

Abstract
A novel strain of Pseudomonas putida LS46 was isolated from wastewater on the basis of its ability to synthesize medium chain-length polyhydroxyalkanoates (mcl-PHAs). P.putida LS46 was differentiated from other P.putida strains on the basis of cpn60 (UT). The complete genome of P.putida LS46 was sequenced and annotated. Its chromosome is 5,86,2556 bp in size with GC ratio of 61.69. It is encoding 5316 genes, including 7 rRNA genes and 76 tRNA genes. Nucleotide sequence data of the complete P. putida LS46 genome was compared with nine other P. putida strains (KT2440, F1, BIRD-1, S16, ND6, DOT-T1E, UW4, W619 and GB-1) identified either as biocontrol agents or as bioremediation agents and isolated from different geographical region and different environment. BLASTn analysis of whole genome sequences of the ten P. putida strains revealed nucleotide sequence identities of 86.54 to 97.52%. P.putida genome arrangement was LS46 highly similar to P.putida BIRD1 and P.putida ND6 but was markedly different than P.putida DOT-T1E, P.putida UW4 and P.putida W619. Fatty acid biosynthesis (fab), fatty acid degradation (fad) and PHA synthesis genes were highly conserved among biocontrol and bioremediation P.putida strains. Six genes in pha operon of P. putida LS46 showed >98% homology at gene and proteins level. It appears that polyhydroxyalkanoate (PHA) synthesis is an intrinsic property of P. putida and was not affected by its geographic origin. However, all strains, including P. putida LS46, were different from one another on the basis of house keeping genes, and presence of plasmid, prophages, insertion sequence elements and genomic islands. While P. putida LS46 was not selected for plant growth promotion or bioremediation capacity, its genome also encoded genes for root colonization, pyoverdine synthesis, oxidative stress (present in other soil isolates), degradation of aromatic compounds, heavy metal resistance and nicotinic acid degradation, manganese (Mn II) oxidation. Genes for toluene or naphthalene degradation found in the genomes of P. putida F1, DOT-T1E, and ND6 were absent in the P. putida LS46 genome. Heavy metal resistant genes encoded by the P. putida W619 genome were also not present in the P. putida LS46 genome. Despite the overall similarity among genome of P.putida strains isolated for different applications and from different geographical location a number of differences were observed in genome arrangement, occurrence of transposon, genomic islands and prophage. It appears that P.putida strains had a common ancestor and by acquiring some specific genes by horizontal gene transfer it differed from other related strains.
Keywords: Pseudomonas putida, Comparative bioinformatics analysis, Comparative genome analysis, Polyhydroxyalkanoates, Pan-Genome, Insertion sequences, Metabolic diversity
Introduction
The genus Pseudomonas consists of a very heterogeneous group of microorganisms isolated from diverse environments and belonging to the gamma (γ)-proteobacteria (Palleroni [1984]). The genus has approximately 100 named species grouped into different subgroups based on multi-locus analysis (Winsor et al. [2011]). The genus Pseudomonas contains human pathogens, plant pathogens, and heterotrophic bacteria prevalent in soil, water, and on plant surfaces. Most pseudomonads are free-living saprophytic organisms in soil or water. Because of their great metabolic diversity they play an important role in decomposition, biodegradation, and the carbon and nitrogen cycles. Pseudomonas putida is non-pathogenic soil bacteria and is capable of degrading xenobiotic and promoting plant growth after root colonization as well as simultaneously providing protection for the plant from pests and other harmful bacteria (Compant et al. [2005]; Singleton [1994]).
Pseudomonas putida KT2440, P. putida BIRD1, P. putida UW4, P. putida S11 were isolated from rhizosphere soil and developed as plant growth promoting rhizobacteria (Glick [1995]; Matilla et al. [2011]; Nakazawa [2002]; Ponraj et al. [2012]). Pseudomonas putida F1 was isolated from polluted soil and developed as a bioremediation agent (Choi et al. [2003]; Eaton [1997]). Pseudomonas putida S16 was isolated in China and identified as a nicotine degrading strain (Tang et al. [2012]). Pseudomonas putida ND6 was identified as a naphthalene degrading bacterium, in which naphthalene degradation genes were located on a plasmid (Dennis et al. [2004]; Li et al. [2002]). Pseudomonas putida DOT-T1E was isolated as a toluene tolerant bacterium (Ramos et al. [1995]), while P. putida W619 was isolated as an endophyte of poplar (Taghavi et al. [2005]). Pseudomonas putida GB-1 was isolated from fresh water and identified as manganese oxidizer (Rosson and Nelson [1982]). None of P.putida strain was isolated for PHAs production; however, P.putida KT2440 was reported as a PHAs producer (Haywood et al. [1989]; Huisman et al. [1989]).
Polyhydroxyalkanoates (PHAs) are the largest group of biologically produced biodegradable biopolyesters providing different properties and applications (Huijbert and Eggink [1996]; de Smet et al. [1983]). A wide variety of microorganisms accumulate PHAs as a carbon and energy storage material in carbon-excess conditions when a major nutrient (typically nitrogen or phosphorus) is limiting (Elbahloul and Steinbüchel [2009]; Rehm [2010]; Brandl et al. [1988]). Accumulation of excess carbon is a general mechanism used by Pseudomonas and is essential for resource balancing (de Eugenio et al. [2010a]; Escapa et al. [2012]). De Smet et al. ([1983]) detected the inclusion bodies in Pseudomonas olevorans for first time when grown on octane and identified is as polymer of 3-hydroxyoctanoate. Huisman et al. ([1989]) studied the PHAs synthesis in fluorescent pseudomonads and confirmed that P.putida, P.fluorescens, P.aeruginosa, P.tetosteroni and P. oleovorans have the capacity to produce PHAs. Among the P.putida strains only P.putida KT2440, which was isolated as a plant growth promoting bacterium, has been studied for PHAs production.
Pseudomonas putida LS46 was isolated from wastewater on the basis of its ability to synthesize novel medium chain-length polyhydroxyalkanoates (mcl-PHAs), which accumulated 22% - 56% of cell dry weight when cultured, under batch culture conditions in media containing glucose, waste vegetable fryer oils or fatty acids (Sharma et al. [2011]). Analysis of P. putida LS46 16S rDNA displayed more than 99% nucleotide sequence identity to other P. putida strains. Complete genome sequence of other P.putida strains which were developed for plant growth promotion or degradation of xenobiotic, are available. Last genome comparison of P. putida strain W619 identified genes for aromatic pathways, heavy metal resistance and plant growth promotion in P.putida strain KT2440, F1 and GB-1 (Wu et al. [2011]). Since then six new P. putida strains, including P. putida LS46 genomes have been added to the database. The objective of this work is to see how the genomes of P.putida identified as either as biocontrol or bioremediation agents differ from each other due to their geographic origin, environment. Keeping this in mind the genome sequence of P. putida LS46 was compared with the genomes of nine other P. putida strains for general genome features, shared gene analysis, occurrence of insertion sequences, prophages, genomic islands and for metabolic diversity including PHAs production. The present study revealed more similarity than variability among the genome of P. putida strains isolated from different ecological niches in different locations.
Materials and methods
Genome sequences and accession numbers
P putida LS46 was isolated from a municipal wastewater treatment plant in Winnipeg, Manitoba, Canada, and the strain was deposited with International Depository Authority of Canada (IDAC) at the National Microbiology Laboratory, Health Canada Culture Collection (NML-HCCC), WDCM number 840 (Sharma et al. [2011]). The P. putida LS46 genome was sequenced by the Genome Canada sequencing facility at McGill University, Montreal, Quebec, using a combination of Illumina Gaii (Bennett [2002]) and 454 pyro-sequencing technology (Margulies et al. [2005]). The draft genome sequence of P.putida LS46 was deposited in the NCBI GenBank database with Accession number ALPV02000000 (Sharma et al. [2013]). The complete genomes of nine P. putida strains KT2440 (NC002947), F1 (NC009512), BIRD1 (NC017530), GB-1 (NC010322) S16 (CP003734), ND6 (CP_003589), DOT-T1E (NC_018222), UW4 (CP_003890) and W619 (NC010501) were obtained from the Joint Genome Institute database (http://img.jgi.doe.gov) and compared with the newly sequenced P. putida LS46. General genome features like genome size, total number of genes, number of coding sequences (CDS), number of Clusters of Orthologous Groups (COGs) were compared among the genomes of the 10 P. putida strains.
Phylogenetic relationship among Pseudomonas strains
The P. putida LS46 chaparonin gene (cpn60) universal target (UT) sequence was obtained from the P. putida LS46 genome sequence and aligned with the cpn60 UT sequences of 29 Pseudomonas strains available in the Integrated Microbial Genome database Markowitz et al. [2006]). The sequences were aligned using Bioedit (Thompson et al. [1994]) and phylogenetic analysis was conducted using MEGA5 (Tamura et al. [2011]). Phylogenetic tree was constructed using the neighbor-joining method (Saitou and Nei [1987]).
Occurrence of prophage, insertion sequence and genomic islands
The ten P. putida genomes were analyzed for presence of prophages using the PHAST (PHAge Search Tool) prophage finder database (Zhou et al. [2011]). Insertion Sequence (IS) elements in the P. putida genomes were identified using the ISfinder tool (Siguier et al. [2006]; Varani et al. [2011]). Genomic islands in P.putida genomes were identified using Island finder web based tools (Langille and Brinkman [2009]).
Whole genome alignments, BLASTn and Pan-genome analysis
Whole genome identity was calculated from pair-wise comparison of genomes using BLASTn analysis. Comparative synteny Dot plot analysis of the P. putida strains was carried out and nine plots of comparison between P. putida LS46 and nine P. putida strains were obtained (Huang and Zhang [2004]). Maps showing P. putida strains were prepared using the Gview server P. putida LS46 was compared with the other eight P. putida strains using Pan genome analysis from Gview server (Petkau et al. [2010]). Shared genes in different strains were calculated using the Phylogenetic profile program using a single gene profile from the Integrated Microbial Genome server. The number of homologous genes in genome 1 in comparison to genome 2 was counted. Reciprocally, the numbers of homologous genes in genome 2 in comparison to genome 1 were counted. Percent shared genes were calculated by adding the homologous genes in genome 1 and 2, dividing by the total number of genes in genome 1 + 2, and multiplied by 100 (Palmer et al. [2012]).
Comparison of metabolic genes
Genes related with different metabolic functions were identified by searching for homologous genes in P.putida genomes by using BLAST analysis of find gene in IMG web page using default parameter (Integrated Microbial Genome). Homologous genes for aromatic compound degradation, heavy metal resistance, manganese oxidation, nicotinic acid degradation, iron scavenging were identified using BLASTn analysis of IMG web page. Likewise TonB dependent receptor and dioxygenase genes were identified in different P.putida strains.
Results
Phylogenetic relationship of P. putida LS46 to other Pseudomonas strains
A polyhydroxyalkanoate producing bacterium was isolated from wastewater and was identified as a strain of Pseudomonas putida on the basis of 16S rRNA gene sequence. The16S rRNA gene sequence analysis revealed more than 99% nucleotide sequence identity to other P. putida strains. Protein-encoding genes are known to provide higher levels of taxonomic resolution than non-protein-encoding genes like 16S rRNA gene. Therefore, a protein coding gene cpn60 (Hsp60 or GroEL) was used for phylogenetic analysis of Pseudomonas species and strains. Neighbor joining trees based on cpn60 genes divided the Pseudomonas species into two clades, one containing P. putida, P. entomophila, P. mendocina, P. fulva P. aeruginosa and P. stutzeri. The other clade included P. fluorescens and P. syringae strains. Although P.putida strains were clustered in one subclade yet minor differences in cpn60 gene sequence separated these strains from each other. Pseudomonas putida LS46 was closely related to P. putida strains ND6, F1, DOT-T1E, BIRD1, KT2440 and clustered with these strains forming a sub-clade (Figure 1). Pseudomonas putida strains LS46 was more distantly related to P.putida strains GB1, S16 and W619. Pseudomonas putida UW4 was not related to any other P. putida strains and clustered with P. fluorescens.
Figure 1.
