

Abstract
Background
Ivabradine (IVBD), a novel I(f)-channel inhibitor and specific heart rate-lowering agent, is known to have anti-oxidative activity that promotes endothelial function. However, the molecular mechanism through which IVBD acts on cardiac function has yet to be elucidated, especially in experimental diabetic animals.
Methods
For this reason, twenty diabetic mice were randomly assigned to IVBD-treated (10 mg/kg/day) and control (saline) groups. After a 3-month treatment, microarray assay was performed to identify differentia expressed genes, and cardiac function was measured by echocardiography, with subsequent immunohistochemistry analysis and western blotting.
Results
Our results showed that ivabradine treatment attenuated the expression and staining score of matrix metalloproteinase (MMP)-2, induced the dephosphorylation of caspase 3, BAX and MMP-2, and enhanced the phosphorylation of NF-κB. Ivabradine treatment led to a significant improvement in cardiac function.
Conclusion
Ivabradine significantly improved cardiac function by attenuating apoptosis and inhibiting the expression and activity of MMP-2 in diabetic mice, which underscored the novel clinical implications of ivabradine for diabetic patients.
Keywords: Diabetes, Gene, Microarray, Signal pathway, Immunohistochemistry, Apoptosis
Background
In 2007, roughly 6% of people were affected worldwide by diabetes and it is estimated that this will increase to 7.3% by 2025 [1]. Diabetes, 90% of which is type 2 diabetes (T2D), is characterized by impaired glucose homeostasis and decreased insulin activity and insulin resistance [2], which lead to elevated blood glucose levels and multiple system complications [3]. Endothelial dysfunction in T2D leads to obstructive coronary arterial stenosis, which could be partially prevented and minimized by the administration of beta-blockers because of their heart rate (HR)-lowering effect [4]. However, severe side effects from beta-blockers, including reduced heart function and blood pressure, limit its use in certain patients.
Ivabradine (IVBD), a novel If-channel inhibitor and specific HR-lowering agent, acts on the sinoatrial node but does not alter ventricular contractility and vascular tone, and has been used for certain patients with angina pectoris or heart failure who are intolerable to beta-blockers [5]. Theoretically, IVBD treatment should be beneficial in T2D, unfortunately, this has not been tested in diabetic state.
Furthermore, previous studies about the mechanisms and effect of IVBD on endothelial protection in non-diabetic animals or cells introduced conflict results [6–9], as such favoring eNOS expression and/or the prevention of NO or H2O2 degradation [6], inhibiting NADPH-oxidase activity, superoxide release and the renin-angiotensin-aldosterone system (RAAS) in ApoE-/- mice [7] ,and not up-regulating the aortic PI3K/Akt/eNOS signaling system [8] which is different to the finding by Walcher et al. [9] who reported that IVBD inhibits the chemokine-induced migration of CD4-positive lymphocytes by limiting both PI-3 kinase activity and the phosphorylation of AKT.
Notably, the biological and molecular effects ofIVBD on the heart in the diabetic state has not been studied previously. Accordingly, the present study aims to determine the improvement in cardiac function after administering IVBD to diabetic mice and to explore the mechanisms by which IVBD acts on.
Methods
Preparation of animals
All diabetic mice were male mices purchased from Nanjing Experimental Center (Nanjing, China) when they were 11–12 weeks old and were housed in a temperature and humidity controlled animal facility with a 12:12 hr light–dark cycle. All rats were fed Teklad Global 18% Protein Rodent Diet (Harlan Laboratories Asia, Hongkong, China) and had free access to water. All protocols in this study were conducted in accordance with the National Health guidelines and were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (Nanjing, China).
Grouping of animals
Twenty diabetic mice were randomly divided into two groups (of 10 rats each): an IVBD-10 group (ivabradine was added in the diet at a dosage of 10 mg/kg/day for three months), and a control group (ivabradine was replaced with saline, which was administered for 3 months).
Measurement of cardiac function by echocardiography
Before and after 3 months of IVBD or saline administration, transthoracic echocardiography (TTE) was performed (MyLab25, Esaote, Italy) with a 13 MHz linear array transducer. Under light anesthesia (ketamine HCl and xylazine, 75 and 3.5 mg/kg body weight, respectively, delivered intraperitoneally), the left ventricular (LV) wall thickness and end-diastolic (LVDd) and end-systolic diameters (LVDs) were determined from the short-axis view at the midpapillary level. The total LV mass (LVM) and corrected LVM were calculated. The LV end-diastolic and end-systolic volumes were planimetered from the parasternal long-axis view. The LV ejection fraction (LVEF) was calculated as (LV diastolic volume – LV systolic volume)/LV diastolic volume. All echocardiographic investigations were performed according to the recommendations of the American Society of Echocardiography [10]. In the current study, the intra- and inter-observer error(s) were calculated according to s = SD/√2. The intra-observer variability for the measurement of LVEF was <5%.
Tissue samples
At 3 months, 5 animals in each group were sacrificed by pentobarbital overdose. The hearts were harvested and rinsed in PBS. Homogenize tissue samples in 1 ml of TRIZOL Reagent per 50–100 mg of tissue using a power homogenizer. The sample volume should not exceed 10% of the volume of TRIZOL Reagent used for homogenization. The cells were then washed and incubated in SmBM + 0.5% FBS. The reactions were performed in quadruplicates.
Cells grown in monolayer
Lyse cells directly in a culture dish by adding 1 ml of TRIZOL Reagent to a 3.5 cm diameter dish, and passing the cell lysate several times through a pipette. The amount of TRIZOL Reagent added is based on the area of the culture dish (1 ml per 10 cm2) and not on the number of cells present.
