

Abstract
Autophagy is a highly conserved degradation process by which intracellular components, including soluble macromolecules (e.g. nucleic acids, proteins, carbohydrates, and lipids) and dysfunctional organelles (e.g. mitochondria, ribosomes, peroxisomes, and endoplasmic reticulum) are degraded by the lysosome. Autophagy is orchestrated by the autophagy related protein (Atg) composed protein complexes to form autophagosomes, which fuse with lysosomes to generate autolysosomes where the contents are degraded to provide energy for cell survival in response to environmental and cellular stress. Autophagy is an important player in cardiovascular disease development such as atherosclerosis, cardiac ischemia/reperfusion, cardiomyopathy, heart failure and hypertension. Autophagy in particular contributes to cardiac ischemia, hypertension and diabetes by interaction with reactive oxygen species generated in endoplasmic reticulum and mitochondria. This review highlights the dual role of autophagy in cardiovascular disease development. Full recognition of autophagy as an adaptive or maladaptive response would provide potential new strategies for cardiovascular disease prevention and management.
Keywords: autophagy, oxidative stress, atherosclerosis, cardiomyopathy, hypertension, heart failure
1. Introduction
Autophagy, first coined by Belgian biochemist Christian de Duve in 1966 [1], is characterized by the lysosome-dependent degradation of cytoplasm and damaged organelles such as mitochondria, endoplasmic reticulum and peroxisomes, as well as eliminating intracellular pathogens. In addition, autophagy serves as a dynamic recycling system to provide energy and building material for new protein and membrane production to promote survival under conditions of starvation [2]. Three types of autophagy have been defined: macroautophagy, microautophagy, and chaperone-mediated autophagy, each of which promotes proteolytic degradation of intracellular cargo at the lysosome. Autophagy related protein (Atg) composed protein complexes coordinate the formation of autophagosomes which fuse with lysosomes to generate autolysosomes. The contents are then degraded, and the breakdown products are released into the cytosol for synthetic and metabolic pathways[2]. Autophagy has been linked to cardiovascular diseases, as it is triggered by inflammation, hypoxia, oxidized lipoprotein, endoplasmic reticulum (ER) stress and reactive oxygen species (ROS), which are involved in atherogenesis [3-5]. Autophagy also promotes atherosclerotic plaque cell survival by degrading damaged intracellular components and protecting cells from apoptosis [6]. However, excessive autophagy may cause cell death. The observation that autophagy is activated during ischemia/reperfusion injury leads to the hypothesis that autophagy is a critical regulator of ischemia/reperfusion injury [7]. Indeed, autophagy promotes cell survival by eliminating dysfunctional mitochondria, which may otherwise release ROS leading to cell death [8]. Moreover, autophagy has diverse effects on cardiomyopathy - whereas augmented autophagy ameliorates dilated cardiomyopathy; autophagy activation promotes diabetic cardiomyopathy [9-11]. Furthermore, autophagy may antagonize ventricular hypertrophy by increasing protein degradation and decreasing tissue mass. Thus, autophagy may be an adaptive response in heart failure [12]. Autophagy also protects the heart by maintaining contractile function, as autophagosome dysfunction is associated with increased apoptosis, mitochondrial injury, intracellular Ca2+ dysregulation and cardiac dysfunction [13]. Lastly, autophagy mediates oxidative stress-elicited myocardial injury in response to ischemia/reperfusion [14]. This review will address the above points in detail.
1.1. Definition of autophagy
Autophagy, literally “self-eating” in Greek, is the major intracellular degradation system by which cellular components, including organelles and protein aggregates, are delivered to and degraded in the lysosome [2, 15]. The purpose of autophagy is not limited to elimination of cell cargo waste, but instead, autophagy serves as a dynamic recycling system to provide energy and building material for new protein and membrane production. Thus, autophagy helps maintain the health of cells and tissues by replacing impaired cellular components with fresh ones. In addition, it supplies endogenously-derived nutrients for energy generation to promote survival under conditions of starvation [2].
1.2. Induction of autophagy
In eukaryotic cells, autophagy is induced by starvation, hypoxia, hormones, ER stress, redox stress, mitochondrial damage, pathogen-associated molecular patterns or danger-associated molecular patterns to degrade protein aggregates, oxidized lipids, damaged organelles, and even intracellular pathogens [16-18]. In addition, autophagy promotes cellular senescence [19], cell surface antigen presentation [20, 21], lipid metabolism [22], protects against genome instability [23-25] and prevents necrosis [26]. Hence, autophagy plays a key role in preventing disease. Dysregulation of autophagy is linked to cancer[27], neurodegeneration [28], metabolic syndrome [29], liver disease [30], autoimmune diseases and infections [31].
1.3. Substrates of autophagy
Autophagy can be classified as nonselective or selective. As an adaptive response to nutrient starvation, nonselective autophagy promotes bulk degradation of cytoplasm and organelles to provides amino acids and lipid substrates for intermediary metabolism [2]. Deficiencies in autophagy result in maladaptation to starvation and poor survival due to insufficient amino acid production and protein synthesis. Autophagy also occurs at low levels to mediate global turnover of cytoplasmic materials under nutrient-rich conditions [15]. Selective autophagy involves degradation of organelles and protein aggregates. Selective autophagy of protein aggregates and peroxisomes [32] involves ubiquitination of target proteins, which are recognized by autophagy receptors, such as p62 or Nbr1, which bind both ubiquitin and microtubule-associated protein 1 light chain 3 (LC3) to deliver cargo to autophagosomes [2, 33, 34]. Under specific conditions, selective autophagy of organelles such as mitochondria (mitophagy) [35], ribosomes (ribophagy) [36], endoplasmic reticulum (reticulophagy) [37], peroxisomes (pexophagy) [38], and lipids (lipophagy) [39] occurs. For instance, mitophagy is triggered by mitochondrial permeability transition pore opening and loss of mitochondrial membrane potential [40].
1.4. Types of autophagy
Three types of autophagy have been defined: macroautophagy, microautophagy, and chaperone-mediated autophagy, each of which promotes proteolytic degradation of intracellular cargo at the lysosome. Macroautophagy transfers soluble cytoplasmic materials and organelles to the lysosomes by utilizing the intermediate organelle called the autophagosome, which is a product of an isolation membrane (termed phagophore) enclosed within a small portion of the cytoplasm. The autophagosome then fuses with the lysosome to form an autolysosome and degrade the materials contained within [15] (Fig.1). In microautophagy, lysosomes directly engulf small pieces of the cytoplasm by inward invagination of the lysosomal membrane [15]. In chaperone-mediated autophagy, substrate proteins containing a KFERQ-like pentapeptide sequence are first bound with cytosolic Hsc70 then translocate into the lysosomal lumen, where they are recognized by the lysosomal membrane receptor lysosomal-associated membrane protein 2A, resulting in their unfolding and degradation [41]. Both macroautophagy and microautophagy are able to consume large structures through selective or non-selective mechanisms. The degradation products of all three types of autophagy can be used for new protein synthesis, energy production or gluconeogenesis. Among these, macroautophagy is the predominant form and the most extensively studied [42]. This review will focus on the role of macroautophagy (autophagy hereafter) in cardiovascular diseases.
Figure 1. Intracellular pathways regulating macroautophagy.
