

The role of phosphorylation in ADAM17-dependent shedding is controversial. We show that the A1 adenosine receptor stimulates exocytosis in umbrella cells by a pathway that requires phosphorylation of ADAM17–Ser-811, followed by HB-EGF shedding and EGF receptor transactivation. Preventing ADAM17 phosphorylation blocks these downstream events.
Abstract
Despite the importance of ADAM17-dependent cleavage in normal biology and disease, the physiological cues that trigger its activity, the effector pathways that promote its function, and the mechanisms that control its activity, particularly the role of phosphorylation, remain unresolved. Using native bladder epithelium, in some cases transduced with adenoviruses encoding small interfering RNA, we observe that stimulation of apically localized A1 adenosine receptors (A1ARs) triggers a Gi-Gβγ-phospholipase C-protein kinase C (PKC) cascade that promotes ADAM17-dependent HB-EGF cleavage, EGFR transactivation, and apical exocytosis. We further show that the cytoplasmic tail of rat ADAM17 contains a conserved serine residue at position 811, which resides in a canonical PKC phosphorylation site, and is phosphorylated in response to A1AR activation. Preventing this phosphorylation event by expression of a nonphosphorylatable ADAM17S811A mutant or expression of a tail-minus construct inhibits A1AR-stimulated, ADAM17-dependent HB-EGF cleavage. Furthermore, expression of ADAM17S811A in bladder tissues impairs A1AR-induced apical exocytosis. We conclude that adenosine-stimulated exocytosis requires PKC- and ADAM17-dependent EGFR transactivation and that the function of ADAM17 in this pathway depends on the phosphorylation state of Ser-811 in its cytoplasmic domain.
INTRODUCTION
Protein ectodomain shedding, a process regulated by proteolysis, is a fundamental mechanism for the release of cytokines, growth factors, and cell adhesion molecules (Reiss and Saftig, 2009) and is altered in cancer, autoimmune and inflammatory diseases, cardiovascular disease, and neurodegeneration (Murphy, 2008). The best-understood sheddases include the “a disintegrin and a metalloproteinase” (ADAM) family members ADAM10 and ADAM17 (also known as TACE), both of which shed a variety of substrates, including the transmembrane ligands for the epidermal growth factor receptor (EGFR). Whereas ADAM10 targets betacellulin, EGF, and neuregulin, ADAM17 is the principal sheddase for transforming growth factor (TGF) α, amphiregulin, epiregulin, epigen, and heparin-binding (HB) EGF (Jackson et al., 2003; Sahin et al., 2004; Sahin and Blobel, 2007; Blobel, 2005; Horiuchi et al., 2005; Sternlicht et al., 2005; Hassemer et al., 2010; Luo et al., 2011). The physiological cues that trigger shedding remain to be specified; however, ADAM17-elicited shedding occurs in response to some G protein–coupled receptor (GPCR) ligands as well as treatment with ionomycin or phorbol-12-myristate-13-acetate (PMA), a diacylglycerol (DAG) mimic and activator of classical protein kinase C (PKC) isoforms (Horiuchi et al., 2007; Le Gall et al., 2009; Dang et al., 2011). In contrast, ADAM10-dependent shedding responds to Ca2+ ionophores (e.g., ionomycin) but generally not PMA (Horiuchi et al., 2007; Le Gall et al., 2009; Dang et al., 2011). In addition, our understanding of the signaling and associated effector pathways that act downstream of these stimuli remains incomplete. The extracellular signal-regulated kinase (ERK), as well as the related p38 mitogen-activated protein kinase (MAPK), PKCα, PKCδ, and the PKC-regulated protein phosphatase inhibitor 14D, has also been linked to ADAM17-dependent shedding, and PKCδ is required for shedding of neuregulin (Bell and Gooz, 2010; Killock and Ivetic, 2010; Dang et al., 2011, 2013; Hall and Blobel, 2012).
An additional unresolved question is how these effectors promote ADAM-dependent shedding of EGFR ligands. Data indicate that they work through multiple mechanisms and perhaps in a cell- and stimulus-dependent manner (Fan et al., 2003; Mifune et al., 2005; Xu and Derynck, 2010). Potential regulatory steps include membrane trafficking of the ADAM or its ligands (Soond et al., 2005), effects on the ADAM17 dimer–monomer equilibrium, and association with tissue inhibitor of metalloproteinases (TIMP) 3 (Xu et al., 2012) or changes in the redox potential of the extracellular protein disulfide isomerase (Willems et al., 2010), which is hypothesized to alter ADAM17 activity by rearrangement of its disulfide bonds. An additional regulatory mechanism may be phosphorylation of EGFR receptor substrates (Dang et al., 2013) or the ADAM metalloproteinase itself. For example, phosphorylation of ADAM17 cytoplasmic residues Tyr-702, Ser-791, Ser-819, and Thr-735 have been reported (Fan et al., 2003; Hall and Blobel, 2012; Niu et al., 2013). Tyr-702 is reportedly phosphorylated by the Src kinase when myogenic precursor cells are stretched (Niu et al., 2013). In contrast, Ser-791 is phosphorylated before stimulation, and mutations of Ser-819 do not appear to affect shedding (Fan et al., 2003). In the case of Thr-735, very high doses of PMA (1 μM) are reported to promote ERK-dependent phosphorylation of this residue (Soond et al., 2005). However, others report that phosphorylation of this residue is mediated by the p38 MAPK and is required for ADAM17-dependent shedding in response to various forms of stress but not PMA (Xu and Derynck, 2010). A significant argument against the role for phosphorylation relies on reports that show that stimulus-evoked shedding of ADAM17 occurs in cells expressing truncated versions of this metalloproteinase that lack a C-terminus (Reddy et al., 2000; Le Gall et al., 2010; Hall and Blobel, 2012).
A potentially useful and physiologically relevant model system in which to study the signals, effector pathways, and mode of ADAM activation is the uroepithelium, a tissue that can be studied both ex vivo and in vivo (Khandelwal et al., 2008, 2010, 2013). Bladder filling triggers the exocytosis of an abundant subapical pool of discoidal- and/or fusiform-shaped vesicle (DFVs) in the outer umbrella cell layer (Wang et al., 2003a, b). Stretch-induced exocytosis progresses in two phases. “Early-phase” exocytosis occurs during bladder filling, as the epithelium is bowing outward, and is triggered by apical Ca2+ entry, likely conducted by a nonselective cation channel (Yu et al., 2009). In contrast, “late-phase” exocytosis is initiated once the tissue is maximally bowed outward (i.e., in response to a full bladder) and requires metalloproteinase-dependent cleavage of HB-EGF, leading to “transactivation” of apical EGFRs (Balestreire and Apodaca, 2007). The latter initiates a downstream ERK pathway that culminates in protein synthesis and exocytosis (Balestreire and Apodaca, 2007). We now report that adenosine, which is released from the epithelium in response to stress (Prakasam et al., 2012), spurs a late phase–like response in umbrella cells. We also find that a Gi-, Gβγ-, phospholipase C (PLC)-, and PKC-dependent effector cascade is initiated downstream of the adenosine A1 receptor (A1AR). Finally, PKC likely promotes the phosphorylation of a previously unreported canonical PKC site centered at Ser-811 in the cytoplasmic domain of rat ADAM17. This phosphorylation event is required for adenosine-induced and ADAM17-dependent HB-EGF cleavage, EGFR transactivation, and exocytosis. These studies lend support to the hypothesis that posttranslational modification of ADAM17—phosphorylation in particular—can control its activity in a physiologically relevant setting.
RESULTS
A1AR-stimulated apical exocytosis occurs through transactivation of the EGFR
Late-phase exocytosis is triggered when uroepithelial tissue is maximally stretched (Balestreire and Apodaca, 2007; Yu et al., 2009), but it is unknown whether other stimuli promote a similar response. Because adenosine release is significantly increased as the bladder reaches its capacity (Prakasam et al., 2012), and because extracellular adenosine also triggers exocytosis (Yu et al., 2006), we tested the hypothesis that adenosine stimulates exocytosis by way of EGFR transactivation. Adenosine can stimulate exocytosis when added to either the serosal or mucosal surfaces of isolated uroepithelium; however, in this study, we focused our attention on the mucosal events, as these are primarily mediated by the A1AR, and EGFR transactivation occurs at this surface (Yu et al., 2006; Balestreire and Apodaca, 2007). Consistent with our previous reports (Yu et al., 2011; Prakasam et al., 2012), the A1AR was localized to the apical pole of rat umbrella cells (as well as in the underlying lamina propria; Figure 1A). When adenosine was added to the mucosal surface of isolated rabbit tissue, it stimulated increased tissue capacitance (CT; 1 μF ≈ 1 cm2; Figure 1B), which in this tissue correlates well with other measures of apical exocytosis (Truschel et al., 2002; Wang et al., 2003a). Similar results were obtained when the highly selective and high-affinity A1AR agonist 2-chloro-N6-cyclopentyladenosine (CCPA) was used. Unlike adenosine, CCPA is not rapidly converted to inosine or to AMP (Manjunath and Sakhare, 2009; Prakasam et al., 2012) and was thus used in our subsequent studies. We next determined whether A1AR-stimulated exocytosis, like that observed in response to excess stretch, is dependent on protein synthesis or secretion. Indeed, treatment with cycloheximide or brefeldin A significantly blocked CCPA-mediated exocytosis (Figure 1C). The effect of the latter was particularly pronounced.
FIGURE 1:
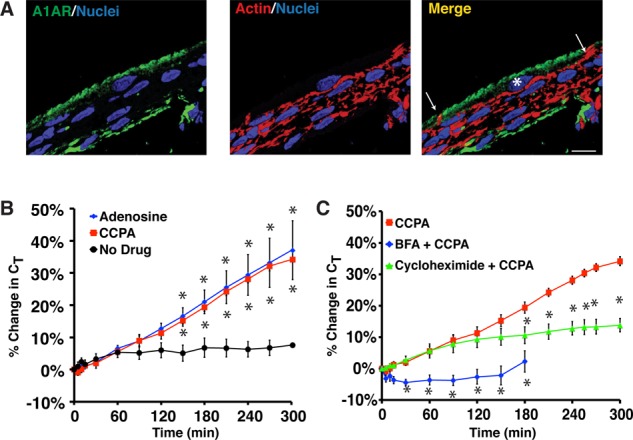
A1AR-stimulated exocytosis is dependent on protein synthesis and secretion. (A) Cryosection of rat bladder epithelium labeled with an A1AR-specific antibody (green), rhodamine–phalloidin to label the actin cytoskeleton (red), and TOPRO-3 (blue) to label the nucleus. Right, merge. An umbrella cell is marked with an asterisk and the white arrows mark the apicolateral borders of the cell. Scale bar, 12 μm. (B) Rabbit uroepithelium was mounted in Ussing chambers and after equilibration left untreated (no drug), or 1 μM adenosine or 500 nM CCPA was added to the mucosal hemichamber. Percentage changes in CT were monitored. (C) Rabbit tissue was left untreated, pretreated with 5 μg/ml brefeldin A (BFA) for 30 min, or pretreated with 100 ng/ml cyclohexamide for 60 min. CCPA (500 nM) was then added to the mucosal hemichamber and CT recorded. Data for CCPA alone are reproduced from B. (B, C) Mean changes in CT ± SEM (n ≥ 4). Significant differences (p < 0.05), relative to no drug in A or CCPA in B, are indicated with an asterisk.
