

Abstract
Purpose of review
This review focuses on current and future strategies to modulate neuroinflammation while reducing residual viral burden in the central nervous system (CNS). This has been realized by targeted long acting antiretroviral nano- and adjunctive therapies being developed for HIV infected people. Our ultimate goal is to eliminate virus from its CNS reservoirs and, in so doing, reverse the cognitive and motor dysfunctions seen in HIV-associated neurocognitive disorders (HAND).
Recent findings
Herein, we highlight our laboratories development of adjunctive and nanomedicine therapies for HAND. An emphasis is placed on drug-drug interactions that target both the viral life cycle and secretory pro-inflammatory neurotoxic factors and signaling pathways.
Summary
Antiretroviral therapy (ART) has improved the quality and duration of life for people living with HIV-1. A significant long-term comorbid illness is HAND. Symptoms, while reduced in severity, are common. Disease occurs, in part, through continued low-level viral replication inducing secondary glial neuroinflammatory activities. Our recent works and those of others have seen disease attenuated in animal models through the use of adjunctive and long-acting reservoir targeted nanoformulated ART. The translation of these inventions from animals to humans is the focus of this review.
Keywords: HIV-associated neurocognitive disorders, nanoformulated antiretroviral therapy, neuroinflammation, mixed lineage kinase
Introduction
While the medical treatment for HIV infection with potent combination antiretroviral therapy (ART) has reduced disease morbidities and mortalities, the prevalence of neurologic disease associated with viral infection has continued almost unabated [1]. Elimination of HIV-1 associated neurocognitive disorders (HAND), while important in the overall treatment strategies for HIV/AIDS, has found a secondary position in therapeutic design for several reasons. First and foremost is the acceptance that HAND has shifted to a more indolent disease phenotype compared to the profound dementia and motor and behavioral abnormalities that have all but vanished but were previously commonplace [2]. Second, the progressive encephalopathy seen in children has been virtually eliminated by ART. This has lulled people living with HIV and their healthcare providers into a mindset of the problem is gone and for “watchful waiting” in disease elimination all together. Even more telling, practitioners do not screen for HAND. This is based on the lack of awareness of subtle neurologic disease in the ART era coupled with inadequate information about its prevalence and limitations in screening tool use and consensus [3]. Though the incidence of the most severe forms of HAND has plummeted, prevalence of all forms of HAND has steadily increased with estimates that range between 40–70% in infected individuals [4], regardless of the ART regimen used and whether or not the drugs target the central nervous system (CNS) [5]. Perhaps more surprisingly, reliance on a CNS penetration score (CPE) for selection of drugs that have favorable CNS profiles (and high CPE scores) has led to the disturbing finding that high CPE scores correlate with a diagnosis of cognitive impairment in infected people [6]. Possible reasons for this include drug-CNS toxicity, patient adherence with high CPE score regimens or even selection bias based on patients with pre-existing HAND who choose such regimens [6]. Regardless, these observations, over 30 years after the AIDS epidemic started, are dispiriting and warrant a renewed push towards developing strategies that can both re-establish homeostasis between CNS immune effector cells and functional synapses as well as eliminate HIV CNS reservoirs that initiate the neuroinflammation that leads to HAND. To these ends, our recent works have sought to merge studies of adjunctive anti-inflammatory and neuroprotective therapies with new delivery schemes for ART. A mixed lineage kinase 3 (MLK3) inhibitor, URMC-099, uncovered for HAND treatments was tested in conjunction with long acting nanoformulated ART (nanoART) and was found to affect drug levels and speed viral elimination in experimental animal models of HIV/AIDS. These effects, interestingly enough, were associated with enhanced particle trafficking in macrophage recycling endosomes. These activities of URMC-099 and those of others provide the means to significantly boost efforts to clear persistent viral infection and cognitive function in HIV infected people [7].
Notwithstanding, the current review focuses on these and other current and future therapeutic strategies to achieve reduction or resolution of neuroinflammation with disease modifying outcomes for HAND with the ultimate goal of eliminating HIV from CNS reservoirs. The overarching idea is to harness novel nanomedicine combating strategies that facilitate ART virucidal activities during viral infection. Prior developed adjunctive therapies for HAND formed the foundation for these works and are reviewed elsewhere [1, 8].