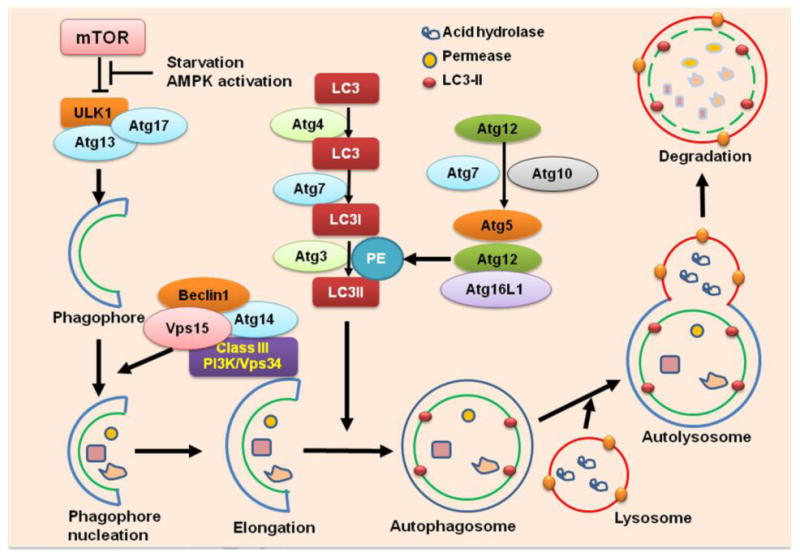
Macroautophagy transfers soluble cytoplasmic materials and organelles to the lysosomes by utilizing the intermediate organelle called the autophagosome, which is a product of an isolation membrane (termed phagophore) enclosed within a small portion of the cytoplasm. The autophagosome then fuses with the lysosome to form an autolysosome and degrades the materials contained within. Autophagy is orchestrated by the Atg-composed protein complexes. Starvation or AMPK activation inhibits mTOR, leading to the release and activation of the ULK1 complex (ULK1/Atg7/Atg13), which stimulates phagophore formation, and activates the Beclin1 complex (Beclin1/Class III PI3K/Vps34/Vps15/Atg14). The Beclin1 complex promotes phagophore nucleation and elongation. Two ubiquitin-like molecules, Atg12 and Atg8 (called LC3 in mammals) are involved in expansion of autophagosome membranes. Conjugated Atg5–Atg12 complexes bind Atg16L1 to form pre-autophagosomal structures which induce the recruitment of processed LC3-II. LC3-II remains on mature autophagosomes until after fusion with lysosomes to generate autolysosomes. The contents are then degraded by proteases, lipases, nucleases, and glycosidases, and the breakdown products such as amino acids, lipids, nucleosides, and carbohydrates are released into the cytosol for synthetic and metabolic pathways. PE: phosphatidylethanolamine.
1.5. Process of autophagy
The first Atg gene was identified in 1997 [43]. Genetic deletion of Atg genes revealed the critical roles of autophagy in physiological adaptation to stress and also a connection between defective autophagy and various diseases. Atg proteins are comprised of four major functional groups: (1) the Atg1/unc-51-like kinase (ULK) complex (Atg1/ULK1–Atg13–Atg17) that responds to upstream signals; (2) the Beclin 1/class III phosphatidylinositol 3-kinase (PI3K) complex (Atg6/Beclin1, Atg14, Vps34/PI3KC3, and Vps15) that mediates vesicle nucleation [44]; (3) two ubiquitin-like protein (Atg12 and Atg8/LC3) conjugation systems that mediate vesicle expansion; (4) Atg9 and its cycling system which provide lipids to the isolation membrane. These Atg-composed protein complexes coordinate the formation of autophagosomes. The Atg1/ULK1 complex (Atg1 in yeast and ULK1 in mammals) is an initial regulator of autophagosome formation [34]. Under nutrient-rich conditions, ULK1 is bound by mammalian target of rapamycin (mTOR) complex 1(mTORC1), which phosphorylates ULK1 and inhibits autophagy initiation. Whereas in starvation, mTORC1 dissociates from the ULK1 complex, releasing it to trigger autophagosome nucleation and elongation. In addition, ULK1 inhibits the kinase activity of mTORC1 through binding with raptor to induce its phosphorylation [45]. Adenosine monophosphate–activated protein kinase (AMPK) mediates the upstream signaling of mTORC1 which controls the cellular energy–sensing pathway [34]. In response to high concentrations of AMP and its effects on energy depletion, AMPK is activated, which, in turn, inhibits mTORC1 and promotes autophagy [2]. Autophagy induction promotes ULK1 complex activation, which phosphorylates Beclin-1 on Ser14, thereby enhancing the activity of Beclin1-Atg14-Vps34-Vps15 class III PI3K core complexes to promote autophagosome nucleation [46]. Additionally, under nutrient-rich conditions, Bcl-2 binds with the BH3 domain of Beclin 1 and inhibits autophagy. The phosphorylation of Bcl-2 and Beclin 1 disrupts this interaction and releases Beclin 1 [47]. When nutrients are abundant, Atg9 shuttles between the Golgi and endosomes. In starvation, Atg9 distributes to endosomal compartments to supply a membrane source for forming autophagosomes. Recent findings suggest Atg9 plays an essential role in regulating oxidative stress-induced JNK activation and autophagy through interaction with tumor necrosis factor receptor-associated factor 6 [48-50]. Two ubiquitin-like molecules, Atg12 and Atg8 (called LC3 in mammals) are involved in expansion of autophagosome membranes. Atg12 is first activated by Atg7 which acts like an E1 ubiquitin activating enzyme in an ATP-dependent manner. Atg12 is then covalently linked to Atg5 by Atg10, an E2-like ubiquitin carrier protein. Conjugated Atg5–Atg12 complexes bind Atg16L1 to form pre-autophagosomal structures that induce a curvature into the growing phagophore through asymmetric recruitment of processed LC3B-II. LC3B is proteolytically cleaved by protease Atg4 togenerate LC3B-I. The carboxy terminal glycine exposed by Atg4-dependent cleavage is activated by the E1-like Atg7 and then transferred to Atg3. As a result, phosphatidylethanolamine is conjugated to the carboxyl glycine to generate LC3B-II. Both Atg5–Atg12 and LC3B-II contribute recruitment and integration of LC3B-II into the growing phagophore. The synthesis and processing of LC3 are commonly used to monitor the progress of autophagy because LC3-II remains on mature autophagosomes until after fusion with lysosomes to generate autolysosomes. The contents are then degraded by proteases, lipases, nucleases, and glycosidases, and the breakdown products such as amino acids, lipids, nucleosides, and carbohydrates are released into the cytosol for synthetic and metabolic pathways [2] (Fig.1).
