), activation of the 20 pS channel was more strong and consistent. As expected, high external KCl also elevated [Ca2+]i in glomus cells (Fig.3E). At the basal level when the bath [KCl] was 5 mm, [Ca2+]i was 110 ± 14 nm. [Ca2+]i changed to 119 ± 22 nm (7 mm KCl), 210 ± 60 nm (10 mm KCl), 359 ± 103 nm (12 mm KCl), 504 ± 64 nm (15 mm) and 798 ± 58 nm (20 mm KCl), as bath [KCl] was increased (n = 12 each). The results from these experiments suggest that the 20 pS channel becomes active when the cell depolarizes ∼20 mV from the resting Em level of −60 mV and the increase in [Ca2+]i becomes evident.
Under the conditions of our experiments (i.e. pipette potential of 0 mV), we did not observe activation of BK with 5–15 mm KCl even though BK channels were present in the patch, as judged by BK openings upon forming inside-out patches with 1 mm Ca2+ in the bath solution. With higher KCl (20 mm), activation of BK was observed in 2 of 9 patches along with the 20 pS channel. These results suggest that activation of the 20 pS channel occurs earlier than BK as the cell depolarizes. Other than the occasional openings of a small conductance channel (∼12 pS) described previously (Kim, 2013), we did not observe ion channels with kinetics distinctly different from those of TASK, BK and the 20 pS channel during normoxia, hypoxia and high external KCl perfusion.
Intracellular Ca2+ activates the 20 pS channel
Because depolarization increases [Ca2+]i, we tested the role of Ca2+ in 20 pS channel activation using 1 μm FPL64176, a Ca2+ channel agonist that acts on the L-type Ca2+ channel (Baxter et al. 1993). FPL64176 elevated [Ca2+]i from a basal level of 104 ± 14 to 880 ± 110 nm (n = 8; Fig.4A). The rise in [Ca2+]i by FPL64176 was abolished in the presence of 1 μm nifedipine, a Ca2+ channel blocker, showing that FPL64176 opens Ca2+ channels in isolated glomus cells, just as KCl does (n = 16; Fig.4A). FPL64176 reversibly activated the 20 pS channel when applied to the external solution perfusing the cell-attached patches (20 of 26 patches; Fig.4B). In the presence of FPL64176, the 20 pS channel closed when Ca2+ was removed from the bath solution and reopened when Ca2+ was added back (5 of 5 patches; Fig.4C). Interestingly, FPL64176 reduced the amplitude of TASK channels in many patches (n = 12), but not in others (n = 8). The reduction of TASK amplitude varied among patches ranging from 1.2 to 2.2 pA from a control level of 2.2 ± 0.1 pA, suggesting that FPL64176 produces varying degrees of depolarization in glomus cells.
Figure 4. Activation of the 20 pS channel by FPL64176 and Ca2+.
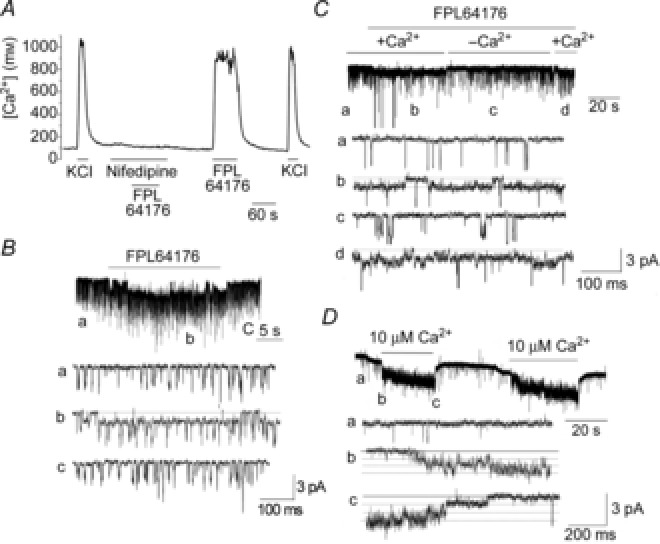
A, tracing showing changes in [Ca2+]i in response to 20 mm KCl and 1 μm FPL64176 in the presence and absence of 1 μm nifedipine in isolated rat glomus cells. B, perfusion of a cell-attached patch showing TASK openings (tracing a) with 1 μm FPL64176 activated the 20 pS channel (tracing b). Washout of the agonist closed the 20 pS channel (tracing c). Pipette potential was 0 mV. C, application of FPL64176 (1 μm) to a cell-attached patch activated the 20 pS channel only when Ca2+ was present in the bath solution (tracings b and d). Perfusion with Ca2+-free solution closed the channel (tracing c). Pipette potential was 0 mV. D, Ca2+ applied to inside-out patch activated the 20 pS channel reversibly.
Direct application of 10 μm Ca2+ to inside-out patches also activated the 20 pS channel (n = 6; Fig.4D). Application of 0.5 and 1 μm Ca2+ did not activate the 20 pS channel (n = 7 each). Single channels activated by 10 μm Ca2+ exhibited long openings, a conductance of 19 ± 1 pS (n = 3) and an approximately linear I–V relationship, similar to the properties of the 20 pS channel observed in cell-attached patches. These results show that Ca2+ directly gates the 20 pS channel. Consistent with the result that a rise in [Ca2+]i is necessary for 20 pS channel activation, hypoxia failed to activate the 20 pS channel when cells were perfused with Ca2+-free solution in all eight patches tested. Modulators of cell redox state such as glutathione (1 mm; n = 7), reduced glutathione (1 mm; n = 7), NAD (1 mm; n = 7), NADH (1 mm; n = 7), hydrogen peroxide (32 mm; n = 7) and xanthine (50 μm)/xanthine oxidase (50 mU) mixture (n = 6) applied to inside-out patches did not activate the 20 pS channel.
Voltage dependence of the 20 pS channel
Does depolarization itself in the absence of a rise in [Ca2+]i affect the 20 pS channel activity? In our initial test for voltage dependence, the pipette potential was held at −120 or −150 mV to depolarize the cell-attached membrane, but this did not activate the 20 pS channel (n = 12). To further study the voltage dependence of the 20 pS channel, we incubated glomus cells with 10 μm BAPTA-AM for 30 min to deplete intracellular free Ca2+. When cell-attached patches were formed on these cells with the pipette containing 140 mm KCl, and 20 mm KCl applied to the bath perfusion solution, no 20 pS channel currents were activated in all six patches (Fig.5A). As predicted, the amplitude of TASK openings decreased markedly, consistent with the strong depolarization of glomus cells produced by 20 mm KCl. In control glomus cells incubated with 0.1% DMSO and no BAPTA-AM for 30 min, 20 mm KCl activated the 20 pS channel in all five patches. In cells incubated with 10 μm BAPTA-AM for 30 min, bath application of 20 mm KCl failed to elicit an increase in [Ca2+]i above the threshold level for activation of the 20 pS channel, as shown by the mean [Ca2+]i response from 28 cells (Fig.5B). Therefore, these results show that depolarization itself without an increase in [Ca2+]i does not open the 20 pS channel.
Figure 5. Lack of voltage dependence of the 20 pS channel.