Challenges to viral eradication in the CNS
One of the greatest challenges in the treatment of HIV-1 infection rests in the abilities of ART to target viral cellular reservoirs in barrier compartments. This includes the gut, lymphoid tissues and the CNS. The latter poses a number of inherent problems: (1) hijacking pro-inflammatory signaling in macrophages/microglia despite combination ART (cART) -mediated suppression of the viral life cycle; (2) bystander toxicity to target cells such neurons and oligodendrocytes with limited or no capacity for self-renewal; (3) eradication of virus may paradoxically lead to more inflammation when the end point is cell death of reservoir populations. Since many of the deleterious substrates for HAND such as destruction of normal synaptic architecture in the CNS are associated with neuroinflammation, a focus on molecular targets such as the MLKs, and in particular MLK-3, that serve as kinase control hubs for mediating inflammation may be key to disease-modifying therapies.
Mixed lineage kinases
MLKs are mitogen activated protein kinase kinase kinases (MKKKs) with features of both serine-threonine and tyrosine kinases (hence the nomenclature “mixed lineage”) that regulate the c-Jun N-terminal kinase (JNK) mitogen activated protein kinase (MAPK) signaling cascade (Fig. 1), and also regulate the other two major MAPK pathways, p38 and extracellular signal-regulated kinase (ERK) [9–11]. MLK3 (aka MAP3K11) is the most widely expressed MLK family member [9–11] and is known to be expressed in neurons [12], dendritic cells [13, 14], and many other cell types. At the cellular level, MLK3 is activated by cellular/metabolic stress, including reactive oxygen species, ceramide and TNF-α [15, 16]. At the molecular level, it is activated by Cdc42 and Rac, which interact with MLK3, and can cause it to dimerize via a leucine zipper interface, resulting in autophosphorylation at Thr277 and Ser281 within the protein activation loop, and enzyme activation [17, 18]. Our research group at the University of Rochester Medical Center has shown that HIV-1 Tat also leads to phosphorylation at these same residues in primary rat neurons [19] and that HIV-1 Tat also leads to activation of glycogen synthase kinase (GSK)-3β in neurons [20, 21]. Interestingly, these seemingly disparate events may be causally connected, since recent findings have shown that MLK3 can be activated as a result of direct phosphorylation by GSK-3β [22]. Endogenous inhibitors of MLK3 include the pro-survival protein kinase, Akt [23, 24]. Finally, in addition to its conventional kinase activity, MLK3 also possesses noncatalytic functions that contribute to activation of the Raf/ERK pathway and induction of cell proliferation; these effects are negatively regulated by the tumor suppressor merlin [25].
Figure 1.

Mixed lineage kinase 3 regulates the JNK and p38 pathways.
Role of MLK3 in neurodegenerative diseases
MLK3 has been implicated in neuronal apoptosis leading to neurodegenerative diseases [19, 22, 26, 27]. In the context of Parkinson’s disease (PD), the first-generation MLK3 inhibitor, CEP-1347 (Fig. 2), has been shown to prevent the induction of neuronal cell death, motor deficits and neuronal degeneration in the MPTP model of Parkinsonism [28–31]. CEP-1347-mediated neuroprotection has also been demonstrated in an in vitro model for PD, using methamphetamine-exposed human mesencephalic-derived neurons[31]. This has led to the use of CEP-1347 in a large Phase II study in patients with PD (see below).
Figure 2.
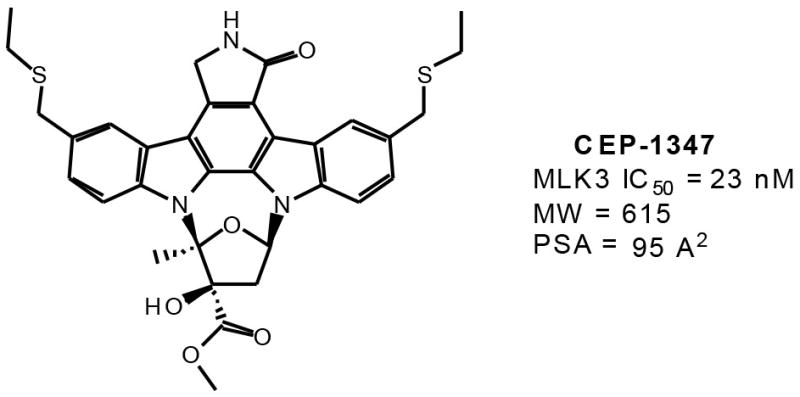
Chemical structure, MLK31C50, and physical properties of CEP-1347.