We also assessed whether CCPA triggered apical exocytosis by transactivating the EGFR. We first tested the effect of treating the mucosal surface of the tissue with CRM197, a mutant version of diphtheria toxin that binds selectively and with high affinity to membrane-bound HB-EGF and prevents its cleavage (Uchida et al., 1973). Indeed, we observed that CRM197 almost completely blocked CCPA-mediated apical exocytosis (Figure 2A). Next we pretreated tissue with AG1478, a small-molecule inhibitor of EGFR, which also significantly blocked CCPA-mediated apical exocytosis (Figure 2B). As further evidence that the EGFR was transactivated, we generated lysates from CCPA-treated epithelium and then probed Western blots with an antibody that detects phosphorylation of the EGFR at Tyr-1173. This residue promotes assembly of a MAPK signaling cascade downstream of other GPCRs (Balestreire and Apodaca, 2007). Phosphorylation of Tyr-1173 was maximally stimulated 10 min after continuous treatment with CCPA, decreased at 30 min posttreatment, and returned to control levels by 60 min (Figure 2, C and D). The Tyr-1173 phosphorylation was prevented when the tissue was pretreated with the EGFR inhibitor AG1478 before CCPA treatment (Figure 2, C and D), confirming that phosphorylation of Tyr-1173 likely results from autophosphorylation. Finally, we observed that U0126, an inhibitor of MEK activity, also caused a significant decrease in CCPA-stimulated changes in CT (Figure 2A).
FIGURE 2:
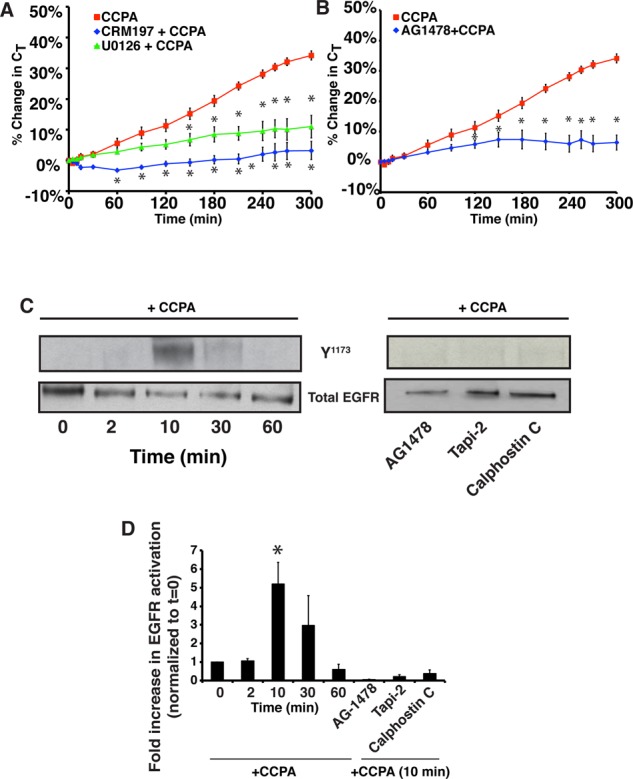
The A1AR transactivates the EGF receptor. (A) The mucosal surface of rabbit uroepithelium was pretreated with 25 ng/ml CRM197 for 25 min or with 10 μM U0126 for 60 min. CCPA (500 nM) was then added to the mucosal hemichamber, and CT was recorded. (B) Rabbit uroepithelium was pretreated with 1 μM AG1478 for 30 min, and then CCPA (500 nM) was added to the mucosal hemichamber and CT was recorded. (A, B) CCPA control data are reproduced from Figure 1B. Mean changes in CT ± SEM (n ≥ 3). Statistically significant differences (p < 0.05), relative to CCPA treatment alone, are marked with an asterisk. (C, D) Rabbit uroepithelium was either left untreated (left) or treated with AG1478 (1 μM) for 25 min, Tapi-2 (15 μM) for 90 min, or calphostin C (500 nM) for 60 min (right). CCPA (500 nM) was then added to the mucosal hemichamber. Left, cells were lysed at the indicated time points. Right, cells were lysed at the 10-min time point. Equal amounts of proteins were resolved by SDS–PAGE and immunoblots probed with a rabbit anti–EGFR-phospho-Y1173 antibody or rabbit anti-EGFR antibody. (D) Quantification of Y1173 phosphorylation. Data (mean ± SEM, n ≥ 3) are reported as fold increase above untreated tissue samples at t = 0. Statistically significant values (p < 0.05) above t = 0 are marked by an asterisk.
Taken together, these data indicate that, like the previously described late-phase response (Balestreire and Apodaca, 2007), A1AR activation stimulates apical exocytosis by promoting transactivation of the EGFR downstream of HB-EGF cleavage, leading to MEK/ERK activation and protein synthesis.
ADAM17 is localized to the apical surface of umbrella cells, where it stimulates CCPA-induced EGFR transactivation and exocytosis
In vivo studies implicate ADAM17 as the physiologically relevant sheddase in HB-EGF–mediated transactivation (Jackson et al., 2003; Sahin et al., 2004). Consistent with these observations, we observed that ADAM17 was localized to small vesicular elements under the apical surface of the rat umbrella cells (Figure 3A), the likely site of HB-EGF cleavage and EGFR transactivation during the late-phase response (Balestreire and Apodaca, 2007). Rat tissues were used in these experiments because our antibody was produced in rabbits. ADAM17 was also detected in the intermediate and basal cell layers of the uroepithelium, as well as in cells in the underlying lamina propria (Figure 3, A and C). The signal for ADAM17 was diminished when the anti-ADAM17 antibody was preincubated with immunizing peptide, confirming the specificity of the antibody (Figure 3B). ADAM17 showed a high degree of colocalization with uroplakin 3a (Manders coefficient of colocalization, 0.92); this is associated with DFVs and the apical surface of umbrella cells (Figure 3C; Apodaca, 2004). Consistent with a role for ADAM17 in DFV trafficking, we observed that the broad-spectrum metalloproteinase inhibitor GM6001 and the ADAM17-selective inhibitor Tapi-2 (Balestreire and Apodaca, 2007; Kveiborg et al., 2011) both significantly inhibited the CCPA-mediated increases in CT (Figure 3D). Moreover, Tapi-2 blocked CCPA-mediated phosphorylation of EGFR Tyr-1173 (Figure 2, C and D).
FIGURE 3:
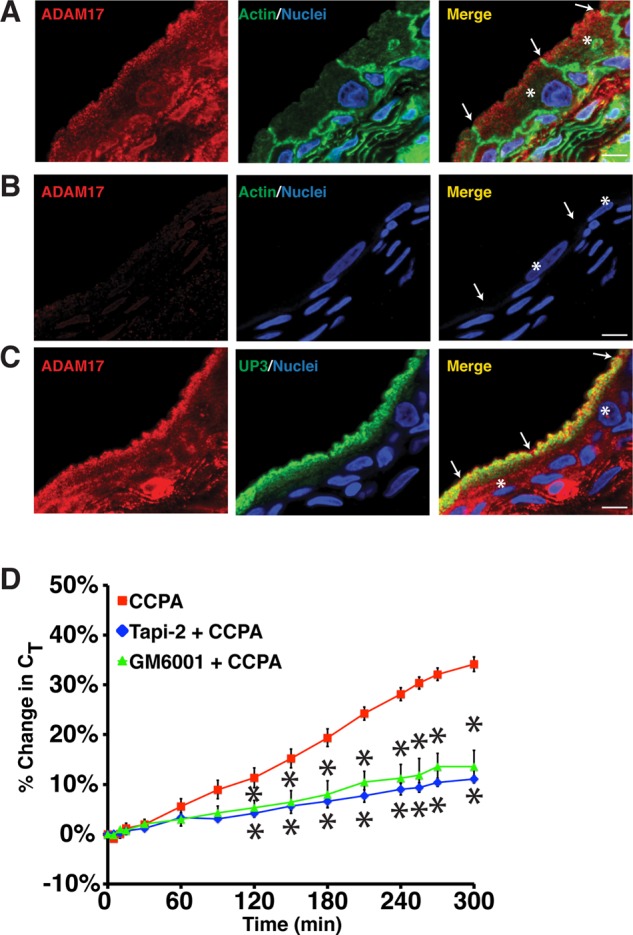
ADAM17 expression in the uroepithelium. (A–C) Cryosections of rat bladder uroepithelium were reacted with antibodies specific for ADAM17 (A), a mixture of antibody and inhibitory peptide (B), or antibodies specific for ADAM17 and uroplakin 3a (C). After incubation with fluorophore-labeled secondary antibodies, the samples were analyzed using confocal microscopy. Where indicated, actin was labeled with phalloidin and nuclei were labeled with TOPRO-3. The umbrella cells are marked with the asterisk in the merged images and the apicolateral junction is indicated by arrowheads; scale bar, 10 μm. (D) Rabbit uroepithelium was pretreated with Tapi-2 (15 μM) or GM6001 (15 μM) for 90 min, CCPA (500 nM) was added, and CT was recorded. Control CCPA data are reproduced from Figure 1B. Data are mean ±SEM (n ≥ 3), and values significantly different (p < 0.05) from CCPA alone are marked with an asterisk.
To provide further evidence that ADAM17 is required for EGFR transactivation, we exploited our previously described in situ viral transduction approach (Khandelwal et al., 2008, 2010), in this case to express ADAM17-specific short hairpin RNAs (shRNAs) or scrambled shRNAs in the rat bladder uroepithelium. We used rat bladders in these studies because the volume capacity of the bladder, and therefore the number of virus particles needed, was relatively small (∼500 μl) compared with the volumes required to fill the rabbit bladder (∼60–100 ml). This technique targets the umbrella cell layer and achieves transduction efficiencies of 70–95% (Khandelwal et al., 2008, 2010). Indeed, we were able to achieve >90% knockdown of ADAM17 expression (Figure 4, A and B). On examining the expression and distribution of ADAM17 in cross sections of uroepithelium, we observed that knockdown of ADAM17 was largely confined to the umbrella cells, and expression of ADAM17 in the underlying cell layers was largely undisturbed (Figure 4C).
FIGURE 4:
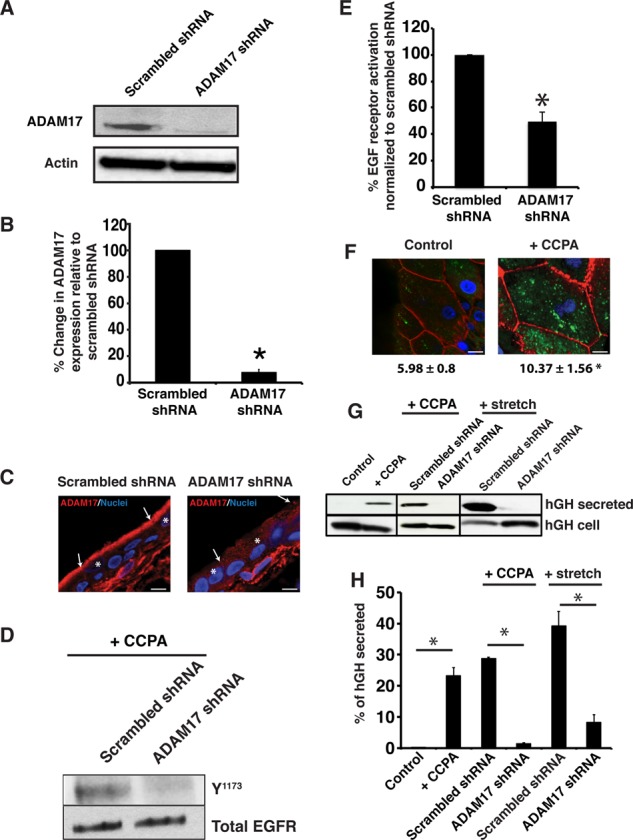
A1AR-stimulated exocytosis is dependent on ADAM17. (A) Uroepithelial lysates obtained from rat tissues transduced in situ with scrambled shRNA or ADAM17 shRNA were resolved by SDS–PAGE and Western blots probed with rabbit-anti ADAM17 antibody (top) or mouse anti–β-actin (bottom). (B) Quantification of ADAM17 expression in rat tissue treated with scrambled shRNA or ADAM17 shRNA. Data are mean ± SEM (n ≥ 4). The means of the two treatment groups are significantly different (p < 0.05). (C) Immunolocalization of ADAM17 expression (red) in rat tissue treated with scrambled or ADAM17-specific shRNAs. Nuclei (blue) are labeled with TOPRO-3. Scale bar, 13 μm. The cell junctions are marked with arrows and the umbrella cells with an asterisk. (D) CCPA (500 nM) was added to rat tissue treated with scrambled or ADAM17 shRNA and total EGFR or receptor phosphorylated at Y1173 was detected by Western blot. (E) Quantification of effects of scrambled or ADAM17-specific shRNA treatment on EGFR activation. Data are mean ± SEM (n = 4). The two treatment groups were significantly different (p < 0.05). (F) WGA-FITC (50 μg/ml) was added to the mucosal hemichamber of untreated rat tissue (control) or that treated with 500 nM CCPA. After 120 min, the tissue was incubated at 4°C with N-acetylglucosamine to remove surface lectin, fixed, and then processed for immunofluorescence. In these whole-mounted tissues, internalized WGA-FITC is shown in green, phalloidin-labeled actin in red, and TOPRO-3-labeled nuclei in blue. Bar, 15 μm. Quantification of the fluorescence intensity of WGA-FITC below the apical membrane. Data are mean ± SEM (n ≥ 8). The two treatment groups were significantly different (p < 0.05). (G) Rat tissues were transduced with hGH alone (control) or transduced with hGH and either scrambled or ADAM17 shRNAs. The excised bladder tissue was mounted in an Ussing stretch chamber and left untreated (control), treated with CCPA (500 nM), or stretched by filling the mucosal hemichamber. The mucosal fluid was collected and concentrated, the tissues were lysed, and hGH was detected in the secreted fraction (1/20 of total) or tissue lysates (1/13 of total) using Western blot. (H) Quantification of effects of ADAM17-specific shRNAs on hGH secretion. Data are mean ± SEM (n ≥ 3). Statistically significant effects (p < 0.05) are indicated with an asterisk.