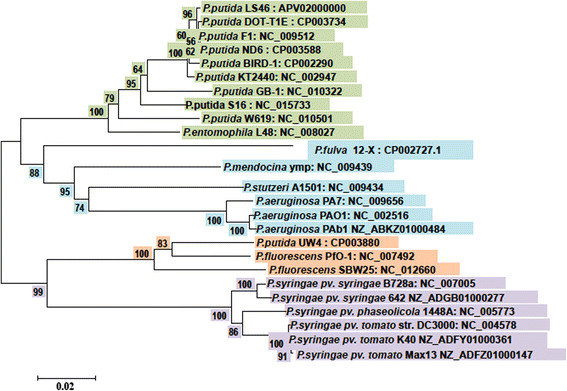
Phylogenetic tree depicting the relationship amongPseudomonasspecies. The tree is based on cpn60 gene sequences, which were aligned by ClustalW and a neighbor-joining tree was generated using MEGA5 program. Bootstrap values are mentioned at the node.
Genome features of P. putida LS46
The complete genome P.putida LS46 was assembled in 32 contigs and consists of 5,874,759 bp DNA with the G + C content of 61.69%. It was predicted to encode 5346 genes, with 5248 CDSs (98.17% of predicted genes). Coding region covers 88.48% of the whole genome. Out of 5248 CDSs, 3889 were assigned functions and 1359 CDSs encoded hypothetical proteins. Among these CDSs, 4270 (79.87%) genes could be classified into COG families composed of 22 categories. A total of seven rRNA operons including seven 5S rRNAs, seven 16S rRNAs, and eight 23S rRNAs were present on the chromosome. In addition, 76 tRNA genes that represent all 20 amino acids, and a tRNA for selenocysteine, were identified (Table 1). No plasmid was detected in P.putida LS46. BLASTn analysis identified 181 unique genes (less than 30% minimal identity and maximum e-value of 1 e-5) in P. putida LS46, which were not present in other nine other P. putida strains. Most of the unique genes were predicted to code hypothetical proteins.
Table 1.
General features of the genomes of P. putida strains
| Strain | BIRD1 | DOT-T1E | F1 | GB-1 | KT2440 | LS46 | ND6 | S16 | UW4 | W619 |
|---|---|---|---|---|---|---|---|---|---|---|
| Isolated in |
Spain |
Spain |
USA |
USA |
Japan |
Canada |
China |
China |
Canada |
USA |
| Genome size |
5731541 |
6260702 |
5959964 |
6078430 |
6181863 |
5862556 |
6304310 |
5984790 |
6183388 |
5774330 |
| % G + C |
61.74 |
61.43 |
61.86 |
61.94 |
61.52 |
61.69 |
61.62 |
62.32 |
60.05% |
61.44 |
| Gene count |
5046 |
5802 |
5423 |
5515 |
5481 |
5316 |
6484 |
5307 |
5517 |
5292 |
| Homologous genes (%)a |
92.62 |
91.39 |
93.75 |
90.17 |
91.32 |
100 |
85.57 |
89.97 |
82.02 |
89.14 |
| % Homologyb |
95.89 |
97.67 |
97.52 |
90.86 |
95.94 |
100 |
97.51 |
91.41 |
86.59 |
87.06 |
| CDS count |
4960 |
5721 |
5300 |
5417 |
5350 |
5219 |
6391 |
5218 |
5423 |
5194 |
| CDS % |
98.30 |
98.60 |
97.73 |
98.22 |
97.61 |
98.17 |
98.57 |
98.32 |
98.30 |
98.15 |
| 16S rRNA |
7 |
0 |
6 |
7 |
7 |
7 |
6 |
6 |
7 |
7 |
| tRNA |
64 |
58 |
76 |
74 |
74 |
76 |
74 |
70 |
72 |
75 |
| COG count |
4139 |
4509 |
4171 |
4267 |
4199 |
4260 |
4519 |
4313 |
4541 |
4089 |
| % COG |
82.02 |
77.71 |
76.91 |
77.37 |
76.61 |
78.61 |
69.69 |
81.27 |
82.31 |
77.27 |
| Protein W FPc |
75.23 |
71.35 |
73.45 |
74.75 |
66.30 |
79.50 |
64.24 |
76.37 |
75.37 |
74.79 |
| Protein w/o FP d |
23.07 |
27.25 |
24.29 |
23.37 |
31.31 |
17.29 |
34.33 |
21.95 |
22.93 |
23.26 |
| Reference | Matilla et al. [2011] | Udaondo et al. [2012] | Wu et al. [2011] | Wu et al. [2011] | Nelson et al. [2002] | Sharma et al. [2013] | Li et al. [2012] | Yu et al. [2011] | Duan t et al. [2013] | Wu et al. [2011] |
a 80% minimal nucleotide sequence identity; e-value 1 e−5.
bBLASTn of whole genome.
c Proteins with function prediction (%).
d Proteins w/o predicted function (%).
Insertion Sequence in P. putida LS46
Blast analyses of the P. putida LS46 genome with IS finder database identified three complete (ISPa41, ISPs1, ISPa42) and one partial (ISPpu9) IS elements (Additional file 1: Table S1). IS element ISPa41, which belongs to IS5 family, was earlier identified in P. aerugonosa DK2, P. putida, and P. syringae DC3000 (IS finder database). Blasting of ISPa41 sequence of P.putida LS46 identified homologous sequences in P.putida strain GB-1, KT2440 and ND6 but not in P.putida BIRD1, DOT-T1E and UW4. Partial sequences of ISPa41 were also present in P.putida F1, S16 and W619. ISPs1 was originated from P. syringae pv. syringae plasmid pPSR1 and P. syringae pv. savastanoi. ISPs1 from P. putida LS46 had 84% nucleotide sequence identity to ISPs1 from P. syringae plasmid pSR1. Complete ISPsI was also present in P.putida GB-1 and ND6. Incomplete ISPsI was also detected in P.putida strain S16, F1 and W619. Complete ISPpu9 from P. putida KT2440 was 2043 bp but in P.putida LS46 partial ISPpu9 was present (1697bp). It showed 98% sequence identity to ISPpu9 from P. putida KT2440. The ISPpu9 integration target site is 23 bp sequence, which is a part of REP identified in P. putida KT2440 (Aranda-Olmedo et al. [2002]). ISPpu9 was also present in P.putida BIRD1 but was absent in P.putida F1, GB1, DOT, ND6 UW4 and W619.
ISPa42, which is synonymous to ISPsy10, encodes a Tn3-like transposable element, which is thought to originate from P. aeruginosa DK3. ISPa42, a complete Tn3 element of 16941 bp in P. aeruginosa DK2, was detected in P. putida LS46, but was disrupted by an intergenic region of 19922 bp. It bifurcated it into two segments of 4765 and 12176 bp. This 17 kb transposon was a deletion derivative of Tn4661 toluene degradation transposon Tn4651. The 19.9kb inergenic region of Tn3 carried heavy metal resistance genes like czcA efflux protein (PPUTLS46_008564) copA (PPUTLS46_008564), copB (PPUTLS46_08569), copper translocating P-type ATPase (PPUTLS46_008644), blue copper domain (PPUTLS46_008654), two component metal regulator sensor signal (PPUTLS46_8669) and heavy metal sensor signal transduction histidine kinase (PPUTLS46_008674) along with sterol desaturase (PPUTLS46_008539). This transposon was unique and was not present in any other P.putida strains. Tn4651, a toluene transposon, carried genes for toluene degradation (xyl) genes (Tsuda et al. [1989]). P.putida LS46 did not have toluene degradation ability due to deletion of toluene genes in Tn4661. A number of catabolic transposons have been identified in Pseudomonas species for toluene, naphthalene, chlorobenzoate, chlorobenzene and halogenated alkanoates (Wyndham et al. [1994]). Absence of these catabolic transposons in P.putida LS46 limited it metabolic diversity.
Prediction of prophages
Prophage finder (PHAST) identified one intact prophage and one putative phage genes in P. putida LS46 genome. The intact prophage sequence was was present on Contig ALPV02000012 (18373 to 68052) and was 49.6 kb in size with G + C ratio of 59.64%. A total of 61 CDSs which included 40 phage proteins, 21 hypothetical proteins and one tRNA were identified in this prophage. P.putida LS46 prophage showed upto to 73% nucleotide sequence identity to Pseudomonas aeruginosa phage Pseudomonas vB_PaeS_PMG1 which is a virulent phage with lytic infection cycle (Krylov et al. [2012]). A 79 bp attachment sites (attL and attR) were also present. This phage was also present in P.putida BIRD1, P.putida KT2440 and P.putida W619 genomes (Additional file 1: Table S2). However the possible prophages are decided on the basis of number of CDS shared with other prophages. P.putida LS46 shared 15 CDS with Pseudomonas vB_PaeS_PMG1 while P.putida BIRD1, P.putida KT2440 and P.putida W619 had 8, 14 and 15 CDS respectively. The organization of prophage Pseudomonas vB_PaeS_PMG1 genes was unique to P.putida LS46. The putative prophage was only 8.4 kb and had only 9 protein coding sequences. It was similar to SXt2_1717. Other P. putida genomes (from PHAST database http://phast.wishartlab.com) were predicted to contain 1–6 (intact, incomplete, and questionable) prophages (Additional file 1: Table S2). No prophage was detected in P. putida UW4.
Genomic islands in P.putida LS46
Genomic island finder using integrated method identified 22 genomic islands (GIs) in P.putida LS46 (Figure 2). The size of smallest genomic island was 4505 bp while largest was 78290 bp (Figure 2, Additional file 1: Table S3). Genomic island 3 was 26639 bp in size and it showed 96% homology to P.putida H8234 genome (Molina et al. [2013]). This GI carried a transposon Tn4652 along with heavy metal resistance genes. P. putida H8234, a clinical isolate from France, showed low pathogenic potential compared with P. aeruginosa and was resistant to commonly used antibiotics. P.putida LS46 was also resistant to ampicillin, choloramphenicol, gentamycin and tetracycline. GI 18 contains M.XmaI and R.XmaI genes, which are component of XmaI restriction system. Two unique genes PPUTLS46_017749, which encodes an N-4 cytosine-specific methyltransferase, and PPUTLS46_017754, which encode a Type II restriction enzyme, encoded this system. This restriction system is isoschizomer of XcyI of Xanthomonas campestris pv cynopsidis. The XcyI restriction-modification system recognizes the sequence, CCCGGG, and cleaves after C1 and (Withers et al. [1992]). These genes are present on the plasmid AG1 of Xanthomonas axonopodis pv glycine as well as on the plasmid pRA2 of Pseudomonsa alcaligenes NCIB 9867. This plasmid carried two mobile elements, Tn5563 and IS1633 along with Pac25I (XcyI) restriction-modification system (Kwong et al. [2000]). This restriction system did not affect the pRA2 plasmid stability in heterologous Pseudomonas hosts. The other GIs carried a number hypothetical protein.
Figure 2.
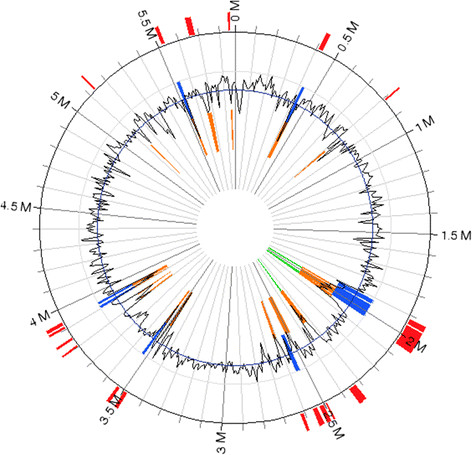
Occurrence of genomic islands inP.putidaLS46 genomes. Genomic islands were identified using IslandViewer programme using Genomic island predictions were calculated for using Integrated method of IslandPick, IslandPath-DIMOB, and SIGI-HMM (Langille and Brinkman ([2009])).
Comparative genome analysis of P.putida strains
The genomes of nine P. putida strains range in size from 5.73 to 6.3 Mb, with an average GC content of 62% (Table 1). Among P. putida strains only strain ND6 carried two plasmids of 117 kb and 101 kb, which encoded an additional 136 and 102 genes, respectively (Table 1). P. putida strains KT2440, BIRD1, GB1, and LS46 had seven copies of 16S rRNA genes, while six copies of 16S rRNA genes were present in strains F1, ND6, UW4, and S16 (Table 1). P. putida strains carried 4171 to 4519 COG counts, representing 69.7% to 82.0% of their genes.
