

Abstract
BACKGROUND
Internal tandem duplication of FMS-like tyrosine kinase (FLT3-ITD) is well known to be involved in acute myeloid leukemia (AML) progression, but FLT3-ITD–negative AML cases account for 70% to 80% of AML, and the mechanisms underlying their pathology remain unclear. This study identifies protein tyrosine phophatase PRL-3 as a key mediator of FLT3-ITD–negative AML.
METHODS
A total of 112 FLT3-ITD–negative AML patients were sampled between 2010 and 2013, and the occurrence of PRL-3 hyperexpression in FLT3-ITD–negative AML was evaluated by multivariate probit regression analysis. Overexpression or depletion of endogenous PRL-3 expression with the specific small interfering RNAs was performed to investigate the role of PRL-3 in AML progression. Xenograft models were also used to confirm the oncogenic role of PRL-3.
RESULTS
Compared to healthy donors, PRL-3 is upregulated more than 3-fold in 40.2% of FLT3-ITD–negative AML patients. PRL-3 expression level is adversely correlated to the overall survival of the AML patients, and the AML relapses accompany with re-upregulation of PRL-3. Mechanistically, aberrant PRL-3 expression promoted cell cycle progression and enhanced the antiapoptotic machinery of AML cells to drug cytotoxicity through downregulation of p21 and upregulation of Cyclin D1 and CDK2 and activation of STAT5 and AKT. Depletion of endogenous PRL-3 sensitizes AML cells to therapeutic drugs, concomitant with apoptosis by upregulation of cleaved PARP (poly ADP ribose polymerase) and apoptosis-related caspases. Xenograft assays further confirmed PRL-3’s oncogenic role in leukemogenesis.
CONCLUSIONS
Our results demonstrated that PRL-3 is a novel independent crucial player in both FLT3-ITD–positive and FLT3-ITD–negative AML and could be a potential therapeutic target. Cancer 2014;120:2130–2141. © 2014 The Authors. Cancer published by Wiley Periodicals, Inc. on behalf of American Cancer Society.
FLT3-ITD–negative acute myeloid leukemia (AML) accounts for up to approximately 70% to 80% of all cases. This study demonstrates that PRL-3, an independent driver in FLT3-ITD–negative AML, is adversely correlated to patient survival. Mechanistically, PRL-3 can promote AML cell cycle progression and render antiapoptosis features to AML cells, suggesting it could be an independent factor for AML diagnosis and therapy.
Keywords: acute myeloid leukemia, PRL-3, FLT3-ITD–negative, apoptosis, drug resistance, cell proliferation
INTRODUCTION
Acute myeloid leukemia (AML) is characterized by a differentiation block and the accumulation of immature cells, resulting in the failure of normal hematopoiesis1 with series of somatic genetic alterations leading to a complicated and heterogeneous disease.2,3 It is believed that the pathogenesis of AML typically requires at least 2 genetic lesions: one which impairs differentiation, such as the AML1-ETO mutation; and the other which promotes proliferation, for instance such as that afforded by the activated KIT or FMS-like tyrosine kinase (FLT3) mutation which is detected in approximately 20% of AML.3 Although insights into the genetic alterations could lead to the improved diagnosis and therapy for AML patients with poor prognosis, the therapeutic outcomes in AML still remain unsatisfactory. Most patients ultimately relapse and long-term survival is seen in approximately 30% to 40% of younger AML patients aged less than 60 years, but only 5% to 10% in the elderly patients aged 60 years and older.4–6 The conventional cytarabine-based therapy in conjunction with stem cell transplantation is not suitable to all patients, due to the unselective damage to normal tissue cells and the immunological rejection problems, respectively.7 To improve the therapeutic efficiency to AML patients, it is necessary to thoroughly understand the underlying mechanism and to identify more genes as potential biomarkers and targets for the individualized AML diagnosis and treatment.