Cells grown in suspension
Pellet cells by centrifugation. Lyse cells in TRIZOL Reagent by repetitive pipetting. Use 1 ml of the reagent per 5–10 × 106 of animal, plant or yeast cells, or per 1 × 107 bacterial cells. Washing cells before addition of TRIZOL Reagent should be avoided as this increases the possibility of mRNA degradation. Disruption of some yeast and bacterial cells may require the use of a homogenizer.
Phase separation
Incubate the homogenized samples for 5 minutes at 15 to 30°C to permit the complete dissociation of nucleoprotein complexes. Add 0.2 ml of chloroform per 1 ml of TRIZOL Reagent. Cap sample tubes securely. Shake tubes vigorously by hand for 15 seconds and incubate them at 15 to 30°C for 2 to 3 minutes. Centrifuge the samples at 12,000 × g for 15 minutes at 4°C. Following centrifugation, the mixture separates into a lower red, phenol-chloroform phase, an interphase, and a colorless upper aqueous phase. RNA remains exclusively in the aqueous phase. The volume of the aqueous phase is about 60% of the volume of TRIZOL Reagent used for homogenization.
Rna precipitation
Transfer the aqueous phase to a fresh tube. Precipitate the RNA from the aqueous phase by mixing with isopropyl alcohol. Use 0.5 ml of isopropyl alcohol per 1 ml of TRIZOL Reagent used for the initial homogenization. Incu4bate samples at 15 to 30°C for 10 minutes and centrifuge at 12,000 × g for 10 minutes at 4°C. The RNA precipitate, often invisible before centrifugation, forms a gel-like pellet on the side and bottom of the tube.
Rna wash
Remove the supernatant. Wash the RNA pellet once with 75% ethanol, adding at least 1 ml of 75% ethanol per 1 ml of TRIZOL Reagent used for the initial homogenization. Mix the sample by vortexing and centrifuge at 7,500 × g for 5 minutes at 4°C.
Redissolving the rna
At the end of the procedure, air-dry the RNA pellet for 5–10 minutes. Do not dry the RNA by centrifugation under vacuum. It is important not to let the RNA pellet dry completely as this will greatly decrease its solubility. Partially dissolved RNA samples have an A260/280 ratio <1.6. Dissolve RNA in RNase-free water by passing the solution a few times through a pipette tip, and incubating for 10 minutes at 55 to 60°C. RNA can be stored at -70°C.
Isolation of small quantity rna
Isolation of RNA from small quantities of tissue (1 to 10 mg) or Cell (102 to 104) Samples: Add 800 μl of TRIZOL to the tissue or cells. Following sample lysis, add chloroform and proceed with the phase separation as described in step 2. Prior to precipitating the RNA with isopropyl alcohol, add 5-10 g RNase-free glycogen as carrier to the aqueous phase. To reduce viscosity, shear the genomic DNA with 2 passes through a 26 gauge needle prior to chloroform addition. The glycogen remains in the aqueous phase and is co-precipitated with the RNA. It does not inhibit first-strand synthesis at concentrations up to 4 mg/ml and does not inhibit PCR.
Denaturing agarose gel electrophoresis
Heat 1 g agarose in 72 ml water until dissolved, then cool to 60°C. Add 10 ml 10X MOPS running buffer, and 18 ml 37% formaldehyde (12.3 M). Pour the gel and allow it to set. The wells should be large enough to accommodate at least 25 μl. Remove the comb, and place the gel in the gel tank. Add enough 1X MOPS running buffer to cover the gel by a few millimeters. To 3 μg RNA, add 3X volumes Formaldehyde Load Dye. Ethidium bromide can be added to the Formaldehyde Load Dye at a final concentration of 10 μg/ml. Heat denature samples at 65–70°C for 15 min.
Load the gel and electrophorese at 5–6 V/cm until the bromophenol blue (the faster-migrating dye) has migrated at least 2–3 cm into the gel.
The 28S and 18S ribosomal RNA bands should be fairly sharp, intense bands (size is dependent on the organism from which the RNA was obtained). The intensity of the upper band should be about twice that of the lower band. Smaller, more diffuse bands representing low molecular weight RNAs (tRNA and 5S ribosomal RNA) may be present. It is normal to see a diffuse smear of ethidium bromide staining material migrating between the 18S and 28S ribosomal bands, probably comprised of mRNA and other heterogeneous RNA species. DNA contamination of the RNA preparation (if present) will be evident as a high molecular weight smear or band migrating above the 28S ribosomal RNA band. Degradation of the RNA will be reflected by smearing of ribosomal RNA bands (Table 1, Figure 1).
Table 1.
RNA yield and quality before and after DNase I treatment
| Samples before Dnase I treatment | OD 260 | OD 280 | OD 260/ OD 280 | RNA concentration(ng/μl) |
|---|---|---|---|---|
| IVBD-1 | 17.079 | 8.765 | 1.95 | 683.14 |
| IVBD-2 | 15.494 | 7.962 | 1.95 | 619.77 |
| IVBD-3 | 5.603 | 3.829 | 1.46 | 224.11 |
| IVBD-4 | 11.815 | 5.889 | 2.01 | 472.58 |
| IVBD-5 | 17.626 | 8.540 | 2.06 | 705.05 |
| Control-1 | 13.908 | 7.215 | 1.93 | 556.32 |
| Control-2 | 9.294 | 4.841 | 1.92 | 371.75 |
| Control-3 | 11.153 | 5.965 | 1.87 | 446.14 |
| Control-4 | 14.234 | 7.344 | 1.94 | 569.36 |
| Control-5 | 14.797 | 7.684 | 1.93 | 591.89 |
| Samples after DNase I treatment | OD 260 | OD 280 | OD 260 / OD 280 | RNA concentration(ng/ul) |
| IVBD-1 | 7.994 | 3.939 | 2.03 | 319.76 |
| IVBD-2 | 6.840 | 3.493 | 1.96 | 273.59 |
| IVBD-3 | 5.417 | 2.769 | 1.96 | 216.68 |
| IVBD-4 | 8.579 | 4.371 | 1.96 | 343.16 |
| IVBD-5 | 12.352 | 6.073 | 2.03 | 494.08 |
| Control-1 | 4.455 | 2.307 | 1.93 | 178.20 |
| Control-2 | 4.807 | 2.405 | 2.00 | 192.27 |
| Control-3 | 10.141 | 5.244 | 1.93 | 405.63 |
| Control-4 | 10.424 | 5.408 | 1.93 | 416.96 |
IVBD, ivabradine.