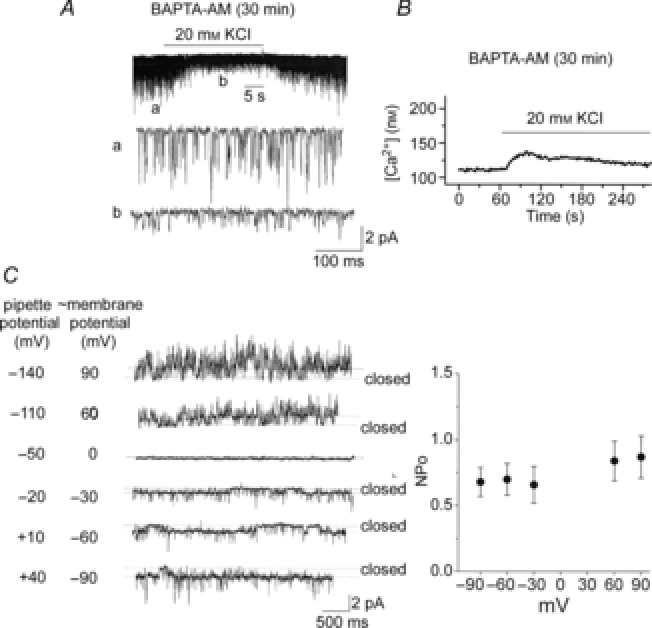
A, glomus cells were incubated with 10 μm BAPTA-AM for 30 min. Current tracings show TASK openings from cell-attached patches with pipette solution containing 140 mm KCl and externally perfused with physiological solution containing 5 mm KCl first, then 20 mm KCl and then back to 5 mm KCl. B, glomus cells were incubated with 10 μm BAPTA-AM for 30 min, and changes in [Ca2+] were measured in response to 20 mm KCl. An averaged tracing from 28 cells is shown. C, cell-attached patches were formed on rat glomus cells with pipette solution containing 140 mm KCl and externally perfused with physiological solution. FPL64176 (1 μm) was applied to the bath to activate the 20 pS channel. Current tracings are shown at various pipette potentials. In this particular patch, the current reversed at −50 mV pipette potential (membrane potential of ∼0 mV). In four other patches, the current reversed at pipette potential between −60 and −40 mV. On the right, the graph plots the 20 pS channel activity as a function of the adjusted cell membrane potential. Each point is the mean ± SD from five patches. No significant differences were present between data points (P > 0.05).
The voltage-dependent change in channel activity was also assessed in cell-attached patches after activating the channel with 1 μm FPL64176. The pipette potential at which TASK reversed the current direction was determined and taken as the cell resting membrane potential. In the current tracings shown in Fig.5C, for example, the zero current was observed at pipette potential of −50 mV, and this value (−50 mV) was taken as the resting membrane potential. A plot of the 20 pS channel activity as a function of membrane potentials adjusted for each cell showed no significant voltage dependence for this channel between −90 and +90 mV (P > 0.05; Fig.5C). Thus, the depolarization-induced activation of the 20 pS channel is mainly due to the rise in [Ca2+]i.
Ion selectivity of the 20 pS channel
The experiments described thus far used K+ as the permeant ion for the 20 pS channel. To test for ion selectivity, cell-attached patches were formed with pipette solution containing 140 mm NaCl, and channel openings were recorded in response to hypoxia and FPL64176. In normoxic solution, cell-attached patches showed no TASK activity as expected, but channel-like spikes with small amplitude persisted throughout the experiment (Fig.6A; tracing a). Under these conditions, hypoxia and FPL64176 activated channels with a single channel conductance of 20 ± 1 pS and with a long open time duration (n = 7; Fig.6A, B). With 140 mm CsCl in the pipette, FPL64176 activated channels with a single channel conductance of 22 ± 1 pS (n = 3; Fig.6C). TASK and the 20 pS channel opened in the outward current direction when the pipette potential was held at −120 mV (patch membrane potential of ∼ +60 mV) with Na+ or Cs+ in the pipette, and the channel currents reversed near a pipette potential of −60 mV (patch potential of ∼0 mV); in these patches, the outward currents are presumably carried by K+.
Figure 6. Ion selectivity of the 20 pS channel.
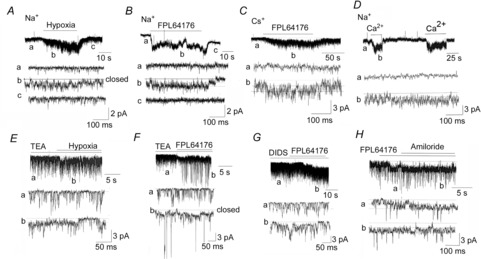
A and B, cell-attached patch was formed with 140 mm NaCl in the pipette and extracellularly perfused with physiological solution. Pipette potential was 0 mV. Hypoxia (A) and FPL64176 (B) activated the 20 pS channel. C, cell-attached patch was formed with 140 mm CsCl in the pipette. FPL64176 activated a 22 pS channel. D, inside-out patch was formed with 140 mm NaCl in the pipette. Bath solution contained 140 mm NaCl with 1 mm EGTA and no Ca2+. Pipette potential was +60 mV. Ca2+ (10 μm) added to the bath solution reversibly activated 20 pS channels. E, cell-attached patch was formed with 5 mm TEA in the pipette. TEA (5 mm) applied to bath solution did not activate the 20 pS channel, whereas hypoxia did. F, FPL64176 applied to the bath solution activated the 20 pS channel as well as BK in this cell-attached patch. TEA was present only in the bath solution. G, DIDS (100 μm) applied to the cell-attached patch for ∼2 min did not activate the 20 pS channel, whereas FPL64176 did. DIDS was present in the pipette solution. H, FPL64176 was added to the bath solution to activate the 20 pS channel in a cell-attached patch. Amiloride (100 μm) addition did not inhibit the 20 pS channel. Amiloride was present in the pipette solution.
With 140 mm NMDG+ in the pipette and FPL64176 in the bath solution, inward 20 pS channel currents were absent at pipette potentials from −60 to 0 mV, but outward 20 pS channel currents were clearly observed at pipette potentials from −60 to −120 mV (n = 5). Thus, NMDG+ is an impermeant ion for the 20 pS channel and does not block the 20 pS channel. In inside-out patches with 140 mm NaCl in the pipette and bath solutions, application of 10 μm Ca2+ to the cytosolic side of the membrane reversibly activated channels with long open time durations and a single channel conductance of 20 ± 1 pS, similar to the kinetics of the hypoxia-activated channel (Fig.6D). These results show clearly that the 20 pS channel is a Ca2+-activated non-selective cation channel.
Effects of ion channel blockers on the 20 pS channel activation by hypoxia or FPL64176 were then studied. Cell-attached patches were formed with pipette solution containing 140 mm KCl and 5 mm TEA. The patches were then extracellularly perfused with physiological bath solution containing 5 mm TEA for ∼1 min. Hypoxia still activated the 20 pS channel in the presence of TEA (n = 9; Fig.6E). Application of FPL64176 to the perfusion solution activated the 20 pS channel whether TEA was present in the pipette and bath solutions (n = 12), or only in the bath solution (n = 12; Fig.6F). When TEA was not present in the pipette, opening of the BK channel was also observed in a few patches along with the 20 pS channel (2 of 12 patches; Fig.6F; tracing b).
Similarly, 4-AP (2 mm; n = 7), iberiotoxin (100 nm; n = 9), apamin (100 nm; n = 8) and 2- APB (0.1 mm; n = 6) added to the pipette and bath solutions did not block the activation of the 20 pS channel by FPL64176. Inhibitors of anion transporters and channels such as DIDS (Fig.6G), 9-AC (0.1 mm) and niflumic acid (0.1 mm) added to the pipette and bath solutions also failed to inhibit FPL64176-induced activation of the 20 pS channel (n = 7 each). In seven cell-attached patches, amiloride (0.1 mm), an inhibitor of Na+ transporters and channels, added to the pipette and bath solutions did not block activation of the 20 pS channel by FPL64176 (Fig.6H). Cytosolic application of 0.1 mm amiloride also failed to block the 20 pS channel in three inside-out patches in the presence of 30 μm Ca2+ in the bath solution. NS-309 (10 μm), an activator of small conductance K+ (SK) channels and glucose-free solution (15 min) present in both pipette and bath solutions did not elicit opening of the 20 pS channel. Taken together, these results further show that the 20 pS channel in glomus cells is a non-selective cation channel.