Genome of nine P.putida strains including P.putida LS46 was blasted against P.putida KT2440 (type strain) as the reference strain. Whole genome BLAST identified several homologous regions as well as diverse region in the P. putida strains in comparison to P.putida KT2440. On the basis of whole genome BLASTn analysis of nine P. putida genomes had 86.6% to 97.7% nucleotide sequence identity to the P. putida LS46 genome (Table 2). P. putida DOT-T1E displayed the greatest sequence identity with P. putida LS46, while P. putida UW4 had the least sequence identity. P.putida DOT-T1E was isolated from Spain after enrichment with 1% toluene while P.putida UW4 was isolated as PGPR from Canada (Ramos et al. [1995]; Glick [1995]).
Table 2.
Percent shared genes among different Pseudomonas putida strains
| Strain | LS46 | BIRD1 | DOT-T1E | F1 | GB1 | KT2440 | ND6 | S16 | UW4 | W619 |
|---|---|---|---|---|---|---|---|---|---|---|
| LS46 |
100 |
|
|
|
|
|
|
|
|
|
| BIRD |
92.54 |
100 |
|
|
|
|
|
|
|
|
| DOT-T1E |
91.60 |
91.90 |
100 |
|
|
|
|
|
|
|
| F1 |
93.67 |
92.72 |
93.99 |
100 |
|
|
|
|
|
|
| GB1 |
90.13 |
89.36 |
92.16 |
89.83 |
100 |
|
|
|
|
|
| KT2440 |
91.28 |
89.38 |
89.96 |
90.32 |
87.90 |
100 |
|
|
|
|
| ND6 |
85.67 |
85.13 |
86.96 |
86.90 |
83.13 |
84.31 |
100 |
|
|
|
| S16 |
89.85 |
89.15 |
88.14 |
90.55 |
90.12 |
89.11 |
83.86 |
100 |
|
|
| UW4 |
82.05 |
82.54 |
79.64 |
81.77 |
81.82 |
81.91 |
75.32 |
82.20 |
100 |
|
| W619 | 89.07 | 88.78 | 88.22 | 89.69 | 87.74 | 87.42 | 83.29 | 89.52 | 81.84 | 100 |
Genome arrangement of P. putida genomes
Genome arrangement of P.putida LS46 was compared with 9 other P.putida strains using Dot plot analysis (Figure 3). Dot plot analysis of P.putida LS46 with other P.putida genomes indicated the similarities in genome arrangement at the nucleotide level. The diagonal line showed the co-linearity DNA strands. The blue block on the left hand indicated the translocation and inversions in the genomes. The red blocks represented translocations in anti parallel strands of the genome. The high degrees of genome similarity as well as differences in the arrangement were apparent among P.putida genomes. The genome wide distribution of conserved region of P. putida strains varied from strain to strain. Organization of the P. putida LS46 genome was clearly different than other strains and a number of inversions and translocations were observed the genome in comparison to other P.putida genomes (Figure 3). On the basis of genome arrangement P.putida strains could be divided into two groups. In first group P.putida strains LS46, ND6, F1, KT2440, GB-1, S16 and BIRD1 can be placed which had significantly similarity to P.putida LS46 genome arrangement while in second group comprises of P.putida W619, DOT-T1E and UW4 which had low similarity to P.putida LS46. P.putida LS46 genome arrangement was markedly different from P.putida DOT-T1E, W619 and UW4 with large number translocations and inversions. Pseudomonas putida LS46 genome showed large conserved blocks that are also present in P. putida BIRD1, F1, and ND6 strains, while P. putida DOT-T1E had a large number of small conserved blocks. In comparison to P. putida LS46, the genomes of P.putida KT2440 and S16 genomes had more inversions.
Figure 3.
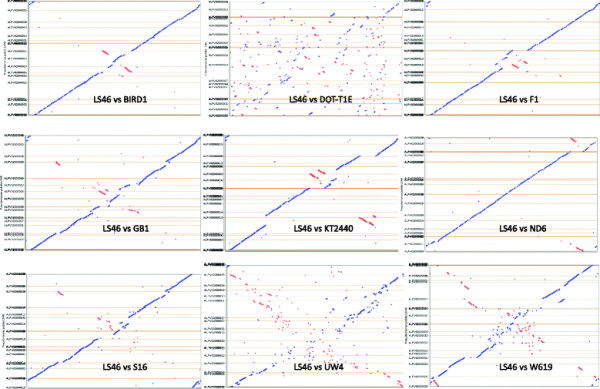
Comparative Synteny Dot plot ofP.putidastrains showing orthologous relationship ofP. putidaLS46 with nineP.putidastrains. The analysis was carried out using the Dot plot from Integrated Microbial Genome (IMG) website.
Homologous and shared gene among P.putida strains
BLASTn analysis of all genes of P. putida LS46 (80% minimal identity e value 1−5) against nine genome identified 82.02- 93.75% homologous genes encoded by the P. putida strains were shared by the ten genomes. P.putida F1 shared highest number (93.7%) of genes with P.putida LS46 while P.putida ND6 (85%) and P.putida UW4 (82%) least number of genes with P.putida LS46 (Table 1). Using single gene profiler 3271 genes were identified which were present in present in all P.putida strains. A total of 8786 core and unique genes were represented the pangenome of nine P. putida strains (excluding P.putida UW4). Unique region in P. putida genomes were identified using pangenome analysis, which identified unique genes present in only one strain (Figure 4). However, all genomes showed higher functional identity (presence of COGs) reflected by high correlation coefficients among the different genomes (r2 = 0.94), although the distribution of different COGs categories among the different genomes was different and represented the functional diversity. The number and percentage of different COG categories varied greatly among ten P. putida strains. P.putida LS46 had highest number of COGs with unknown function. P.putida LS6 genome arrangement was strikingly different from P.putida DOT-T1E however both the strains shared higher percentage of genes (91.6%). Inversely P.putida LS46 and P.putida ND6 had significantly similar genome arrangement but % of shared genes between two genomes was low (85%).
Figure 4.
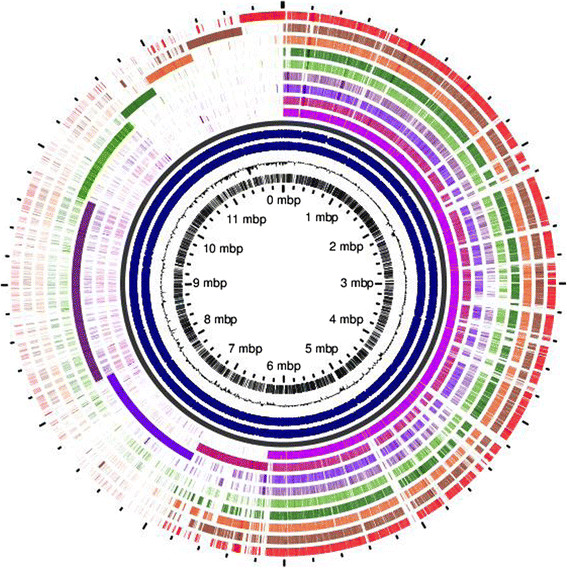
Pangenome analysis of nineP. putidastrains withP. putidaKT2440 as a reference. From inside to outside the circles. COG categories, GC content, backbone, COG in positive strand of pangenome, COG in negative strand in pangenome, P. putida KT2440, P. putida F1, P. putida GB-1, P. putida W619, P. putida S16, P. putida BIRD-1, P. putida ND6, P. putida DOT-T1E and P. putida LS46.
House keeping genes
The 16S rDNA is an universal marker and has been widely used for comparison between divergent bacteria. However, the resolution of 16S rRNA gene sequences at the intrageneric level is low Anzai et al. [2000]; Yamamoto et al. [2000]). Gene sequences for ‘housekeeping’ proteins provide better phylogenetic resolution and have been used to differentiate the genomes of closely related strains. Zeigler ([2005]) identified some house keeping genes for studying genome relatedness among different strains. Comparison of 33 house keeping genes of P. putida LS46 with other nine P.putida strains revealed a high degree of homology among these genes (Additional file 1: Table S4). However, some house keeping genes like dnaX (PPUTLS46_019951), lig (PPUTLS46_022046), pgi (PPUTLS46_007236), uvrC (PPUTLS46_012340,) glyA (PPUTLS46_024688), trpS (PPUTLS46_013403) and trmE (PPUTLS46_016619) were highly diverse among P. putida strains (Additional file 1: Table S4).
Fatty acid and Polyhydroxyalkanoate synthesis
Polyhydroxyalkanoates are produced Pseudomonas putida LS46 from glucose, glycerol, biodiesel glycerol, fatty acids and waste fryer oil (Sharma et al. [2011]). Major components of PHAs produced by P.putida LS46 are either 3-hydroxyoctonoate or 3-hydroxydecanoate depending on the carbon substrate used for PHAs production (Sharma et al. [2011]). There are six genes in the polyhydroxyalkanoate (PHA) synthesis operon (pha) in P.putida. These are phaC1, phaZ, phaC2, phaD, phaF and phaI. The phaC1 and phaC2 are PHA synthases (type II) that incorporate (R)-3-hydroxyacyl-CoA monomers into the PHA polymer (Huisman et al., [1989]). The phaZ encodes a PHA depolymerase, which hydrolyses the PHA monomers, which can be fed into central metabolism for growth (Galán et al. [2011]; de Eugenio et al., [2010a]). The other three genes (phaD, phaF and phaI) are regulatory genes (Arias et al. [2013]; de Eugenio et al. [2010b]; Galán et al. [2011]). The organization of pha operon was identical in all the P. putida strains. The PHA synthesis gene products were highly conserved among all the P. putida strains, with more than 90% amino acid sequence identity (Table 3). The two-polyhydroxyalkanoate synthase genes (phaC1 and phaC2) had only 71% nucleotide identity and 55% aa sequence identity.
Table 3.