Figure 1.
RNA denaturing agarose gel electrophoresis for ivabradine and control groups. Animals in the ivabradine group were numbered as 1–5 and as 11–15.
Real-time polymerase chain reaction (RT-PCR) analysis
The extracted RNA was first DNase-treated with RQ1 RNase-Free DNase (Promega, USA) and heat inactivated according to the manufacturer’s protocol. The threshold cycle (CT) value for amplification of each gene was determined by the auto threshold function of the software. Prior to the amplification, the PCR efficiency and primers compatibility for the gene of interest and a reference gene were validated via the standard curve method [10]. A melting curve analysis with temperature ramping from 55°C–99°C was carried out for each run to confirm the specificity of the PCR amplifications. β-actin, which served as a reference gene, was used for the normalization of the cDNA input. Actb, B2M and Gusb served as housekeeping (HK) genes.
Microarray analysis
Calculate the ΔCt for each pathway-focused gene in each treatment group [11, 12]: ΔCt (group 1) = average Ct – average of HK genes’ Ct for group 1 array; ΔCt (group 2) = average Ct – average of HK genes’ Ct for group 2 array. Calculate the ΔΔCt for each gene across two PCR Arrays (or groups): ΔΔCt = ΔCt (group 2) - ΔCt (group 1), where group 1 is the control and group 2 is the experimental. Finally, the fold-change for each gene from group 1 to group 2 as 2-ΔΔCt was calculated.
Immunohistochemical study
Animals hearts were fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, and cross-sectioned into 10-mm slices. Sections were stained with hematoxylin and eosin (H&E) for cell alignment according to the manufacturer’s instructions. Based on the results of the microarray analysis, staining was performed to determine the expression of MMP-2, TGF-β, TIMP and P53. The staining score was calculated based on the percent positive area (no positive staining = 0; less than 25% = 1 point; 25–50% = 2 points; 51–75% = 3 points; and more than 75% = 4 points) multiplied by the staining intensity (weak = 1; moderate = 2; strong = 3, and very strong = 4). Five fields of view were randomly selected for each sample, and the average positive points for each case were reported as the actual positive points.
Western blot analysis
Cardiomyocytes were gently washed twice with ice-cold PBS and lysed in a cocktail of RIPA buffer, proteinase inhibitor and phosphatase inhibitor. After 20 min on ice, cells were scraped and lysates were clarified by centrifugation at 12,000 x g for 15 min at 4°C. Protein concentrations were quantified by BCA protein assay according to the manufacturer’s instructions. In total, 60 μg of protein were separated by a 10% SDS-PAGE gel and transferred to a PVDF membrane before being incubated overnight at 4°C with a primary antibody (caspase 3, BAX, NF-κB, MMP-2 and β-actin). After that the membrane was washed and incubated with a horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. After a second wash, the membranes were developed using an enhanced chemiluminescence substrate and the band intensities were analyzed using ImageJ (National Institutes of Health, Bethesda, USA).
Statistical analysis
Data are presented as mean ± SD for continuous variables and frequency for categorical variables. Comparison of continuous variables was performed using the t test or Mann–Whitney test as appropriate. The chi-square test or Fisher's exact test was used to analyze categorical variables. All statistical analyses were performed using SPSS® software, version 17.0 (SPSS Inc., USA). Statistical significance was set as P < 0.05.
Results
Measurement of cardiac function by echocardiography
Indices of cardiac function were comparable between the ivabradine and control groups (Table 2 and Figure 2). Importantly, at the 3 month follow-up, IVSTs, LVDs, LVPWs, eject fraction (EF) and fractional shortening (FS) improved significantly in the ivabradine group (1.59 ± 0.03 mm, 1.65 ± 0.08 mm, 1.42 ± 0.09 mm, 75.7 ± 3.43% and 49.33 ± 3.01 %, respectively) compared with the control group (1.41 ± 0.21 mm, p = 0.022; 2.22 ± 0.53 mm, p = 0.017; 1.26 ± 0.14 mm, p = 0.032; 54.1 ± 8.3 %, p = 0.018; and 27.34 ± 4.87%, p < 0.001, respectively). Compared with the baseline measurements, at 3 months after administration of ivabradine, cardiac function was significantly improved with the exception of the left ventricular volume at end-diastole. However, there were no significant differences in echocardiographic measurements between the baseline and the 3 month follow-up in the control group.
Table 2.