Reduction of the 20 pS channel conductance by high external Ca2+
Does the 20 pS channel also contribute to Ca2+ influx? To test for Ca2+ permeability, 100 mm CaCl2 was added to the pipette solution and inward current was recorded with pipette potential set at 0 mV. In all eight cell-attached patches, we were unable to observe activation of the 20 pS channel in response to hypoxia and FPL64176 (Fig.7A). The 20 pS channel was also not present at pipette potentials between −120 and +100 mV, suggesting that Ca2+ could be blocking the channel and inhibiting K+ efflux even at depolarized potentials. Therefore, a concentration-dependent effect of extracellular Ca2+ on the 20 pS channel was further studied using cell-attached patches in the presence of 1 μm FPL64176.
Figure 7. Inhibition of the 20 pS channel conductance by extracellular Ca2+.
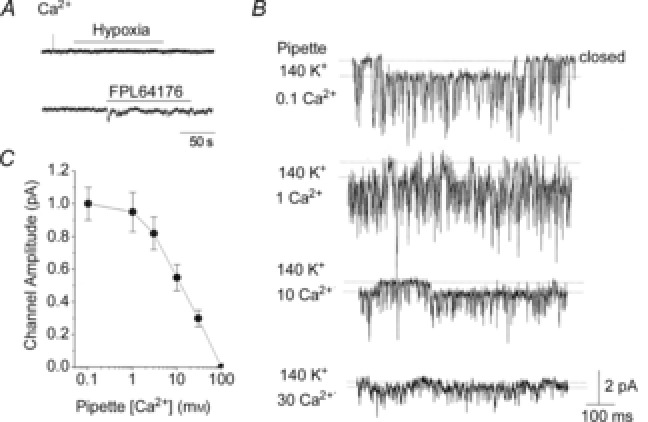
A, cell-attached patches were formed on rat glomus cells with pipette containing 100 mm CaCl2. In physiological bath solution, no channels were activated by hypoxia or FPL64176. The current tracings shown were obtained when the pipette potential was 0 mV. No channels were present with pipette potentials ranging from −120 to +100 mV. B, cell-attached patches were formed with varying levels of Ca2+ in the pipette together with 140 mm KCl. FPL64176 was added to the bath solution to activate the 20 pS channel. Current tracings were obtained at pipette potential set at 0 mV. C, amplitude of the 20 pS channel plotted as a function [Ca2+] in the pipette. Each point is the mean ± SD (n = 5).
With 0.1 and 1 mm Ca2+ in the pipette also containing 140 mm KCl, the 20 pS channel was still active and the single channel conductance was not affected (Fig.7B; n = 5). However, higher [Ca2+] in the pipette (with 140 mm KCl) produced a strong reduction of the single channel conductance of the 20 pS channel opening in the inward direction (Fig.7C; n = 5). With 10 and 30 mm Ca2+ in the pipette, and the pipette potential held at −120 mV, the single channel amplitudes of the 20 pS channel currents in the outward direction were also reduced by 56 ± 5 and 72 ± 5%, respectively (n = 4 each). Thus, high extracellular [Ca2+] inhibited both inward and outward K+ permeation through the 20 pS channel. Although high extracellular [Ca2+] reduced the single-channel conductance, it did not affect the reversal potential for the 20 pS channel (∼ 0 mV membrane potential). At 100 mm Ca2+ in the pipette with 140 mm KCl, no clear channel openings were present. Together, these results suggest that Ca2+ does not permeate the 20 pS channel but modifies the channel structure to reduce the single channel conductance. Although channel activities were not determined, Ca2+ (10 and 30 mm) in the pipette did not appear to shorten the open time duration of the 20 pS channel in all patches tested. These effects of Ca2+ on the 20 pS channel are similar to that reported for TASK, as only the single channel conductance is reduced with a minimal effect on channel activity (Kim et al. 2009a).
Reversal potential of the 20 pS channel current
To determine the direction of the 20 pS channel current under physiological conditions, we estimated the reversal potential of the current by single channel recording in cell-attached patches using physiological solutions containing 5 mm KCl and 135 mm NaCl in both pipette and bath solutions. Under this condition, TASK channel openings only in the outward current direction were observed at pipette potentials ranging from −120 to +60 mV (corresponding to cell Em of approximately +60 mV to −120 mV, assuming a resting cell Em of −60 mV). In all five cells, we could record outward currents at membrane potentials positive to −90 mV, but found no clearly measurable current at and below −90 mV, as predicted of the K+-selective TASK channel.
To obtain the I–V relationship for the 20 pS channel, we applied 1 μm FPL64176 to the bath solution to activate the channel. Under this condition, openings of both TASK and the 20 pS channels were observed. Mean amplitudes of the single channel openings belonging to TASK-1/3 were determined by identifying open and closed levels using pClamp software. I–V relationships for both TASK and the 20 pS channels were then plotted using data from three cell-attached patches that showed outward TASK currents at cell potentials positive to −90 mV and no measurable current at −90 mV and below (Fig.8A; open circles). In these cells, the 20 pS channel current reversed at −28 ± 2 mV, as assessed by linear lines drawn between two points above and below the zero current level. In three other cells that were partially depolarized (i.e. with resting Em levels between −50 and −40 mV, as judged by the reduced amplitude of TASK current compared to control cells), plot of the I–V relationships showed that the 20 pS channel current reversed at −26 ± 2 mV (n = 3), when appropriately adjusted for the cell resting potentials. Thus, these results indicate that activation of the 20 pS channel would produce an inward current when the cell membrane potential is more negative than ∼ −28 mV.
Figure 8. Determination of reversal potential of the 20 pS channel, and whole-cell currents with and without K+ channel inhibitors.
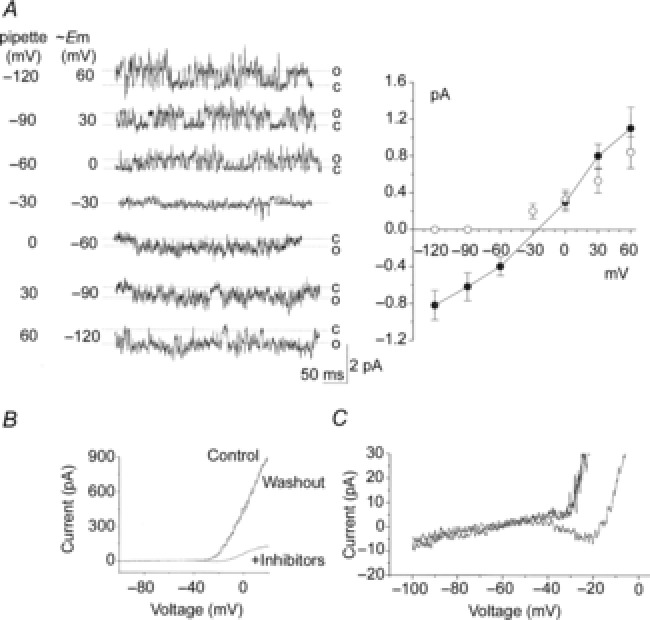
A, tracings of channel currents from a cell-attached patch from a rat glomus cell formed with pipette and bath solutions containing 5 mm KCl and 135 mm NaCl (physiological solution). FPL64176 was added to the bath solution to activate the 20 pS channel. Pipette potential was varied as shown. Amplitude levels of both TASK and the 20 pS channels were determined at each pipette potential. Cell membrane potential corresponding to each pipette potential was calculated as described in the text. Current amplitude levels were then plotted as function of cell potential. Straight lines were drawn between data points to determine the reversal potential for the 20 pS channel. Each point is the mean ± SD (n = 3). B, whole-cell currents were obtained before and after application of K+ channel inhibitors (TEA, 4-AP, iberiotoxin and E4301), and after washout. Whole-cell potential was held at −80 mV, and a 600 ms duration ramp voltage applied (−100 to +20 mV) every 5 s. Control current and the current after washout of inhibitors overlapped in all recordings. C, expanded current tracing showing the negative slope in the I–V relationship when K+ channel inhibitors are present.