Homology of proteins associated with polyhydroxyalkanoate production
| Name | Locus tag | Enzyme | Product size (aa) |
P.putida
strains |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BIRD1 | DOT | F1 | GB1 | KT2440 | ND6 | S16 | W619 | ||||
| FadD |
07414 |
Acyl-CoA synthetase |
565 |
99 |
100 |
99 |
94 |
99 |
99 |
94 |
87 |
| FadA |
04404 |
3-ketoacyl-CoA thiolase |
392 |
99 |
100 |
100 |
99 |
99 |
100 |
99 |
100 |
| FadAx |
07424 |
Acetyl-CoA acetyltransferases |
380 |
99 |
99 |
99 |
98 |
99 |
99 |
98 |
96 |
| FadB |
00695 |
3-hydroxyacyl-CoA dehydrogenase/epimerase |
715 |
98 |
93 |
99 |
94 |
94 |
99 |
93 |
95 |
| FadB2 |
07419 |
3-hydroxyacyl-CoA dehydrogenase |
255 |
99 |
100 |
100 |
97 |
100 |
99 |
94 |
96 |
| FadD2 |
21046 |
Acyl-CoA synthetase |
562 |
99 |
99 |
99 |
98 |
99 |
99 |
96 |
93 |
| FadE |
00145 |
Acyl CoA dehydrogenase |
601 |
98 |
93 |
99 |
94 |
94 |
99 |
93 |
95 |
| FadE2 |
05901 |
Acyl CoA dehydrogenase |
592 |
95 |
99 |
99 |
95 |
98 |
99 |
95 |
94 |
| PhaC1 |
05621 |
Poly(R)-hydroxyalkanoic acid synthase |
560 |
99 |
99 |
98 |
99 |
99 |
99 |
98 |
94 |
| PhaZ |
05616 |
Poly(3-hydroxyalkanoate) depolymerase |
283 |
99 |
100 |
100 |
99 |
99 |
99 |
97 |
95 |
| PhaC2 |
05611 |
Poly(R)-hydroxyalkanoic acid synthase |
559 |
99 |
99 |
99 |
96 |
99 |
100 |
99 |
95 |
| PhaD |
05606 |
Transcriptional regulator |
204 |
100 |
99 |
96 |
99 |
99 |
99 |
97 |
95 |
| PhaF |
05601 |
PHA granule associated protein |
253 |
99 |
100 |
100 |
97 |
99 |
100 |
98 |
94 |
| PhaI |
05596 |
PHA granule associated protein |
139 |
99 |
100 |
96 |
93 |
96 |
96 |
91 |
91 |
| PhaG |
13888 |
Hydroxyacyl-ACP:CoA transacylase |
295 |
99 |
99 |
99 |
98 |
99 |
100 |
94 |
89 |
| FabD |
00250 |
malonyl CoA-acyl carrier protein transacylase |
312 |
99 |
99 |
100 |
96 |
99 |
100 |
98 |
92 |
| FabA |
12710 |
3 hydroxyacyl(decanyl)-ACP dehydratase |
171 |
100 |
100 |
100 |
99 |
100 |
100 |
98 |
92 |
| FabB |
12715 |
2 -Oxoacyl carrier protein synthase |
406 |
100 |
99 |
99 |
99 |
99 |
99 |
99 |
99 |
| FabG |
23353 |
3-ketoacyl-(acyl-carrier-protein) reductase |
450 |
99 |
99 |
99 |
96 |
99 |
99 |
95 |
90 |
| FabG_2 |
00255 |
3-oxoacyl-(acyl-carrier-protein) reductase |
450 |
99 |
100 |
99 |
99 |
100 |
99 |
100 |
99 |
| FabG_3 |
15924 |
3-oxoacyl-(acyl-carrier-protein) reductase |
450 |
100 |
100 |
100 |
97 |
99 |
99 |
94 |
91 |
| FabZ | 14589 | 3-hydroxyacyl-[acyl carrier protein] dehydratase | 146 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | 95 |
The phaC1 and phaC2 of different Pseudomonas species formed different clusters in neighbor joining tree. The phaC1 and phaC2 in P.putida strains were highly conserved but were different phaC1 and phaC2 from other Pseudomonas species. The phaC1 and phaC2 genes of P. putida, P.aeruginosa, P.fluorescens, P.stutzeri. P.entomophila and P.mendocina formed different cluster in neighbor joining tree (Figure 5). Further fatty acid biosynthesis and fatty acid degradation proteins were highly conserved among P.putida strains. Fatty acid biosynthesis (fab) and fatty acid degradation (fad) gene products provide the precursor for PHAs synthesis. Most of the fatty acid synthesis and degradation proteins of P.putida LS46 had multiple genes coding isozymes i.e. FadB had 4 isologs, FadA had 5 isologs and FadD had 7 isologs for short and long chain fatty acids. Likewise FadE (acyl-CoA dehydrogenase) had six isologs specific for small, medium and long chain fatty acids. Fab and Fad proteins of P.putida LS46 showed high homology to Fab and Fad proteins of other P.putida strains. P.putida LS46 can utilize fatty acids (C5-C18) for PHAs production. Two fatty acid transporters (FadL), one for short chain fatty acid (PPUTLS46_007654) and other for long fatty acid (PPUTLS46_015009) were present in P.putida LS46. However, it preferentially used long chain fatty acids (C6-C18) than short chain fatty acid C3-C5) for PHAs synthesis. The specificity of FadD and FadL these proteins are not known but transfer of FadD from E.coli and FadL from P.putida into Aeromonas hydrophila improved its ability to utilize C6 and C8 fatty acids. (Jian et al. [2010]). The intermediate 3hydroxyacyl-ACP of de novo fatty acid synthesis is converted to 3hydroxyacyl-CoA for polymerization to PHAs with help of (R)-3-hydroxyacyl-ACP:CoA transacylase enzyme PhaG. PhaG was present in all P.putida strains. The absence of PhaG in PHB producers limits their ability to produce mcl-PHAs. Transfer and expression of phaG of P.putida in Ralstonia eutropha or Aeromonas hydrophila producer may confer mcl-PHAs production ability into PHB producers.Recently, a PHAs granule-associated acyl-CoA-synthetase (Acs1) has been identified which is highly conserved among P.putida strains. It directs the carbon flux of these central metabolites towards PHA accumulation and converts 3hydroxyalkanoic acids to 3 hydroxyacyl-CoA thioesters (Ruth et al. [2008]).
Figure 5.
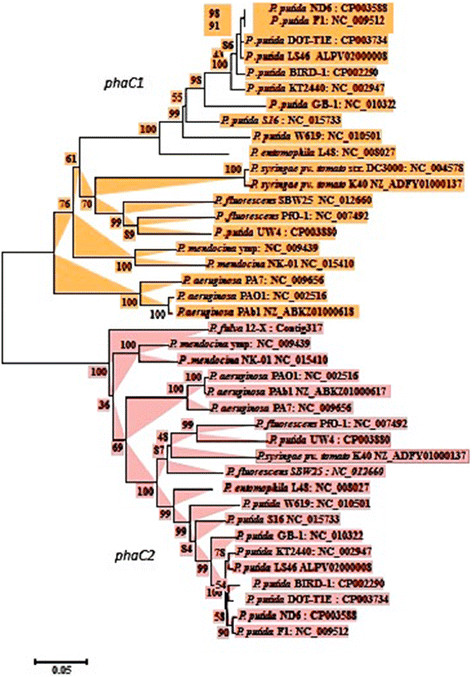
Phylogenetic tree depicting the relationship ofphaC1andphaC2genes amongPseudomonasspecies. The phaC gene sequences were aligned by ClustalW and a neighbor-joining tree was generated using MEGA5 program. Bootstrap values are mentioned at the node.
Comparison of common metabolic pathways of P. putida strains
Metabolic diversity
The compared P. putida strains were isolated from diverse environment for bioremediation of xenobiotics or decomposition of other materials. Different P. putida strains have unique genes associated with specific functions and these genes are not present in other strains. P. putida KT2440 has been considered as a metabolically diverse saprophytic bacterium. Analysis of the P. putida KT2440 genome identified 18 dioxygenase, 51 putative hydrolases, 40 dehydrogenases, and more than 62 transferases (Jimenez et al. [2002]; Molina-Henares et al. [2009]; Nelson et al. [2002]). By way of comparison, the P. putida LS46 genome had 22 dioxygenases (Additional file 1: Table S5), 75 hydrolases, 149 dehydrogenases, and 43 transferases. All the three dioxygenases, benzoate dioxygenase (PPUTLS46_007854, PPUTLS46_007859), catechol dioxygenase PPUTLS46_007879), and protochatuate dioxygenase (PPUTLS46_010694) were present in P. putida LS46. These dioxygenases were earlier identified in P.putida KT2440 and are involved in metabolism of aromatic compounds (Nelson et al. [2002]). P.putida LS46 had 22 dioxygenases genes. One of the dioxygenase genes (PPUTLS46_007879 glyoxalase/bleomycin resistance protein/dioxygenase) was unique to P.putida LS46 and was not present in other P.putida strains (Additional file 1: Table S5).
The majority of the genes for degradation of ferulate, coniferyl, and coumuryl alcohol, aldehydes and acids, p-hydroxybenzoate, and protocatechuate present in P. putida KT2440 were also present in P. putida LS46 (Additional file 1: Table S6). These genes were reported to have role in degradation of aromatic/aliphatic sulphonate, benzoate, toulate, catechol, hippurate, maleate, phenylalanine, phenylacetic acidprotocatechuate, quinate, taurine etc. The notable exceptions in P. putida LS46, however, were ferulic acid transferase (PP_3354), transferuloyl CoA hydratase (PP_3358), and vanillate dehydrogenase (PP_3357). These genes were present in P. putida F1 (Pput_2404, Pput_2400, Pput_2401) and P. putida W619 (PputW619_2051, PputW619_2047, PputW619_2048), and are associated with transformations of ferulic acid to vanillin and vanillin to protocatechuate. Another gene, which was only reported in P. putida KT2440, and that is missing from all other P. putida strains, was propanediol dehydrogenase (PP_2803).
P. putida F1 is another metabolically diverse strain that has been demonstrated to oxidize toluene, 3-hydroxyphenyl propionate, and cymene (Finette and Gibson [1988]; Zylstra et al. [1988]). The genes that encode the enzymes for these reactions were also present in P. putida DOT-T1E, which encodes the genes (TIE_4277-T1E-4240) for degradation of toluene, 3- hydroxyphenylpropoinate, and cymene (Additional file 1: Table S8), but absent in P. putida LS46 and 7 other P. putida strains. As reported earlier the genes for degradation of 3 hydroxyphenylpropionate were also present in P.putida W619 (Wu et al. [2011]).
P. putida KT2440 can use aromatic or aliphatic sulphonates as sulphur sources. The genes for degradation of aromatic or aliphatic sulphonates are encoded in the ssuFBCDEA operon (PP_0241-PP_0235) in P. putida KT2440. This operon was present in P. putida LS46 (PPUTLS46_024748, PPUTLS4_025163, 025168, 025173, 025178, 025183) and all other P. putida strains (Additional file 1: Table S6). Three chlorohyrolases i.e. atrazine chlorohydrolase (PP5036, PP2584) and hydroxydechloroatrazine ethylaminohydrolase (PP3209) detected in P. putida KT2440 were also identified in P. putida LS46 (PPUTLS46_10994, PPUTLS46_005456, and PPUTLS46_008014). However, chloride channel protein of P. putida KT2440 (PP3959) was absent in P. putida LS46. Pak et al. ([2000]) identified xenobiotic reductase from P. fluorescens for the transformation of 2, 4, 6-trinitritoluene (TNT). Its homologue was identified in P. putida KT2440 (PP_0920) (Nelson et al. [2002]) and P. putida LS46 (PPUTLS46_022986). These genes was also present in other eight P. putida strains, but was absent from P. putida UW4.
Pseudomonas putida DOT-T1E is a solvent-tolerant strain able to grow in the presence of > 1% (v/v) toluene in the culture medium. Its multidrug efflux pumps have been found to play a major role in toluene-tolerance (Rojas et al. [2001]). This ability is mainly conferred by an efflux pump encoded in a self-transmissible 133 kb plasmid named pGRT1 (Molina et al. [2011]). No plasmid was detected in P. putida LS46, but the genes encoding an efflux pump (sepA, sepB, sepC), were present in the P. putida LS46 (PPUTLS46_0133778, 013773, 013768) genome, as reported earlier for P. putida strains KT2440, GB1 and W619 (Wu et al. [2011]).
Nicotinic acid and nicotine degradation
Nicotinic acid (NA) is a carboxylic derivative of pyridine that is widely distributed in nature as part of pyridine cofactors (NAD and NADP) and alkaloids (e.g., nicotine and anabasine), and it is essential (vitamin B3) for those organisms that are not able to carry out its synthesis. In bacteria and fungi, NA is used as a carbon source. The biochemical pathways involved in the degradation of NA was elucidated (Kaiser et al. [1996]). Jiménez et al. ([2008]) identified a nic gene cluster (PP_3939 - PP_3948) in P. putida KT2440 responsible for aerobic nicotine degradation. These genes were also present in P. putida strain F1, GB1, and W619 as reported earlier Jiménez et al. ([2008]). All the genes of nic operon were also present in P. putida LS46, BIRD1, ND6, DOT-T1E, and these genes were homologous to genes (89.6% to 100% nucleotide identity) from P. putida KT2440 (Additional file 1: Table S7).