Protein tyrosine phosphatase of regenerating liver 3 (PRL-3, PTP4A3) is one of the members of the cysteine-based protein tyrosine phosphatases,8 and is highly expressed in various solid tumors as a biomarker, including colon, breast, ovary, liver, stomach tumors.9–14 Moreover, PRL-3 has been demonstrated to play important roles in cancer cell proliferation, motility, metastasis, invasiveness, and tumor angiogenesis.15–18 Growing evidence has indicated that high PRL-3 expression is an adverse prognostic factor for recurrence, overall survival, and disease-free survival.19–21 Our previous study suggested that PRL-3 as a metastasis-associated phosphatase, is involved in reinforcing PI3K/Akt activation, consequently promoting epithelial-mesenchymal transition.22 PRL-3 is also reported as a regulator of H3K9 methylation by affecting the activities of JMJD1B and JMJD2B in colorectal tumorigenesis.23 PRL-3 overexpression is also seen in hematopoietic malignancies, including acute lymphoblastic leukemia and multiple myeloma.24,25 In addition, PRL-3 is involved in leukemogenesis of chronic myeloid leukemia as a downstream target of BCR-ABL signaling pathway.26 Besides, PRL-3 protein is upregulated in 47% of AML cases with internal tandem duplication of FLT3 (FLT3-ITD), as a downstream effector of FLT3-ITD to confer therapeutic resistance through upregulation of STAT and antiapoptotic Mcl-1 protein,27 or by FLT3-ITD-STAT5 signaling pathways.28 However, whether PRL-3 independently participates in FLT3-ITD–negative AML progression is not known. FLT3-ITD–negative leukemia is the major subgroup of AML and accounts for up to 70% to 80% of all cases,29 the effective treatment to this subgroup will definitely improve the overall AML therapy efficiency.
In this study, we screened PRL-3 expression in clinical FLT3-ITD–negative AML samples and analyzed the relationship of PRL-3 expression in clinical variables and evaluated the prognostic value of PRL-3 in FLT3-ITD–negative AML. Meanwhile, the effects of PRL-3 in cell proliferation, cell cycle, cellular apoptosis, drug resistance, and leukemogenesis were thoroughly investigated.
MATERIALS AND METHODS
Patients
AML patients treated at the First Affiliated Hospital of Sun Yat-sen University from February 2010 to June 2013 were sampled and followed up. A total of 112 FLT3-ITD–negative AML diagnoses were conducted on the basis of WHO Guideline and confirmed to be FLT3-ITD–negative by multiplex RT-PCR at the Tissue Typing Center of the hospital. All patients were categorized into 2 groups, including the de novo group of 86 cases and the relapsed/refractory group of 26 cases. The main clinical features of the patients are listed in Table 1. Meanwhile, 16 healthy donors were recruited, which was approved by the medical ethical committee of the University. Complete remission is defined as ≤ 5% leukemic blast cells in the bone marrow. Relapse is defined as the recurrence of ≥ 5% blasts in the bone marrow. Disease-free survival (DFS) was considered from the diagnosis to the relapse or death for any cause, with observation censored for patients last known to be alive without report of relapse.
Table 1.
Characteristics of Patients Enrolled in the Study
| Characteristics | No. of Patients (%) |
|---|---|
| Median age, y (range) | 35 (15-74) |
| Sex ration, male/female | 63/49 |
| Blast cell in bone marrow, % (range) | 58 (25-91) |
| FAB category | |
| M0 | 9 (8.04%) |
| M1 | 10 (8.93%) |
| M2 | 18 (16.07%) |
| M3 | 19 (16.96%) |
| M4-M5 | 50 (44.64%) |
| M6-M7 | 6 (5.36%) |
| Median white blood cell, ×109/L (range) | 23 (1.5-312) |
| Cytogenetics | |
| Favorable risk | 24 (21.4%) |
| Intermediate risk | 55 (49.1%) |
| Poor risk | 33 (29.5%) |
Cell Proliferation, Cytotoxicity, Colony Formation, Cell Cycle and Apoptosis Assays
The protocols of all above assays are fully described in the Supporting Materials and Methods (see online supporting information). All experiments were carried out in triplicate each time, and 3 independent experiments were performed.
Western Blots
PRL-3 monoclonal antibodies (clones 318) were used as described.30 Beta-actin antibodies was obtained from CW Biotech (China), the other indicated antibodies used were purchased from Cell Signaling Technology (Beverly, Mass). The detailed protocol of western blotting is described in the Supporting Experimental Procedures.
Tumor Models in Immunodeficient Mice
All experiments using nude mice were strictly performed in accordance to the guidelines of the Institutional Animal Care and Use Committee (IACUC) at Sun Yat-sen University, Guangzhou, China. The detailed protocol is described in the Supporting Experimental Procedures.