Measurements of cardiac function by echocardiography
| Ivabradine (n = 7) | Control(n = 6) | P value | |
|---|---|---|---|
| IVSTd (mm) | |||
| Baseline | 0.95 ± 0.12 | 0.89 ± 0.18 | 0.539 |
| At 3 months | 1.09 ± 0.12 | 0.94 ± 0.15 | 0.089 |
| IVSTs (mm) | |||
| Baseline | 1.37 ± 0.17 | 1.29 ± 0.21 | 0.468 |
| At 3-month | 1.59 ± 0.03 | 1.24 ± 0.21 | 0.022 |
| LVDd (mm) | |||
| Baseline | 3.30 ± 0.31 | 3.33 ± 0.27 | 0.932 |
| At 3-month | 2.99 ± 0.14 | 3.24 ± 0.25 | 0.497 |
| LVDs (mm) | |||
| Baseline | 2.09 ± 0.17 | 2.26 ± 0.50 | 0.436 |
| At 3-month | 1.65 ± 0.08 | 2.22 ± 0.53 | 0.017 |
| LVPWd (mm) | |||
| Baseline | 0.92 ± 0.18 | 0.84 ± 0.12 | 0.353 |
| At 3-month | 1.09 ± 0.09 | 0.94 ± 0.16 | 0.067 |
| LVPWs (mm) | |||
| Baseline | 1.21 ± 0.09 | 1.18 ± 0.11 | 0.577 |
| At 3-month | 1.42 ± 0.09 | 1.26 ± 0.14 | 0.032 |
| EF (%) | |||
| Baseline | 67.1 ± 6.96 | 61.6 ± 7.6 | 0.202 |
| At 3-month | 75.7 ± 3.43 | 54.1 ± 8.3 | 0.018 |
| FS (%) | |||
| Baseline | 36.42 ± 5.56 | 31.66 ± 4.96 | 0.135 |
| At 3-month | 49.33 ± 3.01 | 27.34 ± 4.87 | <0.001 |
| LVM (g) | |||
| Baseline | 107.55 ± 10.75 | 99.81 ± 15.88 | 0.596 |
| At 3-month | 85.85 ± 13.04 | 98.39 ± 27.92 | 0.309 |
| LVM-corrected(mm) | |||
| Baseline | 86.04 ± 8.59 | 79.99 ± 28.77 | 0.605 |
| At 3-month | 66.65 ± 10.51 | 78.60 ± 22.30 | 0.230 |
| LVDd (ml) | |||
| Baseline | 44.84 ± 10.25 | 51.99 ± 25.65 | 0.509 |
| At 3-month | 48.07 ± 6.69 | 49.32 ± 28.40 | 0.911 |
| LVSd (ml) | |||
| Baseline | 14.42 ± 2.67 | 19.79 ± 10.24 | 0.206 |
| At 3-month | 11.97 ± 2.02 | 19.82 ± 11.06 | 0.091 |
IVST, inter-ventricular septal thickness; d, end-diastolic; s, end-systolic; LVD, left ventricular dimension; LVPW, left ventricular posterior wall; EF, eject fraction; FS, fraction of shortness; LVM, left ventricular mass.
Figure 2.
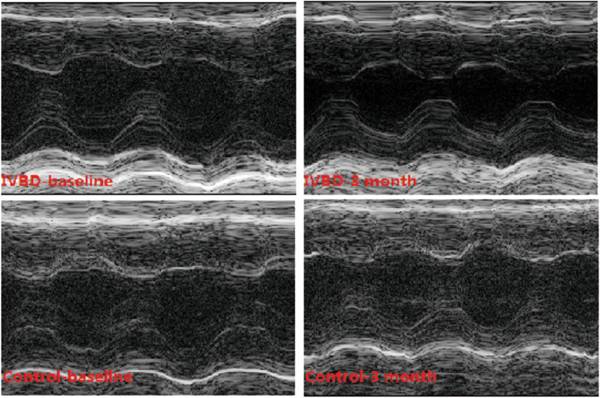
Comparison of cardiac function between ivabradine and control groups. M-model echocardiography showed a significant increase of ventricular wall movement after ivabradine treatment (upper column); however, there was no significant difference in cardiac function in the control group (lower column).
Differentially expressed genes in the myocardium of diabetic mice
According to microarray analysis, 15 genes were differentially expressed in diabetic mice after ivabradine treatment. Of these 15 genes, MMP-2 was down-regulated (fold-change, defined as the ratio of control/ivabradine, 2.459, p = 0.031), and the remaining 14 genes were up-regulated (Table 3).
Table 3.
Genes with significantly higher expression (>2 times) in the ivabradine group than in the control group
| Gene name | Function | IVBD (mean) | Control (mean) | FC | P |
|---|---|---|---|---|---|
| MMP -2(matrix metallopeptidase- 2) | ECM | 0.027 | 0.067 | 0.40 | 0.031 |
| MIP-3 β(macrophage inflammation protein-3 β, CCL 19) | Lymphoid T cells | 0.319 | 0.144 | 2.22 | 0.014 |
| 6Ckine(chemokine with 6 cysteines,CCL21) | Lymphoid organs | 0.421 | 0.177 | 2.37 | 0.024 |
| ACE/CD143(angiogensin I Converting Enzyme/CD 143) | RAAS | 0.433 | 0.189 | 2.29 | 0.006 |
| ALK-1(activin receptor-like kinase) | TGF- β | 0.327 | 0.123 | 2.66 | 0.047 |
| CT-1(cardiotrophin-1) | apoptosis | 0.292 | 0.086 | 3.39 | 0.016 |
| CD27(CD-27) | apoptosis | 0.245 | 0.089 | 2.75 | 0.021 |
| CD27 Ligand(CD70) | TNF | 0.212 | 0.067 | 3.16 | 0.020 |
| Endoglin(endoglin) | TGF- β | 0.333 | 0.159 | 2.09 | 0.005 |
| Epigen(epigen) | EGF-like | 0.173 | 0.0845 | 2.05 | 0.039 |
| IL-17E(interlukin-17E) | T cells | 0.327 | 0.161 | 2.03 | 0.001 |
| IL-17 F(interlukin-17 F) | T cells | 0.385 | 0.163 | 2.36 | 0.001 |
| IL-1ra/IL-1 F3(interlukin-1ra/-1 F3) | T cells | 0.266 | 0.121 | 2.19 | 0.003 |
| IL-2 Rα(interlukin-2 Rα) | T cells | 0.298 | 0.143 | 2.08 | 0.001 |
Fold changes are relative to normal samples.