Recently, the genome sequence of another nictotine degrading P. putida, strain S16, was released (Tang et al. [2012]). P. putida S16 degrades nicotine through pyrrolidine pathway. A gene cluster containing six genes encoding PPS_0380 HSP hydroxylase (hspB), PPS_4060 maleate isomerase (iso), PPS_4059 NFM deformylase (nfo), PPS_4058 DHP dioxygenase (hpo), PPS_4057 maleamate amidase (ami), and PPS_4061 6-hydroxynicotinate 3-monooxygenase (hna) was identified in P. putida S16 and this gene cluster is designated as nic2 cluster. This gene cluster was present on a large genomic island. Both nicotine and nicotinic acid produced 2,5- dihydroxypyridine (2,5-DHP) as an intermediate, which is converted to N-formylmalaemic acid. The N-formylmalaemic acid is degraded to maleic acid and then to fumaric acid as in nicotinic degradation pathway (Wang et al. [2007]). BLAST analysis indicated that these genes were also present in P. putida LS46 (PPUTLS46_011665 (ami), 11670 (iso), 11675 (nfo), 11685 (hpo) and showed 36.63, 69.35, 55.64 43.73% homology to respective proteins of P.putida S16. Other P.putida strains showed low homology to the P. putida S16 nic2 cluster (36.6-65-3%). No homologue of PPS_0380 HSP hydroxylase (hspB) was present in P.putida LS46 (Additional file 1: Table S8). The nic cluster genes of P. putida KT2440 showed high nucleotide sequence identity (>95%) to corresponding genes of P.putida LS46, as reported earlier in P. putida strain W619, F1, and GB1 (Molina et al. [2011]) [59]. P. putida KT2440, however, is not able to degrade nicotine. The nic cluster of P. putida S16 had a GC content of only 48% compared with the 62% GC content of the rest of the P. putida genome sequence. The nic gene cluster for the nicotinic acid degradation pathway of P. putida KT2440 was also detected in P.putida BIRD1, P. putida ND6, and P. putida DOT-T1E, but was absent in the genome of P.putida strain UW4.
Heavy metal resistance
The level of heavy metal tolerance is very high in P. putida W619 in comparison to other P. putida strains (Canovas et al. [2003]; Taghavi et al. [2009]). In P. putida W619, copper, cobalt, zinc, cadmium, arsenate, mercury, nickel chromate, and molybdenum resistance genes are present in two genomic regions (1 and 31). Heavy metal tolerance of P. putida LS46 has not been investigated, but this strain encodes genes for heavy metal resistance present in genomic region 1 of P. putida W619. These genes are involved in copper, cobalt, zinc, and cadmium resistance. However, the genes located on genomic region 31 of P. putida W619, involved in copper and chromate resistance, were absent in the majority of P. putida strains, including P. putida LS46 (Additional file 1: Table S8). All P. putida strains lacked the genes present in genomic region 18 of P. putida W619, which were associated with mercury and nickel resistance.
Manganese oxidation
P. putida GB1 is a Mn (II) oxidizing bacterium (Buzzo [2011]) and attempts have been made to identify the genes related with Mn (II) oxidation by transposon mutagenesis. However, the role of different genes in Mn (II) oxidation is not clear (Caspi et al. [1998]; Brouwers et al. [1999]). Recently, using transposon mutagenesis, Geszvain et al. ([2013]) and Geszvain and Tebo ([2009]) identified two genes, which are homologous to multi-copper oxidases (PputGB1_2447 and PputGB1_2665) in P. putida GB1. The genes encoding multi-copper oxidase homologues of P.putida GB1 were also present in P. putida LS46 (PPUTLS46_002532 and PPUTLS46_006964), as well as in P. putida strains KT2440, F1, ND6, W619, S16, UW4, and DOT-T1E. Only one multi-copper oxidase gene was identified in P. putida BIRD1 (Additional file 1: Table S8).
Iron scavenging genes in P. putida strains
It is well known that iron deficiency in bacteria limits growth Andrew et al. ([2003]). Pseudomonas produces low molecular weight iron-chelating compounds termed ferri-siderophores to scavenge iron (Wiener [2005]). Ferri-siderophores are taken up via outer membrane receptors, which function as gated porin channels (Koebnik [2005]; Ratledge and Dover [2000]). After binding of the ferri-siderophores, transport is mediated by a complex of inner membrane-anchored proteins TonB (Wiener [2005]). Fluorescent pseudomonads respond to iron-deficiency by secreting the yellow-green fluorescent peptidic siderophores called pyoverdines (Meyer [2000]; Cornelis et al. [2009]). Pyoverdines have a conserved chromophore with a variable peptide chain, and each Pseudomonas species produces a different pyoverdine (Ravel and Cornelis [2003]; Visca et al. [2007]). Pyoverdine is synthesized by non-ribosomal peptide synthetase (NRPS). Like other P. putida strains, genes for pyoverdine synthesis were also present in P. putida LS46 (PPUTLS46_025659 and PPUTLS46_012775). The pvdE gene (PPUTLS46_012775) encoded a PVD ABC transporter, as in other P. putida strains. P. putida LS46, like the other P.putida strains did not carry the genes for pseudomonine production, which was reported for P. entomophila L48 (Vodovar et al. [2006]).
The number and type of TonB siderophore receptors show their diversity to survive in the different environment (Cornelis and Bodilis [2009]). TonB-dependent receptor genes are not constitutively expressed and are regulated by the iron availability (Koebnik et al. [2000]; Bodilis et al. [2009]) and their expression provides additional mechanisms to survive under different niches. Twenty-six (26) TonB siderophore receptor genes were present in the P. putida LS46 genome (Additional file 1: Table S9). Across all of the P. putida strains, 19–53 TonB genes are present. Comparison of TonB genes (at 80% nucleotide sequence identity) of P. putida LS46 identified 26, 28, 26 20, 24, 26, 19, 1 and 15 homologous TonB genes in P. putida LS46 strain BIRD1, DOT-T1E, F1, GB1, KT2440, ND6, S16, UW4 and W619, respectively. P.putida strain UW4 had 11 TonB genes, which showed low homology (50% nuclotide sequence identity) to other TonB genes of P. putida strains. The greatest number of TonB genes (55) was present in P. putida strain GB1.
Discussion
Pseudomonas putida strains have been identified as root colonizing bacterium and developed as biocontrol agent. Other P.putida strains due to their diverse metabolic potential have been developed as bioremediation agents. Potential of P.putida as polyhydroxyalkanoates (PHAs) producer was identified in 1989 (Huisman et al. [1989]). However, PHAs producing P.putida strain KT2440 was originally isolated as a root colonizing bacterium (Nelson et al. [2002]). A number of P.putida strains were isolated and developed as bioremediation agents but no other P.putida strain was isolated for PHAs production. P.putida LS46 was isolated and screened for PHAs production and it was proved as a good PHAs producer as P.putida KT2440. Complete genome of this bacterium was sequenced to know the similarities and differences among biocontrol, bioremediation and PHAs producing P.putida strains.
P. putida LS46 strain showed more than 99% identity to 16S rRNA gene of P.putida strains irrespective of their geographic origin or their application. P.putida classifications based on protein-encoding genes like gyrB, rec, rpoB, recN, and cpn60 alongwith 16S rRNA gene have earlier been proposed (Adékambi et al. [2009]; Arahal et al. [2008]; Zeigler [2005]; Mulet et al. [2010,2013]). Cpn60 (Hsp60 or GroEL), a highly conserved protein found in bacteria, has been widely used for phylogeny, microbial identification, microbial ecology and evolution (Hill et al. [2002,2004]). The phylogeny of P. putida strains based on cpn60 confirmed the close relationship among P. putida strains, strengthening the 16S rRNA gene based phylogeny. However, the cpn60 gene phylogeny was more robust and separated strains that were clustered together in the 16S rRNA gene phylogeny. The cpn60 (UT) analysis differentiated P. putida W619, P. putida S16, and P. putida UW4 from other P. putida strains. P. putida W619 was isolated as an endophyte of Poplar, P. putida UW4 was isolated as a plant growth promoting species, and P. putida S16 was isolated as a nicotine degrading species. Recently cpn60 (UT) was used for studying the phylogenetic relationship among Aeromonas, Themoanaerobacter, and vaginal microbiota (Miñana-Galbis et al. [2009], Verbeke et al. [2011]; Schellenberg et al. [2011]).
The phylogeny of P. putida UW4 was described on the basis of 16S rRNA genes and a multilocus approach that used four concatenated housekeeping genes (16S rRNA, gyrB, rpoD and rpoB). P. putida UW4 was found to be closely related to P. fluorescens rather P. putida strains including P. putida LS46 (Duan et al. [2013]). It confirmed our observations on the phylogeny of P. putida UW4 based on the cpn60 gene that P. putida UW4 was clustered with P. fluorescens. Earlier, Loper et al. ([2012]) on the basis of ten concatenated house keeping genes, showed similar phylogenetic relationships among Pseudomonas species. The cpn60 gene phylogeny placed the ten sequenced P. putida strains and P. entomophila in the same clade. The phylogeny of the newly isolated P. putida LS46 based on cpn60 differentiated this strain from other closely related P.putida strains, and based on both the 16S rRNA and cpn60 analyses, P. putida LS46 was closely related to type strain P. putida KT2440. No plasmids have been detected in P. putida LS46, but two plasmids were present in P. putida ND6 and one plasmid was identified in P. putida DOT-T1E (Li et al. [2002]; Udaondo et al. [2012]).
Variation in genome content is thought to be a key factor in the evolution of bacteria and variation in genome arrangement may also improve the fitness of the bacteria (Silby et al. [2011a,b]). Presence of duplicated genes like rRNA operons, multiple transposons, insertion sequences, prophages and genomic islands leads to genome rearrangements in bacteria that contribute to evolution (Tillier and Collins [2000]; Klockgether et al. [2010]). Rearrangements in genome are not random, but predominantly occur at end-points either at the origin or at the terminus of replication (Eisen et al. [2000]).
Overall, strains of P. putida isolated from different geographical regions and from varied ecological niches had high similarity in genome structure and functions (Molina et al. [2011]; Li et al. [2012]; Nelson et al. [2002]; Wu et al. [2011]). All the P. putida genomes had higher level of genome similarity, but differed from other on the basis of insertion sequence, presence of prophage and genomic islands. However, Dot plot analyses of the P. putida strain genomes identified a number of rearrangements, which are possibly due to presence of these genetic elements, such as the presence of ISPa42 in P. putida LS46, which originated from P. aeruginosa DK3. ISPa42 may be associated with the acquisition of genes for heavy metal resistance.
In comparison to P. putida LS46, genome inversions were observed in P. putida BIRD-1 and P. putida W619 at the origin and terminus of replication in P. putida S16 and P. putida ND6, respectively. These observations strongly support the contention that bacterial genomes are not static and significant variations are observed even among strains within the same species. These variations are the result of genome deletions and/or gene acquisitions by horizontal gene transfer of elements such as transposon and genomic islands (Hacker and Carniel [2001], Mackiewicz et al. [2001]).
P. putida LS46 and P. putida DOT-T1E had a maximum sequence identity of 97.7%, but their genome arrangements were very diverse (Figure 2). It appears that the genome of P. putida DOT-T1E has undergone a number of rearrangements without losing any major function. P. putida DOT-T1E was isolated as toluene degrading species, and has acquired genes for toluene degradation that are absent in P. putida LS46. Using BLAST analysis 81.84-93.99% genes were identified which were shared among the 10 P. putida strains, while the rest of the genes were unique to the strain. Ballerstedt et al. ([2007]), using specific gene probe of P. putida KT2440 in a microarray identified 67.8-100% identical genes in six P. putida strains. Acquisition of specific groups of genes conferred specialized functions to specific strains, but their core genomes were identical.
Three P. putida strains, KT2440, BIRD1, and UW4, were isolated from soil and developed as plant growth promoting bacteria. P. putida strains F1, ND6, DOT-T1E, S16, GB1, and W619 were identified as bioremediation agents on the basis of their capacity to degrade pollutants. P. putida strain LS46 was isolated from wastewater for PHA production. This strain was able to use glucose, glycerol, fatty acids, and waste fryer oil and accumulates PHAs to 20-58% of the cell dry weight. Like other P. putida strains, central metabolic pathways for utilization of these substrates were also present in P. putida LS46. The application based classification of P. putida as biocontrol, bioremediation, or PHA producer is not relevant because biocontrol agent BIRD-1, or bioremediation strains F1, ND6, and DOT-T1E also encode the genes for PHA synthesis and homology among these genes is very high. The most of the genes present in the manganese oxidizing strain P. putida GB-1 or the endophyte of poplar stain P. putida W619 are also present in P. putida LS46.
Complete genome sequence of P. putida LS46 was compared with P. putida strains isolated from different geographical regions and different niches. Two strains, P. putida ND6 and P. putida DOT-T1E isolated from China, and P. putida F1 isolated from USA, had the highest sequence identities to P. putida LS46, which was isolated in Canada. P. putida strains GB-1 isolated from USA, UW4 isolated in Canada, and S16 isolated from China, had low sequence identities with P. putida LS46. P. putida W619, which was isolated as an endophyte of poplar had only 87.1% sequence identity with the P. putida LS46 genome, while P. putida UW4 showed only 82.0% identity with P. putida LS46. Our earlier result on phylogeny of P.putida UW4 has clearly demonstrated that this strain is related to P. fluorescens rather than P. putida. The low genome identity of P. putida strains with P. putida UW4 is not surprising. High genome similarities among P. putida strains isolated from different regions indicate their common ancestry (Biello [2008]).