Presence of an inward current in voltage-clamp whole-cell recording
As glomus cells depolarize from the resting potential, the rise in [Ca2+]i produced by opening of Ca2+ channels is predicted to begin activation of the 20 pS channel, and influx of both Ca2+ and Na+ is expected to produce an inward current. To test for the presence of an inward current, whole-cell currents were recorded in physiological solution in response to voltage ramps in the presence and absence of external TEA (3 mm), 4-AP (5 mm), iberiotoxin (0.1 μm) and E-4031 (1 μm) that should eliminate most, but not all, outward K+ currents. Under control conditions before the application of the inhibitors, I–V curves showed two main phases: a near flat phase between −100 and −30 mV, and a rapidly rising phase that begins at ∼ −30 mV. When the inhibitors were applied to the extracellular solution, the large outward current was strongly reduced (Fig.8B). In the presence of the K+ channel blockers, whole-cell currents revealed a negative slope in the I–V curve between ∼ −45 and ∼ −20 mV in 60% of cells (7 of 12 cells). A representative whole-cell current tracing exhibiting a negative slope in the presence of inhibitors is shown in Fig.8C, which is expanded from Fig.8B.
The negative slope indicates the presence of an inward current that probably comprises Ca2+ and Na+ currents. At present, the current contributed by the 20 pS channel cannot be isolated, as blocking the Ca2+ channel would inhibit the 20 pS channel current. The height of the negative slope based on the results obtained from seven cells was −11 ± 2 pA (n = 7). In two successful perforated whole-cell experiments, whole-cell currents were qualitatively similar to those observed with conventional recordings, and a negative slope (mean height of 13 pA) was also observed. We suspect that the size of the inward current recorded would be larger if the background K+ currents such as TASK were also inhibited. The variability in the expression of channels that give rise to inward and outward currents may explain the different magnitudes of the negative slope (i.e. inward current) in glomus cells.
Is the 20 pS channel a TRP ion channel?
Among TRP ion channels reported in the literature, the properties of TRPM4 and TRPM5 are most similar to those of the 20 pS channel, as the two TRP channels are activated by an increase in [Ca2+]i, are permeable to monovalent cations and show single-channel conductance levels of 16–25 pS (Launay et al. 2002; Hofmann et al. 2003; Liu & Liman, 2003; Yoo et al. 2010). Expression of TRPM4 and TRPM5 mRNA was tested by RT-PCR using cDNA prepared from clusters of isolated cells. Figure9A shows cell clusters exposed to fluorescein-conjugated peanut agglutinin (5 μg ml−1) that has been reported to bind glomus cells (Kim et al. 2009b). Two sets of TRPM4 primer pairs amplified the expected regions of the TRPM4 mRNA sequence as indicated by the arrows (Fig.9B left), but two sets of TRPM5 primer pairs failed to show expression of TRPM5. The same two pairs of TRPM5 primers amplified the correct regions of TRPM5 when pcDNA3.1 containing TRPM5 DNA sequence was used as PCR template (Fig.9B right). Two additional pairs of TRPM5 primers (see sequences in the Methods) failed to amplify the expected regions of TRPM5 sequence from glomus cells. These results suggested TRPM4 as a possible gene candidate for the 20 pS channel.
Figure 9. Expression of TASK and the 20 pS channel in mouse glomus cells.
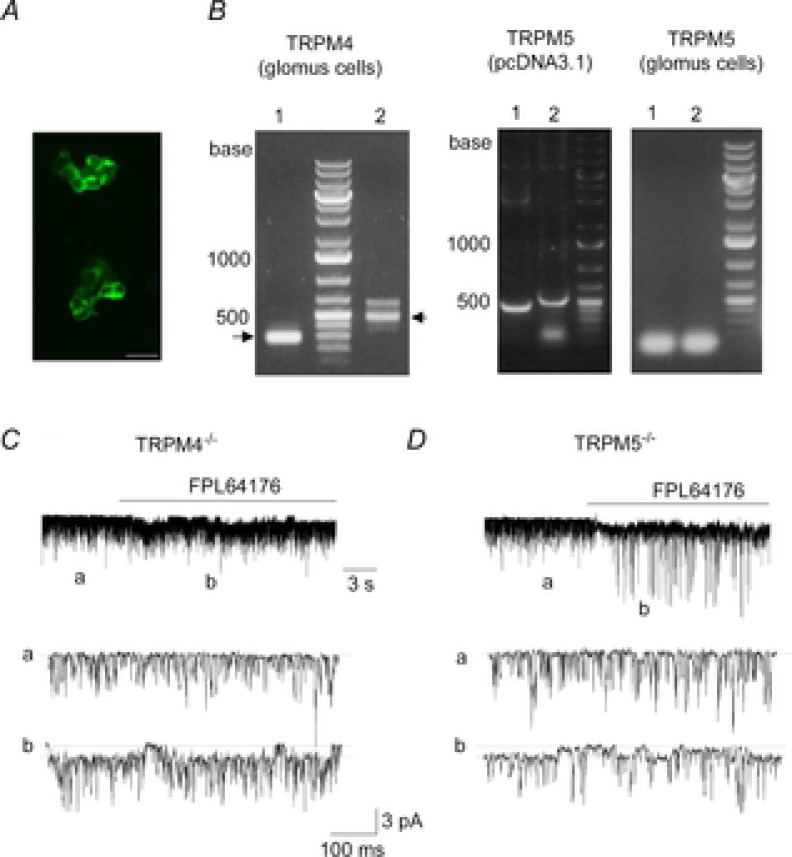
A, clusters of isolated rat glomus cells stained with fluorescein peanut agglutinin, which is used as a marker of rat glomus cells. Such clusters were collected and used for RNA isolation. Scale bar = 20 μm. B, RT-PCR analysis showing 240 bp and 460 bp PCR products for TRMP4 (glomus cells). No PCR products were obtained for TRPM5 (glomus cells), although PCR products were obtained with HEK293 cells expressing TRPM5 (441 bp and 471 bp). Lanes 1 and 2 represent results using two different pairs of primers. PCR products were sequenced for confirmation. C and D, cell-attached patches formed on glomus cells isolated from TRPM4−/− (C) and TRPM5−/− mice (D) shows TASK channels (tracing a). Pipette potential contained 140 mm KCl and was held at 0 mV. FPL64176 activated the 20 pS channels (tracing b) in glomus cells from both mice.
To test whether the 20 pS channel is TRPM4, glomus cells were isolated from TRPM4−/− mice and channel openings recorded from cell-attached patches. TASK-like channels with kinetic properties similar to those of rat glomus cells were active (Fig.9C, tracing a). Similar to TASK channels recorded from glomus cells from rat carotid body, TASK currents from mouse glomus cells reversed direction when the pipette potential was approximately −60 mV (range: −50 to −65 mV). Application of FPL64176 to the bath perfusion solution activated a 20 ± 1 pS channel with long-lasting openings in glomus cells from wild-type mice (n = 3), from TRPM4−/− mice (n = 5; Fig.9C) and from TRPM5−/− mice (n = 5; Fig.9D). Flufenamic acid (0.1 mm), an inhibitor of TRPM4 and TRPM5 (Ullrich et al. 2005), applied to cell-attached patches did not block FPL64176-induced activation of the 20 pS channel (n = 7). Thus, the 20 pS channel does not appear to be TRPM4 or TRPM5, and its molecular identity remains to be determined.