1.6. Autophagic flux measurement
Because the accumulation of autophagosomes may indicate autophagy induction or impairment of autophagolysosomal maturation, autophagic flux assays are necessary to distinguish between autophagy induction and a block in downstream steps [51]. Several of these assays are discussed here. (1) LC3 turnover assay: based on the observation that LC3-II is degraded in autolysosomes, LC3 turnover is one principal method currently used to measure autophagic flux. Inhibition of autophagosome-lysosome fusion and thereafter protein degradation by lysosomotropic reagents or lysosomal proteases inhibitors will result in the accumulation of LC3-II, and the elevated levels of LC3-II indicate that autophagic flux is increased [52]. (2) Degradation of LC3 and its substrates: LC3-II increases transiently upon autophagy induction, and deceases during prolonged autophagy activation. Thus the amount of total cellular LC3 and its substrate p62, quantified by immunoblot analysis or flow cytometry, inversely correlates with autophagic flux [53]. (3) Delivery of mRFP-GFP-LC3 to the lysosome: based on the concept that the low pH inside the lysosome quenches the fluorescent signal of GFP, but not RFP. Therefore, an mRFP-GFP-LC3 tandem construct labels autophagosomes with yellow and autolysosomes with red. The increase of both yellow and red punctae indicates increased autophagic flux. Notably, each of these three methods has its own limitations. For example, autophagic flux can be detected even under basal conditions, LC3 and p62 can be transcriptionally regulated during autophagy, and sometimes autolysosomes are observed as yellow due to different acidification and degradation capacities of the lysosome. Given the limitations of the individual assays discussed above, a combination of these methods would provide the most precise prediction of autophagic flux in different cell types and experimental contexts [51].
2. Autophagy and cardiovascular diseases
Autophagy plays dual roles in cardiovascular diseases through adaptive or maladaptive regulation. Physiological autophagy serves as a protective mechanism to maintain normal cardiovascular function. However, impaired autophagy contributes to disease development. A better understanding of the function of autophagy in the cardiovascular system could provide new therapeutic avenues for disease prevention or control. As a consequence, the role of autophagy in the cardiovascular system is currently under intense investigation.
2.1. Autophagy in atherosclerosis and cardiac ischemia
2.1.1. Autophagy in atherosclerosis
The discovery of autophagy-like ultrastructural features such as vacuolization and formation of myelin figures by transmission electron microscopy and expression of the autophagy marker LC3-II by western blot analysis of human carotid plaques reveals that autophagy is one major component in the process of atherosclerosis [54]. It is well established that inflammation, hypoxia, oxidized lipoprotein, ER stress and ROS are all involved in atherogenesis. In vitro studies have demonstrated that these factors present in atherosclerotic plaques could also serve as the triggers of autophagy [3-5]. Notably, autophagy in atherosclerotic plaques could be either beneficial or detrimental. On one hand, basal autophagy promotes plaque cell survival by successful degradation of damaged intracellular components, and thus protects cells against oxidative stress [6]. In addition, the engulfment of defective mitochondria by autophagosomes limits the release of cytochrome C into the cytosol and protects cells from apoptosis [55]. Autophagy is induced in endothelial cells (ECs) or smooth muscle cells (SMCs) in response to oxidized lipoprotein or lipid peroxidation products to promote cell survival [5]. Autophagy also may mediate some anti-inflammatory effects of resveratrol in EC [56]. Another study shows upregulation of autophagy attenuates 7-ketocholesterol-(a major component of oxidized lipoproteins) induced cell death in SMCs [57]. Together these findings indicate that autophagy might be protective in atherogenesis. Indeed, recent studies suggest autophagy favors plaque stabilization by regulating lipid metabolism. Autophagy delivers lipid droplets to lysosomes, where they are hydrolyzed by lysosomal acid lipase to generate free cholesterol for efflux. Therefore, macrophage foam cell cholesterol efflux is mediated by autophagy [58]. Wild-type p53-induced phosphatase 1(Wip1) is considered to control autophagy-dependent macrophage cholesterol efflux [59]. Autophagy in macrophages is also protective in that disruption of autophagy in macrophages in advanced plaques increases apoptosis and oxidative stress, thus worsening efferocytosis and promoting plaque necrosis [60]. Loss of macrophage autophagy increases plaque formation in part through inflammasome hyperactivation and increased cholesterol crystal formation, suggesting that intact autophagy suppresses the inflammasome, which is essential in atheroprotection [61]. As a consequence, successful autophagy could stabilize the atherosclerotic plaques and reduce adverse vascular events [3, 62].
On the other hand, excessive autophagy in SMCs or ECs may cause cell death. SMC death destabilizes plaques due to the decreased synthesis of collagen which results in a thinner fibrous cap. Autophagy mediated EC death is also the result of sustained ER stress in the atherosclerotic lesion [4]. EC death may be detrimental by promoting thrombosis and clinical events. However, macrophage death is considered a promising approach for plaque stabilization [63]. Recent evidence suggests that phagocytosis of macrophages dying through autophagy results in inflammasome activation and inflammatory factor release [64]. The above findings suggest that appropriate manipulation of autophagy would foster its beneficial effects in stabilizing the plaque by promoting cell survival and reducing cell death.
2.1.2. Autophagy in cardiac ischemia/reperfusion
The direct effect of atherosclerosis is cardiac ischemia, and autophagy activation in response to myocardial ischemia was documented 30 years ago [7]. Hypoxia triggers significant formation of autophagosomes adjacent to swollen and fragmented mitochondria [7], suggesting there may be interaction between autophagy and mitochondria during cardiac ischemia. The findings that autophagy inhibitors promoted cardiac myocyte death under glucose starvation suggested that autophagy could be protective by replenishing depleted energy stores and removing the toxic dysfunctional organelles. Going further, this study indicates that AMPK may participate in ischemia-induced autophagy that promotes cell survival by eliminating dysfunctional mitochondria, which would otherwise become a source of ROS and pro-apoptotic mediators [8].
A second wave of autophagy activation is triggered by reoxygenation following ischemia. More autophagosomes are detected after ischemia/reperfusion as compared to hypoxia alone [7]. Autophagy could be either adaptive or maladaptive in the context of ischemia/reperfusion injury. Some studies suggest autophagic flux is impaired during ischemia/reperfusion at the level of autophagosome formation and lysosome degradation. In addition, impaired autophagosome clearance is associated with increased ROS and mitochondrial permeabilization, resulting in cell death. This process is accompanied by ROS-mediated Beclin-1 upregulation and a decline in lysosome-associated membrane protein-2 and impaired autophagosome processing [65]. In contrast, other studies show that overexpression of Beclin1 enhances autophagic flux and protects cardiac cells from ischemia/reperfusion injury [66]. Consistent with this result, Beclin 1+/− mice manifested attenuated autophagy, with less infarction and suppressed apoptosis [8]. Additionally, new signaling involving BCL-2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) is a recently identified player in ischemia/reperfusion injury. As a downstream target of hypoxia-inducible factor 1α, BNIP3 permeabilizes cardiac mitochondria and promotes mitochondrial fission, causing mitochondrial dysfunction and cardiomyocyte death during ischemia/reperfusion injury. This process is related to autophagosome formation. Forced expression of BNIP3 stimulates autophagy in cardiac myocytes [67]. Autophagy serves as a protective mechanism in the setting of BNIP3 expression in cardiac myocytes, as inhibition of autophagy increases BNIP3-induced cardiomyocyte death by preventing removal of damaged mitochondria [67]. Conversely, BNIP3 expression appears to trigger autophagy and leads to a decline in lysosome abundance in cardiac myocytes. The resultant autophagosome accumulation prevents its pro-survival role and triggers cardiomyocyte death [37].