P. putida LS46 shares a number of metabolic features with P. putida KT2440, P. putida F1, P. putida BIRD1, and P. putida GB-1, such as metabolism of aromatic compounds, manganese oxidation, root colonization, and PHAs production. All P. putida genomes have the ability to synthesize PHA irrespective of their applications, either as biocontrol agents or bioremediation agents. P. putida LS46 differs from other P. putida strains in number of dioxygenase genes, TonB dependent receptors, transferases, hydrolases, dehydrogenases and transferases. P. putida LS46, like other P. putida strains metabolize glucose, glycerol, and fatty acid by glycolysis, the tricarboxylic acid cycle, the pentose pathway, and β oxidation (Figure 6). At least three different metabolic pathways provide the precursors for the synthesis of PHAs. (i) Fatty acid de novo biosynthesis is the main route during growth on carbon sources that are metabolized to acetyl-CoA, like glucose, gluconate, glycerol etc. (ii) β-oxidation is the main pathway for PHAs production when fatty acids are used as carbon source, (iii) chain elongation reactions in which acetyl-CoA moieties are condensed to 3-hydroxyacyl-CoA is involved in the PHA synthesis when small chain length fatty acids like C6 and C7 are used. Intermediates of fatty acid de novo synthesis (3 hydroxylacyl-ACP) as well as fatty acid degradation pathways (3-hydroxylacyl-CoA) are used as precursors for PHAs production. Rehm et al. ([1998]) identified a link between fatty acid degradation and fatty acid synthesis by confirming the PHAs biosynthesis in β-oxidation defection mutants (fadB). This enzyme converts 3hydroxyacyl-ACP to 3-hydroxyacyl-CoA, which is a substrate for polymerization to PHAs. Glucose and glycerol are transported and metabolized to acetyl-CoA, which is further used for production of various fatty acids using fatty acid biosynthesis (fab) genes. Fatty acids or waste fryer oil containing long chain fatty acids (C16 and C18) are utilized by β-oxidation and intermediates are used for PHAs production (Wang et al. [2012]). Manipulation of fatty acid synthesis and degradation genes is known to improve PHAs production with altered monomer composition (Fiedler et al. [2002]). PHAs production in P.putida is a part of central metabolic pathway and it was evident from high level of identity among PHAs production proteins and proteins of feeding pathways like fatty acid biosynthesis and degradation.
Figure 6.
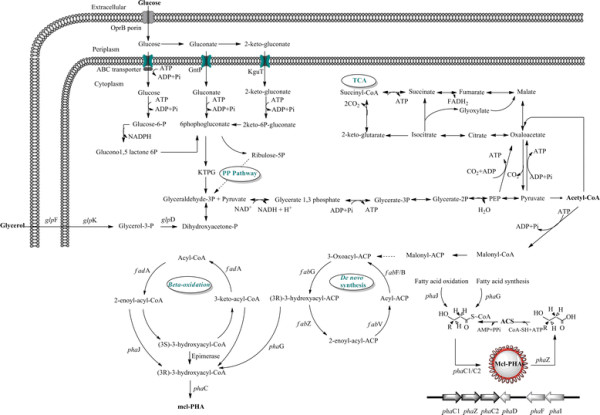
Proposed PHA synthesis and degradation pathways forP. putidaLS46. Metabolic pathways involved carbon metabolism, fatty acid synthesis and degradation, and PHA synthesis are summarized. PHA can be synthesized from glucose, glycerol, or fatty acids via the PHA synthesis pathway.
P. putida LS46 does not match, however, the metabolic diversity of either P. putida F1 or P. putida DOT-T1E, which encode genes for degradation of toluene, cymene, and 3-hydroxyphenylpropionate, or P. putida ND6, which encodes genes for naphthalene degradation. These genes may have been acquired during the evolution process. A number of transposons and insertion sequences were present in P. putida genomes indicating their labile nature. ISPpu10, a transposon present in REP sequence of P. putida KT2440 has been implicated in genome rearrangement (Ramos-Gonzalez et al. [2006]). But this transposon was absent from all P. putida strain except P. putida S16. Mg (II) oxidation (P. putida GB1), pyoverdine production, aromatic/aliphatic sulphonate degradation (P. putida KT2440), and polyhydroxyalkanoate production (P. putida strains KT2440 and LS46) are core genes that are present in all P. putida strains whether these strains were isolated as biocontrol agents or as bioremediation agents (Wu et al. [2011]). The genome of P. putida LS46 had a majority of the genes involved in plant growth promotion even it was not isolated as plant growth promoter and in this regards it was similar to P. putida KT2440 and P. putida BIRD1. Several unique genes were identified in the P. putida LS46 genome. The presence of prophages, insertion sequences, and genomic islands in different genomes indicate that the P. putida genome is not static, but changes by acquiring new genes from related and unrelated species.
Competing interests
The authors declare that they have no competing interests, financial or otherwise.
Authors' contributions
The nucleotide sequence alignments, genome analyses, and preparation of the manuscript draft were carried out by PKS. JF assisted PKS with the genome analyses. The genome assembly and annotation, and uploading of files to IMG were carried out by XLZ, who was supervised by BF. RS and DBL are the co-PIs for the Genome Canada funded research program, and DBL served as PKS’s direct supervisor. DBL also prepared the final draft of the manuscript for submission. All authors read and approved the final manuscript.
Additional file
Supplementary Material
Occurrence of IS elements in different P.putida strains. Table S2: Predicted prophages in P. putida genomes. Table S3: Homology of house keeping genes encoded in the genomes of different P. putida strains. Table S4: Homology of house keeping genes encoded in the genomes of different P. putida strains. Table S5: Occurrence of different dioxygenases encoded in the P.putida LS46 genome and their homologues encoded in the genomes of other P. putida strains. Table S6: Presence of genes encoded in the P. putida KT2440 genome associated with different metabolic pathways in other P.putida strains. Table S7: Presence of genes encoded by the P. putida F1 genome involved in aromatic compound degradation and their homologues encoded in the genomes of other P. putida strains. Table S8: Occurrence of heavy metal tolerance genes encoded by P. putida W619 in different P. putida strains. Table S9: Identification of TonB receptors in P. putida LS46 and their homologues in other P. putida strains.
Contributor Information
Parveen K Sharma, Email: pksharmajan11@yahoo.com.
Jilagamazhi Fu, Email: umfu23@myumanitoba.ca.
Xiangli Zhang, Email: xiangLi.zhang@umanitoba.ca.
Brian Fristensky, Email: brian.fristensky@umanitoba.ca.
Richard Sparling, Email: richard.sparling@umanitoba.ca.
David B Levin, Email: david.levin@umanitoba.ca.
Acknowledgements
This work was supported by funds provided by the Natural Sciences and Engineering Research Council of Canada (NSERC), through a Strategic Programs grant (STPGP 306944–04), by Genome Canada, through the Applied Genomics Research in Bioproducts or Crops (ABC) program for the grant titled, “Microbial Genomics for Biofuels and Co-Products from Biorefining Processes”, and by the Province of Manitoba, through the Manitoba Research Innovation Fund (MRIF).
References
- Adékambi T, Drancourt M, Raoult D. The rpoB gene as a tool for clinical microbiologists. Trends Microbiol. 2009;17:37–45. doi: 10.1016/j.tim.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Andrews SC, Robinson AK, Rodriguez-Quinones F. Bacterial iron homeostasis. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- Anzai Y, Kim H, Park J, Wakabayashi H, Oyaizu H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int J Syst Evol Microbiol. 2000;50:1563–1589. doi: 10.1099/00207713-50-4-1563. [DOI] [PubMed] [Google Scholar]
- Arahal DR, Sanchez E, Macian MC, Garay E. Value of recN sequences for species identification and as a phylogenetic marker within the family “Leuconostocaceae”. Int Microbiol. 2008;11:33–39. [PubMed] [Google Scholar]
- Aranda-Olmedo I, Tobes R, Manzanera M, Ramos JL, Marques S. Species-specific repetitive extragenic palindromic (REP) sequences in Pseudomonas putida. Nucleic Acids Res. 2002;15:1826–1833. doi: 10.1093/nar/30.8.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias S, Bassas-Galia M, Molinari G, Timmis KN. Tight coupling of polymerization and depolymerization of polyhydroxyalkanoates ensures efficient management of carbon resources in Pseudomonas putida. Microbial Biotech. 2013. 10.1111/1751-7915.12040 [DOI] [PMC free article] [PubMed]
- Ballerstedt H, Volkers RJM, Mars AE, Hallsworth JE, dos Santos VA, Puchalka JJ, Eggink G, Timmis KN, de Jan AM, Bont JAM, Wery J. Genomotyping of Pseudomonas putida strains using P. putida KT2440-based high-density DNA microarrays: implications for transcriptomics studies. Appl Microbiol Biotechnol. 2007;75:1133–1142. doi: 10.1007/s00253-007-0914-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett S. Solexa Ltd. Pharmacogenomics. 2002;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- Biello D. Do microbes make snow? Scientific America 28Feb2008. 2008.
