

Abstract
Our aim was to study the capacity of an immortalized cell line (AMJ2-C11) to sustain aerobic cell respiration at decreasing oxygen concentrations under continuous sulfide exposure. We assumed that the capacity of the pathway metabolizing and eliminating sulfide, which is linked to the mitochondrial respiratory chain and therefore operates under aerobic conditions, should decrease with limiting oxygen concentrations. Thus, sulfide’s inhibition of cellular respiration would be dependent of the oxygen concentration in the very low range. The experiments were performed with an O2K-oxygraph (Oroboros Instruments) by suspending 0.5 – 1 × 106 cells in 2 ml of continuously stirred respiration medium at 37°C and calculating the oxygen flux (JO2) as the negative derivative of the oxygen concentration in the medium. The cells were studied in two different metabolic states, namely under normal physiologic respiration (1) and after uncoupling of mitochondrial respiration (2). Oxygen concentration was controlled by means of a titration-injection pump, resulting in average concentration values of 0.73 ± 0.05 μM, 3.1 ± 0.2 μM, and 6.2 ± 0.2 μM. Simultaneously we injected a 2 mM Na2S solution at a continuous rate of 10 μl/s in order to quantify the titration-time required to reduce the JO2 to 50% of the initial respiratory activity. Under the lowest oxygen concentration this effect was achieved after 3.5 [0.3; 3.5] and 11.7 [6.2;21.2] min in the uncoupled and coupled state, respectively. This time was statistically significantly shorter when compared to the intermediate and the highest O2 concentrations tested, which yielded values of 24.6[15.5;28.1] min (coupled) and 35.9[27.4;59.2] min (uncoupled), as well as 42.4 [27.5;42.4] min (coupled) and 51.5 [46.4;51.7] min (uncoupled). All data are medians [25%, and 75% percentiles]. Our results suggest that elimination of sulfide in these cells is limited by oxygen availability when approaching the anoxic condition. This property may contribute to the physiological role of sulfide as an oxygen sensor.
Introduction
Albeit toxic at high concentrations, hydrogen sulfide (H2S) has been recently recognized as a further “gasotransmitter” involved in the regulation of various physiological functions [1]. In particular, it is thought to play a role as an intracellular oxygen sensor. In most species and in humans [2] H2S is released in the cytoplasm by the enzymes cystathionine-γ-lyase (CSE) and cystathionine-β-synthase, as well as in mitochondria by the 3-mercaptopyruvate sulphur transferase (3-MST). Recently, thiosulfate has been recognized as a further source of sulfide, which is particularly involved in the oxygen sensing role exerted by this molecule [3]. Once released, however, H2S is quickly degraded by a strictly aerobic pathway linked to the mitochondrial respiratory chain. This sulfide eliminating pathway is comprised by two enzymes: the sulfide-quinone oxidoreductase (SQR) and a dioxygenase. The former enzyme oxidises the sulfide and passes the electrons to the ubiquinone pool situated in the inner mitochondrial membrane, whereas the dioxygenase, which also requires oxygen as electron acceptor, further oxidises the two disulfides temporarily generated by the SQR [4]. The SQR’s high capacity for sulfide degradation was recently proven and further suggested to play a crucial role in maintaining a non-toxic concentration of H2S within the cells [5,6]. The putative oxygen sensing role of H2S is linked to the oxygen dependency of its degradation pathway [2]. Specifically, under low oxygen conditions, the diminished efficiency of the SQR and dioxygenase would lead to an accumulation of sulfide at the cellular level. As demonstrated by Olson et al hypoxic pulmonary vasodilation was associated with sulfide production in pulmonary artery smooth muscle cells [7]. Albeit under hypoxic conditions cells may still eliminate sulfide in other ways to compensate for the increased concentration. Therefore, a switch between aerobic and anaerobic conditions in sulfide elimination may serve as an indicator for a limiting oxygen concentration. Thus we studied the capacity of immortalised cells derived from alveolar macrophages (AMJ2-C11) to maintain a constant rate of aerobic respiration under a continuous exogenous sulfide exposure at low oxygen concentrations nearing anoxia. Our goal was to determine the oxygen concentration range at which the accumulation of sulfide in the cells would inhibit the mitochondrial respiratory chain thereby fulfilling the putative role of oxygen sensing. We hypothesize that in case of decreasing efficiency of the SQR and dioxygenase, the continuous addition of sulfide would lead to a more rapid accumulation and ultimately an earlier inhibition of aerobic respiration. In order to test this assumption, we used a recently established experimental model [8] based on a high-resolution respirometry [9], which had been used to study the relationship between sulfide toxicity and infusion rate. In the present experiment the setup is modified to maintain stable low oxygen conditions throughout the sulfide titration.