Another important signaling protein in ischemia/reperfusion is glycogen synthase kinase-3β (GSK-3β), which regulates autophagy during prolonged ischemia in a time-dependent manner. In the initial phase of ischemia, activation of GSK-3β stimulates autophagy, whereas during subsequent reperfusion, inactivation of GSK-3β inhibits autophagy. Such time-dependent regulation of endogenous GSK-3β adapts the cardiac myocyte to ischemia and protects against reperfusion injury [68]. In another pathway, ischemia-induced autophagy activation is dependent on AMPK activation. In contrast, reperfusion-augmented autophagy is dependent on Beclin1 instead of AMPK. Collectively, these results indicate the distinct roles of autophagy during ischemia and reperfusion. Autophagy may play an adaptive role during ischemia and a maladaptive role during reperfusion [8]. Thus, precise control of GSK-3β or AMPK at different stages of ischemia/reperfusion would provide a novel strategy to enhance survival under ischemic conditions and protect against cardiac myocyte death from reperfusion injury.
2.2. Autophagy in cardiomyopathy
Cardiomyopathy is classified as dilated, hypertrophic or restrictive cardiomyopathy. Lamin A/C gene (LMNA) mutation-related dilated cardiomyopathy is characterized by left ventricular enlargement and decreased systolic function accompanied by arrhythmias and other systemic diseases. Defective autophagy is one major feature in LMNA cardiomyopathy, which could be caused by overexpression of dual specificity phosphatase 4 in the heart and upregulated AKT-mTOR signaling. Pharmacological interventions improve cardiac function, which is correlated with enhanced autophagy [9-11]. A recent finding is that mammalian target of rapamycin complex 1 mediates LMNA deficiency-induced dilated cardiomyopathy. Elevated mTORC1 signaling impairs autophagy, and mTORC1 inhibition with rapalogs extends survival in the LMNA deficient mice through restoration of autophagic flux and improved cardiac function [69]. The desmin-related cardiomyopathy (DRC) characterized by accumulation of misfolded proteins is triggered by a missense mutation in the alphaB-crystallin (CryAB) gene. Autophagy is considered an adaptive response in this proteo-toxic form of cardiomyopathy, which is evidenced by accelerated heart failure and early mortality in DRC mice with genetic ablation of beclin1, a gene required for autophagy [70]. In addition, transgenic overexpression of the mutant desmin or CryAB (R120G) in mice in vivo as well as in vitro upregulates p62 mRNA and protein levels which protects cardiomyocytes from misfolded protein induced cell injury and death by maintaining responsive autophagosome formation and autophagy [71]. Atg7 induces basal autophagy. Sustained Atg7 expression rescues impaired autophagy in the CryAB (R120G) hearts with decreased cardiac hypertrophy and prolonged survival, suggesting autophagy activation would be a viable therapeutic strategy for ameliorating desmin-related cardiomyopathy [12, 72]. Dilated cardiomyopathy is linked with suppressed mitophagy, which is the result of deficient mitofusin-2. Ablation of mitofusin-2 prevented the translocation of Parkin to the mitochondria and Parkin-mediated ubiquitination [73]. The proapoptotic kinase Mst1 phosphorylates Beclin1 at Thr-108, which enhances its interaction with Bcl-2 and/or Bcl-xL, and inhibits the phosphatidylinositide 3-kinase activity of the Atg14L-Beclin1-Vps34 complex, thus suppressing autophagy and providing a mechanism for the development of dilated cardiomyopathy in man [74]. Very recently, the cardioprotective effects of macrophage migration inhibitory factor (MIF) against doxorubicin-induced cardiomyopathy has been recognized to function through augmented autophagy [75]. Knock down of myeloid cell leukemia-1 (MCL-1), an anti-apoptotic BCL-2 protein, in the adult heart led to rapid development of cardiomyopathy and death, which was associated with impaired induction of autophagy in the heart [76]. The above studies imply that defective autophagy contributes to cardiac dysfunction and cardiomyopathy development; therefore, pharmacologic interventions to augment autophagy might improve cardiac function and ameliorate cardiomyopathy.
In contrast, autophagy may promote cardiomyopathy. For instance, histone deacetylases (HDACs) regulate cardiac plasticity. HDAC activity is required for stress-induced cardiomyocyte autophagy which is linked to load-induced cardiac hypertrophy. HDAC inhibitors have antihypertrophic effects due to the unique action of inhibiting augmented autophagic flux [77]. Diabetic cardiomyopathy, first introduced by Rubler in 1972 [78], has been defined as ventricular dysfunction that occurs independently of coronary artery disease and hypertension. Changes in myocardial structure, Ca2+ signaling and metabolism have been implicated in the pathogenesis of diabetic cardiomyopathy. Recently, the essential role of autophagy in diabetic cardiomyopathy has been investigated intensively. Diabetes induces cardiomyocyte apoptosis and suppresses cardiac autophagy, which is linked with diabetic cardiomyopathy. Activation of AMPK by metformin restores cardiac autophagy and prevents cardiomyopathy presumably through disruption of Beclin1-Bcl-2 complex and protects against cardiac apoptosis [79, 80]. Interestingly, deficiency in autophagy protects cardiac function in type 1 diabetes through upregulation of Rab9 regulated alternative autophagy and mitophagy [81]. Another study shows that cardiac hypertrophy and dysfunction in type-2 diabetes is dependent on saturated fatty acid and sphingolipid synthesis. In particular, ceramide synthase-5 is involved in lipid-induced autophagy and lipotoxic cardiomyopathy [82]. Due to the important effects of autophagy in metabolic disease-induced cardiomyopathy, autophagy could serve as a biomarker for cardiac dysfunction and cardiomyopathy in diabetes.
2.3. Autophagy in heart failure
To date, heart failure remains one of the leading causes of death in the United States. An estimated 5 million Americans have heart failure with a mortality rate of approximately 50% in 5 years [83]. Heart failure is a progressive disease characterized by adverse ventricular remodeling which involves changes in the balance between cardiomyocyte protein synthesis and degradation. It is recognized that the autophagy–lysosome pathway is a house-keeper in cardiomyocytes under physiological conditions. However, the role of autophagy in heart failure is controversial. For instance, autophagy may antagonize ventricular hypertrophy by increasing protein degradation and decreasing tissue mass. As a result, autophagy may be an adaptive response to heart failure. In the mouse heart, autophagy induced by sustained expression of Atg7 ameliorates ventricular dysfunction, decreases cardiac hypertrophy, and prolongs survival. These findings suggest that activation of autophagy may be a viable therapeutic strategy for improving cardiac performance under proteotoxic conditions [12]. However, the efficiency of protective autophagy declines with age, leading to abnormal intracellular protein aggregates, which result in enhanced oxidative stress, decreased ATP production, and cell death. Oxidative stress sensitizes the heart to the renin-angiotensin-aldosterone system, inducing autophagic type-II programmed cell death and increasing the propensity for adverse cardiac remodeling, diastolic dysfunction and heart failure [84]. Angiotensin II (AngII) increases mitochondrial ROS in cardiomyocytes, concomitant with increased autophagy in hearts of angiotensin II-treated mice [47]. AngII type I (AT1) receptor mediates autophagosome formation in response to AngII stimulation. This response, however, is blocked by co-expression of the AngII type 2 (AT2) receptor in neonatal cardiomyocytes [85]. A robust autophagic response in cardiomyocytes is elicited by pressure overload stress, this response is maladaptive, as excessive autophagy in load-induced heart failure leads to autophagic cell death, loss of cardiomyocytes, and may contribute to the worsening of heart failure. Mechanical unloading with a left ventricular assist device attenuated autophagy, reduced energy demand and improved function of the failing human heart [86].