Discussion
In this study, we report the discovery of a 20 pS, non-selective cation channel that is activated by hypoxia in rat carotid body glomus cells. We provide evidence that activation of the 20 pS channel is due to an increase in [Ca2+]i produced by hypoxia-induced inhibition of K+ channels such as TASK. Consistent with this mechanism, acid and high extracellular [K+] that elevate [Ca2+]i also opened the 20 pS channel. Depolarization in the absence of a rise in [Ca2+]i was not effective in activating the 20 pS channel. Under our experimental conditions, the 20 pS channel is active when the level of hypoxia is 1–5% O2 and [Ca2+]i increases more than 100 nm above the basal level, suggesting that the channel is sensitive to moderate and severe levels of hypoxia (<5% O2), but not to mild hypoxia (>5% O2). Our study suggests that this cation channel is a component of the signalling network involved in the excitation of glomus cells in response to moderate to severe hypoxia.
Na+ current in glomus cells
The presence of a standing Na+ current in rat glomus cells has been reported previously (Buckler, 1997; Carpenter & Peers, 2001). In these studies, substitution of Na+ in the external solution by NMDG+ caused hyperpolarization and decreased the holding current to near zero (Carpenter & Peers, 2001). The standing inward current is still present even in the absence of external Ca2+ (Buckler, 1997), suggesting that the Ca2+-sensitive 20 pS channel does not contribute to the standing inward Na+ current at rest. This is in agreement with our observation that the 20 pS channel is mostly in the closed state in normoxia. In human glomus cells, a rapidly activating and inactivating Na+ current has been recorded upon cell depolarization, but it is probably a tetrodotoxin-sensitive Na+ channel (Ortega-Saenz et al. 2013). Whether the 20 pS channel is also expressed in human glomus cells remains to be determined.
In previous studies, hypoxia was found to have no effect on the whole-cell holding current of glomus cells when pipette and bath did not contain K+ (Buckler, 1997), or the pipette contained 1 mm Ca2+ (Carpenter & Peers, 2001). In one study, whole cells were clamped at −70 mV and currents were recorded in either K+/Na+-free or K+/Ca2+-free external solutions. Under these ionic conditions, the 20 pS channel would not be activated, because either Na+ or Ca2+ is absent. In the second study, whole cells were clamped at −80 mV and currents were recorded with pipette solution containing 1 mm Ca2+. Under this ionic condition, the 20 pS channel would already be fully active, inactivated or desensitized, because [Ca2+]i is extremely high. Due to the dependence of the 20 pS channel on [Ca2+]i, it would be difficult to isolate the whole-cell Na+ current generated by the 20 pS channel activity under physiological conditions without a specific inhibitor of the channel.
Sensitivity of the 20 pS channel to hypoxia and [Ca2+]i
Under our experimental conditions, the O2 content of the perfusion solution that activated the 20 pS channel was 1–5%. In an earlier study using glomus cells isolated from 8- to 14-day-old rats, O2 pressure of 19–38 mmHg (1–2.5%) elicited a rise in [Ca2+]i from ∼100 to ∼200–400 nm (Buckler & Vaughan-Jones, 1994b). In another study, [Ca2+]i was 200–300 nm at 5% O2, and ∼400 nm at 2% O2, compared to the basal level of ∼50 nm in 21% O2 in glomus cells from ∼2-week-old rats (Wasicko et al. 1999). These studies, together with our own findings, suggest that consistent activation of the 20 pS channel would occur at moderate to severe levels of hypoxia (<5%O2) when [Ca2+]i rises beyond ∼200 nm.
In inside-out patches, 0.5 and 1 μm Ca2+ did not activate the 20 pS channel, suggesting that the sensitivity of the channel may be reduced under excised conditions compared to the cell-attached conditions. This could be due to loss of intracellular factors such as a Ca2+-sensitive protein that is associated with the 20 pS channel and regulates the channel function. However, global [Ca2+] used to activate the channel in inside-out patches may not correlate with the actual [Ca2+] that activates the cell in intact cells, if Ca2+ and 20 pS channels are located close to each other within microdomains. Further studies are needed to understand this difference in Ca2+ sensitivity.
Molecular identity of the 20 pS channel
A number of ion channels are activated by elevation of [Ca2+]i. These include Ca2+-activated K+ channels (e.g. BK and SK), Ca2+-activated Cl− channels (e.g. Ano-1) and Ca2+-activated or -dependent TRP ion channels (e.g. TRPA1, TRPM2, TRPM4, TRPM5, TRPC5). Ca2+-activated Cl− channels have single-channel conductance levels that are less than 5 pS (Piper & Large, 2003). TRPC channels such as TRPC3 and TRPC6 have been detected at the plasma membrane of rat glomus cells by immunochemistry (Buniel et al. 2003). However, TRPC channels are generally Ca2+ permeable, blocked by 2-APB and not activated by intracellular Ca2+ (Putney, 2005; Owsianik et al. 2006; Ramsey et al. 2006). TRPA1 and TRPC5 have high single-channel conductance levels (100-pS and ∼40 pS, respectively) (Kim & Cavanaugh, 2007; Gross et al. 2009). Glomus cells express Na+-permeable acid-sensing ion channels (ASICs) (Tan et al. 2007), and ASICs are blocked by amiloride, which has no effect on the 20 pS channel.
Of the Ca2+-activated channels published in the literature, TRPM4 and TRMP5 were the most likely candidates to be the 20 pS channel, as they possess biophysical properties most similar to those of the 20 pS channel. Our results show, however, that the 20 pS channel is functionally expressed and activated by Ca2+ channel agonist in glomus cells of TRPM4−/− and TRPM5−/− mice. As some functional native TRP channels are heteromeric, the possibility that the 20 pS channel could be a heteromer composed of different TRP channel subunits will need to be tested in future.
Role of the 20 pS channel in hypoxia-induced depolarization
Single channel studies in cell-attached patches show that under physiological conditions hypoxia activates the 20 pS cation channel and the single channel current carried by Na+ is in the inward direction. This alone indicates that opening of the 20 pS channel would contribute to depolarization. This agrees well with our assessment of the reversal potential of the 20 pS channel (∼ −28 mV) under physiological conditions. Our measurement of the reversal potential of the 20 pS channel could be off by a few millivolts, due to minor errors in the measurement of the cell resting membrane potential based on TASK amplitude and interpolation of the zero current potential using a straight line between data points. Nevertheless, we used a conservative approach such that the values that we obtained would be an underestimate of the true reversal potential. For example, if we found our estimate of the cell resting membrane potential to be between −55 and −60 mV, we used −55 mV for our calculation, which should shift the I–V curve of the 20 pS channel to the left. Therefore, the actual reversal potentials could be more positive than −28 mV. We therefore think that activation of the 20 pS channel should definitely contribute to the inward current between the cell membrane potential of −60 and −30 mV.
In our recent study, severe hypoxia (0% O2) produced ∼35 mV depolarization of glomus cells and an increase in [Ca2+]i similar to that produced by 20 mm KCl (Kim et al. 2011a,b2011b). This suggests that the 20 pS channel contributes to the inward current until the cell depolarizes to levels close to that produced by severe hypoxia. At present, it is difficult to know how much of the depolarization is actually contributed by Na+ influx via the 20 pS channel, because we do not know the magnitude of this cation current. Our findings suggest that Ca2+ and Na+ channels support each other's activity, and therefore both channels are likely to be important for cell depolarization.
In earlier studies, whole-cell currents were recorded from glomus cells using voltage ramps from −100 to −40 mV while holding the cell membrane potential at −70 or −80 mV to study the current near the resting Em of glomus cells (Buckler, 1997; Wasicko et al. 2006). Hypoxia (or anoxia) caused flattening of the whole-cell I–V relationship, and the hypoxia-sensitive current reversed near −80 mV (Buckler, 1997). These findings have led to the idea that most or all of the hypoxia-sensitive current is carried by K+. Because the 20 pS channel becomes active when the membrane potential is near −40 mV (see Fig.3), the involvement of the 20 pS channel should be minimal when the membrane potential is between −100 and −40 mV, as [Ca2+]i would be below the threshold for activation of the 20 pS channel. As the cell depolarizes further and [Ca2+]i rises beyond ∼200 nm, the 20 pS channel is expected to become active and begin to contribute to the inward current and further depolarization.