Statistical Analysis of Clinical Samples
The SPSS 16.0 software (SPSS, Chicago, Ill) were adopted to perform the statistical analysis. Disease-free survival (DFS) curves were plotted by the Kaplan-Meier method. Univariate and multivariate analyses comprising common prognostic factors for DFS were performed using the Cox regression analysis. Differences were considered to be statistically significant at P < .05.
RESULTS
PRL-3 Is Upregulated in FLT3-ITD–Negative AML Patients and Cells
PRL-3 was shown to be upregulated in FLT3-ITD positive AML as its downstream effector.27 In order to explore whether PRL-3 aberrantly expresses in the FLT3-ITD–negative AML, we evaluated PRL-3 mRNA expression in 112 FLT3-ITD–negative AML patients by quantitative RT-PCR. Compared to the 16 normal healthy donors, the PRL-3 mRNA expression was overall increased in the bone marrow blast cells of AML patients (median 13.464 versus 5.542, P = .002, Fig. 1A), and the averaged value of PRL-3 expression of 16 healthy donors was 6.223, compared to the averaged value of 18.670 seen in the 112 patients. We defined the average expression level (18.670) of AML cases as a cutoff threshold to classify the patients into 2 groups, designated as low and high PRL-3 expression. Of them, 45 individual cases (40.2%) showed high PRL-3 expression. Additional analysis indicated that the 26 relapsed/refractory AML patients expressed higher PRL-3 than did the 86 newly diagnosed patients (median of 21.110 versus 11.076, P < .001, Fig. 1B). Furthermore, in 30 AML patients, the PRL-3 expression level at their first complete remission was significantly lower than that observed when they were initially diagnosed (median, 12.115 versus 20.717, paired t test, P < .001, Fig. 1C). Accordingly, PRL-3 protein expression level is obviously higher in a majority of AML patients than that observed in healthy donors (Fig. 1D), confirming the results of PRL-3 mRNA expression. Meanwhile, we observed that PRL-3 protein was not only detected in FLT3-ITD–positive cell lines MOLM-13 and MV4-11, but also in FLT3-ITD–negative cell lines, especially in ML-1 (Fig. 1E). PRL-3 protein in ML-1 cells was further stained by immunofluorescence (Fig. 1F). Thereafter, endogenous PRL-3 in ML-1 was silenced to characterize its function in this study. All above results demonstrated that PRL-3 is generally upregulated in FLT3-ITD–negative AML.
Figure 1.

PRL-3 highly expresses in FLT3-ITD–negative AML patients and cells. (A-C) Comparison of PRL-3 mRNA expression in bone marrow of the 112 FLT3-ITD–negative AML patients to that of the 16 normal healthy donors (A), or the relapsed/refractory AML patients to the initially diagnosed patients (B), or the patients with complete remission to that of the initially diagnosed patients (C) by quantitative RT-PCR analysis. Statistical analysis of PRL-3 mRNA expression level was performed with SPSS, version 16.0, software. The data are indicated as mean ± standard error of the mean. (D) Detection of PRL-3 protein expression in bone marrow of the indicated FLT3-ITD–negative AML patients and healthy donors by western blotting. The standardized relative PRL-3 expression to beta-actin (ST) is indicated below each lane. (E) PRL-3 expression in the indicated human AML cell lines by western blotting. (F) PRL-3 protein expression in ML-1 cells viewed by confocal microscope (magnification, 40× times 10×).
PRL-3 Promotes FLT3-ITD–Negative Cell Proliferation
To examine the possible roles of PRL-3 in AML occurrence, especially in the AML cell proliferation, we ectopically expressed PRL-3 and its phosphatase-dead mutant (C104S) in FLT3-ITD–negative cell lines U937 and ML-1. Meanwhile, the endogenous PRL-3 was silenced in ML-1 cells. Immunoblotting results confirmed the ectopic expression of PRL-3 in these 2 stable cell lines (Fig. 2A,B); furthermore, depletion of endogenous PRL-3 was also successful (Fig. 2C). Cell proliferation assays showed that PRL-3 rather than its phosphatase-dead mutant PRL-3 (C104S) promoted the proliferation of U937 and ML-1 cells, compared with the control cells (Vector) (P < .05; Fig. 2D,E). Conversely, depletion of endogenous PRL-3 significantly suppressed ML-1 cell proliferation (P < .05; Fig. 2F), indicating that PRL-3 can enhance AML cell proliferation.