FC, fold change; ECM, extracellular matrix; RAAS, renin angiotensin aldosterone system; TGF, tissue growth factor; TNF, tumor necrosis factor; EGF, epidermal growth factor.
Functional gene sets that discriminate between ivabradine and control groups
From these significant diabetic development genes, we have been able to identify some specific functional groups. MMP-2 is one member of the matrix metalloproteinase family, which modulates the inflammatory system and apoptosis. MMP-2 targeting at the extracellular matrix was significantly inhibited by ivabradine. Lymphocyte proliferation induced by inflammation and immunity was markedly promoted by ivabradine, and this was confirmed by the significant up-regulation of 6Ckine, ACE/CD143, ALK-1, CT-1, CD27, endoglin, MIP-3β, epigen, IL-17E/F, IL-1ra/IL-1 F3, and IL-2 Rα.
Immunohistochemical study
Based on the microarray analysis, the up-regulation of MMP-2 expression was analyzed by immunostaining, and the results are shown in Table 4 and Figure 3. The mean staining score and staining intensity in the ivabradine group were significantly improved (2.1 ± 0.2 scores and 1.9 ± 0.3 grade), compared with the control group (3.6 ± 0.3 scores, p = 0.038 and 3.9 ± 0.3 grade, p = 0.022, respectively).
Table 4.
Immunohistochemistry analysis
| ivabradine (n = 6) | Control (n = 6) | P value | |
|---|---|---|---|
| Matrix metalloproteinase-2 | |||
| Staining score | |||
| Median (75% quartile)* | 0.75 (1.00) | 1.5 0(1.25) | 0.048 |
| Mean (95%CI) | 0.75 (0.29-0.92) | 1.2 (0.64-1.76) | 0.032 |
| Staining intensity, grade | 1.9 ± 0.3 | 3.9 ± 0.3 | 0.022 |
| Week + moderate, n(%) | 4 (66) | 1 (17) | 0.043 |
| Strong + very strong, n(%) | 2 (14) | 5 (83) | 0.043 |
*Non-normal data distribution. Nonparametric analysis was used for the comparison between the two groups.
Figure 3.
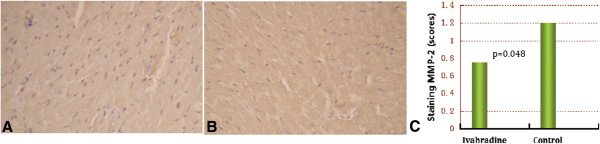
Comparison of MMP-2 staining before (A) and 3 months after (B) ivabradine treatment. The difference in staining score reached significance (p = 0.048) (C).
Western blot analysis
Based on the results of the microarray, we investigated the phosphorylation of MMP-2 and several other proteins involved in apoptosis, including NF-κB, caspase 3 and BAX. Phosphorylation of caspase 3 and BAX was significantly reduced by ivabradine (Figure 4 and Figure 5), consistent with the increased phosphorylation of NF-κB after treatment by ivabradine (Figure 6). In line with the microarray analysis, MMP-2 phosphorylation was inhibited in ivabradine-treated rats (Figure 7) compared with the control group.
Figure 4.
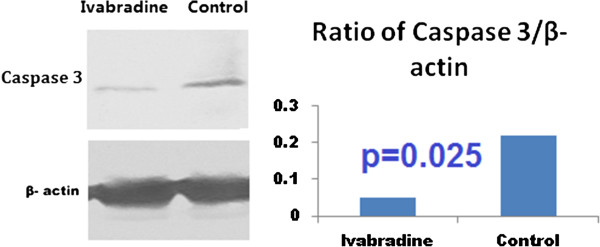
Western blot analysis of the phosphorylation state of caspase 3. Caspase 3 was dephosphorylated significantly in the ivabradine group, compared with the control group.
Figure 5.
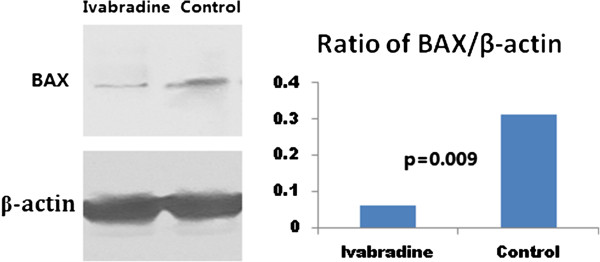
Western blot analysis of the phosphorylation of BAX. BAX was dephosphorylated significantly in the ivabradine group, compared with the control group.
Figure 6.
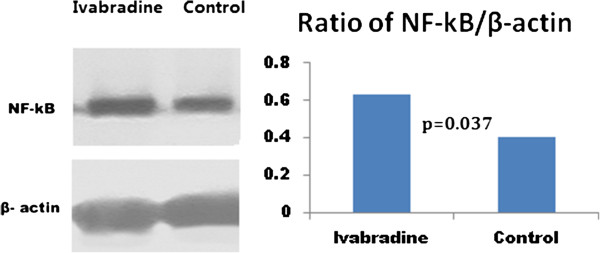
Western blot analysis of the phosphorylation of NF-κB. Phosphorylation of NF-κB was increased significantly in the ivabradine group, compared with the control group.
Figure 7.