Materials and Methods
1. Cell cultures
AMJ2-C11 cells were cultured for 36–48 h at 37° C in a standard medium (DMEM, Gibco, Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal calf serum, 1% non-essential amino acids, 1% glutamine, and 0.5% gentamicin and an atmosphere containing 95% air and 5% CO2. We used this cell line for our experiments because they provided a reliable model to study the effects of sulfide in our previous investigation [8].
2. High-resolution respirometry
Cellular oxygen uptake was quantified by high-resolution respirometry using the Oroboros® Oxygraph-2K (Oroboros Instruments, Innsbruck, Austria). This device allows for simultaneous recording of the O2 concentration in two parallel chambers calibrated for 2 ml of respiration medium equilibrated with 21% O2 in N2 at 37 °C. For our present scope the medium was supplemented with catalase, which allowed to maintain the oxygen concentration by adding small volumes of hydrogen peroxide without reopening the chamber (see below for more details). The partially adherent cultured cells were scraped, centrifuged at 150 × g for 5 min, and then suspended in the respiration medium at a concentration of 0.5 to 1* 106 cells/ml. The cell suspensions were continuously stirred at 750 rpm. Cellular respiration was quantified in terms of oxygen flux (JO2) based on the rate of change of the O2 concentration in the chambers.
The continuous sulfide titration as well as the control of the oxygen concentration in the chamber were performed using a TIP-2K® titration-injection micropump (Oroboros Instruments). Control for the TIP-2K® as well as data acquisition and analysis were performed with the DatLab® software, version 4.3 (Oroboros Instruments). This enables continuous monitoring and recording of the oxygen concentration in the chambers as well as of the derived oxygen flux over time, normalized for the amount of cells at rates of 0.5–1 Hz.
3. Respiration under sulfide-exposure
Aerobic respiration under Na2S exposure was assessed using a similar experimental setup as previously described [8]. Briefly, this technique consists of measuring the JO2 while continuously injecting a 2 mM Na2S solution at a rate of 10 nl/s into the oxygraph chambers containing the cell suspension. We performed the experiments on intact cells under normal physiologic, coupled respiration (1) and under uncoupled respiration (2). The latter condition was achieved by sequentially injecting 2.5 μM oligomycine to inhibit the ATP-synthase, and 1 – 1.5 μM of the uncoupler p-trifluoromethoxy-carbonyl-cyanide phenylhydrazone (FCCP) into the respiration medium before starting the sulfide-injection. The reason to test both coupling states was to exclude the influence of the added sulfide on ATP-consuming processes, which may indirectly modify the level of cellular respiration independent from any effect on the cytochrome-c oxidase. Regardless of the coupling state, in both cases the cells were intact, and cell respiration was therefore sustained by endogenous substrates without the need for exogenous supplementation. Simultaneous to the sulfide injection, specific oxygen concentrations were maintained in the oxygraph chambers by a closed loop task of the DatLab® software. This tool allowed controlling the TIP-2K® for titrating 50 – 100 nl of a 20 mM hydrogen peroxide solution, thus maintaining the oxygen level in the respiration medium close within three predefined concentration ranges yielding mean values of 6.2 ± 0.2 μM, 3.1 ± 0.2 μM, and 0.73 ± 0.04 μM, and oscillating between average minima of 5.8 ± 0.1 μM, 2.7 ± 0.3 μM, and 0.62 ± 0.05 μM, and average maxima of 6.5 ± 0.5 μM, 3.3 ± 0.4 μM, and 0.82 ± 0.05 μM, respectively (see figure 1). The JO2 throughout the sulfide-injection was then obtained calculating the linear decay of