Activation of MLK3 and downstream kinases (JNK, p38) has also been implicated in the pathogenesis of several other neurodegenerative diseases [11], including HIV-associated dementia [19, 32], Alzheimer’s disease (AD) [33, 34], and ischemic injury/stroke [35–40]. MLK3’s role in ischemic injury may extend to other tissues, such as heart, where a first-generation MLK3 inhibitor (CEP11004) has been shown to reduce myocardial cell death and restore post-ischemic contractile function [41].
In addition, MLK3 has been implicated as playing a causal role in peripheral neuronal degeneration, including the development of HIV-associated peripheral neuropathy, which can be induced both by soluble HIV-1 gene products and also by the antiviral drugs used to treat HIV-1 [42, 43]. Finally, the first generation MLK3 inhibitor, CEP-1347, has been shown to prevent the death of vestibular and cochlear hair cells in models for ototoxicity caused by exposure to aminoglycoside antibiotics [44–46].
Role of MLK3 in inflammation and immunity
One contributing factor to the neuroprotective efficacy of MLK3 blockade is the fact that MLK3 activation plays an essential role in the activation of microglia and astrocytes, and their subsequent release of proinflammatory cytokines [47, 48]. Thus, MLK3 likely plays an important role in inflammation and immunity. Consistent with this, MLK3 is expressed in dendritic cells [13, 14] and regulates CD3/CD28-mediated signaling events in T cells [49].
Development and clinical evaluation of first-generation MLK3 inhibitors
Cephalon’s CEP-1347 (Fig. 2) is the first and to date, the only inhibitor showing significant MLK3 activity that has been tested in human subjects. The compound is not completely specific for MLK3, and there is no published data that quantify its ability to penetrate the CNS. It is a large molecular weight compound (MW = 615) with high polar surface area (95 square angstroms), properties that are known to limit CNS penetration. CEP-1347 is an ethylthiomethyl analog of K-252a, a natural product indolocarbazole isolated from the bacterium Nocardiopsis species [12]. CEP-1347 demonstrated neuroprotective activity in preclinical models for PD, which were sufficiently compelling to initiate early Phase 1 studies to demonstrate the safety and tolerability of CEP-1347 in patients suffering from PD [50], followed by a larger blinded, placebo-controlled trial of efficacy in patients with early untreated PD (PRECEPT study) [51].
The MLK3 inhibitor CEP-1347 was safe and well tolerated in human subjects [50] but was an ineffective treatment in subjects with early PD [52]. We believe it is likely that early symptomatic PD patients may already have an advancing underlying disease that is not readily amenable to therapeutic intervention [52]. Also, failure of the PRECEPT trial may reflect dosage considerations related to the bell-shaped efficacy curve for CEP-1347 and/or failure to maintain adequate therapeutic levels of CEP-1347 within the CNS. It is also possible that an additional reason for failure of the PRECEPT trial may be related to the fact that MLK3 inhibition has both a cell survival-promoting effect and an inhibitory effect on neuroinflammation. These represent strong synergistic neuroprotective activities in the context of preclinical models for PD, as well as in human neurodegenerative diseases such as neuroAIDS and AD, which are characterized by a combination of neuronal damage/neuronal loss plus a profound neuroinflammatory reaction associated with the release of neurotoxic inflammatory mediators. In neuroAIDS and AD, the synapse is likely to be the primary locus of dysfunction that is the substrate for neurologic disease and thus MLK3 inhibition may be a particularly valuable strategy for therapeutic intervention. In the case of early PD, neuroinflammation does not appear to be a major component of nigrostriatal degeneration. Thus, this may not have been an ideal target population for evaluation of the neuroprotective efficacy of CEP-1347.
MLK3 inhibitors and microglial inflammation
Researchers at Lundbeck A/S demonstrated that CEP-1347 reduced cytokine production in human and murine microglial cell cultures, and in monocyte/macrophage-derived cell lines that were stimulated with various endotoxins or the plaque forming peptide Aβ1-40. CEP-1347 inhibited brain TNF-α production induced by intracerebroventricular injection of LPS (lipopolysaccharide) in mice. The authors postulated that MLKs may function as significant modulators of microglial inflammation and demonstrated anti-inflammatory potential for an MLK3 inhibitor [48].