Figure 2.
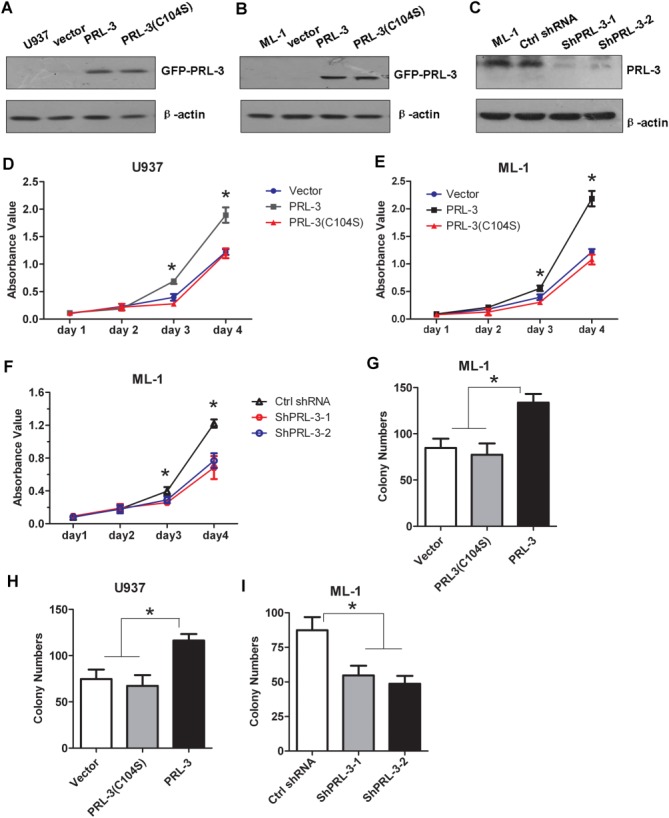
The effect of PRL-3 on FLT3-ITD–negative AML cell proliferation. (A,B) Detection of the ectopic GFP-PRL-3 expression in U937 cells (A) and ML-1 cells (B) stably transduced with either pLVX-puro-EGFP-PRL-3 wt or EGFP-PRL-3 (C104S) expression constructs by western blotting. (C) Depletion of endogenous PRL-3 expression in ML-1 cells transfected by either 2 PRL-3–specific shRNAs (shPRL-3-1 and shPRL-3-2), respectively, or by control (Ctrl) shRNA by western blotting. (D,E) The effects of PRL-3 overexpression on the proliferation of U937 (D) and ML-1 cells (E) assayed by CCK8 cell proliferation methods. (F) The effects of endogenous PRL-3 depletion on ML-1 cell proliferation by CCK8 cell proliferation methods. (G-I) Colony formation of ML-1 (G) and U937 (H) cells transfected with either PRL-3 or its mutant, or ML-1 cells (I) with endogenous PRL-3 silencing by specific shRNA1 and shRNA2. Data were shown as mean ± standrad deviation of triplicate experiments. *P < .05; **P < .01.
Overexpressing PRL-3 led to clearly more colonies, compared to cells expressing either PRL-3(C104S) or the empty vector (Vector) in both U937 and ML-1 cells (Fig. 2G,H), whereas PRL-3 depletion in ML-1 cells significantly blocked colony formation (P < .01, Fig. 2I), demonstrating that PRL-3 can promote cell proliferation and tumorigenesis.
PRL-3 Enhances FLT3-ITD–Negative AML Cell Cycle
To evaluate the effect of PRL-3 on the FLT3-ITD–negative cell cycle progression, flow cytometry analysis was performed. PRL-3 overexpression clearly enhanced cell cycle progression by lowering the cell population in G0/G1 phase and increasing that in S phase in U937 and ML-1 cells (Fig. 3A,B). By contrast, knockdown of PRL-3 arrested cell progression in G0/G1 phase and decreased cell population in the S phase (P < .05, Fig. 3C). These results indicate that silencing PRL-3 in the AML cells could inhibit cell growth. To explore the underlying mechanism of PRL-3 in AML cell proliferation, we checked the expression of the cell cycle regulatory molecules. The results showed that overexpression of PRL-3 upregulated CDK2 and Cyclin D1 expression, and decreased p21 expression (Fig. 3D), indicating that PRL-3 may be an important player in cell cycle regulation.