the oxygen concentration from each maximum to the next minimum (see figure 1 for more details), which corresponds to the standard method used for respirometry as mentioned above. The oxygen concentration ranges chosen for the experiments were determined based on the average results of five preliminary experiments performed according to previously published methods [9] recording the JO2 of the AMJ2-C11 cells throughout the full range between normoxia and anoxia (“anoxic transition”), and allowed to limit mitochondrial respiration to 95%, 90%, and 70% of the maximum JO2, respectively. Finally, each sulfide titration experiment was quantified in terms of the mean duration of the sulfide injection required to reduce the JO2 to 50% of its value preceding the addition of sulfide. Since the titration rate was constant, this corresponds exactly to the total amount of sulfide injected into the chamber. For each specific oxygen concentration and cell respiration state we performed five separate experiments in order to obtain average results for mathematical and statistical calculations. Furthermore, we performed two additional experiments in order to demonstrate in principle the effects of low oxygen concentration on sulphide degradation. For these experiments we used intact AMJ2-C11 cells in the FCCP-induced uncoupled state, and added of 0.5 μM rotenone to inhibit mitochondrial respiration by blocking complex I. Then we injected two sequential 4 μM boluses of the sulfide donor Na2S. Since complex I had been blocked before, the increase in JO2 induced by Na2S was now exclusively attributable to sulfide turnover. After the second sulfide bolus the medium in the chamber was re-oxygenated to determine the JO2 after restoration of the initial oxygen concentration.
Figure 1.
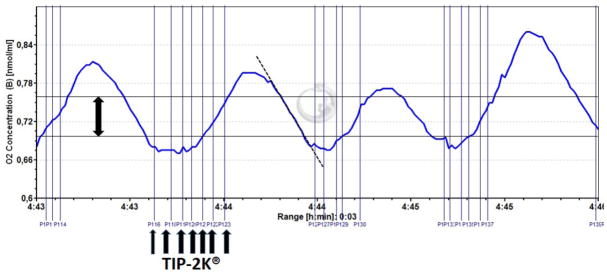
The plot shows an original registration of the oxygen concentration during a titration experiment at low oxygen. The method used to calculate the oxygen flux (JO2) in terms of the decay of oxygen concentration over time corresponds to the standard in respirometry. However, it was modified for the actual experiment to account for the need to maintain the oxygen concentrations within narrow ranges. Therefore, the titration pump (TIP-2K®, see arrows in the botton) was used to automatically inject small volumes (50 – 100 nl) of peroxide to the catalase supplemented respiration medium, in order to restore the oxygen concentration once it felled due to the cell’s respiration below the lower limit set for the control algorithm of the pump (lower horizontal line). In that way, oxygen concentration re-increased up to the upper limit of the control algorithm (upper horizontal line), and was then allowed to fall again. This linear fall of the oxygen concentration, which is the result of the cell’s respiration, is used to calculate the JO2 (dotted line). In summary, the basic principle of calculating the JO2 is that normally used in respirometry, but slightly modified to account for the necessity to keep the oxygen within very small concentration ranges.
4. Mathematical data processing and statistics
The resulting data obtained from each titration experiment were fitted to polynomial functions mathematically describing the O2 flux as a function of the length of time of the H2S administration for the three oxygen concentrations tested. The polynomial coefficients were determined using a previously described regression method [10]. The different time required by the sulfide injection to inhibit mitochondrial respiration by 50% at each experimental condition were tested for statistical significance by means of the signed rank ANOVA, and, when significant, by pairwise multiple comparison procedures using the Dunn’s test. Statistical significance was assumed with p<0.05.