Efficacy of CEP-1347 in murine HIVE model
In our own experiments, HIV infected monocyte derived macrophages were stereotactically injected into the basal ganglia of CB17 severe combined immunodeficient mice (murine HIVE model) [53]. Daily intraperitoneal injections of CEP-1347 (up to 15 mg/kg) produced a dose dependent reduction in microgliosis measured by post mortem staining with Iba-1, suggesting the value of further evaluation of the neuroprotective properties of MLK3 inhibitors with better blood brain barrier penetration [54].
Development of novel MLK3 inhibitors
The human clinical experience with CEP-1347 demonstrated that MLK3 blockade is safe and well tolerated [50]. This is consistent with the fact that MLK3 knockout mice have no discernable phenotype, other than a selective reduction in tumor necrosis factor (TNF)-stimulated JNK activation [55]. We believe that there are compelling reasons to develop new, second-generation inhibitors of MLK3. CEP-1347 is based on the molecular scaffold of a staurosporine analog (K252a), which imposes a concern with respect to kinase selectivity (staurosporine being a very broadly active kinase inhibitor [56]). Moreover, CEP-1347 has complex biological effects that include not only inhibition of MLK3 [12] but also unrelated and/or off-target effects such as activation of Akt and Erk [57]. Finally, CEP-1347 exhibits a bell-shaped efficacy curve in vivo and is effective only over a relatively narrow dose concentration [28, 29]. For example, a dose of 0.3 mg/kg/day was highly effective in attenuating the loss of substantia nigra TH immunoreactive neurons after MPTP lesion in mice, but a dose of 3 mg/kg/day was markedly less effective and a dose of 0.03 mg/kg/day was completely ineffective [28]. Similar findings have been reported in a nonhuman primate model for PD [29]. In light of these considerations, we have developed improved second-generation MLK3 inhibitors with novel molecular structures distinct from CEP-1347 and proven CNS penetrance in pharmacokinetic studies. Thus, our initial goal was to derive inhibitors with enhanced specificity, reduced off-target effects, and a more favorable window of efficacy compared to CEP-1347. We achieved this goal after a focused approach to design drug-like compounds with favorable CNS profiles and nanomolar potency for MLK3, recently reporting on a small molecule MLK3 inhibitor, URMC-099, as a potential first-in-class adjunctive therapy for HAND [58, 59]. Further, in our goal to establish compatibility of URMC-099 with current cART, we serendipitously discovered that URMC-099 boosts antiviral activities of long acting antiretroviral therapy. Specifically, URMC-099 potentiates antiretroviral actions of nanoformulated ritonavir-boosted atazanavir (nanoATV/r). This drug combination led to a marked reduction of residual HIV-1 infection. URMC-099 facilitated nanoATV/r therapeutic effects by affecting the expression of the Rab family proteins that regulate endosomal vesicle trafficking, augmenting interactions between nanoATV/r and the viral life cycle. This combination of a MLK3 inhibitor with anti-inflammatory and neuroprotective properties with nanoART has led to the concept to treat persistent HIV-1 infection. The observation supports the idea that MLK3 inhibition uniquely targets sites of viral maturation with the ability to synergistically act with ART to prevent the viral life cycle from successfully achieving productive infection without accompanying cell death or inflammation.
Conclusion
Future strategies to achieve eradication of HIV infection in its CNS sanctuary must both target the virus and its life cycle and achieve resolution of associated neuroinflammation. We posit that one way this may be achieved is by improved targeting of ART with nanoformulations and accompanying ART treatment with disease modifying adjunctive therapies.
Key points.