Figure 3.

The effect of PRL-3 on cell cycle and apoptosis. (A,B) The effect of PRL-3 overexpression on cell cycle progression of U937 (A) and ML-1 (B) cells with the typical histograms. The indicated cells were fixed and analyzed by flow cytometry after propidium iodide staining. The statistical analyses are indicated at the right. Data were shown as mean ± SD of triplicate experiments. *P < .05, n=3. (C) The effect of endogenous PRL-3 silenced by either 2 PRL-3–specific shRNA-1,-2, or by control shRNA on ML-1 cell cycle progression. The analysis is similar to that in (A) and (B). (D) The effect of PRL-3 overexpression on the expression of cell cycle–related CDK2, CyclinD1 and p21 by western blotting in ML-1 and U937 whole-cell lysates.
PRL-3 Exerts Antiapoptosis and Drug Resistance
To further investigate the effect of PRL-3 in drug therapy of the AML, PRL-3 overexpressing U937 and ML-1 cells were treated with Ara-C for 72 hours. PRL-3 overexpression counteracted the drug sensitivity of AML cells to Ara-C, but the PRL-3 mutant (C104S) did not (Fig. 4A,B). In addition, knockdown of the endogenous PRL-3 obviously sensitized cells to the cytotoxicity of the drug (Fig. 4C). PRL-3 effects on drug resistance and cell apoptosis was further explored. Aberrant upregulation of wild-type PRL3 but not its mutant form sufficiently neutralized Ara-C and daunorubicin (DNR)-induced apoptosis (P < .05, Fig. 4D,E). As expected, PRL-3 depletion significantly sensitized cells to death by this drug (P < .05, Fig. 4F). Given that PRL-3 is implicated in antiapoptosis,31 the expression of apoptosis-related proteins were checked. Upon Ara-C treatment, the apoptotic proteins PARP, caspase-9, and caspase-3 were clearly activated in ML-1 cells where the expression of endogenous PRL-3 was silenced by siRNA (Fig. 4G). Because PRL-3 can activate STAT5 to confer antiapoptosis effect in FLT3-ITD positive AML cell,27 we asked if PRL-3 functions in FLT3-ITD–negative cells in a similar manner. Our immunoblotting results showed that PRL-3 can effectively enhance STAT5 phosphorylation (Fig. 4H). Moreover, activation of PI3K/AKT pathway is well known to be involved in survival, the phosphorylation status of AKT and its downstream effectors were checked in these FLT3-ITD–negative AML cells. In line with our observation in other solid tumor cells,22 PRL-3 overexpression could activate AKT to some extent, rather than the further downstream effectors, including 4E-BP1 and p70S6K in both ML-1 and U937 cells (Fig. 4H). Taken together, all above results demonstrate that PRL-3 exerts its antiapoptotic effects on drug resistance in FLT3-ITD–negative AML via regulating phosphorylation of STAT5 and AKT.
Figure 4.

The effect of PRL-3 on drug sensitivity and drug-induced apoptosis. (A-C) Cytotoxicity analysis of U937 (A) and ML-1 (B) cells with ectopic expression of PRL-3 or its mutant, or with silenced endogenous PRL-3 by the specific shRNAs as shown (C). The cells were treated with Ara-C or doxorubicin (DNR) in the indicated concentrations for 72 hours, and assayed by CCK8 methods. (D-F) Annexin V/7-AAD staining and flow cytometry analysis of U937 (D) and ML-1 (E,F) in the indicated conditions. Cells were treated with 0.4 μM Ara-C or 0.04 μM DNR for 24 hours and analyzed with flow cytomery. The representive histograms and statistical analysis are indicated. Data were shown as mean ± SD of triplicate experiments. *P < .05; **P < .01, n=3. (G) Western blots of the apoptosis-related proteins as indicated in ML-1 whole-cell lysates. The indicated cells were treated with 0.4 μM Ara-C for 24 hours and lyzed for immunoblotting. (H) Western blots of the survival-related proteins as indicated in ML-1 and U937 whole-cell lysates.