Results
The results of the anoxic transition experiments conducted in order to determine the JO2 in the low oxygen range are shown in figure 2. The average JO2 curves recorded during the sulfide titrations and under normal physiologic respiration as well as under uncoupled maximum respiration at the three oxygen concentrations chosen for the experiment are shown in figure 3. At 95% of JO2 max we obtained in the coupled cells similar titration curves to those previously reported without regulation of oxygen concentration [8], thus validating the model. Under the average oxygen concentration in the respiration medium of 6.21 ± 0.22 μM, the mitochondrial respiratory activity was close to 95% of the JO2 max. A tenfold reduction of the O2-concentration down to 0.73 ± 0.04 μM led to a decrease of the routine respiratory activity approximating 70% of the JO2 max, and, simultaneously, to a quick decline in the time required to reduce aerobic respiration to 50% under the continuous sulfide titration (53.4 [47.6;55.9] min at the higher O2 concentration compared to 17.9 [2,8;24.6] min at the lower O2 concentration in the uncoupled condition, and 42.4 [27.5;42.4] min at high oxygen compared to 3.5 [0.3;3.5] min at low oxygen in the coupled state, p<0.05). The intermediate concentration of 3.1 ± 0.25 μM reduced routine respiratory activity 90% of the JO2 max, and aerobic respiration decreased to 50% with an average time for sulfide titration of 34.0 [26.7;50.3] min in the uncoupled condition and 24.6 [15.5;28.1] min in the coupled. At 95% and 70% of JO2 max the average time to achieve a 50% decrease in aerobic respiration was statistically significantly lower in the coupled state in comparison to the uncoupled (p<0.05). At 90% of JO2 max the difference between the two states was not statistically significant. The original tracings of the two additional experiments demonstrating the effects of oxygen concentration on sulfide degradation are shown in figure 4 (see legend for more details on the experiments). They show that two sequential 4 μM boluses of sulfide produced a largely differing response in JO2, which was pronounced after the first bolus, but only weak after the second one. In order to exclude that the minor response to the second bolus was due to a residual inhibition of mitochondrial respiration already induced by the first injection, the medium in the chamber was allowed to re-oxygenate. This immediately restored the sulfide supported respiration.
Figure 2.
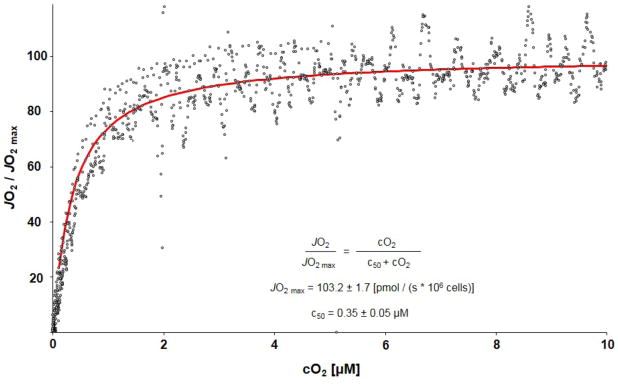
The lower plot shows the anoxic transition in the AMJ2-C11 cells determined form the results of five separate experiments based on methods described elsewhere [9]. The dots are the data from the single experiments and the line the average result fitted to the hyperbolic equation indicated in the diagram using the Levenberg-Marquardt algorithm.
Figure 3.

The plots show the JO2 on Y-Axis versus time on the X-Axis under the continuous sulfide injection at the oxygen concentrations decreasing from the top to the bottom. Note that the JO2 declines over the course of the sulfide exposure, but that this decline is faster at the lowest oxygen concentration (bottom diagrams). Moreover, the left plots show the average results in the coupled state, whereas the plots on the right relate to the uncoupled condition. The plotted curves show the average results of 5 – 8 experiments performed under each condition, and the 95% confidence intervals calculated based on the methods described in the text.
Figure 4.