Next we used ADAM17-specific shRNAs to test whether ADAM17 was required for CCPA-stimulated EGFR transactivation and exocytosis in rat epithelium. Treatment with ADAM17-shRNA, but not scrambled shRNA, decreased EGFR phosphorylation (Figure 4, D and E). We show later in Figure 6, F and G, that the effects of the ADAM17-shRNA are specific, and ADAM17 function is restored when the shRNA is coexpressed with shRNA-resistant variant of wild-type ADAM17. We also attempted to measure changes in CT in the rat bladders treated with apical CCPA; however, we did not observe a response. Because CT is dependent on the rates of membrane addition and removal, we reasoned that one possible explanation was that CCPA stimulated both exocytosis and endocytosis in rat epithelium, thus obviating any change in apparent CT. Indeed, we observed that wheat germ agglutinin–fluorescein isothiocyanate (WGA-FITC), added to the apical hemichamber of mounted rat bladders, was endocytosed in CCPA-treated tissue but much less so in control, untreated tissue (Figure 4F). To circumvent the effects of endocytosis, we instead measured release of exogenously expressed human growth hormone (hGH), which we and others previously showed is packaged into DFVs and released from the luminal surface of the bladder into the urinary space (Kerr et al., 1998; Khandelwal et al., 2008). Because of rapid dilution, there is little secreted hGH that is endocytosed. Compared to control bladders, CCPA stimulated a large increase in the mucosal release of hGH from the tissue (Figure 4, G and H). Moreover, expression of ADAM-17-specific shRNA, but not scrambled shRNA, caused a large inhibition (>90%) in mucosal hGH release (Figure 4, G and H).
FIGURE 6:
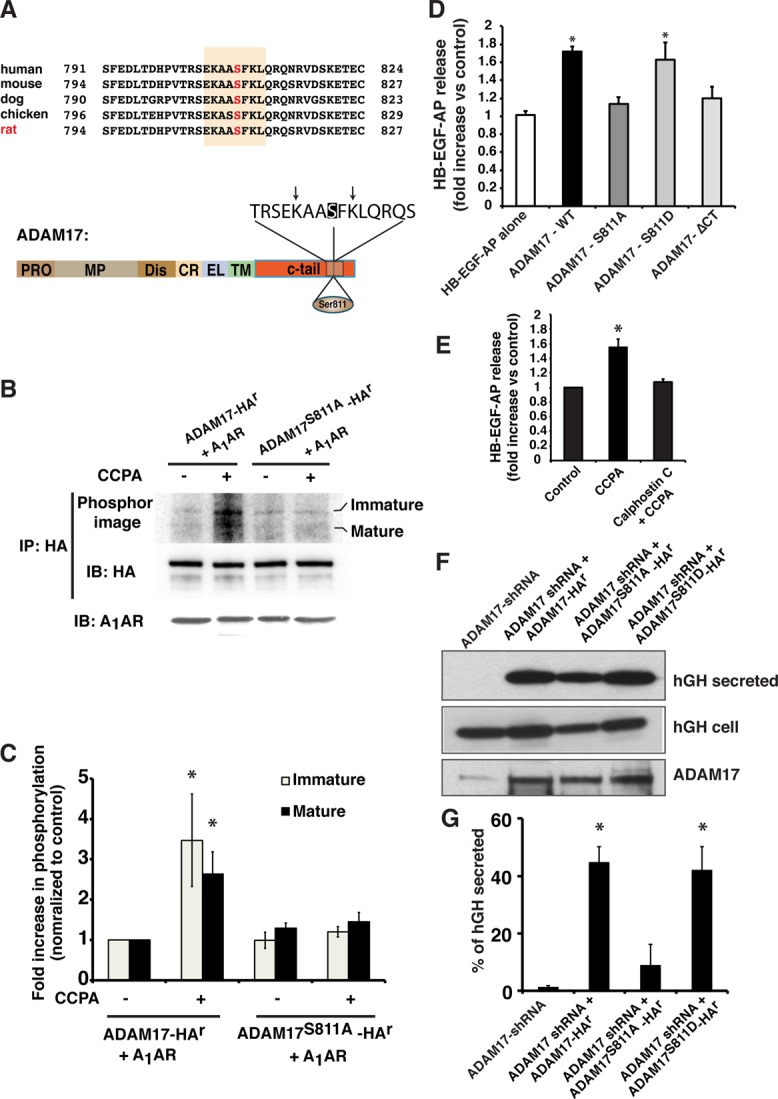
Effect of Ser-811 phosphorylation on the function of ADAM17. (A) Top, alignment of the C-termini of ADAM17 from different species. Numbers indicate the amino acids involved. The canonical PKC phosphorylation site is shaded, and the position of the conserved Ser residue (Ser-811 in rat) is indicated in red. Bottom, domain structure of ADAM17. CR, cysteine-rich domain; C-tail, cytoplasmic domain; Dis, disintegrin domain; EL, EGF-like; MP, metalloproteinase domain; Pro, propeptide; TM, transmembrane domain. The consensus PKC phosphorylation motif is shown in an expanded view. Ser-811 is shaded, and the critical, flanking basic residues at the −3 and + 2 positions are marked with arrows. (B) HEK-293FT cells expressing the A1AR in combination with ADAM17-HAr or ADAM17S811A-HAr were labeled with 32P-orthophosphate and then left untreated or treated with CCPA (500 nM). HA-tagged ADAM17 constructs were immunoprecipitated and Western blots probed with an anti–HA-HRP- conjugated secondary antibody to detect total ADAM17 (middle) and subsequently exposed to a PhosphorImager screen to detect 32P-labeled ADAM17 (top). Total A1AR was detected by Western blot (bottom). (C) Data (mean ± SEM; n = 3) were quantified, and values significantly different from ADAM17-HAr + CCPA are indicated with an asterisk. (D) HEK-293FT cells were cotransfected with the A1AR, HB-EGF-AP, and ADAM17-HAr, ADAM17S811A-HAr, or ADAM17S811D-HAr. Fold stimulation of HB-EGF-AP release in CCPA-treated cells vs. control, untreated cells. Data are mean ± SEM, n = 5. Data significantly different from HB-EGF-AP alone are indicated with an asterisk. (E) HEK-293FT cells were cotransfected with the A1AR, HB-EGF-AP, and ADAM17-HAr. Cells were pretreated with calphostin C (500 nM) for 60 min, and the fold stimulation of HB-EGF-AP release in CCPA-treated cells vs. control, untreated cells is reported. Data are mean ± SEM, n = 7. Values significantly different from the control, as assessed by ANOVA, are indicated (*p < 0.05). (F) Rat tissues were transduced in situ with hGH and ADAM17 shRNA alone or in combination with ADAM17-HAr, ADAM17S811A-HAr, or ADAM17S811D-HAr. The excised bladder tissue was mounted in an Ussing stretch chamber, and CCPA (500 nM) was added to the mucosal hemichamber. After 60 min, the mucosal fluid was collected and concentrated, the tissues were lysed, and hGH detected using Western blot. (G) Quantification of hGH secretion. Data are mean ± SEM (n = 5). Values that are statistically different from ADAM17 shRNA alone (p < 0.05) are indicated with an asterisk.
Finally, because stretch-mediated, late-phase exocytosis is dependent on EGFR transactivation and sensitive to metalloproteinase inhibitors (Balestreire and Apodaca, 2007), we determined whether ADAM17 also played a role in stretch-mediated exocytosis. We observed that shRNA-mediated ADAM17 knockdown significantly reduced the stretch-induced apical release of hGH by ∼80% compared with tissues transduced with scrambled shRNA (Figure 4, G and H). Taken together, our results provide strong evidence that in rat tissues, ADAM17 is critical for CCPA- and stretch-induced EGFR transactivation and exocytosis.
Gi, PLC, and PKC act upstream of ADAM17 and HB-EGF to promote A1AR-mediated EGFR transactivation
We next addressed how ADAM17 activity is coupled to A1AR activation. Previous studies showed that A1AR signals through Giα to inhibit the activity of adenylyl cyclase, whereas the βγ subunits of Gi increase the activity of phospholipase C-β (PLCβ), which hydrolyzes phosphatidylinositol 4,5-bisphosphate to generate inositol trisphosphate (IP3) and diacylgycerol (Freund et al., 1994; Bucheimer and Linden, 2004; Chang et al., 2008). The latter stimulates the activity of PKC, a well-known regulatory kinase that was previously implicated in ADAM17 activation (Dang et al., 2011; Kveiborg et al., 2011; Lemjabbar-Alaoui et al., 2011). We found that pertussis toxin–mediated inhibition of Gi or inhibition of Gβγ subunit activity by the inhibitor M119K (Kirui et al., 2010) significantly impaired CCPA-mediated apical exocytosis in rabbit bladder umbrella cells (Figure 5A). Furthermore, the PLC-selective antagonist U73122 caused marked inhibition of CCPA-induced changes in CT (Figure 5A). Thus ADAM17 activation downstream of A1AR likely occurs by way of a classical Gi-stimulated signaling cascade involving Gβγ and PLCβ.