Autophagy in cardiomyocytes is affected by a variety of stimuli, such as lipid and glucose. High fat diet (HFD) disrupts autophagosome maturation at the step of autophagosomes fusion with lysosomes. This impaired autophagic flux is associated with increased apoptosis, mitochondrial injury, intracellular Ca2+ dysregulation and cardiac dysfunction [13]. Akt2 was considered to play a predominant role in HFD-induced cardiac hypertrophy and contractile dysfunction. Akt2 ablation has cardioprotective effect in that it rescues HFD-induced disruption of the autophagosome maturation process and facilitated the transition from autophagosomes to autophagolysosomes[13]. Conversely, compared with normal glucose (5.5 mM), high glucose (17 or 30 mM) decreases autophagic flux in cardiomyocytes. In addition, high glucose-induced cardiomyocyte death is attenuated by suppression of autophagy by 3-methyladenine or silencing of the Becn1 or Atg7 gene. In contrast, augmentation of autophagy with rapamycin or overexpression of Becn1 or Atg7 predisposes cardiomyocytes to high glucose toxicity. These results indicate that reduced autophagic flux is an adaptive response that serves to limit high glucose-induced cardiac toxicity [87]. Autophagy also protects the heart from intermittent hypoxia-induced systolic dysfunction by maintaining contractile function [88]. miR-212 and miR-132, two important miRNA involved in cardiac hypertrophy, are upregulated in hypertrophic cardiomyocytes. Overexpression of miR-212 and miR-132 leads to impaired autophagic response to starvation, hypertrophy and heart failure. MiR-212/132 deletion protects mice from pressure-overload-induced heart failure [89]. The cytokine MIF elicits cardioprotective effects through AMPK activation. As an illustration of its importance, MIF deficiency exacerbates left-ventricular dysfunction following starvation. This process is mediated by interrupted starvation-induced autophagic vacuole formation and exacerbated starvation-induced cell death. These results indicate that MIF preserves cardiac contractile function under starvation by regulating autophagy [90]. A recent study showed that mitochondrial homeostasis and autophagy are dependent on MCL-1 which is based on the observation that cardiomyocyte-specific MCL-1 knockout mice developed rapid cardiomyopathy and heart failure [76]. Finally, the regulatory associated protein of mTOR (Raptor) is an mTOR binding partner that mediates mTOR signaling to downstream targets. Ablation of raptor reduces myocardial mTORC1 activity, leading to heart failure, dilated cardiomyopathy and high mortality, which is associated with apoptosis and augmented autophagy in cardiomyocytes [91]. In summary, autophagy, controlled by a variety of factors, may antagonize ventricular hypertrophy and alleviate heart failure by increasing protein degradation and decreasing tissue mass. On the other hand, excessive autophagy under certain conditions such as pressure overload leads to cell death, and may contribute to the worsening of heart failure.
2.4. Autophagy in hypertension
Hypertension is classified as primary hypertension and secondary hypertension. Primary hypertension accounts for 90–95% of cases without obvious medical cause. The remaining 5–10% of cases are secondary effects of diseases in kidneys, arteries, heart or endocrine system. The heart exhibits robust hypertrophic growth in response to hypertension, which is regulated by autophagy. Essential cellular elements eliminated by excessive autophagy may provoke cell death and contribute to hypertension-related heart disease [92]. Excessive proliferation and resistance to apoptosis of pulmonary artery smooth muscle cells (PASMCs) are features of pulmonary arterial hypertension. PASMC proliferation facilitates vascular remodeling, leads to narrowed vascular lumen and increased pulmonary vascular resistance, and eventually increased pulmonary arterial pressure. Recently, chloroquine, a widely used antimalarial and antirheumatoid drug, was shown to prevent the development of monocrotaline-induced pulmonary hypertension by inhibition of autophagy. Monocrotaline-induced pulmonary hypertension is characterized by increased expression of LC3B-II and reduced expression of p62 in muscularized small pulmonary arteries, which is accompanied by increased medial thickness and proliferation of PASMCs. Chloroquine inhibited autophagy and restored p62 levels in the media of small pulmonary arteries in vivo, and was associated with inhibition of proliferation and induction of apoptosis in PASMCs in small pulmonary arteries. The underlying mechanism was attributed to chloroquine preventing acidification of the lysosome and subsequent processing of the autophagosome, thus preventing degradation of bone morphogenetic protein type II receptor (BMPR-II). The conserved intact BMPR-II signaling along with impaired autophagy contributes to a pro-apoptotic, anti-proliferative phenotype in PASMCs, indicating that autophagy is involved in pulmonary hypertension [93]. In another model, sympathetic premotor neurons that maintain vasomotor tone in the rostral ventrolateral medulla (RVLM) play a pivotal role in neurogenic hypertension. Drugs inhibiting autophagy in RVLM decreased hypertension in spontaneously hypertensive rats, suggesting autophagy could be a therapeutic target in the control of neurogenic hypertension [94]. Taken together, these studies suggest that autophagy promotes pulmonary hypertension and neurogenic hypertension. Inhibition of autophagy could provide a new therapeutic strategy in the management of hypertension.
2.5. Autophagy and oxidative stress in cardiovascular disease
Emerging evidence indicates oxidative stress plays an important role in cardiovascular disease development. Dysregulation of autophagy renders cardiomyocytes more prone to ischemia-induced injury and cardiac remodeling [95]. H2O2 significantly increased both autophagosomes and autolysosomes and, thus, autophagic flux in cardiac myocytes. N-2-mercaptopropionyl glycine (MTG), an antioxidant, attenuates autophagy in the presence of H2O2 in vitro, as well as autophagic flux in ischemia/reperfusion-induced oxidative stress in vivo. The impaired autophagy in vivo is accompanied by a decrease in the size of myocardial infarction. Moreover, Beclin1+/- mice have reduced myocardial infarction after ischemia/reperfusion, whereas MTG treatment results in no additional reduction of infarct size. These results imply that autophagy mediates myocardial injury by increasing the oxidative stress elicited by ischemia/reperfusion [14]. In cardiac myocytes, ROS are also stimulated by glucose deprivation. Inhibition of ROS disrupted autophagy induced by glucose deprivation, emphasizing the important effects of oxidative stress on the activation of autophagy in cardiac myocytes induced by energy stress [96]. In the context of hypertension and diabetes, oxidative stress sensitizes the heart to the renin-angiotensin-aldosterone system, induces autophagic type-II programmed cell death, and leads to accelerated cardiac remodeling and cardiac dysfunction. Adiponectin, an adipokine known to mediate cardioprotective effects, demonstrates antioxidant potential to attenuate autophagy induced by excessive ROS in cardiomyocytes by inhibiting H2O2-induced AMPK/mTOR signaling [84]. Mitophagy plays an important role in mitochondrial quality control and cellular homeostasis through selective degradation of dysfunctional mitochondria. Dysfunctional mitochondria produce excess ROS which triggers oxidative stress. In the diabetic heart, mitophagy was decreased as evidenced by decreased Pink and Parkin, accompanied by decreased Lamp1 levels. As expected, impaired mitophagy in the diabetic heart increased ROS generation and oxidative protein damage, and the antioxidant enzyme MnSOD was also decreased. Notably, Beclin-1 deficiency restored mitophagy, and the MnSOD level was partially restored, and thereafter, ROS generation and oxidative protein damage was attenuated. These results indicate that inhibition of autophagy improves mitophagy and protects the heart from diabetes-induced oxidative injury [81]. Furthermore, autophagy also mediates inflammatory responses in cardiac myocytes. For instance, monocyte chemotactic protein-1 (MCP-1) promotes the development of heart failure by inducing oxidative stress. MCP-1-induced protein (MCPIP) mediates the downstream effects of oxidative stress, including ER stress, autophagy and cell death, and these processes are inhibited by inhibitors of oxidative stress. In addition, inhibitors of ER stress inhibit autophagy and cell death. These findings indicate that the elevated MCP-1 levels associated with chronic inflammation may contribute to the development of heart failure through oxidative stress-induced ER stress and autophagy [97].