PRL-3 Enhances FLT3-ITD–Negative Tumor Formation and Metastasis
To directly examine the in vivo function of PRL-3 in tumorigenesis and invasiveness/metastasis, we conducted xenograft experiments in immunodeficient mice. Knockdown of endogenous PRL-3 significantly inhibited tumor formation by ML-1 cells when assayed at the 20th day after subcutaneous inoculation (Fig. 5A and Supporting Fig. S1A). To further characterize the molecular events in these cancer cells, wild-type and PRL-3–silenced ML-1 cells were evaluated by immunoblotting with other markers and the results showed that endogenous PRL-3 depletion can efficiently decrease Ki67 expression (Supporting Fig. S2) whereas increases Bax expression (Supporting Fig. S3), indicating that the delayed tumor formation may be due to the blocked cell proliferation in vivo. By contrast, overexpressing PRL-3 markedly enhanced tumorigenesis in the nude mice only on the 14th day after injection of ML-1 cells, as expected (Fig. 5B and Supporting Fig. S1B). Similar results were also obtained in U937 cells (Fig. 5C and Supporting Fig. S1C). To investigate the role of PRL-3 in invasiveness/metastasis, the assayed mice were injected with the stably transfected cells through the tail vein. PRL-3 overexpression in ML-1 cells enhanced their invasive ability to other mouse tissues and this lead to typically enlarged spleens and livers (Fig. 5D,E). In contrast, knockdown of endogenous PRL-3 significantly abrogated the tissue invasion ability of ML-1 cells (Fig. 5D,E). Immunohistofluorescence staining further confirmed the presence of infiltrated xenografted ML-1 cells with GFP label in the spleens of sacrificed mice (Fig. 5F), indicating that the enlargement of the spleens may have been caused by the invasion and the proliferation of the injected cells. In addition, PRL-3 overexpression in U937 cells also induced the intumescentia of livers and spleens in the nude mice (Fig. 5G,H). These results together showed that PRL-3 can efficiently promote AML progression in FLT3-ITD–negative cells by enhancing cell proliferation and antiapoptosis in vivo.
Figure 5.

The function of PRL-3 in tumorigenesis and leukemogenesis. (A,B) Tumor formation induced by ML-1 cells stably transfected with scrambled ShRNAs (Ctrl shRNA) or with PRL-3–specific RNAs (ShPRL-3) on the 20th day post-subcutaneous injection (psi) (A), or with empty vector (Vector) or PRL-3 (PRL-3) on the 14th day psi (B) in nude mice. Upper panel shows the dissected paired tumors from each mouse; Lower panel shows the weight (g) of the indicated tumors. *P < .05; **P < .01; n = 5 (paired Student t test). (C) Tumor formation induced by U937 cells transfected [as in (B)] in nude mice on 14th day psi. **P < .01, n = 5 (paired Student t test). (D,E) Livers (D) and spleens (E) weight (g) of the nude mice injected with the indicated ML-1 cells through tail-vein transplantation. Mice were sacrificed on the 15th day post inoculation, and the organs were dissected and measured. *P < .05; **P < .01, n = 5 (unpaired Student t test). (F) IF staining of GFP on the frozen sections of the spleens as shown in (E) to discriminate the injected GFP-labeled AML cells from mice cells. Nuclei were stained with DAPI. Bar represents 25 μm. (G,H) Livers (G) and Spleens (H) weight (g) of the nude mice injected with U937 cells stably transfected with the indicated constructs as in (D) through (E). Mice were sacrificed and dissected on the 15th day post-inoculation as in (D) through (E). *P < .05;**P < .01, n = 4-5 (unpaired Student t test).
PRL-3 Is an Independent Factor for FLT3-ITD–Negative AML Progression
To confirm our results regarding the clinical relevance of PRL-3 in AML pathogenesis, the relationships of PRL-3 expression to various clinical features indicated and summarized in Table 1, in which patients with poor risk was 29.5% of the total enrolled cases. Further association analysis of PRL-3 to other clinical features was assessed individually. PRL-3 expression is significantly correlated with FAB category (P = .002), CD7 expression (P = .017) (Table 2). PRL-3 was more highly expressed in M4/M5 FAB category than in non-M4/M5. PRL-3 mRNA expression in patients with CD7 positivity was higher than in CD7 negativity. There was no statistically significant difference in PRL-3 expression by WBC count, cytogenetic risk or CD56 expression. Taken together, our results show that PRL-3 could be considered as an independent factor for the diagnosis and prognosis, regardless of other features, including the status of FLT3-ITD.