FIGURE 5:
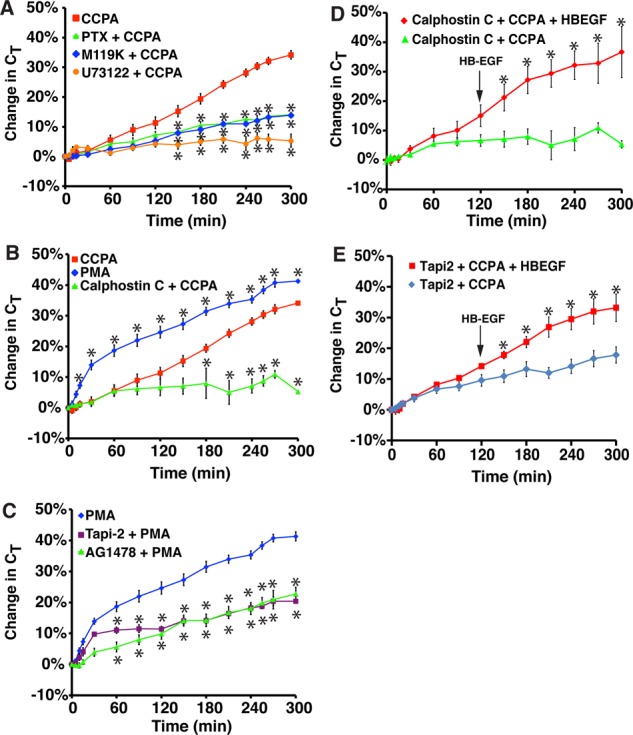
Role for Gi, Gβγ, PLC, and PKC in A1AR-stimulated exocytosis. (A) Rabbit tissue was left untreated or pretreated with 100 ng/ml pertussis toxin (PTX) for 90 min, 10 μM M119K for 60 min, or 10 μM U73122 for 60 min. CCPA (500 nM) was added to the mucosal hemichamber, and CT was recorded. (B) Rabbit tissue was left untreated or pretreated with calphostin C (500 nM) for 90 min. Subsequently, tissue was treated with CCPA (500 nM) or PMA (10 nM). (C) Rabbit tissue was pretreated with 15 μM Tapi-2 for 90 min or 1 μM AG1478 for 30 min and then treated with 10 nM PMA. The data for PMA treatment alone were reproduced from B. (A–C) Data for CCPA treatment alone were reproduced from Figure 1B. (D, E) Rabbit tissue was pretreated with calphostin C (D) or Tapi-2 (E) for 90 min, and then at t = 0, CCPA was added to the mucosal hemichamber. After 120 min, HG-EGF (1 nM) was added to the mucosal hemichamber (indicated with an arrow), and the tissue was incubated for additional 180 min. In D, data for calphostin C + CCPA are reproduced from B. (A–E) Data are presented as mean ± SEM (in A–C, n ≥ 3; in D and E, n ≥ 6). In A–C, values that are significantly different from CCPA alone (p < 0.05) are marked with an asterisk. In D and E, values that are significantly different from calphostin C + CCPA or Tapi-2 + CCPA (p < 0.05) are marked with an asterisk.
Next we examined whether PKC is important for A1AR-dependent EGFR transactivation and exocytosis. Strikingly, the PKC inhibitor calphostin C caused a marked decrease in CCPA-stimulated changes in CT and EGFR transactivation (Figures 2, C and D, and 5B). In contrast, treatment with PMA, an activator of classical PKCs (Nishizuka, 1992), caused robust stimulation of exocytosis in the absence of CCPA (Figure 5B). Of interest, the kinetics of PMA-mediated exocytosis were faster than those mediated by CCPA, particularly during the first 30 min, and then appeared to increase at a similar rate to CCPA-treated tissue. This may indicate that PKC stimulates not only late-phase-like responses, but perhaps early-phase ones as well. To confirm that PMA acted by way of ADAM17, we treated the tissue with Tapi-2, which significantly inhibited PMA-mediated apical exocytosis (Figure 5C). We also observed that AG1478 impaired PMA-mediated exocytosis (Figure 5C). Our data pointed to the possibility that PKC acted upstream of ADAM17, which acted before HB-EGF cleavage. We reasoned, therefore, that the calphostin C–or TAPI-2–mediated inhibition of CT should be relieved by addition of HB-EGF. To test this possibility, we pretreated cells with calphostin C or TAPI-2, added CCPA at t = 0, and then added HB-EGF 2 h later (Figure 5, D and E). In both cases, we observed that HB-EGF significantly stimulated exocytosis, even in the presence of the PKC or ADAM17 inhibitor. Together our data are consistent with a model in which A1AR-mediated transactivation requires Gi, Gβγ, PLC, PKC, and ADAM17, and these effectors likely act upstream of HB-EGF release and EGFR transactivation.
CCPA-stimulated HB-EGF shedding and exocytosis are dependent on phosphorylation of ADAM17 Ser-811
To explore how PKC might act to stimulate ADAM17 activity, we compared the amino acid sequences from multiple vertebrate species using the proteomics tool Scansite and identified a conserved, canonical PKC phosphorylation motif (X-R/K-X-X-S/T-X-R/K-X; Pearson and Kemp, 1991; Nishikawa et al., 1997) in the cytoplasmic tail of the protein centered at Ser-811 (equivalent to Ser-808 in the human protein; Figure 6A). To our knowledge, a functional role for this residue was not explored previously. We first confirmed that ADAM17 was phosphorylated in response to CCPA by expressing the A1AR in combination with epitope-tagged ADAM17-HAr (this variant of ADAM17 is resistant to shRNA; see Figure 6F) in 32P-orthophosphate–labeled HEK cells (Figure 6B). HEK cells were used in these assays because these experiments were difficult to perform in whole tissues. On addition of CCPA, we observed that phosphorylation of both the “immature” proform of ADAM17-HAr (∼120 kDa) and the “mature” cleaved form of the enzyme (∼93 kDa) was stimulated approximately threefold (Figure 6, B and C). In contrast, CCPA had no significant effect when wild-type ADAM17-HAr was substituted with a nonphosphorylatable mutant of ADAM17 in which Ser-811 was changed to an Ala residue (ADAM17S811A-HAr; Figure 6, B and C). These results indicate that in response to A1AR activation, Ser-811 may be a major site of ADAM17 phosphorylation.
Because the physiological role of ADAM17 phosphorylation is a matter of some controversy (Reddy et al., 2000; Horiuchi et al., 2007; Le Gall et al., 2010; Dang et al., 2013), we next determined whether phosphorylation of Ser-811 was biologically relevant. First, we measured shedding of a chimeric protein in which alkaline phosphatase (AP) was fused to the extracellular domain of HB-EGF (HB-EGF-AP; Sahin et al., 2004; Uttarwar et al., 2011), an ADAM17 substrate that was coexpressed in HEK cells along with the A1AR and ADAM17-HAr (Figure 6D). In response to CCPA, we observed a significant ∼70% increase in HB-EGF-AP release. In contrast, when we substituted ADAM17-HAr with ADAM17S811A-HAr, there was no significant increase in CCPA-stimulated HB-EGF-AP release. We also tested a mutant in which Ser-811 was mutated to an Asp residue (ADAM17S811D-HAr). In the absence of CCPA, this variant did not significantly affect constitutive HG-EGF-AP release. However, ADAM17S811D-HAr retained the ability to promote HB-EGF-AP release in response to CCPA (Figure 6D), indicating that under these conditions it acted as a “phosphomimetic.” In addition, we determined whether HB-EGF-AP release was stimulated in cells expressing a mutant of ADAM17 that lacked all but two of its cytoplasmic amino acids (ADAM17-∆CT), This construct was previously used to show that activation of ADAM17-dependent shedding occurred independent of its cytoplasmic domain (Le Gall et al., 2010; Hall and Blobel, 2012). However, this mutant was unable to stimulate HB-EGF-AP release in CCPA-treated cells (Figure 6D). In addition, we assessed whether inhibition of PKC affected HB-EGF release. As predicted, calphostin C caused significant inhibition of CCPA-stimulated HB-EGF-AP release (Figure 6E).
We also tested whether phosphorylation of Ser-811 might be critical for CCPA-induced exocytosis in umbrella cells. We silenced endogenous ADAM17 by transducing rat bladders in situ with adenovirus encoding shRNA and then expressed hGH in conjunction with shRNA-resistant ADAM17-HAr, ADAM17S811A-HAr, or ADAM17S811D-HAr. The expression levels of the three ADAM constructs were approximately equal (e.g., see Figure 6F) and similar to the expression levels of the endogenous protein (∼50–90% of endogenous levels). The exogenous expression of ADAM17-HAr restored CCPA-induced hGH secretion in the shRNA background (Figure 6, F and G). In contrast, expression of ADAM17S811A-HAr caused a significant, ∼80% reduction in hGH release, again indicating a critical role for this amino acid in A1AR-mediated activation of ADAM17 (Figure 6, F and G). Finally, ADAM17S811D-HAr was able to rescue shRNA-mediated inhibition of hGH secretion (Figure 6, F and G).
In sum, our data indicate that activation of the A1AR stimulates phosphorylation of ADAM17 Ser-811, and this phosphorylation event is likely necessary for A1AR-induced HB-EGF cleavage and exocytosis in umbrella cells.
DISCUSSION
ADAM17 is expressed in a variety of tissues, including heart, lungs, brain, kidney, skeletal muscles, and the bladder (Gooz, 2010), and there exists a large body of evidence that implicates it in normal biological phenomena (e.g., migration, adhesion, differentiation), as well as in numerous pathologies (e.g., inflammation, multiple sclerosis, diabetes, and kidney disorders; Plumb et al., 2006; Shah and Catt, 2006; Serino et al., 2007; Arribas and Esselens, 2009). Nonetheless, we still lack sufficient insight into the physiological cues that stimulate ADAM17 activity, the upstream signaling pathways that promote ADAM17 activation, or the mechanisms that control ADAM17-dependent sheddase activity. Our studies show that 1) like stretch, the A1AR stimulates umbrella cell exocytosis by way of EGFR transactivation and ADAM17 is the physiologically relevant proteinase; 2) an A1AR → Gαi → Gβγ → PLC → PKC signaling cascade likely acts upstream of ADAM17 to promote HB-EGF cleavage; and 3) phosphorylation of Ser-811 in the cytoplasmic domain of ADAM17 appears to be required for A1AR-stimulated HB-EGF shedding and exocytosis.
A1AR-mediated exocytosis requires ADAM17 and EGFR transactivation
Similar to the effects of maximal stretch (Balestreire and Apodaca, 2007), we found that activation of the A1AR stimulated a slow and gradual increase in apical exocytosis, which required metalloproteinase-dependent HB-EGF cleavage, EGFR transactivation, protein synthesis, and secretion. Although these two stimuli appear dissimilar at first blush, adenosine is released from the uroepithelium in response to maximal stretch (Yu et al., 2006; Prakasam et al., 2012) and can function as a stress hormone in some settings (Fredholm et al., 2001; Hasko et al., 2008; Wilson and Mustafa, 2009). Thus EGFR transactivation may be a common pathway the uroepithelium uses to cope with stressful stimuli. A critical step in transactivation is the cleavage of EGFR substrates by a metalloproteinase, whose identity in umbrella cells was previously unknown. We focused our studies on ADAM17 in part because it is the major sheddase for several EGFR ligands, including HB-EGF (Sahin et al., 2004; Blobel, 2005; Hassemer et al., 2010). In addition, we found that in umbrella cells, ADAM17 was localized to uroplakin 3a–positive DFVs, the major apically directed vesicle population in these cells. Of importance, these vesicles position ADAM17 at or near the apical surface of the umbrella cells, which we previously showed is a primary site for EGFR- and HB-EGF-dependent receptor transactivation (Balestreire and Apodaca, 2007). Furthermore, our use of adenovirally mediated expression of shRNAs allowed us to significantly decrease ADAM17 expression in the uroepithelium, which caused >90% decrease in CCPA-induced hGH release. Although we cannot rule out a role for other ADAMs in A1AR-mediated exocytosis, the knockdown of ADAM17 in rat umbrella cells was sufficient to block the majority of A1AR- or stretch-stimulated exocytosis in rat uroepithelium.
A Gi → Gβγ → PLC → DAG → PKC pathway promotes ADAM17 activation
Although there are previous reports that adenosine can transactivate the EGFR, these studies did not identify the metalloproteinase or upstream signaling pathways involved (Xie et al., 2009; Williams-Pritchard et al., 2011). Furthermore, many studies of ADAM17 function employ nonphysiological stimuli, such as PMA, and as such, the upstream signaling events that lead to ADAM17 activation are not always well understood. Work has shown roles for ERK, p38 MAPK, and several classical PKC isoforms in ADAM17 function (Soond et al., 2005; Bell and Gooz, 2010; Killock and Ivetic, 2010; Dang et al., 2011, 2013; Hall and Blobel, 2012). For example, in lung cells, PKCε-mediated ADAM17 activation was shown to be necessary for premalignant changes after exposure to tobacco smoke (Lemjabbar-Alaoui et al., 2011), whereas in glioblastoma cells, migration was initiated by PKCα-mediated activation and membrane translocation of ADAM10 and the subsequent cleavage of N-cadherin (Kohutek et al., 2009).