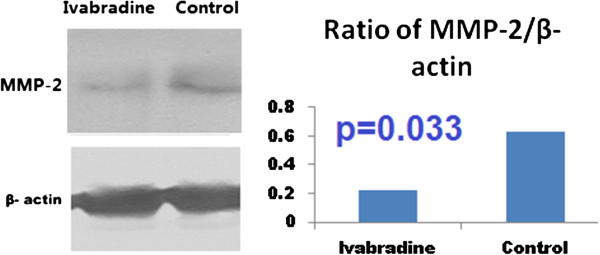
Western blot analysis of the phosphorylation of MMP-2. MMP-2 was dephosphorylated significantly in the ivabradine group, compared with the control group.
Discussion
The major findings of the present study are: (1) Ivabradine treatment significantly inhibits the expression and activity of MMP-2 in diabetic mice, (2) Ivabradine down-regulates the phosphorylation of Caspase 3, BAX, but up-regulats the phosphorylation of NF-kB in mice with diabetes. Taken together, heart function of diabetic nimals is improved significantly during Ivabradine treatment, compared to control group.
A number of genes have been discovered as potential candidates to cause T2D via the modulation of different signal pathways mediated [13–24]. Cross-talk between these signaling systems unravels the complexity of the molecular mechanism of T2D.
Matrix metalloproteinases (MMPs) are a family of zinc-binding proteolytic enzymes that normally remodel the extracellular matrix and pathologically attack substrates as part of an inflammatory response. The major MMP species in the myocardium and vasculature are the gelatinases (MMPs 2 and 9), MMP-1 (interstitial collagenase) and Mt1-MMP. Recently it has been proven that matrix metalloproteinases (MMPs) play an important role in atherosclerosis and the rebuilding of the vascular wall [25]. The alteration of MMP-2 gene expression induced by a 3 month ivabradine treatment, was associated with an improvement of cardiac function in diabetic mices, as confirmed by our immunohistochemistry and Western blot analyses.
It has been found that high glucose concentration promotes TGF-β expression and activates the Jak/STAT signaling cascade in diabetic kidney cells. Activation of this signaling cascade can stimulate excessive proliferation and the growth of glomerular mesangial cells, contributing to diabetic nephropathy [14, 15]. Exposure to high glucose concentrations has also been shown to activate the MAPK signaling pathway in skeletal muscle cells [16]. These findings were in agreement with our finding that ivabradine improved cardiac function by promoting the expression and activity of endoglin. Endoglin is an auxiliary receptor for the TGF-β receptor complex, which functions in related signaling pathways and is mainly expressed in vascular and connective tissues and in endothelial and stromal cells [26, 27]. Up-regulated endoglin expression has been reported during wound healing and tumor vascularization, and in inflammatory tissues and developing embryos [26–28]. Mutations in endoglin have been found to be a causal factor in hereditary hemorrhagic telangiectasia (HHT), a disease characterized by the malformation of vascular structure [29, 30]. We postulated that the up-regulation of endoglin expression by ivabradine would be related to the impairment of malformed vascular structure [29–31].
PI3K/Akt is a key molecule in insulin signaling that is found to be down-regulated in T2D [20]. However, there is a discrepancy in terms of the regulation of the PI3K/Akt transduction pathway by ivabradine: up-regulating eNOS expression independent of PI3K/Akt pathway [5], inhibiting NADPH/ROS/RAAS but regulating PI3K/Akt in ApoE-/- mice [6, 7], limiting PI3K activity and the phosphorylation of AKT in CD4-positive lymphocytes [8]. We found that the expression and activity of the epigen gene was up-regulated by ivabradine treatment in diabetic myocardium. Epigen encodes a protein of 152 amino acids that contains EGF-like features. Epigen exhibits 24–37% sequence identity with EGF, TGF, and epiregulin. EGF exerts insulin-like effects on glucose transport and lipolysis and can increase the tyrosine phosphorylation and activation of IRS-1 and IRS-2. EGF is also capable of activating additional PI3K pools, thereby augmenting the downstream signaling of insulin in insulin-resistant states like T2D [13]. As a result, the modulation of epigen expression and activity by ivabradine via PI3K and MAPK signaling [32] would be predicted to be associated with the improvement of cardiac function. Similarly, MIP 3-β also regulated both the P38/MAPK and PI3K/Akt signaling pathways [33–35]. However, there was a lack of direct evidence of ivabradine regulating the PI3K/Akt signaling system provided by our results, as the change of Akt expression and Combo protein were not significantly different between the ivabradine and control groups.
Caspase 3 and BAX are two signals participating in the process of apoptosis [35, 36]. Our study showed that ivabradine was associated with the significant dephosphorylation of caspase 3 and BAX, indicating the anti-apoptotic effect of ivabradine in diabetic animals. In contrast, NF-κB was activated by ivabradine, and phosphorylated NF-κB would promote protein synthesis and thus inhibit apoptosis [37]. These results would provide additional evidences of anti-ischemic effect by ivabradine for diabetic setting, in line with previously published results confirming the cardioprotective effect by ivabradine for patients with ischemic heart disease [38, 39].
Limitation
Obviously, the current study did not analyze the mechanism attributive to the signal pathways in which ivabradine involved. For example, the question why NF-kB was activated by ivabradine was not studied. Another limitation was small sample size, which would be expanded in our next study. Finally, we did not performed Western Blotting analysis for all differentially expressed genes, which would be at least mask the potential of cross-talking by different signal pathways.
Conclusion
Our study for the time reported the cardioprotective effect by ivabradine in diabetic animal. The major finding would be implied the possible benefits of ivabradine for diabetic patients. As a result, further clinical study is required in order to elucidate the efficacy and safety of ivabradine for Type 2 diabetes.