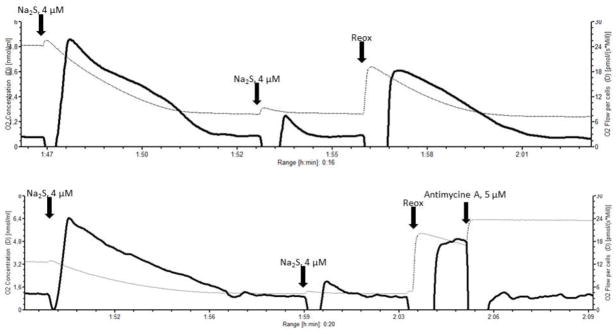
The plots show original tracing of two selective experiments conducted in order to show the effects of decreasing oxygen concentrations on sulfide turnover and thus elimination. The plots report the oxygen concentration (thin dotted line, left Y-axis), and the oxygen flux (bold closed line, right Y-axis) over time. The oxygen concentration was within the same range as for the sulfide titration experiments. For these experiments we used intact AMJ2-C11 cells in the FCCP-induced uncoupled state. 0.5 μM rotenone were added in order to inhibit mitochondrial respiration at the level of complex I. This was done in order ensure that any subsequent increase in oxygen flux following a sulfide injection would be attributable exclusively to sulphide turnover. Then we injected two sequential 4 μM boluses of the sulfide donor Na2S as indicated by the arrows. The first bolus induced a pronounced response in oxygen flux, whereas the response to the second bolus was much weaker. The oxygen concentrations were 4.9 μM (upper plot) and 3.4 μM (lower plot) before the first injection, and 1.6 (upper plot) and 1.1 (lower plot) before the second bolus. To exclude that a residual inhibition of the cytochrome-c oxidase was responsible for this weak response the medium was reoxygenated (Reox, see arrows), to increase the oxygen concentration to 3.8 μM in the experiment shown by the upper plot, and 5.3 μM in the lower plot. Under the higher oxygen concentrations the oxygen flux supported by sulfide immediately re-increased again to values approaching those after the injection of the first bolus. This suggest that the low oxygen flux observed after the second bolus of sulfide was likely due to the low oxygen concentration in the medium, in spite of a sufficient amount of sulfide available as substrate. In the experiment shown in the lower diagram, the complex III inhibitor antimycine A (5 μM) was injected after the final reoxygenation of the medium. This was done to show that after the inhibition of complex III the residual cellular respiration goes back to the values preceding any addition of sulfide. This indicates that the presence of sulfide in the chamber does not interfere with the oxygen sensors of the oxygraph, which could alternatively explain the increase in oxygen flux following the sulfide boluses.
Discussion
The aim of the present study was to determine the oxygen concentration range where sulfide may be implicated in oxygen sensing according to the hypothesis that in increasing hypoxic conditions the capacity of cells to metabolise the compound is progressively reduced. The concept of a finite sulfide eliminating capacity in the cells of higher organisms was first suggested by Haggard [11], who observed that the well-known toxicity of sulfide was strongly dependent on its application rate. Thus he concluded that sulfide probably accumulates in the tissues once it is applied at a rate exceeding the rate of elimination. More recently, the corresponding biochemical processes leading to the elimination of sulfide have been detected in non-vertebrates [13] as well as in mammalian species including humans [14]. Additional in-vivo experimental evidence further demonstrate, that the toxicity of sulfide also depends on body temperature, suggesting that some factors modifying the activity of the biochemical pathways involved in the elimination of the compound [8,12] may modulate the effects on cellular respiration. In our present study we addressed the question, to which extent the oxygen concentration may be another modulator of the capacity to eliminate exogenously applied sulfide, considering the well-known fact that the sulfide eliminating pathway operates under strongly aerobic conditions. Accordingly, the results of the two preliminary experiments shown in figure 4 already show that the sulfide turnover and, consequently, elimination is decreased at low oxygen concentration. Since a reliable method for detecting the sulfide concentration in biological media is not available yet, which is a clear limitation for our study, we used for the further experiments the same approach as in our previous investigation to indirectly study the efficiency of the sulfide eliminating pathway. This approach consists in determining the ability of cells to maintain a steady state aerobic respiration under a constant exogenous sulfide application, and quantifying the time required by the sulfide-titration to suppress respiration by 50%. This suppression indicates the inhibition of the cytochrome-c oxidase presumably due to the accumulating sulfide. Indeed, our results show that the capacity of AMJ2-C11 cells to sustain aerobic respiration is suppressed more rapidly by the exogenous sulfide exposure under decreasing oxygen levels toward anoxia. This observation agrees with the hypothesis that the cells eliminate sulfide less efficiently with decreasing oxygen concentrations, leading to an accumulation of the sulfide when added to the medium at constant rate. Thus the accumulation of sulfide in the cells may contribute to the oxygen sensing effect according as previously postulated [2]. When