Our whole-cell current recordings show that in the presence of TEA, 4-AP, iberiotoxin and E4301, the remaining current reveals a negative slope beginning between −45 and −20 mV, showing that an inward current is present in this range of potentials. A negative slope in the I–V relationship in rat glomus cells has been reported earlier under anoxic conditions only when Ca2+ was present in the bath perfusion solution (Buckler, 1997). It was therefore speculated that hypoxia might be increasing Ca2+ current at relative negative potentials in glomus cells. The negative slope (starting at ∼ −50 mV) in the whole-cell I–V relationship of rat glomus cells has also been observed when Ba2+ was used to block the outward current in normoxia (Wasicko et al. 2006). Our studies suggest that the inward current is probably composed of Na+ influx via the 20 pS channel and Ca2+ influx via the voltage-dependent Ca2+ channel.
An updated model of O2 sensing by ion channels in isolated glomus cell
Figure10 shows a model of O2 sensing that incorporates the functional input of the 20 pS channel. During mild hypoxia (5% O2 and higher), inhibition of background K+ channels such as TASK leads to a mild depolarization (<20 mV) that allows voltage-dependent Ca2+ channels to mildly increase [Ca2+]i. This pathway (steps 1–3) is already well established from earlier studies, except that the relative roles of TASK and BK remain unresolved (Pardal et al. 2000; Buckler, 2007; Peers & Wyatt, 2007). The level of depolarization and rise in [Ca2+] is not sufficient to activate the 20 pS channel, and therefore mild hypoxia does not affect the 20 pS channel.
A model of O2 sensing by glomus cells during moderate to severe hypoxia.
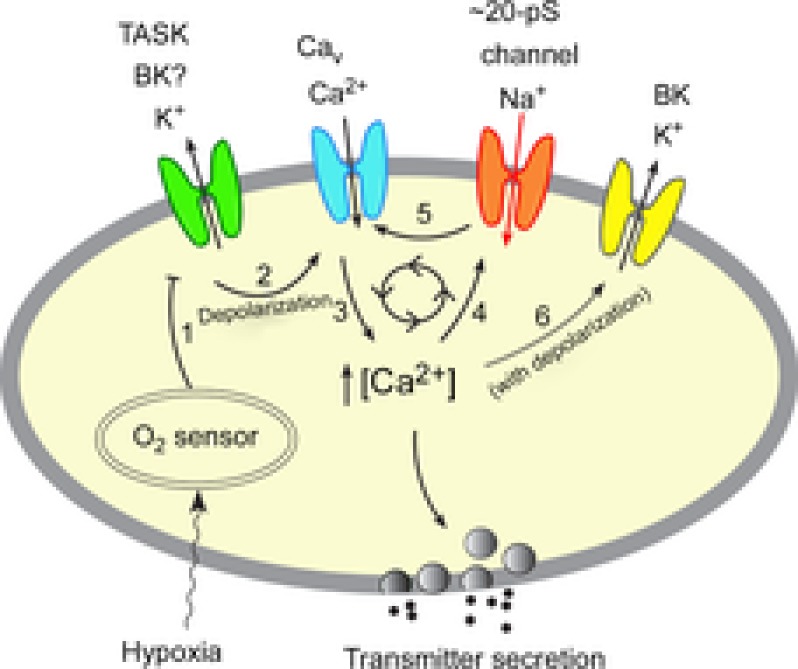
Mild hypoxia (>5% O2) inhibits (step 1) the outward K+ current such as TASK via an O2 sensor. This depolarizes the cell, causing opening of the voltage-gated Ca2+ channels (step 2), influx of Ca2+ and elevation of [Ca2+]i (step 3). The rise in [Ca2+]i causes transmitter secretion. Moderate and severe hypoxia (<5% O2) causes stronger inhibition of the outward K+ current than that produced by mild hypoxia, producing a greater level of depolarization and a higher [Ca2+]i. Rise of [Ca2+]i beyond ∼200 nm from a basal level of ∼100 nm begins to activate the Na+-permeable 20 pS channel (step 4). Na+ influx via the 20 pS channel may initiate a feed-forward mechanism (steps 5→3→4→5) to further increase and maintain the levels of depolarization and [Ca2+]i during moderate to severe hypoxia. Depolarization and the associated increase in [Ca2+]i may activate BK (step 6) and limits the feed-forward mechanism and over-depolarization (see text for further explanation). Thus, our model incorporates the role of both K+- and Na+-permeable channels in the depolarization of glomus cells when hypoxia is moderate to severe.
At levels of O2 less than 5%, the rise in [Ca2+]i is sufficient to activate the 20 pS channel (step 4). Therefore, the Ca2+-sensitive 20 pS channel found in glomus cells can now be added to the earlier model to be part of the overall signalling complex involved in sensing moderate to severe levels of hypoxia. Recruitment of the 20 pS channel should initiate a feed-forward mechanism whereby an increased Na+ influx enhances Ca2+ influx via additional depolarization (steps 5→3→4→5). This mechanism may be important for maintaining the levels of depolarization and [Ca2+]i in response to hypoxia (<5% O2). When the cell depolarizes to levels more positive than ∼ −28 mV and [Ca2+]i rises further, Na+ efflux via the 20 pS channel may oppose further depolarization and help limit over-excitation of glomus cells. Defining the function of this cation channel in glomus cells in a more intact system (i.e. in undissociated carotid body) would be important to further understand its role in hypoxia-mediated excitation of glomus cells in vivo.
Because the BK channel is well expressed in glomus cells, it is likely to serve an important role in regulating the excitability of glomus cells. As the cell depolarizes, the depolarized membrane and the associated rise in [Ca2+]i should activate BK (step 6) at some point. As shown in Fig.8B and as reported by others, of the flat and rapidly rising phases of the I–V relationship obtained from whole-cell recordings, only the rapidly rising phase which begins at ∼ −30 mV is strongly reduced by TEA and K+ channel inhibitors (Hatton et al. 1997; Lopez-Lopez et al. 1997). This suggests that BK begins to activate when the cell depolarization reaches ∼ −30 mV. This is consistent with our finding that 12–15 mm KCl activates the 20 pS channel without opening BK channels (at pipette potential of 0 mV), suggesting that BK comes into play when the membrane potential of glomus cells is depolarized to levels more positive than ∼ −30 mV. If BK is active at rest and hypoxia inhibits BK to contribute to the initial depolarization (Peers & Wyatt, 2007), our model should still hold true as the 20 pS channel would be activated by the rise in [Ca2+]i. When BK is activated by depolarization and the rise in [Ca2+]i, K+ efflux via BK should help limit over-excitation of glomus cells, as suggested recently (Gomez-Nino et al. 2009; Donnelly et al. 2011).
In summary, we have identified and characterized a 20 pS non-selective cation channel that is activated by cytosolic Ca2+. Our data show that hypoxia and other depolarizing stimuli activate a 20 pS channel in isolated glomus cells by a direct action of elevated [Ca2+]i on the channel. The 20-pA channel is recruited by initial depolarization of glomus cells elicited by inhibition of K+ channels (such as TASK) by hypoxia. A revised model of O2 sensing includes the 20 pS cation channel as a part of the feed-forward mechanism to help maintain the levels of depolarization and [Ca2+]i in glomus cells in response to moderate to severe hypoxia. Thus, O2 sensing involves both inhibition of outward K+ current and activation of inward Na+ current. Our study sets the foundation for future studies to further define the role of the 20 pS channel in O2 and glucose sensing, to identify potential interactions with other ion channels, and to determine its contribution to the regulation of the cardiorespiratory function during hypoxia, acidosis and hypercapnia.