Table 2.
Association of PRL-3 Expression to Clinical Features
| Variables | Group | No. | PRL-3 Expression | P | |
|---|---|---|---|---|---|
| Low | High | ||||
| White blood cell count | <50 × 109/L | 79 | 49 | 30 | .462 |
| ≥50 × 109/L | 33 | 18 | 15 | ||
| FAB category | M4/M5 | 50 | 22 | 28 | .002 a |
| Non-M4/M5 | 62 | 45 | 17 | ||
| Cytogenetics risk | Favorable | 24 | 16 | 8 | .277 |
| Intermediate | 55 | 35 | 20 | ||
| Poor | 33 | 16 | 17 | ||
| Expression of CD7 | Positive | 40 | 18 | 22 | .017a |
| Negative | 72 | 49 | 23 | ||
| Expression of CD56 | Positive | 18 | 8 | 10 | .146 |
| Negative | 94 | 59 | 35 | ||
P < .05.
PRL-3 Expression Level Is Inversely Correlated to Patient Survival
To examine the prognostic significance of PRL-3 expression, we performed a follow-up study on 80 patients with de novo AML, with 3-fold of the averaged PRL-3 expression level (18.670) in healthy donors as a cutoff value. Of these 80, 27 patients were classified into the PRL-3 high-expression group (PRL-3 expression ≥ 18.670) and 53 patients in the low-expression group (PRL-3 expression < 18.670). Disease-free survival (DFS) was then performed with the Kaplan-Meier method. The DFS rate of the patients in the PRL-3 high-expression group was only 16.3%, whereas the survival rate in the PRL-3 low-expression group is up to 53.5% in the 40-month period from the first remission (Fig. 6), demonstrating that PRL-3 is inversely well-correlated to the overall survival of FLT3-ITD–negative AML patients (P = .001, n=80).
Figure 6.
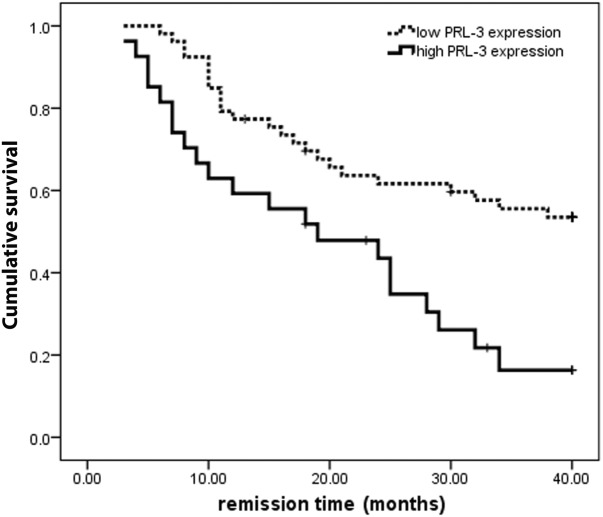
The Kaplan-Meier survival analysis of de novo AML patients with high PRL-3 expression and low PRL-3 expression. P = .001, n = 80.
We also investigated the association between the clinical features and DFS, and performed univariate and multivariate Cox regression analysis. The DFS was statistically significantly correlated with PRL-3 expression, age, CD34 expression, CD7 expression, and cytogenetic risk (Table 3). Multivariate analysis showed that PRL-3 expression (hazard ratio [HR] = 1.609; P = .035), cytogenetic risk (HR = 2.143; P = .014), and CD34 expression (HR = 2.089; P = .000) were also independent predictors for DFS, respectively, but no CD7 expression. Therefore, PRL-3 could be a novel independent prognostic factor.
Table 3.