comparing the results obtained in the coupled and uncoupled state, we noted that the suppression of aerobic respiration occurs more rapidly in the former condition with all three oxygen concentrations tested. This may be explained by the inhibition of ATP-demanding processes in the cells occurring in response to the accumulation of sulfide in addition to the effects on the cytochrome-c oxidase. However, when comparing both states one should be aware that in the uncoupled state mitochondrial respiration is maximally stimulated, whereas it is only about 30%–50% of the maximum in the physiologic coupled state. Furthermore, an alternative explanation for the observed difference between the two states is suggested by our previous data, which showed that under maximum respiratory activity exogenous sulfide was capable of further increasing aerobic respiration in the uncoupled, but not in the coupled state [8]. These data may indicate that sulfide is processed slightly more efficiently in the uncoupled state. This explanation, however, cannot be proven without directly measuring the sulfide concentration in the medium or in the cells, since sulfide is known to be metabolised preferentially by the mitochondria [14]. Hence, albeit the JO2 is not futher increased in the coupled state by the addition of sulfide, we cannot exclude that sulfide is still eliminated efficiently at the expense of other substrates. Despite of these differences between the coupled and the uncoupled state, however, our actual investigation shows that the response of cellular respiration to decreasing oxygen is qualitatively comparable under both conditions.
The titration curves presented in figure 2 further suggest a slight effect of sulfide on non-mitochondrial respiration. In fact, residual JO2 at the end of the titration period is greater at the higher oxygen concentration when compared to the lowest one. Of course, this effect may be simply explained by the fact that even non-mitochondrial respiration is expected to decrease with diminishing oxygen availability. On the other hand, we cannot exclude that sulfide itself additionally suppresses the non-mitochondrial fraction of the JO2, this effect being more pronounced at low oxygen.
As a prerequisite for our experiments, we determined the mitochondrial respiratory activity under oxygen concentrations between 0 and 10 μM. This allowed us to establish a mathematical description of the anoxic transition, which was in close agreement with previously published data [9]. These calculations enabled us to estimate the oxygen concentrations required to achieve 95%, 90%, and 70% of the maximum mitochondrial respiration under routine conditions. Interestingly, the corresponding decrease of the capacity to eliminate the added sulfide was much more pronounced. This result may emphasize the potential contribution of sulfide in oxygen sensing in as much as it suggests that, at least locally, the available sulfide increased already before the lack of oxygen becomes limiting for the aerobic mitochondrial respiration.
The consequences of the supposedly less efficient sulfide elimination at low oxygen, as well as the mechanisms further translating the hypoxic signal remain speculative. The sulfide concentration may rise, at least locally in the hypoxic region, resulting in a partial limitation of the cytochrome c oxidase, i.e. of the mitochondrial respiration. However, in this case the inhibition of mitochondrial respiration should in turn reduce the oxygen consumption in the specific region thus limiting a further decrease in oxygen concentration. In the end, this mechanism may result in slowing down the rate by which the cells becomes more hypoxic or even anoxic. Unfortunately, due to the lack of validated methods for measuring the sulfide concentration, especially in the tissue, this mechanism remains for the most part theoretical. On the other hand, it is also conceivable that simply increasing the availability of sulfide by limiting its decomposition produces biochemical and physiological effects, even without an increase in concentration. In fact, it has been postulated that sulfide signalling may also partially occur through protein sulfhydration [15]. This effect is even predominant compared to the nitrosylation of proteins induced by NO, and seems to be involved in the regulation of inflammation, stress signalling, and vascular tension. Finally, the reducing capacity of the sulfide may also translate into the hypoxic signal; for example, in trout gills sulfide has been recently shown to increase mitochondrial ROS production [16], thus mediating hypoxic vasoconstriction.
In conclusion, our data suggest that aerobic respiration is more rapidly inhibited by a continuous sulfide application at low oxygen conditions, and further show that this effect occurs even before the aerobic metabolism is reduced to the same extent by the lack of oxygen itself. This property in theory agrees with the supposed ability of sulfide to participate in sensing hypoxia, in as much as it responds to a fall in oxygen concentration even before hypoxia per se becomes a limiting factor for cell metabolism.