Acknowledgments
None.
Glossary
- 2-APB
2-aminoethoxydiphenyl borate
- 4-AP
4-aminopyridine
- 9-AC
anthracene-9-carboxylic acid
- ASICs
acid-sensing ion channels
- BK
large-conductance Ca2+-activated channel
- [Ca2+]i
intracellular Ca2+ concentration
- DIDS
4,4′ diisothio-cyanatostilbene-2,2′-disulfonic acid
- DMSO
dimethyl sulfoxide
- NMDG
N-methyl-d-glucamine
- PBS
phosphate-buffered saline
- SK
small conductance K+ channel
- TASK
TWIK-related acid-sensing K+ channel
- TEA
tetraethylammonium
Key points
Hypoxia is thought to depolarize glomus cells by inhibiting the outward K+ current, which sets in motion a cascade of ionic events that lead to transmitter secretion, increased afferent carotid sinus nerve activity and increased ventilation.
Our study of Na+-permeable channels in glomus cells has revealed that hypoxia not only inhibits TASK background K+ channels but also indirectly activates a non-selective cation channel with a single channel conductance of 20 pS. Under physiological conditions, the reversal potential of the cation channel is ∼ –28 mV, indicating that Na+ influx is also involved in hypoxia-induced excitation of glomus cells.
Activation of the 20 pS cation channel is present when the O2 content is 5% or less, indicating that Na+ influx occurs during moderate to severe hypoxia (<5% O2), but not mild hypoxia (>5% O2).
The 20 pS cation channel is directly activated by a rise in intracellular Ca2+. Thus, factors that elevate intracellular Ca2+ such as hypoxia, extracellular acidosis and high external KCl all activate the cation channel. A feed-forward mechanism may be present in which an initial depolarization-induced rise in intracellular Ca2+ opens the Na+-permeable cation channel, and the Na+ influx causes additional depolarization and influx of Ca2+ into glomus cells.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
This work was performed in the Department of Physiology at Chicago Medical School, Rosalind Franklin University, North Chicago, IL, USA. Donghee Kim (Do.K.) conceived the study. Da.K., J.W., J.O.H. and Do.K. were involved in collection of data, analysis and interpretation of data. Do.K. and Da.K. drafted the article for publication. R.V. and M.F. constructed the TRPM4−/− mice. C.W. contributed to the measurement of [Ca2+]i. All authors have read and approved the final version of the manuscript and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by NIH (HL111497 to Do.K.) and National Research Foundation grant of Korea (NRF-2010-0024258 to Da.K.).
Translational perspective
There is increasing evidence showing that altered chemoreceptor glomus cell function is associated with changes in the autonomic nervous activity and cardiorespiratory function. Full understanding of the cellular processes that regulate the function of glomus cells is therefore critical for potential treatment of various diseases related to carotid body dysfunction. This study provides the first description of a Na+-permeable ion channel that is activated by hypoxia in glomus cells, and thus advances our knowledge of the ionic mechanisms that regulate glomus cell excitability.
References
- Baxter AJ, Dixon J, Ince F, Manners CN. Teague SJ. Discovery and synthesis of methyl 2,5-dimethyl-4-[2- (phenylmethyl)benzoyl]-1H-pyrrole-3-carboxylate (FPL 64176) and analogues: the first examples of a new class of calcium channel activator. J Med Chem. 1993;36:2739–2744. doi: 10.1021/jm00071a004. [DOI] [PubMed] [Google Scholar]
- Bin-Jaliah I, Maskell PD. Kumar P. Indirect sensing of insulin-induced hypoglycaemia by the carotid body in the rat. J Physiol. 2004;556:255–266. doi: 10.1113/jphysiol.2003.058321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscoe TJ. Duchen MR. Responses of type I cells dissociated from the rabbit carotid body to hypoxia. J Physiol. 1990;428:39–59. doi: 10.1113/jphysiol.1990.sp018199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type I cells. J Physiol. 1997;498:649–662. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. TASK-like potassium channels and oxygen sensing in the carotid body. Respir Physiol Neurobiol. 2007;157:55–64. doi: 10.1016/j.resp.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. Vaughan-Jones RD. Effects of hypercapnia on membrane potential and intracellular calcium in rat carotid body type I cells. J Physiol. 1994a;478:157–171. doi: 10.1113/jphysiol.1994.sp020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol. 1994b;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buniel MC, Schilling WP. Kunze DL. Distribution of transient receptor potential channels in the rat carotid chemosensory pathway. J Comp Neurol. 2003;464:404–413. doi: 10.1002/cne.10798. [DOI] [PubMed] [Google Scholar]
- Carpenter E. Peers C. A standing Na+ conductance in rat carotid body type 1 cells. Neuroreport. 2001;12:1421–1425. doi: 10.1097/00001756-200105250-00025. [DOI] [PubMed] [Google Scholar]
-
Delpiano MA. Hescheler J. Evidence for a
 -sensitive K+ channel in the type-1 cell of the rabbit carotid body. FEBS Lett. 1989;249:195–198. doi: 10.1016/0014-5793(89)80623-4. [DOI] [PubMed] [Google Scholar]
-sensitive K+ channel in the type-1 cell of the rabbit carotid body. FEBS Lett. 1989;249:195–198. doi: 10.1016/0014-5793(89)80623-4. [DOI] [PubMed] [Google Scholar] - Donnelly DF, Kim I, Yang D. Carroll JL. Role of MaxiK-type calcium dependent K+ channels in rat carotid body hypoxia transduction during postnatal development. Respir Physiol Neurobiol. 2011;177:1–8. doi: 10.1016/j.resp.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AM, Hardie DG, Peers C, Wyatt CN, Viollet B, Kumar P, Dallas ML, Ross F, Ikematsu N, Jordan HL, Barr BL, Rafferty JN. Ogunbayo O. Ion channel regulation by AMPK: the route of hypoxia-response coupling in the carotid body and pulmonary artery. Ann N Y Acad Sci. 2009;1177:89–100. doi: 10.1111/j.1749-6632.2009.05041.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Fernandez M, Ortega-Saenz P, Castellano A. Lopez-Barneo J. Mechanisms of low-glucose sensitivity in carotid body glomus cells. Diabetes. 2007;56:2893–2900. doi: 10.2337/db07-0122. [DOI] [PubMed] [Google Scholar]
- Gomez-Nino A, Obeso A, Baranda JA, Santo-Domingo J, Lopez-Lopez JR. Gonzalez C. MaxiK potassium channels in the function of chemoreceptor cells of the rat carotid body. Am J Physiol Cell Physiol. 2009;297:C715–722. doi: 10.1152/ajpcell.00507.2008. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A. Rigual R. Oxygen and acid chemoreception in the carotid body chemoreceptors. Trends Neurosci. 1992;15:146–153. doi: 10.1016/0166-2236(92)90357-e. [DOI] [PubMed] [Google Scholar]
- Gross SA, Guzman GA, Wissenbach U, Philipp SE, Zhu MX, Bruns D. Cavalie A. TRPC5 is a Ca2+-activated channel functionally coupled to Ca2+-selective ion channels. J Biol Chem. 2009;284:34423–34432. doi: 10.1074/jbc.M109.018192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M. Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hatton CJ, Carpenter E, Pepper DR, Kumar P. Peers C. Developmental changes in isolated rat type I carotid body cell K+ currents and their modulation by hypoxia. J Physiol. 1997;501:49–58. doi: 10.1111/j.1469-7793.1997.049bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Chubanov V, Gudermann T. Montell C. TRPM5 is a voltage-modulated and Ca2+-activated monovalent selective cation channel. Curr Biol. 2003;13:1153–1158. doi: 10.1016/s0960-9822(03)00431-7. [DOI] [PubMed] [Google Scholar]