Conversely, autophagy serves to protect against disease development by mediating protein quality control. AntimycinA (AMA) induces mitochondrial stress and increases mitochondrial superoxide generation, as well as augments nuclear DNA oxidation and cell death in cardiomyocytes. Upregulated autophagy by rapamycin promotes mitochondrial clearance and protects cardiomyocytes from the AMA-induced cell injury. In addition, the accumulation of ubiquitinylated proteins induced by AMA is suppressed by autophagy. Hence, autophagy induction could become a potential therapeutic strategy against oxidative stress-mediated injury in cardiomyocytes [98]. Autophagy also plays a protective role in AngII induced oxidative stress. Atg5+/- mice have impaired autophagy and increased production of ROS, which activates nuclear factor κB (NF-κB) in macrophages and increases cardiac inflammation. As a consequence, Atg5+/- mice are associated with increased cardiac fibrosis. Thus, intact autophagy signaling protects the heart from hypertension-induced inflammation and cardiac injury [99]. In addition to mitochondria, ROS production in cardiomyocytes during glucose deprivation is dependent on NADPH oxidase-4 expressed in the ER. ROS produced in the ER induce autophagy in the cardiomyocytes and promote survival in response to energy stress. These effects are mediated by the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor-2α/activating transcription factor-4 pathway [100]. To summarize, depending on the pathologic environment, autophagy may mediate oxidative stress-induced cell injury and cell death through autophagic type-II programmed cell death or protect against cell injury from ROS produced by mitochondria or the ER.
3. Manipulation of cardiac autophagy
The transition of autophagy from an adaptive phase to a maladaptive phase plays a vital role in driving the development of cardiovascular disease. As described above, autophagy can have either beneficial or detrimental effects in the cardiovascular system. Adaptive autophagy promotes survival in response to hypoxia and oxidative stress by removing damaged organelles as well as recycling macromolecules to maintain energy levels and support protein synthesis. In contrast, prolonged hypoxia and subsequent reperfusion result in maladaptive autophagy which causes cell death through excessive self-digestion of essential organelles and proteins or inducing apoptosis. Thus, manipulation of autophagy may represent a potential therapeutic target to treat or prevent development of heart disease. Lysosomal-associated membrane protein 2 (Lamp-2) is a ubiquitous lysosomal membrane protein required for the proper fusion of lysosomes with autophagosomes [101]. Lamp-2 depletion results in the inhibition of cytoprotective autophagy signaling secondary to the failure of fusion between lysosomes and autophagosomes which is linked to Danon disease, characterized by severe cardiomyopathy [102]}. The failure of autolysosome fusion is a hallmark of the maladaptive autophagy which results in the accumulation of autophagic vacuoles and the autophagosome-associated proteins LC3 and p62, coupled with increased apoptosis [103]. Autophagic flux assays may be used to determine the autophagic status in pathological conditions; however, the lack of suitable assays for measuring the ongoing autophagic flux in humans limits the feasibility of identifying the optimal window of autophagic activation to exploit the cardioprotective effects of macroautophagy without disrupting cardiac homeostatic mechanisms. A combination of measurement of circulating LC3 levels in the blood and in vivo imaging of autophagy markers in the heart would provide better information about disease progression and generate corresponding therapeutic approaches. Indeed, animal studies have yielded promising results. Upregulated autophagy has beneficial effects on cardiovascular disease management. In vivo work indicates sustained expression of autophagy-related 7 in the CryAB (R120G) hearts leads to decreased cardiac hypertrophy, ameliorated ventricular dysfunction, and prolonged survival [12]. In addition, Inhibition of mTOR by everolimus limits infarct size and attenuates adverse left ventricular remodeling after myocardial infarction in mice [8]. Moreover, chronic AMPK activation by metformin prevents cardiomyopathy by upregulating autophagy in diabetic mice [104]. Recently, Xu et al. reported that rapamycin restored autophagy activity in the hearts of cardiomyocyte-phosphatase and tensin homolog knockout mice, reversing hypertrophic cardiomyopathy [105]. In contrast, downregulation of autophagy may also protect cardiac function. For instance, histone deacetylase inhibitors attenuate cardiac hypertrophy and improve cardiac function by suppressing autophagy [77]. In addition to testing autophagy inhibitors or activators such as mTOR inhibitors and AMPK activators, it is worthwhile to explore the effects of current medications already used clinically such as β-blockers, Ca2+ channel blockers, vasodilators and statins on autophagy, as well as safe dose ranges to ensure their positive effects in cardiac autophagy and protect cardiac function.
4. Conclusions
This review summarizes the most current findings on how autophagy is executed and regulated, and how the integration or disruption of autophagy pathways contributes to cardiovascular physiology and disease. Defining the mechanisms of autophagy pathways in different cell types in the cardiovascular system and the importance of autophagosomal degradation of organelles such as endoplasmic reticulum and mitochondria during oxidative stress has the potential to yield novel therapeutic approaches for the treatment of cardiovascular diseases.
Highlights.
This is a revision of review article that highlights:
Introduced types and process of autophagy
Summarized the correlation of autophagy with cardiovascular diseases
Highlighted the interaction of autophagy and oxidative stress in the development of cardiovascular diseases
Acknowledgments
This study was supported by grants from American Diabetes Association award (7-09-JF-69, XYT), the National Institutes of Health (HL031607, HL068758, HL104017, HL105287, RAC) and Martin Luther King, Jr. Fellowship from Boston University (MDT), and a predoctoral T32 training grant (HL07969, MDT).