Highlights.
We describe a test for quantifying the inhibition of cellular respiration by sulfide under very low oxygen conditions
We assess the oxygen concentration range, where the sulfide effects on cellular respiration can be expected to be oxygen dependent
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (KFO 200, DFG RA396/9-2), the Land Baden-Württemberg (Innovationsfond Medizin), and is part of the PHYPODE project, financed by the European Union under a Marie Curie Initial Training Network.
References
- 1.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 2.Olson KR, Perry SF. H2S and O2 sensing. Proc Natl Acad Sci U S A. 2010;107:E141. doi: 10.1073/pnas.1009210107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olson KR, Deleon ER, Gao Y, Hurley K, Sadauskas V, Batz C, Stoy GF. Thiosulfate: a Readily Accessible Source of Hydrogen Sulfide in Oxygen Sensing. Am J Physiol Regul Integr Comp Physiol. 2013 Jun 26; doi: 10.1152/ajpregu.00421.2012. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 4.Bouillaud F, Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid Redox Signal. 2011;15:379–91. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 5.Vitvitsky V, Kabil O, Banerjee R. High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid Redox Signal. 2012 Jul 1;17(1):22–31. doi: 10.1089/ars.2011.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kabil O, Banerjee R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid Redox Signal. 2013 Jun 7; doi: 10.1089/ars.2013.5339. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson KR, Whitfield NL, Bearden SE, St Leger J, Nilson E, Gao Y, Madden JA. Hypoxic pulmonary vasodilation: a paradigm shift with a hydrogen sulfide mechanism. Am J Physiol Regul Integr Comp Physiol. 2010;298:R51–60. doi: 10.1152/ajpregu.00576.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groeger M, Matallo J, McCook O, Wagner F, Wachter U, Bastian O, Gierer S, Reich V, Stahl B, Huber-Lang M, Szabó C, Georgieff M, Radermacher P, Calzia E, Wagner K. Shock. 2012;38:367–74. doi: 10.1097/SHK.0b013e3182651fe6. [DOI] [PubMed] [Google Scholar]
- 9.Gnaiger E. Bioenergetics at low oxygen: dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir Physiol. 2001;128:277–97. doi: 10.1016/s0034-5687(01)00307-3. [DOI] [PubMed] [Google Scholar]
- 10.Vogt JA, Fabinski W, Kappler J, Fischer H, Georgieff M. Response surface calibration of 13CO2-NDIR offset values: A ‘random coefficients’ approach. Chemometrics and Intelligent Laboratory Systems. 2011;2:377–383. [Google Scholar]
- 11.Haggard HW. The fate of sulfides in the blood. J Biol Chem. 1921;49:519–521. [Google Scholar]
- 12.Baumgart K, Wagner F, Gröger M, Weber S, Barth E, Vogt JA, Wachter U, Huber-Lang M, Knöferl MW, Albuszies G, Georgieff M, Asfar P, Szabó C, Calzia E, Radermacher P, Simkova V. Cardiac and metabolic effects of hypothermia and inhaled hydrogen sulfide in anesthetized and ventilated mice. Crit Care Med. 2010;38:588–595. doi: 10.1097/ccm.0b013e3181b9ed2e. [DOI] [PubMed] [Google Scholar]
- 13.Völkel S, Grieshaber MK. Mitochondrial sulfide oxidation in Arenicola marina. Evidence for alternative electron pathways. Eur J Biochem. 1996;235:231–237. doi: 10.1111/j.1432-1033.1996.00231.x. [DOI] [PubMed] [Google Scholar]
- 14.Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Paul BD, Snyder SH. H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 16.Skovgaard N, Olson KR. Hydrogen sulfide mediates hypoxic vasoconstriction through a production of mitochondrial ROS in trout gills. Am J Physiol Regul Integr Comp Physiol. 2012;303:R487–94. doi: 10.1152/ajpregu.00151.2012. [DOI] [PubMed] [Google Scholar]