Our studies showed that A1AR-mediated apical exocytosis required Gi and Gβγ, confirming that the activation of ADAM17 occurs downstream of an A1AR, Gi-protein signaling event. Furthermore, we observed a requirement for PLC, which is known to generate IP3 and DAG, a well-known activator of PKC. Finally, we observed a requirement for PKC in apical exocytosis, noting that the selective PKC inhibitor calphostin C inhibited adenosine-stimulated exocytosis, whereas PMA stimulated it. We do not know whether there are additional effectors in the pathway; however, our results are consistent with the hypothesis that a Gi → Gβγ → PLC → DAG → PKC cascade promotes ADAM17 activation.
PKC may promote HB-EGF cleavage and apical exocytosis by phosphorylation of Ser-811 in the cytoplasmic domain of ADAM17
An important but unresolved question concerns the mechanism(s) by which upstream stimuli promote ADAM17-dependent function. One possible mechanism is phosphorylation, and several cytoplasmic targets have been described, including Tyr-702, Ser-791, Ser-829, and Thr-735 (Fan et al., 2003; Soond et al., 2005; Niu et al., 2013). However, other reports show that ADAM17 lacking its cytoplasmic domain can still trigger stimulus-evoked shedding and EGFR transactivation (Doedens and Black, 2000; Black et al., 2003; Doedens et al., 2003; Le Gall et al., 2010), a finding seemingly at odds with any role for phosphorylation in ADAM17 activation.
We observed that ADAM17 contains a canonical PKC phosphorylation site (which includes Ser-811) that was phosphorylated in response to A1AR activation. Although we cannot completely rule out that PKC acts upstream of a different kinase, as reported for ERK-dependent phosphorylation of Thr-735 (Diaz-Rodriguez et al., 2002), the most straightforward interpretation of our data is that PKC phosphorylates the residue directly. We further showed that the nonphosphorylatable mutant ADAM17S811A blocked CCPA-induced HB-EGF shedding and hGH secretion. In contrast, ADAM17S811D (which also cannot be phosphorylated at Ser-811) was able to promote CCPA-stimulated HB-EGF cleavage and hGH release. However, its inability to stimulate constitutive HB-EGF cleavage likely indicates that phosphorylation of Ser-811 is necessary but not sufficient to promote A1AR-induced events. Finally, we observed that a “tail-minus” construct was unable to stimulate CCPA-induced HB-EGF-AP release. Thus our results are inconsistent with the hypothesis that ADAM17 functions independently of its cytoplasmic domain and phosphorylation. However, it is possible that the mechanism(s) of ADAM17 activation may depend on the stimulus, cell type, or growth conditions. Furthermore, some stimuli may primarily work by directly affecting ADAM17 activity, whereas other stimuli may work through other mechanisms that affect the traffic and/or “activation” of ADAM17 ligands (e.g., Dang et al., 2013).
If phosphorylation of Ser-811 is physiologically relevant, then how might it act? One possibility is that phosphorylation of this residue alters the conformation of ADAM17, thus promoting its activity. This could be a direct effect on ADAM17 or might be indirect; for example, phosphorylation could act by modifying the association of this proteinase with TIMP3, which may modulate ADAM17 activity (Amour et al., 1998; Kwak et al., 2009; Xu et al., 2012). An additional possibility is that phosphorylation or Ser-811 forms a docking site for other proteins to bind and trigger ADAM17 activation or the association of ADAM17 with its substrates. Potential interacting proteins include MAD-2, a component of mitotic spindle assembly (Nelson et al., 1999), the protein tyrosine phosphatase-H1 (PTPH-1), and SAP9. These last two interact with the COOH terminal of ADAM17 and negatively regulate its function (Zheng et al., 2002; Peiretti et al., 2003). Other potential interacting partners include Eve-1 (Tanaka et al., 2004) and the N-arginine dibasic convertase (nardilysin), which interacts with both HB-EGF and ADAM17 and regulates cleavage of the latter (Nishi et al., 2006). Obviously, more work is needed to understand the mechanisms by which ADAM17 activity is regulated.
In summary, we propose the following model for the role of ADAM17 and the role of Ser-811 phosphorylation in A1AR-stimulated exocytosis in umbrella cells. In the quiescent state, before adenosine stimulation (or stretch, in the case of rat umbrella cells), ADAM17 at the apical surface may be kept in its inactive, dimeric state by the inhibitory effects of TIMP3 (Figure 7A; Xu et al., 2012). As a result of its increased production (or decreased turnover), adenosine binds to the A1AR, triggering a Gi → Gβγ → PLC → PKC pathway, which leads to phosphorylation at Ser-811 in the cytoplasmic domain of ADAM17 (Figure 7B). This leads to activation of ADAM17, possibly by altering its conformation and/or its association with regulatory proteins (e.g., TIMP3) and/or its substrates. Although not shown, signaling pathways downstream of the A1AR are also likely to function by regulating other processes (e.g., “activation” of HB-EGF). This could explain why ADAM17S811D-HAr was unable to stimulate HB-EGF release in the absence of CCPA stimulation. Finally, in its activated state, ADAM17 cleaves and releases HB-EGF, which binds to the EGFR, promoting autophosphorylation of EGFR Y1173, ultimately leading to downstream ERK1/2 activation, protein synthesis, and exocytosis (Figure 7B).
FIGURE 7:
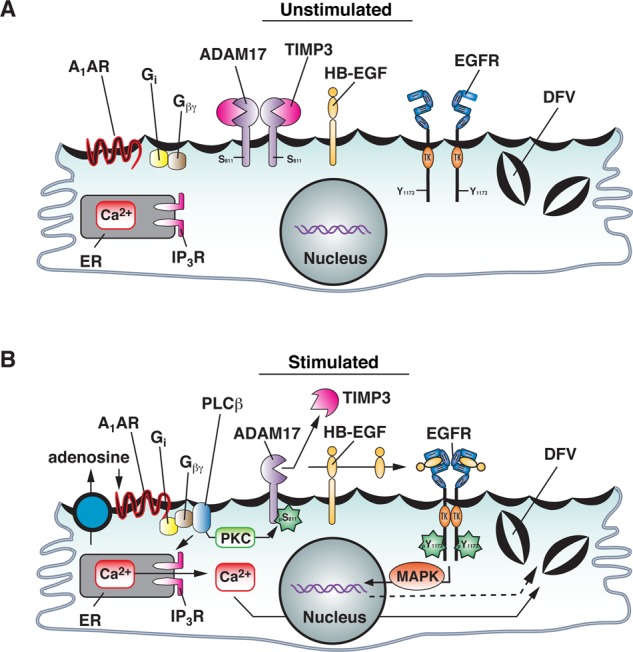
Model for function of ADAM17 Ser-811 phosphorylation in A1AR-stimulated EGFR transactivation and exocytosis. See the text for description.
MATERIALS AND METHODS
Reagents and antibodies
Unless otherwise specified, all chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and were of reagent grade or better. Adenosine was freshly prepared and dissolved in Krebs buffer (110 mM NaCl, 5.8 mM KCl, 25 mM NaHCO3, 1.2 mM KH2PO4, 2.0 mM CaCl2, 1.2 mM MgSO4, 11.1 mM glucose, pH 7.4). The following stock solutions were prepared in dimethyl sulfoxide: AG1478 (10 mM), calphostin C (250 μM), CCPA (10 mM), GM6001 (15 mM), SB203580 (1 mM), Tapi-2 (15 mM; Tocris, Bristol, United Kingdom), and U0126 (10 mM). The following stocks were made in molecular biology–grade water: pertussis toxin (100 μg/ml) and M119K (10 mM; purchased from the National Cancer Institute, Bethesda, MD). A 10 mM stock of PMA was prepared in ethanol. CRM197 (25 ng/ml) was dissolved directly in Krebs buffer. WGA-FITC was purchased from Vector Labs (Burlingame, CA) and used at a final concentration of 50 μg/ml. Beuthanasia-D was purchased from Butler Schein (Dublin, OH). Lidocaine (LMX4) and isoflurane were purchased from Webster Veterinary (Webster, NY). The polyclonal anti-A1AR rabbit antibody was obtained from Abcam (ab82477; Cambridge, MA), anti-ADAM17 rabbit polyclonal antibody from EMD-Millipore (Billerica, MA), anti-EGFR and anti–EGFR-phospho-Y-1173 rabbit polyclonal antibodies from Cell Signaling Technology (Danvers, MA), anti-hemagglutinin (HA) rabbit polyclonal antibody from Covance (Princeton, NJ), and anti–HA-horseradish peroxidase (HRP) rabbit monoclonal antibody from Roche (Mannheim, Germany); fluorophore- or HRP-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA), and tetramethylrhodamine isothiocyanate–labeled phalloidin and TO-PRO3 were from Molecular Probes/Invitrogen (Grand Island, NY). Mouse monoclonal anti–uroplakin 3a antibody was described previously (Truschel et al., 1999).
Animals
Animals used in this study were female New Zealand white rabbits (3–4 kg; Covance) and female Sprague Dawley rats (250–300 g; Harlan Laboratories, Indianapolis, IN). Rabbits were killed by intravenous injection of 300 mg of Buthensia D into the ear vein after the area was numbed using topical lidocaine ointment. After death, the bladders were rapidly excised and processed as described later. Rats were sedated by inhalation of isoflurane and kept under sedation during the adenoviral transduction procedure by constant inhalation of isoflurane. At the end of the procedure, the rats were allowed to revive. At 24 h after infection, the rats were killed by inhalation of 100% CO2, a thoracotomy was performed, and the bladder was excised. All animal studies were carried out with the approval of the University of Pittsburgh Animal Care and Use Committee.
Cell culture
HEK293FT cells were obtained from Invitrogen. A HEK cell variant that has flattened morphology and increased viral transduction, HEK293A cells (Invitrogen), was used to prepare ADAM17- or scrambled-shRNA viruses. The cells were grown in DMEM purchased from Corning Cellgro (Corning, NY) supplemented with 10% (vol/vol) fetal bovine serum (GE Healthcare Life Sciences, Pittsburgh, PA), 1% (vol/vol) MEM nonessential amino acids (Life Technologies, Grand Island, NY), 2 mM glutamate (Sigma-Aldrich), and 1% (vol/vol) penicillin/streptomycin (Lonza, Walkersville, MD). Cre8 cells were grown in DMEM purchased from Sigma-Aldrich and supplemented with 10% (vol/vol) defined fetal bovine serum and 1% (vol/vol) penicillin/streptomycin. HEK293 cells were grown in DMEM supplemented with 10% (vol/vol) fetal bovine serum and 1% (vol/vol) penicillin/streptomycin. When producing viruses, cells were grown in their respective media but without penicillin/streptomycin.
Immunofluorescence labeling, image acquisition, and colocalization analysis
Bladder tissue was fixed and processed as described previously (Truschel et al., 1999; Khandelwal et al., 2008). Briefly, tissues fixed with 4% PFA fixative (4% paraformaldehyde in 100 mM sodium cacodylate buffer, pH 7.4) were embedded in Optimal Cutting Temperature (OCT; Sakura Finetek, Torrance, CA) medium and frozen and stored at −80°C. Frozen sections, 4 μm in thickness, were obtained using a Leica CM1950 cryostat and adhered to Fisher brand Superfrost slides. The sections were washed three times for 5 min each with phosphate-buffered saline (PBS), and the fixation was stopped with quench buffer (20 mM glycine, 75 mM NH4Cl, dissolved in PBS) for 10 min. When staining for ADAM17, the sections were quenched for 7 min and subsequently treated with quench buffer supplemented with 0.05% (wt/vol) SDS for an additional 3 min. After quenching, the tissue was then washed with PBS and transferred to block buffer (7% [wt/vol] fish gelatin, 0.25% saponin [vol/vol], and 0.05% NaN3 [wt/vol] made in PBS) for 1 h at room temperature. A 1:200 dilution of the antibodies was made in the block buffer, applied to the sections, and incubated at room temperature for either 2 h (in the case of ADAM17 antibody) or overnight at 4°C for the A1AR or UP3a antibodies. The primary antibody was removed by three 5-min washes with PBS and detected using Alexa 488– or Cy3–labeled secondary antibodies diluted in block buffer. Additional washes with PBS were performed. Actin and nuclei were stained with either rhodamine or fluorescein phalloidin and TO-PRO3, respectively. After staining, the samples were postfixed for 10 min with 4% PFA fixative and then mounted in medium containing 1% (wt/vol) phenylenediamine, 90% (vol/vol) glycerol, and 20 mM Tris, pH 8.0.