Acknowledgments
The authors would like to thank Yan Liu and Sheng-Xia Zhang for their excellent technical assistance during this study, Mrs. Zhi-Mei Wang for secretarial assistance and Miss Zu-Ling Xu for editorial assistance with the paper.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have read and approved this submission. SLC designed this study, participated in the data analysis, and wrote this manuscript. ZYH carried out animal study, participated in the statistical analysis and writing. GFZ participated in the measuring mRNA and data analysis. MHL carried out microarray analysis and data analysis. BL carried out feeding animal, microarray analysis and signal pathways.
Contributor Information
Shao-Liang Chen, Email: chmengx@126.com.
Zuo-Ying Hu, Email: huzuoying@126.com.
Guang-Feng Zuo, Email: zuogf@126.com.
Ming-Hui Li, Email: limingh@126.com.
Bin Li, Email: libin@126.com.
References
- 1.Van den Oever IAM, Raterman HG, Nurmohamed MT, Simsek S. Mediators Inflamm. 2012. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus; p. 792393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramachandran A, Snehalatha C, Viswanathan V. Burden of type 2 diabetes and its complications- the Indian scenario. Curr Sci India. 2002;83:1471–1476. [Google Scholar]
- 3.Mazzone T, Chait A, Plutzky J. Cardiovascular disease risk in type 2 diabetes mellitus: insights from mechanistic studies. Lancet. 2008;371:1800–1809. doi: 10.1016/S0140-6736(08)60768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fihn SD, Gardin JM, Abrams J, Berra K, Blankenship JC, Dallas AP, Douglas PS, Foody JM, Gerber TC, Hinderliter AL, King SB, 3rd, Kligfield PD, Krumholz HM, Kwong RY, Lim MJ, Linderbaum JA, Mack MJ, Munger MA, Prager RL, Sabik JF, Shaw LJ, Sikkema JD, Smith CR, Jr, Smith SC, Jr, Spertus JA, Williams SV, Anderson JL. ACCF/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American college of cardiology foundation/American Heart Association task force on practice guidelines, and the American college of physicians, American Association for thoracic surgery, preventive cardiovascular nurses association, society for cardiovascular angiography and interventions, and society of thoracic surgeons. Circulation. 2012;126:e354. doi: 10.1161/CIR.0b013e318277d6a0. [DOI] [PubMed] [Google Scholar]
- 5.Borer JS. Drug insight: If inhibitors as specific heart-rate-reducing agents. Nat Clin Pract Cardiovasc Med. 2004;1:103–109. doi: 10.1038/ncpcardio0052. [DOI] [PubMed] [Google Scholar]
- 6.Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg-Gaudin F, Tardif JC. Chronic heart rate reduction by ivabradine prevents endothielial dysfunction in dyslipidaemic mice. Br J Pharmacol. 2008;154:749–757. doi: 10.1038/bjp.2008.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/S0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 8.Custodis F, Baumhäkel M, Schlimmer N, List F, Gensch C, Böhm M, Laufs U. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2008;117:2377–2387. doi: 10.1161/CIRCULATIONAHA.107.746537. [DOI] [PubMed] [Google Scholar]
- 9.Walcher T, Bernhardt P, Vasic D, Bach H, Durst R, Rottbauer W, Walcher D. Mediators Inflamm. 2010. Ivabradine reduces chemokine-induced CD4-positive lymphocyte migration; p. 751313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douglas PS, Garcia MJ, Haines DE, Lai WW, Manning WJ, Patel AR, Picard MH, Polk DM, Ragosta M, Parker Ward R, Weiner RB. ACCF/ASE/AHA/ASNC/HFSA/HRS/SCAI/SCCM/SCCT/SCMR 2011 Appropriate use criteria for echocardiography. A report of the American college of cardiology foundation appropriate use criteria task force, American society of echocardiography, American heart association, American society of nuclear cardiology, heart failure society of america, heart rhythm society, society for cardiovascular angiography and interventions, society of critical care medicine, society of cardiovascular computed tomography, and society for cardiovascular magnetic resonance endorsed by the American college of chest physicians. J Am Coll Cardiol. 2011;57:1126–1166. doi: 10.1016/j.jacc.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 22DDCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 12.Yuan JS, Reed A, Chen F, Stewart CN., Jr Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85–87. doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gogg S, Smith U. Epidermal growth factor and transforming growth factor alpha mimic the effects of insulin in human fat cells and augment downstream signalling in insulin resistance. J Biol Chem. 2002;277:36045–36051. doi: 10.1074/jbc.M200575200. [DOI] [PubMed] [Google Scholar]
- 14.Marrero MB, Banes-Berceli AK, Stern DM, Eaton DC. Role of the JAK/STAT signalling pathway in diabetic nephropathy. Am J Physiol Renal Physiol. 2006;290:F762–F768. doi: 10.1152/ajprenal.00181.2005. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Shaw S, Amiri F, Eaton DC, Marrero MB. Inhibition of the Jak/STAT signalling pathway prevents the high glucose-induced increase in tgf-beta and fibronectin synthesis in mesangial cells. Diabetes. 2002;51:3505–3509. doi: 10.2337/diabetes.51.12.3505. [DOI] [PubMed] [Google Scholar]
- 16.Adhikary L, Chow F, Nikolic-Paterson DJ, Stambe C, Dowling J, Atkins RC, Tesch GH. Abnormal p38 mitogen-activated protein kinase signalling in human and experimental diabetic nephropathy. Diabetologia. 2004;47:1210–1222. doi: 10.1007/s00125-004-1437-0. [DOI] [PubMed] [Google Scholar]