Anti-inflammatory events need be eliminated in order to affect the secondary consequences of CNS viral infections
Chemical eradication of HIV infection in its CNS reservoirs could be achieved by combined approaches that improve delivery of ART to its sites of action
MLK3 inhibitors affect ART efficacy by affect intracellular trafficking of nanoparticles
Acknowledgments
The authors thank Robin Taylor and Jocelyn McMillan for critical reading of the manuscript and helpful suggestions provided. This work was supported by the University of Nebraska Foundation, which includes individual donations from Dr. Carol Swarts and Frances and Louie Blumkin; the Vice Chancellor’s office of the University of Nebraska Medical Center; Vivo Healthcare; and National Institutes of Health grants P01 MH64570 and RO1 MH104147 (HAG) and P01 DA028555, R01 NS36126, P01 NS31492, 2R01 NS034239, P01 NS43985, P30 MH062261 and R01 AG043540 (HEG).
Abbreviations
- AD
Alzheimer’s Disease
- CNS
central nervous system
- JNK
c-Jun N-terminal kinase
- CPE
CNS penetration score
- cART
combination ART
- ERK
extracellular signal-regulated kinase
- GSK
glycogen synthase kinase
- HAND
HIV-associated neurocognitive disorders
- MLK3
mixed lineage kinase 3 inhibitor
- MAPK
mitogen activated protein kinase
- MKKKs
mitogen activated protein kinase kinase kinases
- nanoART
nanoformulated antiretroviral therapy
- nanoATV/r
nanoformulated ritonavir-boosted atazanavir
- PD
Parkinson’s Disease
Footnotes
Conflict of Interest: Authors declare no conflict of interest
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
- 1.Gelbard HA, Dewhurst S, Maggirwar SB, Kiebala M, et al. Rebuilding synaptic architecture in HIV-1 associated neurocognitive disease: a therapeutic strategy based on modulation of mixed lineage kinase. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2010;7(4):392–8. doi: 10.1016/j.nurt.2010.08.001. Epub 2010/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blackstone K, Moore DJ, Heaton RK, Franklin DR, Jr, et al. Diagnosing symptomatic HIV-associated neurocognitive disorders: self-report versus performance-based assessment of everyday functioning. Journal of the International Neuropsychological Society: JINS. 2012;18(1):79–88. doi: 10.1017/S135561771100141X. Epub 2011/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪3.Morley D, McNamara P, Kennelly S, McMahon G, et al. Limitations to the identification of HIV-associated neurocognitive disorders in clinical practice. HIV medicine. 2013;14(8):497–502. doi: 10.1111/hiv.12036. Epub 2013/04/19. An evaluation of how the diagnosis of HAND could be missed in clinical practice. [DOI] [PubMed] [Google Scholar]
- 4.Heaton RK, Clifford DB, Franklin DR, Jr, Woods SP, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75(23):2087–96. doi: 10.1212/WNL.0b013e318200d727. Epub 2010/12/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis RJ, Letendre S, Vaida F, Haubrich R, et al. Randomized trial of central nervous system-targeted antiretrovirals for HIV-associated neurocognitive disorder. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2014;58(7):1015–22. doi: 10.1093/cid/cit921. Epub 2013/12/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪6.Caniglia EC, Cain LE, Justice A, Tate J, et al. Antiretroviral penetration into the CNS and incidence of AIDS-defining neurologic conditions. Neurology. 2014;83(2):134–41. doi: 10.1212/WNL.0000000000000564. Epub 2014/06/08. Covers the relationships between antiretroviral penetration and AIDS-defining neurologic conditions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang G, Dash PK, Guo D, Wiederin JL, et al. NanoART Facilitated HIV-1 Clearance by a Mixed-Lineage Kinase 3 Inhibitor Conference on Retroviruses and Opportunistic Infections; March 3–6, 2014; Boston Massachuetts, USA. 2013. [Google Scholar]
- 8.Tan IL, McArthur JC. HIV-associated neurological disorders: a guide to pharmacotherapy. CNS Drugs. 2012;26(2):123–34. doi: 10.2165/11597770-000000000-00000. Epub 2011/12/29. [DOI] [PubMed] [Google Scholar]