Images were captured using a 63×/1.2 numerical aperture glycerol objective and the appropriate laser lines of a Leica TCS SP5 CW-STED confocal microscope (in normal confocal mode). The photomultipliers were set at 900–1200 V, and images were collected using an average of six line scans. Serial 0.25-μm Z-sections were acquired. The images were imported into Volocity 4-D software (PerkinElmer, Waltham, MA) and, after image reconstruction and contrast correction, exported as TIFF files. Composite images were prepared in Illustrator CS5 (Adobe, San Jose, CA). Colocalization analysis and measurements of fluorescence intensity were performed as described previously, using a fixed threshold of 40 (Khandelwal et al., 2008, 2010).
Mounting rabbit uroepithelium or rat bladders in Ussing stretch chambers and measurement of CT
Isolation of rabbit uroepithelium from the underlying muscle layers and mounting in Ussing stretch chambers was performed as described earlier (Wang et al., 2003a). Briefly, rabbit bladders were excised, slit open vertically along one of the lateral veins, and spread on a custom-made Teflon rack with the uroepithelium facing down. The muscles were carefully removed with the pair of tweezers and sharp scissors. The remaining tissue containing the intact uroepithelium was mounted on the pins at the outer edges of a plastic ring with an opening of 2 cm2. The rings were locked between two Ussing stretch hemichambers, which were clamped into position on a Teflon base. Warm Krebs buffer was simultaneously added to the mucosal (apical facing) and serosal (muscle facing) chambers. The tissue was gassed with 95% air/5% CO2 during a 30- to 45-min equilibration period and treated with the indicated drug added to the mucosal and serosal hemichambers bathing the tissue. However, CCPA, CRM197, or PMA was added only to the mucosal hemichamber. The CT and transepithelial resistance were measured as described previously for a period up to 300 min (Wang et al., 2003a).
Rat bladders were mounted in Ussing stretch chambers as described earlier (Khandelwal et al., 2008, 2010). Briefly, the bladders were excised, cut open along one of the lateral veins and then carefully spread and pinned out on rubber dissection mats. The dissected bladders were then mounted on the pins of a plastic ring with an opening of 0.75 cm2 and the rings clamped between two Ussing stretch hemichambers. The chambers were filled with Krebs buffer and equilibrated. To stretch the tissue, buffer was added to the mucosal hemichamber via Luer ports at a rate of 35 μl/min using a syringe pump (New Era Pump Systems, Farmingdale, NY). Once the chamber was filled, an additional 250 μl was then pumped into the chamber to stretch the tissue.
Preparation of cell lysates and Western blot analysis
To prepare lysates, bladder tissue was placed on a rubber dissection mat with the mucosal surface facing up. The tissue was held in place by pinning it at the four corners using 20-gauge ¾-inch needles (BD Biosciences). An aliquot (35–50 μl) of SDS lysis buffer (50 mM triethanolamine, pH 8.6, 100 mM NaCl, 5 mM EDTA, 0.2% [wt/vol] NaN3, 0.5% [wt/vol] SDS) containing a protease and phosphatase inhibitor cocktail (Cell Signaling Technologies, Boston, MA), as well as 10 mM 1,10-phenanthroline, was added to the mucosal surface and the outer uroepithelium recovered by gently scraping the cells using a rubber cell scraper (Sarstedt, Newton, NC). The cell lysate was transferred to a 1.5-ml Eppendorf tube, vortex shaken at 4°C for 10 min using a model 5432 vortex mixer (Eppendorf, Hauppauge, NY), and centrifuged at 13,000 rpm for 10 min at 4°C in a table-top model 5415D microcentrifuge (Eppendorf). The clear supernatant was collected, flash frozen, and stored at −80°C. Before use, the protein concentration was quantified using the bicinchoninic acid assay (Pierce, Rockford, IL). Equal amounts of protein from the bladder lysates were resolved by SDS–PAGE on 4–15% polyacrylamide gradient gels (Bio-Rad, Hercules, CA) at 200 V and constant current for 30 min. For ADAM17 and hGH, proteins were transferred to Immobilon P membranes (Millipore) at 375 mA constant current using a 100 mM 3-[cyclohexylamino]-1-propanesulfonic acid (CAPS), pH 11.0, transfer buffer. In the case of the EGFR, proteins were transferred to nitrocellulose membranes (GE Healthcare Life Sciences) at 375 mA constant current using a Tris-glycine running buffer (25 mM Tris, 190 mM glycine) containing 0.01% (wt/vol) SDS for 2 h. In either case, the membrane was blocked for 30 min with 5% (wt/vol) nonfat milk made in TBST buffer (Tris-buffered saline + 0.1% Tween 20). After the blocking step, the membrane was incubated with primary antibodies overnight at 4°C. After several washes with TBST buffer, the membrane was incubated with goat anti-rabbit-HRP or goat anti-mouse-HRP secondary antibodies for 1 h at room temperature and washed with TBST buffer. The bands were detected by incubating the membrane with enhanced chemiluminescence (ECL) solution for 2 min (Pierce), followed by film capture on Carestream Kodak Biomax films (Carestream, Rochester, NY). Data were quantified using QuantityOne quantification software (Bio-Rad)
Generation of adenoviruses encoding ADAM17 shRNA
The iRNAi software (Nucleobytes.com) was used to search the Rat ADAM17 cDNA (PubMed accession number NM_020306) for optimal targets containing the sequence AA(N19). Four shRNA sequences were selected. The top and the bottom strands were individually synthesized (IDT, Coralville, IA) and annealed by mixing the two strands in equal amounts, heating to 94°C, and cooling gradually to room temperature. The annealed shRNA sequences were then ligated into the linearized pU6/ENTR vector (Life Technologies). The ability of the different shRNA sequences to silence ADAM17 was determined by cotransfecting SV40 large T antigen–expressing HEK293FT cells with the shRNA-pU6/ENTR-shRNA vectors and rat ADAM17 cDNA. At 24 h later, cells were lysed, the proteins were resolved by SDS–PAGE, and ADAM17 was detected by Western blotting using the techniques described earlier. Of the four, the sequence that had maximum silencing efficiency (>80%; 5′-GGATTAGCTTACGTTGGTTCT-3′) was selected and recombined into the pBLOCKiT Adenovirus System vector (Invitrogen) using an in vitro Clonase-mediated recombination reaction according to the vendor's protocol. The recombined pBLOCKiT vector was linearized by Pac1 restriction digestion and transfected into 293A cells using Lipofectamine 2000 reagent (Life Technologies). At 11 d posttransfection, when the cytopathic effect was >75%, the cells in this first P1 generation were harvested by tituration and lysed by freezing the cells at −70°C and thawing them for 5–10 min in a 37°C water bath. Three freeze–thaw cycles were typically sufficient to lyse the cells. The lysate, containing released virus, was used to infect a new round of cells (P2), a process that was repeated one additional time (P3). To produce large quantities of virus, 10 15-cm Petri dishes (BD Falcon, San Jose, CA) of 293A cells were infected with virus-containing lysate from P3. On the third day, when the cells showed >85% cytopathic effect, they were recovered by tituration, pooled, centrifuged at 3500 × g (Eppendorf 5810 R) for 14 min at 4°C, and mixed in 7 ml of resuspension buffer (100 mM Tris, pH 7.4, 10 mM EDTA). The concentrated cell suspension was lysed by repeated freeze–thaw cycles as described. The lysate was separated from the cell debris by centrifuging at 5000 × g (Eppendorf 5810 R) for 15 min at 4°C. The supernatant was carefully removed and applied to the top of a step gradient containing 2.5 ml of 1.25 g/ml CsCl solution, which was layered on top of 2.5 ml of 1.4 g/ml CsCl loaded into clear 13-ml PET ultracentrifugation tubes (Thermo Scientific). The samples were centrifuged at 35,000 rpm for 1 h at 4°C using a Beckman Coulter centrifuge (Brea, CA) and an SW-41 swinging bucket rotor. The concentrated virus, which appeared as an off-white band at the interface of the two CsCl layers, was collected by piercing the side wall of the tube with an 18-gauge needle and aspirating it into a connected 5-cc syringe (BD Biosciences). The viruses were further purified by passage through a PE10 gel filtration column (GE Healthcare) equilibrated with virus suspension buffer (PBS containing 10% [vol/vol] glycerol). The virus-containing fractions were detected by monitoring the A260, pooled, and stored at −70°C in small aliquots, which were thawed in a 37°C water bath just before use.
Generation of adenoviruses encoding ADAM17-HAr, ADAM17S811A-HAr, ADAM17S811D-HAr, or ADAM17-∆CT-HAr
ADAM17 rat cDNA (from Addgene plasmid 19141; Lemieux et al., 2007) was cloned into pADLOX vector, and an oligonucleotide encoding the HA tag (YPYDVPDYA) was added in-frame to the C-terminus of ADAM17 after a gap of two amino acids. A silent mutation was engineered into the ADAM17 sequence (5′-GGATTAGCGTACGTTG GTTCT) using the QuikChange XL mutagenesis kit (Agilent, Santa Clara, CA), making the construct resistant to the ADAM17 shRNA. This resulting construct, called pADLOX-ADAM17-HAr, was further mutagenized to convert serine at position 811 to an alanine or aspartate residue, generating pADLOX-ADAM17S811A-HAr (TCA to GCC) and pADLOX-ADAM17S811D-HAr (TCA to GAC). In addition, we also generated a tail-minus construct, pADLOX-ADAM17-∆CT-HAr. This was made by deleting the last 127 amino acids of the ADAM17 backbone, leaving two cytoplasmic amino acids that were fused in-frame to an HA tag. The constructs (3 μg) were preincubated with Lipofectamine 2000 Opti-MEM media for 30 min, mixed with 3 μg of Ψ5 adenoviral genomic DNA (Ad5 strain), and added to Cre8 cells (Hardy et al., 1997; Khandelwal et al., 2008). Production of adenoviruses was performed as described, except that Cre8 cells were used throughout.
In situ adenoviral transduction and detection of hGH release
In situ transduction of rat bladder uroepithelium and measurement of hGH release were performed as described (Khandelwal et al., 2008). Briefly, rats were sedated with isoflurane, and a Jelco IV catheter (Smith Medicals, Southington, CT) was introduced into the bladder via the urethra. The bladder was rinsed with PBS and filled with 400 μl of 0.1% (wt/vol) n-dodecyl-β-d-maltoside dissolved in PBS. The urethra was clamped, and after 5 min, it was unclamped to allow the detergent to void. The latter step was facilitated by applying slight pressure to the lower abdomen. The bladder was filled with 400 μl of PBS containing adenoviruses encoding hGH alone or in combination with adenoviruses encoding scrambled-shRNA, ADAM17-shRNA, or the ADAM17-HAr constructs described earlier (2.5 × 108 infectious virus particles, typically in a volume of 2–10 μl). The bladder was then clamped. After 30 min, the clamp was removed, and the virus solution was allowed to void. The bladder was rinsed with PBS, anesthesia was discontinued, and the rats were allowed to revive. At 30 h postinfection, the animals were killed by inhalation of CO2, and the bladder was immediately excised, slit open, and mounted on tissue rings as described. After 90 min of equilibration, the buffer bathing the apical surface was isovolumetrically replaced with fresh buffer and then treated or not with 500 nM CCPA. At 60 min later, the apical buffer was removed and concentrated using a 10K molecular weight cutoff Amicon Centricon (Millipore) to a volume of 250 μl. The corresponding tissue was unmounted from its tissue ring, and a lysate was prepared before Western blot analysis to detect hGH. The fraction of hGH secreted was calculated as secreted hGH/(secreted hGH + cell-associated hGH in lysate).