- Bodilis J, Ghysels B, Osayande J, Matthijs S, Pirnay JP, Denayer S, De Vos D, Cornelis P. Distribution and evolution of ferripyoverdine receptors in Pseudomonas aeruginosa. Environ Microbiol. 2009;11:2123–2135. doi: 10.1111/j.1462-2920.2009.01932.x. [DOI] [PubMed] [Google Scholar]
- Brandl H, Gross RA, Lenz RW, Fuller RC. Pseudomonas oleovorans as a source of poly(3-hydroxyalkanoates) for potential application as biodegradable polyesters. Appl Environ Microbiol. 1988;54:1977–1982. doi: 10.1128/aem.54.8.1977-1982.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers GJ, de Vrind JP, Corstjens PL, Cornelis P, Baysse C, de Jong EW. cumA, a gene encoding a multicopper oxidase, is involved in Mn21 oxidation in Pseudomonas putida GB-1. Appl Environ Microbiol. 1999;65:1762–1768. doi: 10.1128/aem.65.4.1762-1768.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzo J. New insights in Mn(II) oxidation in Pseudomonas putida GB-1. Oregon Health & Science University, Portland, OR, M.S. thesis; 2011. [Google Scholar]
- Canovas D, Cases I, de Lorenzo V. Heavy metal tolerance and metal homeostasis in Pseudomonas putida as revealed by complete genome analysis. Environ Microbiol. 2003;5:1242–1256. doi: 10.1111/j.1462-2920.2003.00463.x. [DOI] [PubMed] [Google Scholar]
- Caspi R, Tebo BM, Haygood MG. C-type cytochromes and manganese oxidation in Pseudomonas putida MnB1. Appl Environ Microbiol. 1998;64:3549–3555. doi: 10.1128/aem.64.10.3549-3555.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EN, Cho MC, Kim Y, Kim CK, Lee K. Expansion of growth substrate range in Pseudomonas putida F1 by mutations in both cymR and todS, which recruit a ring-fission hydrolase CmtE and induce the tod catabolic operon, respectively. Microbiology. 2003;149:795–805. doi: 10.1099/mic.0.26046-0. [DOI] [PubMed] [Google Scholar]
- Compant S, Duffy B, Jerzy Nowak J, Clément C, Barka EA. Use of plant growth-promoting bacteria for biocontrol of plant diseases: principles, mechanisms of action, and future prospects. Appl Environ Microbiol. 2005;71:4951–4959. doi: 10.1128/AEM.71.9.4951-4959.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis P, Bodilis J. A survey of TonB-dependent receptors in fluorescent pseudomonads. Environ Microbiol Rep. 2009;1:256–262. doi: 10.1111/j.1758-2229.2009.00041.x. [DOI] [PubMed] [Google Scholar]
- Cornelis P, Matthijs S, Van Oeffelen L. Iron uptake regulation in Pseudomonas aeruginosa. Biometals. 2009;22:15–22. doi: 10.1007/s10534-008-9193-0. [DOI] [PubMed] [Google Scholar]
- de Eugenio LI, Galán B, Escapa IF, Maestro B, Sanz JM, García J, Prieto MA. The PhaD regulator controls the simultaneous expression of the pha genes involved in polyhydroxyalkanoate metabolism andturnover in Pseudomonas putida KT2442. Environ Microbiol. 2010;12:1591–1603. doi: 10.1111/j.1462-2920.2010.02199.x. [DOI] [PubMed] [Google Scholar]
- de Eugenio LI, Escapa IF, Morales V, Dinjaski N, Galán B, García JL, Prieto MA. The turnover of medium chain-length polyhydroxyalkanoates in Pseudomonas putida KT2442 and the fundamental role of PhaZ depolymerase for the metabolic balance. Environ Microbiol. 2010;12:207–221. doi: 10.1111/j.1462-2920.2009.02061.x. [DOI] [PubMed] [Google Scholar]
- de Smet MJ, Eggink G, Witholt B, Kingma J, Wynberg H. Characterization of intracellular inclusions formed by Pseudomonas oleovorans during growth on octane. J Bacteriol. 1983;154:870–878. doi: 10.1128/jb.154.2.870-878.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis JJ, Zylstra GJ. Complete sequence and genetic organization of pDTG1, the 83 kilobase naphthalene degradation plasmid from Pseudomonas putida strain NCIB 9816–4. J Mol Biol. 2004;341:753–768. doi: 10.1016/j.jmb.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Duan J, Jiang W, Cheng Z, Heikkila JJ, Glick BR. The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. PLoS ONE. 2013;8(3):e58640. doi: 10.1371/journal.pone.0058640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton RW. p -Cymene catabolic pathway in Pseudomonas putida F1: cloning and characterization of DNA encoding conversion of p -cymene to p -cumate. J Bacteriol. 1997;179:3171–3180. doi: 10.1128/jb.179.10.3171-3180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Heidelberg JF, White O, Salzberg SL. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 2000;1(6):RESEARCH0011. doi: 10.1186/gb-2000-1-6-research0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbahloul Y, Steinbüchel A. Large-scale production of poly(3-Hydroxyoctanoic Acid) by Pseudomonas putida GPo1 and a simplified downstream process. Appl Environ Microbiol. 2009;75:643–651. doi: 10.1128/AEM.01869-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escapa IF, García JL, Bühler B, Blank LM, Prieto MA. The polyhydroxyalkanoate metabolism controls carbon andenergy spillage in Pseudomonas putida. Environ Microbiol. 2012;14:1049–1063. doi: 10.1111/j.1462-2920.2011.02684.x. [DOI] [PubMed] [Google Scholar]
- Finette BA, Gibson DT. Initial studies on the regulation of toluene degradation by Pseudomonas putida Fl. Biocatalysis. 1988;2:29–37. [Google Scholar]
- Fiedler S, Steinbüchel A, Rehm BH. The role of the fatty acid beta-oxidation multienzyme complex from Pseudomonas oleovorans in polyhydroxyalkanoate biosynthesis: molecular characterization of the fadBA operon from P. oleovorans and of the enoyl-CoA hydratase genes phaJ from P. oleovorans and Pseudomonas putida. Arch Microbiol. 2002;178:149–160. doi: 10.1007/s00203-002-0444-0. [DOI] [PubMed] [Google Scholar]
- Galán B, Dinjaski N, Maestro B, de Eugenio LI, Escapa IF, Sanz JM. Nucleoid-associated PhaF phasin drives intracellular location and segregation of polyhydroxyalkanoate granules in Pseudomonas putida KT2442. Mol Microbiol. 2011;79:402–418. doi: 10.1111/j.1365-2958.2010.07450.x. [DOI] [PubMed] [Google Scholar]
- Geszvain K, Tebo BM. Identification of a two-component regulatory pathway essential for Mn(II) oxidation in Pseudomonas putida GB-1. Appl Environ Microbiol. 2009;76:1224–1231. doi: 10.1128/AEM.02473-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geszvain K, McCarthy JK, Tebo BM. Elimination of manganese (ii, iii) oxidation in Pseudomonas putida GB-1 by a double knockout of two putative multicopper oxidase genes. Appl Environ Microbiol. 2013;79:357–366. doi: 10.1128/AEM.01850-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BR. The enhancement of plant growth promotion by free-living bacteria. Can J Microbiol. 1995;141:109–117. [Google Scholar]
- Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2001;2:376–381. doi: 10.1093/embo-reports/kve097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood GW, Anderson AJ, Dawes EA. A survey of the accumulation of novel polyhydroxyalkanoates by bacteria. Biotechnol Lett. 1989;11:471–476. [Google Scholar]
- Hill JE, Seipp RP, Betts M, Hawkins L, Van Kessel AG, Crosby WL, Hemmingsen SM. Extensive profiling of a complex microbial community by high-throughput sequencing. Appl Environ Microbiol. 2002;68:3055–3066. doi: 10.1128/AEM.68.6.3055-3066.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JE, Penny SL, Crowell KG, Goh SH, Hemmingsen SM. cpnDB: a chaperonin sequence database. Genome Res. 2004;14:1669–1675. doi: 10.1101/gr.2649204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhang L. Rapid and sensitive dot matrix methods for genome analysis. Bioinformatics. 2004;20:460–466. doi: 10.1093/bioinformatics/btg429. [DOI] [PubMed] [Google Scholar]
- Huijberts GNM, Eggink G. Production of poly(3- hydroxyalkanoates) by Pseudomonas putida KT2442 in continuous cultures. Appl Microbiol Biotechnol. 1996;46:233–239. [Google Scholar]
- Huisman GW, de Leeuw O, Eggink G, Witholt B. Synthesis of poly(3-hydroxyalkanoates) is a common feature of fluorescent pseudomonads. Appl Environ Microbiol. 1989;55:1949–1954. doi: 10.1128/aem.55.8.1949-1954.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, Li ZJ, Ye HM, Yuan MQ, Chen GQ. Metabolic engineering for microbial production of polyhydroxyalkanoates consisting of high 3-hydroxyhexanoate content by recombinant Aeromonas hydrophila. Bioresour Technol. 2010;101:6096–6102. doi: 10.1016/j.biortech.2010.02.089. [DOI] [PubMed] [Google Scholar]
- Jimenez JI, Minambres B, Garcia JL, Diaz E. Genomic analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ Microbiol. 2002;4:824–841. doi: 10.1046/j.1462-2920.2002.00370.x. [DOI] [PubMed] [Google Scholar]
- Jiménez JI, Canales A, Jiménez-Barbero J, Ginalski K, Rychlewski L, García JL, Díaz E. Deciphering the genetic determinants for aerobic nicotinic acid degradation: the nic cluster from Pseudomonas putida KT2440. Pro Nat Acad Sci. 2008;105:11329–11334. doi: 10.1073/pnas.0802273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser JP, Feng Y, Bollag JM. Microbial metabolism of pyridine, quinoline, acridine, and their derivatives under aerobic and anaerobic conditions. Microbiol Rev. 1996;60:483–498. doi: 10.1128/mr.60.3.483-498.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether J, Munder A, Neugebauer J, Davenport CF, Stanke F, Larbig KD, Heeb S, Schöck U, Pohl TM, Wiehlmann L, Tümmler B. Genome diversity of Pseudomonas aeruginosa PAO1 laboratory strains. J Bacteriol. 2010;192:1113–1121. doi: 10.1128/JB.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebnik R. TonB-dependent trans-envelope signalling: the exception or the rule? Trends Microbiol. 2005;3:343–347. doi: 10.1016/j.tim.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Koebnik R, Locher KP, Van Gelder P. Structure and function of bacterial 1225 Q40 outer membrane proteins: barrels in a nutshell. Mol Microbiol. 2000;37:239–253. doi: 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- Krylov SV, Kropinski AM, Pleteneva EA, Shaburova OV, Burkal'tseva MV, Miroshnikov KA, Krylov VN. Properties of the new D3-like Pseudomonas aeruginosa bacteriophage phiPMG1: genome structure and prospects for the use in phage therapy. Genetika. 2012;48:1057–1067. [PubMed] [Google Scholar]
- Kwong SM, Yeo CC, Suwanto A, Poh CL. Characterization of the endogenous plasmid from Pseudomonas alcaligenes NCIB 9867: DNA sequence and mechanism of transfer J. Bacteriol. 2000;182:81–90. doi: 10.1128/jb.182.1.81-90.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MGI, Brinkman FSL. “IslandViewer: an integrated interface for computational identification and visualization of genomic islands”. Bioinformatics. 2009;25:664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Shi J, Wang X, Han Y, Tong W, Ma L, Liu B, Cai B. Complete nucleotide sequence and organization of the naphthalene catabolic plasmid pND6-1 from Pseudomonas sp Strain ND6. Gene. 2002;336:231–240. doi: 10.1016/j.gene.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Li S, Zhao H, Li Y, Niu S, Cai B. Complete genome sequence of the naphthalene-degrading Pseudomonas putida Strain ND6. J Bacteriol. 2012;194:5154–5155. doi: 10.1128/JB.01190-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loper JE, Hassan KA, Mavrodi DV, Davis EW II. Lim CK (2012) Comparative genomics of plant-associated Pseudomonas spp: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 2012;8:e1002784. doi: 10.1371/journal.pgen.1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackiewicz P, Mackiewicz D, Kowalczuk M, Cebrat S. Flip-flop around the origin and terminus of replication in prokaryotic genomes. Genome Biol. 2001;2(12):INTERACTIONS1004. doi: 10.1186/gb-2001-2-12-interactions1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Korzeniewski F, Palaniappan K, Szeto E, Werner G, Padki A, Zhao X, Dubchak I, Hugenholtz P, Anderson I, Lykidis K, Ivanova N, Kyrpides NC. The integrated microbial genomes (IMG) system. Nucleic Acid Res. 2006;34:D334–D348. doi: 10.1093/nar/gkj024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla MA, Pizarro-Tobias P, Roca A, Fernández M, Duque E, Molina L, Wu X, van der Lelie D, Gómez MJ, Segura A, Ramos JL. Complete genome of the plant growth-promoting rhizobacterium Pseudomonas putida BIRD-1. J Bacteriol. 2011;193:1290. doi: 10.1128/JB.01281-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J. Pyoverdines: pigments, siderophores and potential taxonomic markers of fluorescent Pseudomonas species. Arch Microbiol. 2000;174:135–142. doi: 10.1007/s002030000188. [DOI] [PubMed] [Google Scholar]
- Miñana-Galbis D, Urbizu-Serrano A, Farfán M, Fusté CM, Gaspar Lorén JG J. Phylogenetic analysis and identification of Aeromonas species based on sequencing of the cpn60 universal target. Int J Syst Evol Microbiol. 2009;59:1976–1983. doi: 10.1099/ijs.0.005413-0. [DOI] [PubMed] [Google Scholar]
- Molina L, Duque E, Manuel J, Gómez MJ, Krell T, Lacal J, García-Puente A, García V, Matilla MA, Ramos JL, Segura A. The pGRT1 plasmid of Pseudomonas putida DOT-T1E encodes functions relevant for survival under harsh conditions in the environment. Environ Microbiol. 2011;13:2315–2327. doi: 10.1111/j.1462-2920.2011.02492.x. [DOI] [PubMed] [Google Scholar]