- 9.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3(9):663–72. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 10.Silva RM, Kuan CY, Rakic P, Burke RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson’s disease. Mov Disord. 2005;20(6):653–64. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- 11.Wang LH, Besirli CG, Johnson EM., Jr Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu Rev Pharmacol Toxicol. 2004;44:451–74. doi: 10.1146/annurev.pharmtox.44.101802.121840. [DOI] [PubMed] [Google Scholar]
- 12.Maroney AC, Finn JP, Connors TJ, Durkin JT, et al. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276(27):25302–8. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- 13.Handley ME, Rasaiyaah J, Barnett J, Thakker M, et al. Expression and function of mixed lineage kinases in dendritic cells. Int Immunol. 2007;19(8):923–33. doi: 10.1093/intimm/dxm050. [DOI] [PubMed] [Google Scholar]
- 14.Handley ME, Rasaiyaah J, Chain BM, Katz DR. Mixed lineage kinases (MLKs): a role in dendritic cells, inflammation and immunity? Int J Exp Pathol. 2007;88(2):111–26. doi: 10.1111/j.1365-2613.2007.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaeschke A, Davis RJ. Metabolic stress signaling mediated by mixed-lineage kinases. Mol Cell. 2007;27(3):498–508. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sathyanarayana P, Barthwal MK, Kundu CN, Lane ME, et al. Activation of the Drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3. Mol Cell. 2002;10(6):1527–33. doi: 10.1016/s1097-2765(02)00734-7. [DOI] [PubMed] [Google Scholar]
- 17.Leung IW, Lassam N. Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J Biol Chem. 1998;273(49):32408–15. doi: 10.1074/jbc.273.49.32408. [DOI] [PubMed] [Google Scholar]
- 18.Leung IW, Lassam N. The kinase activation loop is the key to mixed lineage kinase-3 activation via both autophosphorylation and hematopoietic progenitor kinase 1 phosphorylation. J Biol Chem. 2001;276(3):1961–7. doi: 10.1074/jbc.M004092200. [DOI] [PubMed] [Google Scholar]
- 19.Sui Z, Fan S, Sniderhan L, Reisinger E, et al. Inhibition of mixed lineage kinase 3 prevents HIV-1 Tat-mediated neurotoxicity and monocyte activation. J Immunol. 2006;177(1):702–11. doi: 10.4049/jimmunol.177.1.702. [DOI] [PubMed] [Google Scholar]
- 20.New DR, Maggirwar SB, Epstein LG, Dewhurst S, et al. HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-D-aspartate receptors by a NFkappaB-independent mechanism. J Biol Chem. 1998;273(28):17852–8. doi: 10.1074/jbc.273.28.17852. [DOI] [PubMed] [Google Scholar]
- 21.Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, et al. Glycogen synthase kinase 3 beta (GSK-3 beta) as a therapeutic target in neuroAIDS. J Neuroimmune Pharmacol. 2007;2(1):93–6. doi: 10.1007/s11481-006-9051-1. [DOI] [PubMed] [Google Scholar]
- 22.Mishra R, Barthwal MK, Sondarva G, Rana B, et al. Glycogen synthase kinase-3beta induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. J Biol Chem. 2007;282(42):30393–405. doi: 10.1074/jbc.M705895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin XH, Zhang QG, Miao B, Zhang GY. Neuroprotective effects of preconditioning ischaemia on ischaemic brain injury through inhibition of mixed-lineage kinase 3 via NMDA receptor-mediated Akt1 activation. J Neurochem. 2005;93(4):1021–9. doi: 10.1111/j.1471-4159.2005.03096.x. [DOI] [PubMed] [Google Scholar]
- 24.Barthwal MK, Sathyanarayana P, Kundu CN, Rana B, et al. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J Biol Chem. 2003;278(6):3897–902. doi: 10.1074/jbc.M211598200. [DOI] [PubMed] [Google Scholar]
- 25.Chadee DN, Xu D, Hung G, Andalibi A, et al. Mixed-lineage kinase 3 regulates B-Raf through maintenance of the B-Raf/Raf-1 complex and inhibition by the NF2 tumor suppressor protein. Proc Natl Acad Sci U S A. 2006;103(12):4463–8. doi: 10.1073/pnas.0510651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mota M, Reeder M, Chernoff J, Bazenet CE. Evidence for a role of mixed lineage kinases in neuronal apoptosis. J Neurosci. 2001;21(14):4949–57. doi: 10.1523/JNEUROSCI.21-14-04949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savinainen A, Garcia EP, Dorow D, Marshall J, et al. Kainate receptor activation induces mixed lineage kinase-mediated cellular signaling cascades via post-synaptic density protein 95. J Biol Chem. 2001;276(14):11382–6. doi: 10.1074/jbc.M100190200. [DOI] [PubMed] [Google Scholar]