Endocytosis of WGA-FITC
Rat bladders were mounted in Ussing chambers, and after equilibration, they were incubated with 25 μg/ml WGA-FITC for 2 h with or without 500 nM CCPA added to the mucosal hemichamber. The tissues were then unmounted from the chambers, washed with ice-cold 100 mM N-acetyl-d-glucosamine four times for 20 min, and then washed with ice-cold PBS three times for 15 min and then fixed with 4% PFA fixative for 30 min at 37°C. The tissue was stained, and the images were captured and processed as described. Because some WGA-FITC remained bound to the apical surface of the umbrella cells even after washing with cognate sugar, the apicalmost sections (∼1–1.5 μm) in the confocal stacks were excluded from the quantitation.
32P-orthophosphate labeling of ADAM17
HEK293FT cells were cotransfected with 3 μg of pcDNA-A1AR and 3 μg of either pADLOX-ADAM17-HAr or pADLOX-ADAM17S811A-HAr using Lipofectamine 2000 reagent. At 48 h posttransfection, the cells were washed two times with PO4 efflux buffer (140 mM NaCl, 2 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 10 mM glucose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, titrated to pH 7.4 using 1 M Tris-base) and then incubated with PO4 efflux buffer supplemented with 300 μCi of 32P-orthophosphate (5 mCi/ml; MP Biomedicals; Santa Ana, CA) for 2 h at 37°C. During the last 15 min of the incubation, the cells were treated or not with 500 nM CCPA. Subsequently, the efflux buffer was aspirated, the cells were washed two times with Tris-buffered saline (pH 7.5), and the cells were dissolved in 500 μl of RIPA lysis buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 50 mM NaF, 0.1% [wt/vol] SDS, 1% [wt/vol] sodium deoxycholate, 1% [vol/vol] Triton X-100, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 mM orthovanadate, 0.1 mg/ml aprotinin, 1 mM ethylene glycol tetraacetic acid, and 10 mM 1,10-phenanthroline). The cell lysate was collected, placed in 1.5-ml Eppendorf tubes, and incubated on ice for 30 min to complete the cell lysis. The lysate was centrifuged at 16,000 × g at 4°C using a Marathon 16km table-top centrifuge (Fisher Scientific, Waltham, MA) for 1 h, and the resulting supernatant was collected in a fresh tube. The volume in each tube was brought up to 900 μl with RIPA lysis buffer, and then 7 μl of rabbit anti-HA antibody (Covance) and 40 μl of 10% SDS were added to each sample. After a 1-h incubation at 4°C with constant rotation, 100 μl of a 20% (wt/vol) slurry of protein G-Sepharose beads (GE Healthcare) was added to each sample and incubated at 4°C overnight with constant rotation. The beads were washed with RIPA lysis buffer three times, resuspended in 40 μl of 2× Laemmli sample buffer, heated for 15 min at 65°C, and centrifuged at 16,000 × g for 10 min, and then the proteins in the supernatant were resolved by SDS–PAGE. The proteins were transferred to nitrocellulose membranes as described and the immobilized HA-labeled proteins reacted with mouse monoclonal anti-HA antibody conjugated to HRP (Roche, Mannheim, Germany) overnight at 4°C. The membrane was then washed three times with TBST buffer and then incubated with ECL chemiluminescence reagent for 5 min (GE Healthcare). The ECL signal was captured using a VersaDoc molecular imager (Bio-Rad). The same membrane was then exposed on a phospho-screen (GE Healthcare) to capture the phosphorylation signal. The phosphorylation signal on the phospho-screen was visualized using a Personal Molecular Imager system (Bio-Rad), and the image was analyzed using Quantity One quantification software (Bio-Rad).
Expression of HB-EGF-AP and monitoring of its activity
An HB-EGF release assay was performed using the pRC/CMV-HB-EGF-AP cDNA construct, kindly provided by J. C. Krepinsky (McMaster University, Hamilton, Canada). HEK293FT cells, seeded in 12-well plates, were transfected with 1.5 μg of pRC/CMV-HB-EGF-AP construct alone or in combination with pcDNA-A1AR and pADLOX-ADAM17-HAr, pADLOX-ADAM17S811A-HAr, pADLOX-ADAM17S811D-HAr, or pADLOX-ADAM17-∆CT-HAr using Lipofectamine 2000 as described. At 48 posttransfection, the cells were washed two times with phenol red– and serum-free DMEM (Invitrogen), which was replaced after 1 h by fresh medium with or without 500 nM CCPA. After 1 h of treatment, the culture supernatant was collected and the cells lysed to quantify total and released AP activity using the SensoLyte pNPP Alkaline Phosphatase Assay Kit (AnaSpec, Freemont, CA) according to the manufacturer's protocols. Briefly, 10 μl of culture supernatant (1 ml total) and 2 μl of cell lysate (500 μl total; prepared in 1× assay buffer containing 0.2% [vol/vol] TX-100) were incubated with the AP substrate (p-nitrophenyl phosphate) for 20 min in the dark at 25°C. After the addition of 50 μl of stop solution, the AP activity was measured at an absorbance of 405 nm. The percentage of HB-EGF-AP release was calculated by dividing the total AP activity in the supernatant by the sum of total activity in the supernatant and the cell lysate. Next the percentage of HB-EGF-AP released in the presence of CCPA was normalized to reactions in which CCPA treatment was omitted. Each experiment was repeated at least five times. No AP activity was present in the supernatant of nontransfected cells.
Statistical analysis
Statistical significance between means was determined by Student's t test or, in the case of multiple comparisons, by analysis of variance (ANOVA). If a significant difference in the means was detected by ANOVA, multiple comparisons were performed using Dunnett's posttest correction. Statistical analyses were performed using Prism 5 software (GraphPad, La Jolla, CA). In the figure legends, n refers to the number of individual animals or experiments.
Acknowledgments
This work was supported by National Institutes of Health Grants R37-DK54425, R01-DK077777, and P30-DK079307 to G.A. and R01-DK075048 to K.R.H. and the Cellular Physiology and Kidney Imaging Cores of the Pittsburgh Center for Kidney Research (P30-DK079307).
Abbreviations used
- A1AR
A1 adenosine receptor
- ADAM
a disintegrin and a metalloproteinase
- CCPA
2-chloro-N6-cyclopentyladenosine
- DFV
discoidal and/or fusiform-shaped vesicle
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- GPCR
G protein–coupled receptor
- MAPK
mitogen-activated protein kinase
- PKC
protein kinase C
- PLC
phospholipase C
- PMA
phorbol-12-myristate-13-acetate
- TIMP
tissue inhibitor of metalloproteinase.
Footnotes
H.S.P., L.I.G., W.G.R., and H.L. designed and executed the experiments and assisted in the writing of the manuscript. K.R.H. and G.A. assisted in the design of the experiments and wrote and prepared the manuscript.
The authors have no conflicts of interest to report.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-03-0818) on September 17, 2014.
REFERENCES
- Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, et al. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- Apodaca G. The uroepithelium: not just a passive barrier. Traffic. 2004;5:1–12. doi: 10.1046/j.1600-0854.2003.00156.x. [DOI] [PubMed] [Google Scholar]
- Arribas J, Esselens C. ADAM17 as a therapeutic target in multiple diseases. Curr Pharm Des. 2009;15:2319–2335. doi: 10.2174/138161209788682398. [DOI] [PubMed] [Google Scholar]
- Balestreire EM, Apodaca G. Apical epidermal growth factor receptor signaling: regulation of stretch-dependent exocytosis in bladder umbrella cells. Mol Biol Cell. 2007;18:1312–1323. doi: 10.1091/mbc.E06-09-0842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell HL, Gooz M. ADAM-17 is activated by the mitogenic protein kinase ERK in a model of kidney fibrosis. Am J Med Sci. 2010;339:105–107. doi: 10.1097/MAJ.0b013e3181cb4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA, Doedens JR, Mahimkar R, Johnson R, Guo L, Wallace A, Virca D, Eisenman J, Slack J, Castner B, et al. Substrate specificity and inducibility of TACE (tumour necrosis factor alpha-converting enzyme) revisited: the Ala-Val preference, and induced intrinsic activity. Biochem Soc Symp. 2003;2003(70):39–52. doi: 10.1042/bss0700039. [DOI] [PubMed] [Google Scholar]
- Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- Bucheimer RE, Linden J. Purinergic regulation of epithelial transport. J Physiol. 2004;555:311–321. doi: 10.1113/jphysiol.2003.056697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SJ, Tzeng CR, Lee YH, Tai CJ. Extracellular ATP activates the PLC/PKC/ERK signaling pathway through the P2Y2 purinergic receptor leading to the induction of early growth response 1 expression and the inhibition of viability in human endometrial stromal cells. Cell Signal. 2008;20:1248–1255. doi: 10.1016/j.cellsig.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Dang M, Armbruster N, Miller MA, Cermeno E, Hartmann M, Bell GW, Root DE, Lauffenburger DA, Lodish HF, Herrlich A. Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proc Natl Acad Sci USA. 2013;110:9776–9781. doi: 10.1073/pnas.1307478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang M, Dubbin K, D’Aiello A, Hartmann M, Lodish H, Herrlich A. Epidermal growth factor (EGF) ligand release by substrate-specific a disintegrin and metalloproteases (ADAMs) involves different protein kinase C (PKC) isoenzymes depending on the stimulus. J Biol Chem. 2011;286:17704–17713. doi: 10.1074/jbc.M110.187823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Rodriguez E, Montero JC, Esparis-Ogando A, Yuste L, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Mol Biol Cell. 2002;13:2031–2044. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens JR, Black RA. Stimulation-induced down-regulation of tumor necrosis factor-alpha converting enzyme. J Biol Chem. 2000;275:14598–14607. doi: 10.1074/jbc.275.19.14598. [DOI] [PubMed] [Google Scholar]
- Doedens JR, Mahimkar RM, Black RA. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem Biophys Res Commun. 2003;308:331–338. doi: 10.1016/s0006-291x(03)01381-0. [DOI] [PubMed] [Google Scholar]
- Fan H, Turck CW, Derynck R. Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-alpha converting enzyme and of an alternatively translated polypeptide. J Biol Chem. 2003;278:18617–18627. doi: 10.1074/jbc.M300331200. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Freund S, Ungerer M, Lohse MJ. A1 adenosine receptors expressed in CHO-cells couple to adenylyl cyclase and to phospholipase C. Naunyn-Schmiedeberg's Arch Pharmacol. 1994;350:49–56. doi: 10.1007/BF00180010. [DOI] [PubMed] [Google Scholar]
- Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45:146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KC, Blobel CP. Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PLoS One. 2012;7:e31600. doi: 10.1371/journal.pone.0031600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Construction of adenovirus vectors through Cre-lox recombination. J Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassemer EL, Le Gall SM, Liegel R, McNally M, Chang B, Zeiss CJ, Dubielzig RD, Horiuchi K, Kimura T, Okada Y, et al. The waved with open eyelids (woe) locus is a hypomorphic mouse mutation in Adam17. Genetics. 2010;185:245–255. doi: 10.1534/genetics.109.113167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179:2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Zhou HM, Kelly K, Manova K, Blobel CP. Evaluation of the contributions of ADAMs 9, 12, 15, 17, and 19 to heart development and ectodomain shedding of neuregulins beta1 and beta2. Dev Biol. 2005;283:459–471. doi: 10.1016/j.ydbio.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, Lee DC. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J. 2003;22:2704–2716. doi: 10.1093/emboj/cdg264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DE, Liang F, Bondioli KR, Zhao H, Kreibich G, Wall RJ, Sun TT. The bladder as a bioreactor: urothelium production and secretion of growth hormone into urine. Nat Biotechnol. 1998;16:75–79. doi: 10.1038/nbt0198-75. [DOI] [PubMed] [Google Scholar]