- Molina L, Bernal P, Udaondo Z, Segura A, Ramos JL. Complete genome sequence of a Pseudomonas putida clinical isolate, strain H8234. Genome Announc. 2013;1(4):E00496-13. doi: 10.1128/genomeA.00496-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Henares MA, Garcıa-Salamanca A, Molina-Henares J, de la Torre AJ, Herrera MC, Ramos JL, Duque E. Functional analysis of aromatic biosynthetic pathways in Pseudomonas putida KT2440. Microbial Biotech. 2009;2:91–100. doi: 10.1111/j.1751-7915.2008.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulet M, Lalucat J, Garcia-Valdes E. DNA based analysis of the Pseudomonas species. Environ Microbiol. 2010;12:1513–1530. doi: 10.1111/j.1462-2920.2010.02181.x. [DOI] [PubMed] [Google Scholar]
- Mulet M, Garcıa-Valdes E, Lalucat J. Phylogenetic affiliation of Pseudomonas putida biovar A and B strains. Res Microbiol. 2013;164:351–359. doi: 10.1016/j.resmic.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Nakazawa T. Travels of a Pseudomonas from Japan around the world. Environ Microbiol. 2002;4:782–786. doi: 10.1046/j.1462-2920.2002.00310.x. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Weinel C, Paulsen IT, Dodson RJ, Hilbert H, Santos VAP M d, Fouts DE, Gill SR, Pop M, Holmes M, Brinkac L, Beanan M, DeBoy RT, Daugherty S, Kolonay J, Madupu R, Nelson W, White O, Peterson J, Khouri H, Hance I, Chris Lee P, Holtzapple E, Scanlan D, Tran K, Moazzez A, Utterback T, Rizzo M, Lee K, Kosack D. et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol. 2002;4:799–808. doi: 10.1046/j.1462-2920.2002.00366.x. [DOI] [PubMed] [Google Scholar]
- Pak JW, Knoke KL, Noguera DR, Fox BG, Chambliss GH. Transformation of 2,4,6-trinitrotoluene by purified xenobiotic reductase B from Pseudomonas fluorescens I-C. Appl Environ Microbiol. 2000;66:4742–4750. doi: 10.1128/aem.66.11.4742-4750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palleroni NJ. In: Bergey’s manual of systematic bacteriology. Vol 2. Krieg NR, Holt JG, editor. The Williams and Wilkins Co, Baltimore, MD, USA; 1984. Genus I Pseudomonas Migula 1894; pp. 323–379. [Google Scholar]
- Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, Cerqueira G, Gevers D, Walker S, Wortman J, Feldgarden M, Haas B, Birren B, Gilmore MS. Comparative genomics of Enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. MBio. 2012;3:00318-11. doi: 10.1128/mBio.00318-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkau A, Stuart-Edwards M, Stothard P, Van Domselaar G. Interactive microbial genome visualization with Gview. Bioinformatics. 2010;26:3125–3126. doi: 10.1093/bioinformatics/btq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponraj P, Shankar M, Ilakkiam D, Rajendhran J, Gunasekaran P. Genome sequence of the plant growth-promoting rhizobacterium Pseudomonas putida. J Bacteriol. 2012;194:6015. doi: 10.1128/JB.01509-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos JL, Duque E, Huertas MJ, Dour AH. Isolation and expansion of the catabolic potential of a Pseudomonas putida strain able to grow in the presence of high concentrations of aromatic hydrocarbons. J Bacteriol. 1995;177:3911–3916. doi: 10.1128/jb.177.14.3911-3916.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-González MI, Campos MJ, Ramos JL, Manuel Espinosa-Urgel M. Characterization of the Pseudomonas putida mobile genetic element ISPpu10: an occupant of repetitive extragenic palindromic sequences J. Bacteriol. 2006;188:37–44. doi: 10.1128/JB.188.1.37-44.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- Ravel J, Cornelis P. Genomics of pyoverdine mediated iron uptake in pseudomonads. Trends Microbiol. 2003;11:195–200. doi: 10.1016/s0966-842x(03)00076-3. [DOI] [PubMed] [Google Scholar]
- Rehm BH. Bacterial polymers: biosynthesis, modifications and applications. Nat Rev Microbiol. 2010;2010(8):578–592. doi: 10.1038/nrmicro2354. [DOI] [PubMed] [Google Scholar]
- Rehm BH, Niels Krueger N, Steinbuechel A. New metabolic link between fatty acid de novo synthesis and polyhydroxyalkanoic acid synthesis: the phaG gene from Pseudomonas putida KT2440 encodes a 3-hydroxyacyl-acyl carrier protein-coenzyme A transferase. J Biol Chem. 1998;273:24044–24051. doi: 10.1074/jbc.273.37.24044. [DOI] [PubMed] [Google Scholar]
- Rojas A, Duque E, Mosqueda G, Golden G, Hurtado A, Ramos JL, Segura A. Three efflux pumps are required to provide efficient tolerance to toluene in pseudomonas putida DOT-T1E. J Bacteriol. 2001;183:3967–3973. doi: 10.1128/JB.183.13.3967-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosson RA, Nealson KH. Manganese binding and oxidation by spores of a marine Bacillus. J Bacteriol. 1982;151:1027–1034. doi: 10.1128/jb.151.2.1027-1034.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruth K, de Roo G, Egli T, Ren Q. Identification of two acyl-CoA synthetases from Pseudomonas putida GPo1: one is located at the surface of polyhydroxyalkanoates granules. Biomacromolecules. 2008;9:1652–1659. doi: 10.1021/bm8001655. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schellenberg J, Links MG, Hill JE, Dumonceaux TJ, Kimani J, Jaoko W, Wachihi C, Mungai JN, Peters GA, Tyler S, Graham M, Severini A, Fowke KR, Ball TB, Plummer FA. Molecular definition of vaginal microbiota in East African commercial sex workers. Appl Environ Microbiol. 2011;77:4066–4074. doi: 10.1128/AEM.02943-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma PK, Fu J, Cicek N, Sparling R, Levin DB. Kinetics of medium-chain-length polyhydroxyalkanoates (mcl-PHAs) production by a novel isolate of Pseudomonas putida LS46. Can J Microbiol. 2011;58:982–989. doi: 10.1139/w2012-074. [DOI] [PubMed] [Google Scholar]
- Sharma PK, Fu J, Zhang XL, Fristensky BW, Davenport K, Chain PSG, Sparling R, Levin DB. Draft genome sequence of medium-chain-length polyhydroxyalkanoate-producing Pseudomonas putida strain LS46. Genome Announc. 2013;1:e00151-13. doi: 10.1128/genomeA.00151-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. Isfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silby MW, Cerdeno-Tarraga AM, Vernikos GS. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 2011;10:R51. doi: 10.1186/gb-2009-10-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silby MW, Winstanley C, Godfrey SAC, Levy SB, Jackson RW. Pseudomonas genome: diverse and adaptable. FEMS Microbiol Rev. 2011;35:652–680. doi: 10.1111/j.1574-6976.2011.00269.x. [DOI] [PubMed] [Google Scholar]
- Singleton I. Microbial metabolism of xenobiotics: fundamental and applied research. J Chem Technol Biotechnol. 1994;59:9–23. [Google Scholar]
- Taghavi S, Barac T, Greenberg B, Borremans B, Vangronsveld J, van der Lelie D. Horizontal gene transfer to endogenous endophytic bacteria from poplar improves phytoremediation of toluene. Appl Environ Microbiol. 2005;71:8500–8505. doi: 10.1128/AEM.71.12.8500-8505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghavi S, Garafola C, Monchy S. Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl Environ Microbiol. 2009;75:748–757. doi: 10.1128/AEM.02239-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Yao Y, Wang L, Yu H, Ren Y, Wu G, Xu P. Genomic analysis of Pseudomonas putida: genes in a genome island are crucial for nicotine degradation. Sci Rep. 2012;2:377. doi: 10.1038/srep00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillier ER, Collins RA. Genome rearrangement by replication-directed translocation. Nat Genet. 2000;26:195–197. doi: 10.1038/79918. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Minegishi K, Iino T. Toluene transposons Tn4651 and Tn4653 are class II transposons. J Bacteriol. 1989;171:1386–1393. doi: 10.1128/jb.171.3.1386-1393.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udaondo Z, Duque E, Fernández M, Molina L, de la Torre J, Bernal P, Niqui J, Pini C, Roca A, Matilla MA, Molina-Henares A, Hortencia Silva-Jiménez H, Gloria Navarro-Avilés G, Busch A, Lacal J, Krell T, Segura A, Juan-Luis Ramos JL. Analysis of solvent tolerance in Pseudomonas putida DOT-T1E based on its genome sequence and a collection of mutants. FEBS Lett. 2012;586:2932–2938. doi: 10.1016/j.febslet.2012.07.031. [DOI] [PubMed] [Google Scholar]
- Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. ISsaga: an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011;12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke TJ, Sparling R, Hill JE, Links MG, Levin D, Dumonceaux TJ. Predicting relatedness of bacterial genomes using the chaperonin-60 universal target (cpn60 UT): Application to Thermoanaerobacter species. Syst Appl Microbiol. 2011;34:171–179. doi: 10.1016/j.syapm.2010.11.019. [DOI] [PubMed] [Google Scholar]
- Visca P, Imperi F, Lamont IL. Pyoverdine siderophores: from biogenesis to biosignificance. Trends Microbiol. 2007;15:22–30. doi: 10.1016/j.tim.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Vodovar N, Vallenet D, Cruveiller S. Complete genome sequence of the entomopathogenic and metabolically versatile soil bacterium Pseudomonas entomophila. Nat Biotechnol. 2006;24:673–679. doi: 10.1038/nbt1212. [DOI] [PubMed] [Google Scholar]
- Wang SN, Liu Z, Tang HZ, Meng J, Xu P. Characterization of environmentally friendly nicotine degradation by Pseudomonas putida biotype A strain S16. Microbiology. 2007;153:1556–1565. doi: 10.1099/mic.0.2006/005223-0. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tappel RC, Zhu C, Nomura CT. Development of a new strategy for production of medium-chain-length polyhydroxyalkanoates by recombinant Escherichia coli via inexpensive non-fatty acid feedstocks. Appl Environ Microbiol. 2012;78:519–527. doi: 10.1128/AEM.07020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener MC. TonB-dependent outer membrane transport: going for Baroque? Curr Opin Struct Biol. 2005;15:394–400. doi: 10.1016/j.sbi.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. Pseudomonas genome database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39(Database issue):D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers BE, Ambroso LA, Dunbar JC. Structure and evolution of the XcyI restriction-modification system. Nucl Acids Res. 1992;20:6267–6273. doi: 10.1093/nar/20.23.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Monchy S, Taghavi S, Zhu W, Ramos J, Van der Lelie D. Comparative genomics and functional analysis of niche specific adaptation in P putida. FEMS Microbiol Rev. 2011;2011(35):299–323. doi: 10.1111/j.1574-6976.2010.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyndham RC, Cashore AE, Nakatsu CH, Peel MC. Catabolic transposons. Biodegradation. 1994;5:323–342. doi: 10.1007/BF00696468. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Kasai H, Arnold DL, Jackson RW, Vivian A, Harayama S. Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology. 2000;2000(146):2385–2394. doi: 10.1099/00221287-146-10-2385. [DOI] [PubMed] [Google Scholar]
- Yu H, Tang H, Wang L, Yao Y, Wu G, Xu P. Complete genome sequence of nicotine-degrading Pseudomonas putida strain S16. J Bacteriol. 2011;193:5541–5542. doi: 10.1128/JB.05663-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigler DR. Application of a recN sequence similarity analysis to the identification of species within the bacterial genus Geobacillus. Int J Syst Evol Microbiol. 2005;55:1171–1179. doi: 10.1099/ijs.0.63452-0. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. A fast phage search tool. Nucleic Acids Res. 2011;39(Web Server issue):W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylstra GJ, McCombie WR, Gibson DT, Finette BA. Toluene degradation by Pseudomonas putida F1: genetic organization of the tod operon. Appl Environ Microbiol. 1988;54:1498–1503. doi: 10.1128/aem.54.6.1498-1503.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Occurrence of IS elements in different P.putida strains. Table S2: Predicted prophages in P. putida genomes. Table S3: Homology of house keeping genes encoded in the genomes of different P. putida strains. Table S4: Homology of house keeping genes encoded in the genomes of different P. putida strains. Table S5: Occurrence of different dioxygenases encoded in the P.putida LS46 genome and their homologues encoded in the genomes of other P. putida strains. Table S6: Presence of genes encoded in the P. putida KT2440 genome associated with different metabolic pathways in other P.putida strains. Table S7: Presence of genes encoded by the P. putida F1 genome involved in aromatic compound degradation and their homologues encoded in the genomes of other P. putida strains. Table S8: Occurrence of heavy metal tolerance genes encoded by P. putida W619 in different P. putida strains. Table S9: Identification of TonB receptors in P. putida LS46 and their homologues in other P. putida strains.