- Kim D. K+ channels in O2 sensing and postnatal development of carotid body glomus cell response to hypoxia. Respir Physiol Neurobiol. 2013;185:44–56. doi: 10.1016/j.resp.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. Cavanaugh EJ. Requirement of a soluble intracellular factor for activation of transient receptor potential A1 by pungent chemicals: role of inorganic polyphosphates. J Neurosci. 2007;27:6500–6509. doi: 10.1523/JNEUROSCI.0623-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Cavanaugh EJ, Kim I. Carroll JL. Heteromeric TASK-1/TASK-3 is the major oxygen-sensitive background K+ channel in rat carotid body glomus cells. J Physiol. 2009a;587:2963–2975. doi: 10.1113/jphysiol.2009.171181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim I, Papreck JR, Donnelly DF. Carroll JL. Characterization of an ATP-sensitive K+ channel in rat carotid body glomus cells. Respir Physiol Neurobiol. 2011a;177:247–255. doi: 10.1016/j.resp.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Papreck JR, Kim I, Donnelly DF. Carroll JL. Changes in oxygen sensitivity of TASK in carotid body glomus cells during early postnatal development. Respir Physiol Neurobiol. 2011b;177:228–235. doi: 10.1016/j.resp.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Yang DJ, Donnelly DF. Carroll JL. Fluoresceinated peanut agglutinin (PNA) is a marker for live O2 sensing glomus cells in rat carotid body. Adv Exp Med Biol. 2009b;648:185–190. doi: 10.1007/978-90-481-2259-2_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. Prabhakar NR. Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol. 2012;2:141–219. doi: 10.1002/cphy.c100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R. Kinet JP. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109:397–407. doi: 10.1016/s0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- Liu D. Liman ER. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc Natl Acad Sci U S A. 2003;100:15160–15165. doi: 10.1073/pnas.2334159100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Lopez-Lopez JR, Urena J. Gonzalez C. Chemotransduction in the carotid body: K+ current modulated by PO2 in type 1 chemoreceptor cells. Science. 1988;241:580–582. doi: 10.1126/science.2456613. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez JR, Gonzalez C. Perez-Garcia MT. Properties of ionic currents from isolated adult rat carotid body chemoreceptor cells: effect of hypoxia. J Physiol. 1997;499:429–441. doi: 10.1113/jphysiol.1997.sp021939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Saenz P, Levitsky KL, Marcos-Almaraz MT, Bonilla-Henao V, Pascual A. Lopez-Barneo J. Carotid body chemosensory responses in mice deficient of TASK channels. J Gen Physiol. 2010;135:379–392. doi: 10.1085/jgp.200910302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Saenz P, Pardal R, Levitsky K, Villadiego J, Munoz-Manchado AB, Duran R, Bonilla-Henao V, Arias-Mayenco I, Sobrino V, Ordonez A, Oliver M, Toledo-Aral JJ. Lopez-Barneo J. Cellular properties and chemosensory responses of the human carotid body. J Physiol. 2013;591:6157–6173. doi: 10.1113/jphysiol.2013.263657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsubo T, Kostuk EW, Balbir A, Fujii K. Shirahata M. Differential expression of large-conductance Ca-activated K channels in the carotid body between DBA/2J and A/J strains of mice. Front Cell Neurosci. 2011;5:1–8. doi: 10.3389/fncel.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owsianik G, Talavera K, Voets T. Nilius B. Permeation and selectivity of TRP channels. Ann Rev Physiol. 2006;68:685–717. doi: 10.1146/annurev.physiol.68.040204.101406. [DOI] [PubMed] [Google Scholar]
- Pardal R. Lopez-Barneo J. Low glucose-sensing cells in the carotid body. Nat Neurosci. 2002;5:197–198. doi: 10.1038/nn812. [DOI] [PubMed] [Google Scholar]
- Pardal R, Ludewig U, Garcia-Hirschfeld J. Lopez-Barneo J. Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci U S A. 2000;97:2361–2366. doi: 10.1073/pnas.030522297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C. Wyatt CN. The role of maxiK channels in carotid body chemotransduction. Respir Physiol Neurobiol. 2007;157:75–82. doi: 10.1016/j.resp.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Peers C, Wyatt CN. Evans AM. Mechanisms for acute oxygen sensing in the carotid body. Respir Physiol Neurobiol. 2010;174:292–298. doi: 10.1016/j.resp.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Piper AS. Large WA. Multiple conductance states of single Ca2+-activated Cl– channels in rabbit pulmonary artery smooth muscle cells. J Physiol. 2003;547:181–196. doi: 10.1113/jphysiol.2002.033688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW. Physiological mechanisms of TRPC activation. Pflugers Arch. 2005;451:29–34. doi: 10.1007/s00424-005-1416-4. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M. Clapham DE. An introduction to TRP channels. Ann Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Tan ZY, Lu Y, Whiteis CA, Benson CJ, Chapleau MW. Abboud FM. Acid-sensing ion channels contribute to transduction of extracellular acidosis in rat carotid body glomus cells. Circ Res. 2007;101:1009–1019. doi: 10.1161/CIRCRESAHA.107.154377. [DOI] [PubMed] [Google Scholar]
- Tepikin A. Calcium: A Practical Approach. Oxford: Oxford University Press; 2001. [Google Scholar]
- Trapp S, Aller MI, Wisden W. Gourine AV. A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing. J Neurosci. 2008;28:8844–8850. doi: 10.1523/JNEUROSCI.1810-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich ND, Voets T, Prenen J, Vennekens R, Talavera K, Droogmans G. Nilius B. Comparison of functional properties of the Ca2+-activated cation channels TRPM4 and TRPM5 from mice. Cell Calcium. 2005;37:267–278. doi: 10.1016/j.ceca.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE, Schmitz F, Weissgerber P, Nilius B, Flockerzi V. Freichel M. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nature Immunol. 2007;8:312–320. doi: 10.1038/ni1441. [DOI] [PubMed] [Google Scholar]
- Wasicko MJ, Breitwieser GE, Kim I. Carroll JL. Postnatal development of carotid body glomus cell response to hypoxia. Respir Physiol Neurobiol. 2006;154:356–371. doi: 10.1016/j.resp.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Wasicko MJ, Sterni LM, Bamford OS, Montrose MH. Carroll JL. Resetting and postnatal maturation of oxygen chemosensitivity in rat carotid chemoreceptor cells. J Physiol. 1999;514:493–503. doi: 10.1111/j.1469-7793.1999.493ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt CN, Mustard KJ, Pearson SA, Dallas ML, Atkinson L, Kumar P, Peers C, Hardie DG. Evans AM. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J Biol Chem. 2007;282:8092–8098. doi: 10.1074/jbc.M608742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JC, Yarishkin OV, Hwang EM, Kim E, Kim DG, Park N, Hong SG. Park JY. Cloning and characterization of rat transient receptor potential-melastatin 4 (TRPM4) Biochem Biophys Res Commun. 2010;391:806–811. doi: 10.1016/j.bbrc.2009.11.142. [DOI] [PubMed] [Google Scholar]
- Zhang M, Buttigieg J. Nurse CA. Neurotransmitter mechanisms mediating low-glucose signalling in cocultures and fresh tissue slices of rat carotid body. J Physiol. 2007;578:735–750. doi: 10.1113/jphysiol.2006.121871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hoon MA, Chandrashekar J, Mueller KL, Cook B, Wu D, Zuker CS. Ryba NJ. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signalling pathways. Cell. 2003;112:293–301. doi: 10.1016/s0092-8674(03)00071-0. [DOI] [PubMed] [Google Scholar]