Abbreviations
- AMA
antimycin A
- AMPK
adenosine monophosphate–activated protein kinase
- AngII
angiotensin II
- AT1
angII type I
- AT2
angII type 2
- Atg
autophagy related protein
- BMPR-II
bone morphogenetic protein type II receptor
- BNIP3
BCL-2/adenovirus E1B 19-kDa interacting protein 3
- CryAB
alphaB-crystallin
- DRC
desmin-related cardiomyopathy
- ECs
endothelial cells
- ER
endoplasmic reticulum
- GSK-3β
glycogen synthase kinase-3β
- HDACs
histone deacetylases
- HFD
high fat diet
- LC3
light chain 3
- LMNA
lamin A/C gene
- MCL-1
myeloid cell leukemia-1
- MCP-1
monocyte chemotactic protein-1
- MCPIP
MCP-1-induced protein
- MIF
macrophage migration inhibitory factor
- MTG
N-2-mercaptopropionyl glycine
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- NF-κB
nuclear factor κB
- PASMCs
pulmonary artery smooth muscle cells
- PI3K
class III phosphatidylinositol 3-kinase
- Redox
reduction-oxidation
- ROS
reactive oxygen species
- RVLM
rostral ventrolateral medulla
- SMCs
smooth muscle cells
- ULK
unc-51-like kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 2.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ouimet M. Autophagy in obesity and atherosclerosis: Interrelationships between cholesterol homeostasis, lipoprotein metabolism and autophagy in macrophages and other systems. Biochim Biophys Acta. 2013;1831:1124–1133. doi: 10.1016/j.bbalip.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, Wang W, Jiang Z, Gao C, Ma B, Chen YG, Cockerill G, Hu Y, Xu Q, Zeng L. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem. 2013;288:859–872. doi: 10.1074/jbc.M112.412783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller C, Salvayre R, Negre-Salvayre A, Vindis C. Oxidized LDLs trigger endoplasmic reticulum stress and autophagy: prevention by HDLs. Autophagy. 2011;7:541–543. doi: 10.4161/auto.7.5.15003. [DOI] [PubMed] [Google Scholar]
- 6.Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal. 2006;8:152–162. doi: 10.1089/ars.2006.8.152. [DOI] [PubMed] [Google Scholar]
- 7.Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98:425–444. [PMC free article] [PubMed] [Google Scholar]
- 8.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 9.Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, Worman HJ. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. 2012;4:102. doi: 10.1126/scitranslmed.3003875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi JC, Worman HJ. Reactivation of autophagy ameliorates LMNA cardiomyopathy. Autophagy. 2013;9:110–111. doi: 10.4161/auto.22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi JC, Wu W, Muchir A, Iwata S, Homma S, Worman HJ. Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J Biol Chem. 2012;287:40513–40524. doi: 10.1074/jbc.M112.404541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, Hua Y, Nair S, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol. 2013;5:61–63. doi: 10.1093/jmcb/mjs055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2013;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13:373–387. doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel KR, Andreadi C, Britton RG, Horner-Glister E, Karmokar A, Sale S, Brown VA, Brenner DE, Singh R, Steward WP, Gescher AJ, Brown K. Sulfate metabolites provide an intracellular pool for resveratrol generation and induce autophagy with senescence. Sci Transl Med. 2013;5:133. doi: 10.1126/scitranslmed.3005870. [DOI] [PubMed] [Google Scholar]
- 20.Tey SK, Khanna R. Host immune system strikes back: autophagy-mediated antigen presentation bypasses viral blockade of the classic MHC class I processing pathway. Autophagy. 2012;8:1839–1841. doi: 10.4161/auto.21860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma J, Becker C, Lowell CA, Underhill DM. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J Biol Chem. 2012;287:34149–34156. doi: 10.1074/jbc.M112.382812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan SH, Shui G, Zhou J, Shi Y, Huang J, Xia D, Wenk MR, Shen HM. Critical role of SCD1 in autophagy regulation via lipogenesis and lipid rafts-coupled AKT-FOXO1 signaling pathway. Autophagy. 2014;10:1–17. doi: 10.4161/auto.27003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Z, Ni D, Ghozalli I, Pirooz SD, Ma B, Liang C. UVRAG: at the crossroad of autophagy and genomic stability. Autophagy. 2012;8:1392–1393. doi: 10.4161/auto.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsui A, Kamada Y, Matsuura A. The role of autophagy in genome stability through suppression of abnormal mitosis under starvation. PLoS Genet. 2012;9:e1003245. doi: 10.1371/journal.pgen.1003245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vessoni AT, Filippi-Chiela EC, Menck CF, Lenz G. Autophagy and genomic integrity. Cell Death Differ. 2013;20:1444–1454. doi: 10.1038/cdd.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15:105–112. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 27.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 29.Jung HS, Lee MS. Role of autophagy in diabetes and mitochondria. Ann N Y Acad Sci. 2010;1201:79–83. doi: 10.1111/j.1749-6632.2010.05614.x. [DOI] [PubMed] [Google Scholar]
- 30.Czaja MJ, Ding WX, Donohue TM, Jr, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, Perlmutter DH, Randall G, Ray RB, Tsung A, Yin XM. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoefkens E, Nys K, John JM, Van Steen K, Arijs I, Van der Goten J, Van Assche G, Agostinis P, Rutgeerts P, Vermeire S, Cleynen I. Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress. Autophagy. 2013;9:2046–2055. doi: 10.4161/auto.26337. [DOI] [PubMed] [Google Scholar]
- 32.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iglewski M, Hill JA, Lavandero S, Rothermel BA. Mitochondrial fission and autophagy in the normal and diseased heart. Curr Hypertens Rep. 2010;12:418–425. doi: 10.1007/s11906-010-0147-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 2010;21:683–690. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Brot S, Auger C, Bentata R, Rogemond V, Menigoz S, Chounlamountri N, Girard-Egrot A, Honnorat J, Moradi-Ameli M. Collapsin Response Mediator Protein 5 (CRMP5) Induces Mitophagy Thereby Regulating Mitochondrion Numbers in Dendrites. J Biol Chem. 2014;289:2261–2276. doi: 10.1074/jbc.M113.490862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraft C, Peter M. Is the Rsp5 ubiquitin ligase involved in the regulation of ribophagy. Autophagy. 2008;4:838–840. doi: 10.4161/auto.6603. [DOI] [PubMed] [Google Scholar]
- 37.Cebollero E, Reggiori F, Kraft C. Reticulophagy and ribophagy: regulated degradation of protein production factories. Int J Cell Biol. 2012:182834. doi: 10.1155/2012/182834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakai Y, Oku M, van der Klei IJ, Kiel JA. Pexophagy: autophagic degradation of peroxisomes. Biochim Biophys Acta. 2006;1763:1767–1775. doi: 10.1016/j.bbamcr.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 39.Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013;20:3–11. doi: 10.1038/cdd.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tolkovsky AM. Mitophagy. Biochim Biophys Acta. 2009;1793:1508–1515. doi: 10.1016/j.bbamcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Orenstein SJ, Cuervo AM. Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol. 2010;21:719–726. doi: 10.1016/j.semcdb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene. 1997;192:245–250. doi: 10.1016/s0378-1119(97)00084-x. [DOI] [PubMed] [Google Scholar]
- 44.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7:1212–1221. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang HW, Liao HM, Peng WH, Lin HR, Chen CH, Chen GC. Atg9 Interacts with dTRAF2/TRAF6 to Regulate Oxidative Stress-Induced JNK Activation and Autophagy Induction. Dev Cell. 2013;27:489–503. doi: 10.1016/j.devcel.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 49.Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23:1860–1873. doi: 10.1091/mbc.E11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reggiori F, Tooze SA. Autophagy regulation through Atg9 traffic. J Cell Biol. 2012;198:151–153. doi: 10.1083/jcb.201206119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 53.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 54.Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009;104:304–317. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- 55.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 56.Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T, Xie Q, Zhou X, Chang H, Fu YJ, Zhu JD, Zhang QY, Mi MT. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013;9:2033–2045. doi: 10.4161/auto.26336. [DOI] [PubMed] [Google Scholar]