- 28.Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons In vivo. J Pharmacol Exp Ther. 1999;288(2):421–7. [PubMed] [Google Scholar]
- 29.Saporito MS, Hudkins RL, Maroney AC. Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem. 2002;40:23–62. doi: 10.1016/s0079-6468(08)70081-x. [DOI] [PubMed] [Google Scholar]
- 30.Mathiasen JR, McKenna BA, Saporito MS, Ghadge GD, et al. Inhibition of mixed lineage kinase 3 attenuates MPP+-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2004;1003(1–2):86–97. doi: 10.1016/j.brainres.2003.11.073. [DOI] [PubMed] [Google Scholar]
- 31.Lotharius J, Falsig J, van Beek J, Payne S, et al. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci. 2005;25(27):6329–42. doi: 10.1523/JNEUROSCI.1746-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, et al. Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol. 2004;164(2):355–62. doi: 10.1016/S0002-9440(10)63125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson GV, Bailey CD. The p38 MAP kinase signaling pathway in Alzheimer’s disease. Exp Neurol. 2003;183(2):263–8. doi: 10.1016/s0014-4886(03)00268-1. [DOI] [PubMed] [Google Scholar]
- 34.Troy CM, Rabacchi SA, Xu Z, Maroney AC, et al. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77(1):157–64. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- 35.Barone FC, Parsons AA. Therapeutic potential of anti-inflammatory drugs in focal stroke. Expert Opin Investig Drugs. 2000;9(10):2281–306. doi: 10.1517/13543784.9.10.2281. [DOI] [PubMed] [Google Scholar]
- 36.Hu WW, Du Y, Li C, Song YJ, et al. Neuroprotection of hypothermia against neuronal death in rat hippocampus through inhibiting the increased assembly of GluR6-PSD95-MLK3 signaling module induced by cerebral ischemia/reperfusion. Hippocampus. 2008;18(4):386–97. doi: 10.1002/hipo.20402. [DOI] [PubMed] [Google Scholar]
- 37.Tian H, Zhang QG, Zhu GX, Pei DS, et al. Activation of c-Jun NH2-terminal kinase 3 is mediated by the GluR6.PSD-95.MLK3 signaling module following cerebral ischemia in rat hippocampus. Brain Res. 2005;1061(1):57–66. doi: 10.1016/j.brainres.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Zhang QG, Wang RM, Yin XH, Pan J, et al. Knock-down of POSH expression is neuroprotective through down-regulating activation of the MLK3-MKK4-JNK pathway following cerebral ischaemia in the rat hippocampal CA1 subfield. J Neurochem. 2005;95(3):784–95. doi: 10.1111/j.1471-4159.2005.03435.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang QG, Wang XT, Han D, Yin XH, et al. Akt inhibits MLK3/JNK3 signaling by inactivating Rac1: a protective mechanism against ischemic brain injury. J Neurochem. 2006;98(6):1886–98. doi: 10.1111/j.1471-4159.2006.04020.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhang QX, Pei DS, Guan QH, Sun YF, et al. Crosstalk between PSD-95 and JIP1-mediated signaling modules: the mechanism of MLK3 activation in cerebral ischemia. Biochemistry. 2007;46(13):4006–16. doi: 10.1021/bi0615386. [DOI] [PubMed] [Google Scholar]
- 41.Liao S, Porter D, Scott A, Newman G, et al. The cardioprotective effect of the low molecular weight isoform of fibroblast growth factor-2: the role of JNK signaling. J Mol Cell Cardiol. 2007;42(1):106–20. doi: 10.1016/j.yjmcc.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bodner A, Maroney AC, Finn JP, Ghadge G, et al. Mixed lineage kinase 3 mediates gp120IIIB-induced neurotoxicity. J Neurochem. 2002;82(6):1424–34. doi: 10.1046/j.1471-4159.2002.01088.x. [DOI] [PubMed] [Google Scholar]