- 17.Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signalling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J Am Coll Cardiol. 2005;46:827–834. doi: 10.1016/j.jacc.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Das UN, Rao AA. Gene expression profile in obesity and type 2 diabetes mellitus. Lipids Health Dis. 2007;6:35–39. doi: 10.1186/1476-511X-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burgermeister E, Seger R. MAPK kinases as nucleo-cytoplasmic shuttles for PPARgamma. Cell Cycle. 2007;6:1539–1548. doi: 10.4161/cc.6.13.4453. [DOI] [PubMed] [Google Scholar]
- 20.Jiang G, Zhang BB. Pi 3-kinase and its up- and down-stream modulators as potential targets for the treatment of type II diabetes. Front Biosci. 2002;7:d903–d907. doi: 10.2741/jiang. [DOI] [PubMed] [Google Scholar]
- 21.Rulifson IC, Karnik SK, Heiser PW, ten Berge D, Chen H, Gu X. Wnt signalling regulates pancreatic beta cell proliferation. Proc Natl Acad Sci U S A. 2007;104:6247–6252. doi: 10.1073/pnas.0701509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho E, Bray TM. Antioxidants, NFkappaB activation, and diabetogenesis. Proc Soc Exp Biol Med. 1999;222:205–213. doi: 10.1046/j.1525-1373.1999.d01-137.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhao J, Zhang N, He M, Yang Z, Tong W. Increased beta-cell apoptosis and impaired insulin signalling pathway contributes to the onset of diabetes in OLETF rats. Cell Physiol Biochem. 2008;21:445–454. doi: 10.1159/000129637. [DOI] [PubMed] [Google Scholar]
- 24.Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag. 2007;3:853–876. [PMC free article] [PubMed] [Google Scholar]
- 25.Beaudeux JL, Giral P, Brukert E. Matrix metalloproteinases and atherosclerosis. Therapeutic aspects. Annu Biol Clinic. 2003;61:147–158. [PubMed] [Google Scholar]
- 26.Bernabeu C, Conley BA, Vary CP. Novel biochemical pathways of endoglin in vascular cell physiology. J Cell Biochem. 2007;102:1375–1388. doi: 10.1002/jcb.21594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu Y, Sun Y, Xie L, Jin K, Sheibani N, Greenberg DA. Hypoxic induction of endoglin via mitogen-activated protein kinases in mouse brain microvascular endothelial cells. Stroke. 2003;34:2483–2488. doi: 10.1161/01.STR.0000088644.60368.ED. [DOI] [PubMed] [Google Scholar]
- 28.Quintanilla M, Ramirez JR, Pérez-Gómez E, Romero D, Velasco B, Letarte M, López-Novoa JM, Bernabéu C. Expression of the TGF-beta coreceptor endoglin in epidermal keratinocytes and its dual role in multistage mouse skin carcinogenesis. Oncogene. 2003;22:5976–5985. doi: 10.1038/sj.onc.1206841. [DOI] [PubMed] [Google Scholar]
- 29.Ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11:79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 30.Fernández-L A, Sanz-Rodriguez F, Blanco FJ, Bernabéu C, Botella LM. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clinic Med Res. 2006;4:66–78. doi: 10.3121/cmr.4.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kopczyńska E, Makarewicz R. Endoglin – a marker of vascular endothelial cell proliferation in cancer. Contemp Oncol. 2012;16:68–71. doi: 10.5114/wo.2012.27340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dreux AC, Lamb DJ, Modjtahedi H, Ferns GA. The epidermal growth factor receptors and their family of ligands: their putative role in atherogenesis. Atherosclerosis. 2006;186:38–53. doi: 10.1016/j.atherosclerosis.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 33.Chen ML, Lin YH, Yang CM, Hu ML. Lycopene inhibits angiogenesis both in vitro and in vivo by inhibiting MMP-2/uPA system through VEGFR2-mediated PI3K-Akt and ERK/p38 signaling pathways. Mol Nutr Food Res. 2012;56:889–899. doi: 10.1002/mnfr.201100683. [DOI] [PubMed] [Google Scholar]
- 34.Zhao ZJ, Liu FY, Li P, Ding X, Zong ZH, Sun CF. CCL19-induced chemokine receptor-7 activates the phosphoinositide-3 kinase-mediated invasive pathway through Cdc42 in metastatic squamous cell carcinoma of the head and neck. Oncol Republ. 2011;25:729–737. doi: 10.3892/or.2010.1109. [DOI] [PubMed] [Google Scholar]
- 35.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baltadzhiev IG, Delchev SD. Changes of Bcl-2, Bax and Caspase-3 expression in the dermal microvascular endothelial cells and the epidermal layers of the eschar (tache noire) in patients with Mediterranean spotted fever. Folia Histochem Cytobiol. 2013;51:121–126. doi: 10.5603/FHC.2013.0019. [DOI] [PubMed] [Google Scholar]
- 37.Wang PH, Gu ZH, Wan DH, Liu BD, Huang XD, Weng SP, Yu XQ, He JG. The shrimp IKK-NF-κB signaling pathway regulates antimicrobial peptide expression and may be subverted by white spot syndrome virus to facilitate viral gene expression. Cell Mol Immunol. 2013;10:423–436. doi: 10.1038/cmi.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Effect of ivabradine in patients with left-ventricular systolic dysfunction: a pooled analysis of individual patient data from the BEAUTIFUL and SHIFT trials. Eur Heart J. 2013;34:2263–2270. doi: 10.1093/eurheartj/eht101. [DOI] [PubMed] [Google Scholar]
- 39.Komajda M, Böhm M, Borer JS, Ford I, Robertson M, Manolis AJ, Tavazzi L, Swedberg K, SHIFT Investigators Efficacy and safety of ivabradine in patients with chronic systolic heart failure according to blood pressure level in SHIFT. Eur J Heart Fail. 2014;16:810–816. doi: 10.1002/ejhf.114. [DOI] [PubMed] [Google Scholar]
Pre-publication history
- The pre-publication history for this paper can be accessed here: http://www.biomedcentral.com/1471-2261/14/150/prepub