- 57.He C, Zhu H, Zhang W, Okon I, Wang Q, Li H, Le YZ, Xie Z. 7-Ketocholesterol induces autophagy in vascular smooth muscle cells through Nox4 and Atg4B. Am J Pathol. 2013;183:626–637. doi: 10.1016/j.ajpath.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le Guezennec X, Brichkina A, Huang YF, Kostromina E, Han W, Bulavin DV. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab. 2012;16:68–80. doi: 10.1016/j.cmet.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 60.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schrijvers DM, De Meyer GR, Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol. 2011;31:2787–2791. doi: 10.1161/ATVBAHA.111.224899. [DOI] [PubMed] [Google Scholar]
- 63.Martinet W, Verheye S, De Meyer GR. Selective depletion of macrophages in atherosclerotic plaques via macrophage-specific initiation of cell death. Trends Cardiovasc Med. 2007;17:69–75. doi: 10.1016/j.tcm.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 64.Petrovski G, Ayna G, Majai G, Hodrea J, Benko S, Madi A, Fesus L. Phagocytosis of cells dying through autophagy induces inflammasome activation and IL-1beta release in human macrophages. Autophagy. 2011;7:321–330. doi: 10.4161/auto.7.3.14583. [DOI] [PubMed] [Google Scholar]
- 65.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 67.Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson AB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18:721–731. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhai P, Sadoshima J. Glycogen synthase kinase-3beta controls autophagy during myocardial ischemia and reperfusion. Autophagy. 2012;8:138–139. doi: 10.4161/auto.8.1.18314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramos FJ, Kaeberlein M, Kennedy BK. Elevated MTORC1 signaling and impaired autophagy. Autophagy. 2013;9:108–109. doi: 10.4161/auto.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105:9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151–160. doi: 10.1161/CIRCRESAHA.110.237339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M, Sadoshima J. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu X, Bucala R, Ren J. Macrophage migration inhibitory factor deficiency augments doxorubicin-induced cardiomyopathy. J Am Heart Assoc. 2013;2:e000439. doi: 10.1161/JAHA.113.000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson AB. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27:1365–1377. doi: 10.1101/gad.215871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 79.He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270–1281. doi: 10.2337/db12-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zou MH, Xie Z. Regulation of interplay between autophagy and apoptosis in the diabetic heart: new role of AMPK. Autophagy. 2013;9:624–625. doi: 10.4161/auto.23577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077–18092. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, Cowart LA. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest. 2012;122:3919–3930. doi: 10.1172/JCI63888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 84.Essick EE, Wilson RM, Pimentel DR, Shimano M, Baid S, Ouchi N, Sam F. Adiponectin modulates oxidative stress-induced autophagy in cardiomyocytes. PLoS One. 2013;8:e68697. doi: 10.1371/journal.pone.0068697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Porrello ER, Delbridge LM. Cardiomyocyte autophagy is regulated by angiotensin II type 1 and type 2 receptors. Autophagy. 2009;5:1215–1216. doi: 10.4161/auto.5.8.10153. [DOI] [PubMed] [Google Scholar]
- 86.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120:S191–197. doi: 10.1161/CIRCULATIONAHA.108.842252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kobayashi S, Xu X, Chen K, Liang Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy. 2013;8:577–592. doi: 10.4161/auto.18980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maeda H, Nagai H, Takemura G, Shintani-Ishida K, Komatsu M, Ogura S, Aki T, Shirai M, Kuwahira I, Yoshida K. Intermittent-hypoxia induced autophagy attenuates contractile dysfunction and myocardial injury in rat heart. Biochim Biophys Acta. 2013;1832:1159–1166. doi: 10.1016/j.bbadis.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 89.Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, Remke J, Caprio M, Jentzsch C, Engelhardt S, Geisendorf S, Glas C, Hofmann TG, Nessling M, Richter K, Schiffer M, Carrier L, Napp LC, Bauersachs J, Chowdhury K, Thum T. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078. doi: 10.1038/ncomms2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu X, Pacheco BD, Leng L, Bucala R, Ren J. Macrophage migration inhibitory factor plays a permissive role in the maintenance of cardiac contractile function under starvation through regulation of autophagy. Cardiovasc Res. 2013;99:412–421. doi: 10.1093/cvr/cvt116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, Krishnan J, Lerch R, Hall MN, Ruegg MA, Pedrazzini T, Brink M. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066. [DOI] [PubMed] [Google Scholar]
- 92.Wang ZV, Rothermel BA, Hill JA. Autophagy in hypertensive heart disease. J Biol Chem. 2010;285:8509–8514. doi: 10.1074/jbc.R109.025023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, Morrell NW. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res. 2013;112:1159–1170. doi: 10.1161/CIRCRESAHA.111.300483. [DOI] [PubMed] [Google Scholar]
- 94.Chao YM, Lai MD, Chan JY. Redox-sensitive endoplasmic reticulum stress and autophagy at rostral ventrolateral medulla contribute to hypertension in spontaneously hypertensive rats. Hypertension. 2013;61:1270–1280. doi: 10.1161/HYPERTENSIONAHA.111.00469. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Y, Ren J. Targeting Autophagy for the Therapeutic Application of Histone Deacetylase (HDAC) Inhibitors in Ischemia-Reperfusion Heart Injury. Circulation. 2014;129:1088–1091. doi: 10.1161/CIRCULATIONAHA.113.008115. [DOI] [PubMed] [Google Scholar]
- 96.Marambio P, Toro B, Sanhueza C, Troncoso R, Parra V, Verdejo H, Garcia L, Quiroga C, Munafo D, Diaz-Elizondo J, Bravo R, Gonzalez MJ, Diaz-Araya G, Pedrozo Z, Chiong M, Colombo MI, Lavandero S. Glucose deprivation causes oxidative stress and stimulates aggresome formation and autophagy in cultured cardiac myocytes. Biochim Biophys Acta. 2010;1802:509–518. doi: 10.1016/j.bbadis.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 97.Younce CW, Kolattukudy PE. MCP-1 causes cardiomyoblast death via autophagy resulting from ER stress caused by oxidative stress generated by inducing a novel zinc-finger protein MCPIP. Biochem J. 2010;426:43–53. doi: 10.1042/BJ20090976. [DOI] [PubMed] [Google Scholar]
- 98.Dutta D, Xu J, Kim JS, Dunn WA, Jr, Leeuwenburgh C. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy. 2013;9:328–344. doi: 10.4161/auto.22971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao W, Li Y, Jia L, Pan L, Li H, Du J. Atg5 Deficiency-Mediated Mitophagy Aggravates Cardiac Inflammation and Injury in Response to Angiotensin II. Free Radic Biol Med. 2014;69:108–115. doi: 10.1016/j.freeradbiomed.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 100.Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ Res. 2013;113:1253–1264. doi: 10.1161/CIRCRESAHA.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26:313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fortunato F, Burgers H, Bergmann F, Rieger P, Buchler MW, Kroemer G, Werner J. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137:350–360. doi: 10.1053/j.gastro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 103.Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, Eskelinen EL, Pierron G, Saftig P, Kroemer G. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–3102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 104.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xu X, Roe ND, Weiser-Evans MC, Ren J. Inhibition of Mammalian Target of Rapamycin With Rapamycin Reverses Hypertrophic Cardiomyopathy in Mice With Cardiomyocyte-Specific Knockout of PTEN. Hypertension. 2014;63:729–739. doi: 10.1161/HYPERTENSIONAHA.113.02526. [DOI] [PubMed] [Google Scholar]