- 43.Bodner A, Toth PT, Miller RJ. Activation of c-Jun N-terminal kinase mediates gp120IIIB- and nucleoside analogue-induced sensory neuron toxicity. Exp Neurol. 2004;188(2):246–53. doi: 10.1016/j.expneurol.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 44.Pirvola U, Xing-Qun L, Virkkala J, Saarma M, et al. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci. 2000;20(1):43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugahara K, Rubel EW, Cunningham LL. JNK signaling in neomycin-induced vestibular hair cell death. Hear Res. 2006;221(1–2):128–35. doi: 10.1016/j.heares.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ylikoski J, Xing-Qun L, Virkkala J, Pirvola U. Blockade of c-Jun N-terminal kinase pathway attenuates gentamicin-induced cochlear and vestibular hair cell death. Hear Res. 2002;166(1–2):33–43. doi: 10.1016/s0378-5955(01)00388-4. [DOI] [PubMed] [Google Scholar]
- 47.Falsig J, Porzgen P, Lotharius J, Leist M. Specific modulation of astrocyte inflammation by inhibition of mixed lineage kinases with CEP-1347. J Immunol. 2004;173(4):2762–70. doi: 10.4049/jimmunol.173.4.2762. [DOI] [PubMed] [Google Scholar]
- 48.Lund S, Porzgen P, Mortensen AL, Hasseldam H, et al. Inhibition of microglial inflammation by the MLK inhibitor CEP-1347. J Neurochem. 2005;92(6):1439–51. doi: 10.1111/j.1471-4159.2005.03014.x. [DOI] [PubMed] [Google Scholar]
- 49.Hehner SP, Hofmann TG, Ushmorov A, Dienz O, et al. Mixed-lineage kinase 3 delivers CD3/CD28-derived signals into the IkappaB kinase complex. Mol Cell Biol. 2000;20(7):2556–68. doi: 10.1128/mcb.20.7.2556-2568.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Investigators TPSGP. The safety and tolerability of a mixed lineage kinase inhibitor (CEP-1347) in PD. Neurology. 2004;62(2):330–2. doi: 10.1212/01.wnl.0000103882.56507.20. [DOI] [PubMed] [Google Scholar]
- 51.Wu SS, Frucht SJ. Treatment of Parkinson’s disease: what’s on the horizon? CNS Drugs. 2005;19(9):723–43. doi: 10.2165/00023210-200519090-00001. [DOI] [PubMed] [Google Scholar]
- 52.Investigators TPSGP. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2007;69(15):1480–90. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- 53.Dou H, Ellison B, Bradley J, Kasiyanov A, et al. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci. 2005;25(37):8375–85. doi: 10.1523/JNEUROSCI.2164-05.2005. Epub 2005/09/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪54.Eggert D, Dash PK, Gorantla S, Dou H, Poluektova L, et al. Neuroprotective activities of CEP-1347 in models of neuroAIDS. J Immunol. 2010;184:746–756. doi: 10.4049/jimmunol.0902962. Evaluates how CEP-1347 could be used as an adjunctive therapy for HIV-associated neurocognitive disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brancho D, Ventura JJ, Jaeschke A, Doran B, et al. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol Cell Biol. 2005;25(9):3670–81. doi: 10.1128/MCB.25.9.3670-3681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23(3):329–36. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 57.Roux PP, Dorval G, Boudreau M, Angers-Loustau A, et al. K252a and CEP1347 are neuroprotective compounds that inhibit mixed-lineage kinase-3 and induce activation of Akt and ERK. J Biol Chem. 2002;277(51):49473–80. doi: 10.1074/jbc.M203428200. [DOI] [PubMed] [Google Scholar]
- ▪58.Goodfellow VS, Loweth CJ, Ravula SB, Wiemann T, et al. Discovery, synthesis, and characterization of an orally bioavailable, brain penetrant inhibitor of mixed lineage kinase 3. Journal of medicinal chemistry. 2013;56(20):8032–48. doi: 10.1021/jm401094t. Epub 2013/09/21. The discovery, synthesis and characterization of URMC-099 is provided. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪▪59.Marker DF, Tremblay ME, Puccini JM, Barbieri J, et al. The new small-molecule mixed-lineage kinase 3 inhibitor URMC-099 is neuroprotective and anti-inflammatory in models of human immunodeficiency virus-associated neurocognitive disorders. J Neurosci. 2013;33(24):9998–10010. doi: 10.1523/JNEUROSCI.0598-13.2013. Epub 2013/06/14. A complete description of the anti-inflammation and neuroprotective properties of URMC-099 is shown. [DOI] [PMC free article] [PubMed] [Google Scholar]