- Khandelwal P, Prakasam HS, Clayton DR, Ruiz WG, Gallo L, van Roekel D, Lukianov S, Peranen J, Goldenring JR, Apodaca G. A Rab11a-Rab8a-Myo5B network promotes stretch-regulated exocytosis in bladder umbrella cells. Mol Biol Cell 24, 1007–1019. 2013 doi: 10.1091/mbc.E12-08-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal P, Ruiz WG, Apodaca G. Compensatory endocytosis in bladder umbrella cells occurs through an integrin-regulated and RhoA- and dynamin-dependent pathway. EMBO J. 2010;29:1961–1975. doi: 10.1038/emboj.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal P, Ruiz WG, Balestreire-Hawryluk E, Weisz OA, Goldenring JR, Apodaca G. Rab11a-dependent exocytosis of discoidal/fusiform vesicles in bladder umbrella cells. Proc Natl Acad Sci USA. 2008;105:15773–15778. doi: 10.1073/pnas.0805636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killock DJ, Ivetic A. The cytoplasmic domains of TNFalpha-converting enzyme (TACE/ADAM17) and L-selectin are regulated differently by p38 MAPK and PKC to promote ectodomain shedding. Biochem J. 2010;428:293–304. doi: 10.1042/BJ20091611. [DOI] [PubMed] [Google Scholar]
- Kirui JK, Xie Y, Wolff DW, Jiang H, Abel PW, Tu Y. Gbetagamma signaling promotes breast cancer cell migration and invasion. J Pharmacol Exp Ther. 2010;333:393–403. doi: 10.1124/jpet.109.164814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohutek ZA, diPierro CG, Redpath GT, Hussaini IM. ADAM-10-mediated N-cadherin cleavage is protein kinase C-alpha dependent and promotes glioblastoma cell migration. J Neurosci. 2009;29:4605–4615. doi: 10.1523/JNEUROSCI.5126-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kveiborg M, Instrell R, Rowlands C, Howell M, Parker PJ. PKCalpha and PKCdelta regulate ADAM17-mediated ectodomain shedding of heparin binding-EGF through separate pathways. PLoS One. 2011;6:e17168. doi: 10.1371/journal.pone.0017168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak HI, Mendoza EA, Bayless KJ. ADAM17 co-purifies with TIMP-3 and modulates endothelial invasion responses in three-dimensional collagen matrices. Matrix Biol. 2009;28:470–479. doi: 10.1016/j.matbio.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol Biol Cell. 2009;20:1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gall SM, Maretzky T, Issuree PD, Niu XD, Reiss K, Saftig P, Khokha R, Lundell D, Blobel CP. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J Cell Sci. 2010;123:3913–3922. doi: 10.1242/jcs.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux GA, Blumenkron F, Yeung N, Zhou P, Williams J, Grammer AC, Petrovich R, Lipsky PE, Moss ML, Werb Z. The low affinity IgE receptor (CD23) is cleaved by the metalloproteinase ADAM10. J Biol Chem. 2007;282:14836–14844. doi: 10.1074/jbc.M608414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemjabbar-Alaoui H, Sidhu SS, Mengistab A, Gallup M, Basbaum C. TACE/ADAM-17 phosphorylation by PKC-epsilon mediates premalignant changes in tobacco smoke-exposed lung cells. PLoS One. 2011;6:e17489. doi: 10.1371/journal.pone.0017489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Prior M, He W, Hu X, Tang X, Shen W, Yadav S, Kiryu-Seo S, Miller R, Trapp BD, et al. Cleavage of neuregulin-1 by BACE1 or ADAM10 protein produces differential effects on myelination. J Biol Chem. 2011;286:23967–23974. doi: 10.1074/jbc.M111.251538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunath S, Sakhare PM. Adenosine and adenosine receptors: newer therapeutic perspective. Indian J Pharmacol. 2009;41:97–105. doi: 10.4103/0253-7613.55202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifune M, Ohtsu H, Suzuki H, Nakashima H, Brailoiu E, Dun NJ, Frank GD, Inagami T, Higashiyama S, Thomas WG, et al. G protein coupling and second messenger generation are indispensable for metalloprotease-dependent, heparin-binding epidermal growth factor shedding through angiotensin II type-1 receptor. J Biol Chem. 2005;280:26592–26599. doi: 10.1074/jbc.M502906200. [DOI] [PubMed] [Google Scholar]
- Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev. Cancer. 2008;8:929–941. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- Nelson KK, Schlondorff J, Blobel CP. Evidence for an interaction of the metalloprotease-disintegrin tumour necrosis factor alpha convertase (TACE) with mitotic arrest deficient 2 (MAD2), and of the metalloprotease-disintegrin MDC9 with a novel MAD2-related protein, MAD2beta. Biochem J. 1999;343:673–680. [PMC free article] [PubMed] [Google Scholar]
- Nishi E, Hiraoka Y, Yoshida K, Okawa K, Kita T. Nardilysin enhances ectodomain shedding of heparin-binding epidermal growth factor-like growth factor through activation of tumor necrosis factor-alpha-converting enzyme. J Biol Chem. 2006;281:31164–31172. doi: 10.1074/jbc.M601316200. [DOI] [PubMed] [Google Scholar]
- Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Niu A, Wen Y, Liu H, Zhan M, Jin B, Li YP. Src mediates the mechanical activation of myogenesis by activating TNFalpha-converting enzyme. J Cell Sci. 2013;126:4349–4357. doi: 10.1242/jcs.125328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RB, Kemp BE. Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol. 1991;200:62–81. doi: 10.1016/0076-6879(91)00127-i. [DOI] [PubMed] [Google Scholar]
- Peiretti F, Deprez-Beauclair P, Bonardo B, Aubert H, Juhan-Vague I, Nalbone G. Identification of SAP97 as an intracellular binding partner of TACE. J Cell Sci. 2003;116:1949–1957. doi: 10.1242/jcs.00415. [DOI] [PubMed] [Google Scholar]
- Plumb J, McQuaid S, Cross AK, Surr J, Haddock G, Bunning RA, Woodroofe MN. Upregulation of ADAM-17 expression in active lesions in multiple sclerosis. Mult Scler. 2006;12:375–385. doi: 10.1191/135248506ms1276oa. [DOI] [PubMed] [Google Scholar]
- Prakasam HS, Herrington H, Roppolo JR, Jackson EK, Apodaca G. Modulation of bladder function by luminal adenosine turnover and A1 receptor activation. Am J Physiol. Renal Physiol. 2012;303:F279–F292. doi: 10.1152/ajprenal.00566.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem. 2000;275:14608–14614. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- Reiss K, Saftig P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20:126–137. doi: 10.1016/j.semcdb.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Sahin U, Blobel CP. Ectodomain shedding of the EGF-receptor ligand epigen is mediated by ADAM17. FEBS Lett. 2007;581:41–44. doi: 10.1016/j.febslet.2006.11.074. [DOI] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serino M, Menghini R, Fiorentino L, Amoruso R, Mauriello A, Lauro D, Sbraccia P, Hribal ML, Lauro R, Federici M. Mice heterozygous for tumor necrosis factor-alpha converting enzyme are protected from obesity-induced insulin resistance and diabetes. Diabetes. 2007;56:2541–2546. doi: 10.2337/db07-0360. [DOI] [PubMed] [Google Scholar]
- Shah BH, Catt KJ. TACE-dependent EGF receptor activation in angiotensin-II-induced kidney disease. Trends Pharmacol Sci. 2006;27:235–237. doi: 10.1016/j.tips.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Soond SM, Everson B, Riches DW, Murphy G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci. 2005;118:2371–2380. doi: 10.1242/jcs.02357. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Sunnarborg SW, Kouros-Mehr H, Yu Y, Lee DC, Werb Z. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development. 2005;132:3923–3933. doi: 10.1242/dev.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Nanba D, Mori S, Shiba F, Ishiguro H, Yoshino K, Matsuura N, Higashiyama S. ADAM binding protein Eve-1 is required for ectodomain shedding of epidermal growth factor receptor ligands. J Biol Chem. 2004;279:41950–41959. doi: 10.1074/jbc.M400086200. [DOI] [PubMed] [Google Scholar]
- Truschel ST, Ruiz WG, Shulman T, Pilewski J, Sun TT, Zeidel ML, Apodaca G. Primary uroepithelial cultures. A model system to analyze umbrella cell barrier function. J Biol Chem. 1999;274:15020–15029. doi: 10.1074/jbc.274.21.15020. [DOI] [PubMed] [Google Scholar]
- Truschel ST, Wang E, Ruiz WG, Leung SM, Rojas R, Lavelle J, Zeidel M, Stoffer D, Apodaca G. Stretch-regulated exocytosis/endocytosis in bladder umbrella cells. Mol Biol Cell. 2002;13:830–846. doi: 10.1091/mbc.01-09-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida T, Pappenheimer AM, Jr, Harper AA. Diphtheria toxin and related proteins. 3. Reconstitution of hybrid “diphtheria toxin” from nontoxic mutant proteins. J Biol Chem. 1973;248:3851–3854. [PubMed] [Google Scholar]
- Uttarwar L, Peng F, Wu D, Kumar S, Gao B, Ingram AJ, Krepinsky JC. HB-EGF release mediates glucose-induced activation of the epidermal growth factor receptor in mesangial cells. Am J Physiol. Renal Physiol. 2011;300:F921–F931. doi: 10.1152/ajprenal.00436.2010. [DOI] [PubMed] [Google Scholar]
- Wang E, Truschel S, Apodaca G. Analysis of hydrostatic pressure-induced changes in umbrella cell surface area. Methods. 2003a;30:207–217. doi: 10.1016/s1046-2023(03)00027-6. [DOI] [PubMed] [Google Scholar]
- Wang EC, Lee JM, Johnson JP, Kleyman TR, Bridges R, Apodaca G. Hydrostatic pressure-regulated ion transport in bladder uroepithelium. Am J Physiol. Renal Physiol. 2003b;285:F651–F663. doi: 10.1152/ajprenal.00403.2002. [DOI] [PubMed] [Google Scholar]
- Willems SH, Tape CJ, Stanley PL, Taylor NA, Mills IG, Neal DE, McCafferty J, Murphy G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem J. 2010;428:439–450. doi: 10.1042/BJ20100179. [DOI] [PubMed] [Google Scholar]
- Williams-Pritchard G, Knight M, Hoe LS, Headrick JP, Peart JN. Essential role of EGFR in cardioprotection and signaling responses to A1 adenosine receptors and ischemic preconditioning. Am J Physiol. Heart Circ Physiol. 2011;300:H2161–H2168. doi: 10.1152/ajpheart.00639.2010. [DOI] [PubMed] [Google Scholar]
- Wilson CN, Mustafa SJ. Adenosine receptors in health and disease. Preface. Handb Exp Pharmacol. 2009;193(v–vi) [PubMed] [Google Scholar]
- Xie KQ, Zhang LM, Cao Y, Zhu J, Feng LY. Adenosine A(1) receptor-mediated transactivation of the EGF receptor produces a neuroprotective effect on cortical neurons in vitro. Acta Pharmacol Sin. 2009;30:889–898. doi: 10.1038/aps.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell. 2010;37:551–566. doi: 10.1016/j.molcel.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Liu J, Sakaki-Yumoto M, Derynck R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci Signal. 2012;5:ra34. doi: 10.1126/scisignal.2002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Hill WG, Apodaca G, Zeidel ML. Expression and distribution of transient receptor potential (TRP) channels in bladder epithelium. Am J Physiol Renal Physiol. 2011;300:F49–F59. doi: 10.1152/ajprenal.00349.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Khandelwal P, Apodaca G. Distinct apical and basolateral membrane requirements for stretch-induced membrane traffic at the apical surface of bladder umbrella cells. Mol Biol Cell. 2009;20:282–295. doi: 10.1091/mbc.E08-04-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Zacharia LC, Jackson EK, Apodaca G. Adenosine receptor expression and function in bladder uroepithelium. Am J Physiol Cell Physiol. 2006;291:C254–C265. doi: 10.1152/ajpcell.00025.2006. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Schlondorff J, Blobel CP. Evidence for regulation of the tumor necrosis factor alpha-convertase (TACE) by protein-tyrosine phosphatase PTPH1. J Biol Chem. 2002;277:42463–42470. doi: 10.1074/jbc.M207459200. [DOI] [PubMed] [Google Scholar]