Univariate and Multivariate Analysis of Disease-Free Survival in FLT3-ITD–Negative Acute Myeloid Leukemia
| Risk Parameter | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| PRL-3+ | 2.538 | 1.403-4.592 | .002a | 1.609 | 0.932-3.668 | .035a |
| Age ≥50 y | 2.233 | 1.257-3.582 | .003a | 2.142 | 0.643-6.058 | .169 |
| WBC ≥50 × 109/L | 1.63 | 0.873-3.044 | .125 | |||
| Unfavorable cytogenetic risk | 2.489 | 1.590-3.895 | .000a | 2.143 | 0.778-3.985 | .014a |
| CD7+ | 1.48 | 0.784-2.794 | .047a | |||
| CD34+ | 3.102 | 1.691-5.691 | .000a | 2.089 | 0.731-3.970 | .000a |
| CD56+ | 0.903 | 0.434-1.880 | .785 | |||
P < .05.
Abbreviations: CI, confidence interval; HR, hazard ratio; WBC, white blood cell.
DISCUSSION
FLT3-ITD–negative AML accounts for 70% to 80% of all AML cases,29 but the mechanism of its occurrence and progression still remain elusive. In this study, we provided several lines of evidence demonstrating that like FLT3-ITD, phosphatase PRL-3 alone can also enhance the AML progression by promoting leukemia cell proliferation and antiapoptosis ability even under cytotoxic drug stress. In vivo xenograft results of PRL-3 in AML pathogenesis also confirmed this conclusion, because PRL-3 depletion significantly blocked the invasiveness of the assayed AML cells (Fig. 5D,E), indicating that PRL-3 plays an important role in AML progression. In line with this, the relevance analysis of the clinical AML cases revealed that PRL-3 expression level is adversely correlated to patients’ overall survival, emphasizing its independent role in AML progression.
Accumulated evidence indicates that FLT3-ITD functions in approximately 20% of AML as driver mutations, and screening FLT3-ITD could benefit to select the more accurate prognostic criteria and therapeutic strategies.32,33 PRL-3 as a downstream effector of FLT3-ITD and STAT5 is upregulated in approximately 47% of this positive AML subgroup, rather than the FLT3-ITD–negative group,27,28 indicating that PRL-3 just functions in a supplementary or accessory alliance with FLT3-ITD to drive AML pathogenesis. By contrast, in our study, we found that PRL-3 mRNA and protein expression are upregulated more than 3-fold in 40.2% of the FLT3-ITD–negative AML compared to that of the healthy donors, and PRL-3 attenuation obviously delayed the AML progression in nude mice, indicating that PRL-3 could be another novel promoter of AML pathology, independent of FLT3-ITD status. The observation of higher PRL-3 expression in refractory/relapsed AML reinforced this notion (Fig. 1B). Moreover, PRL-3 mRNA expression is associated with a poor prognosis in both univariate and multivariate analysis for DFS, because the survival rate in PRL-3 high group is much less than that in the low group, which are consistent with the function of PRL-3 in FLT3-ITD–positive cases.28 Therefore, the notion that PRL-3 expression was unchanged in FLT3-ITD–negative AML cases28 seems invalid, due to the distinct analysis. Ignoring the key function of PRL-3 would not benefit FLT3-ITD–negative AML prognosis and therapy.
Mechanistically, PRL-3 promotes cell proliferation and migration by downregulating Csk, leading to Src activation.34 We have also demonstrated that PRL-3 initiates tumor angiogenesis by recruiting vascular endothelial cells instead of its mutant.15 In this study, we reproducibly showed that elevated PRL-3 expression promotes FLT3-ITD negative AML cell proliferation, rather than the catalytically inactive PRL-3 (C104S) mutant, uncovering PRL-3’s phosphatase-dependent function in AML progression. PRL-3 depletion induces G1 arrest and sensitizes the AML cells to anticancer drugs. By contrast, PRL-3 upregulation decreased the sensitivity to cytotoxic agents and shielded the cell apoptosis even under direct cytotoxic stress. Therefore, our results further suggest that PRL-3 per se can be another novel causal factor for AML progression, as PRL-3 silencing can succeed against the AML pathogenesis in vivo, which is consistent with the clinical relevance of PRL-3 to AML prognosis (Fig. 6 and Table 3). Collectively, our findings indicate that PRL-3 may play critical roles in cell proliferation and drug resistance by antiapoptosis and enhancement of cell proliferation in FLT3-ITD–negative AML progression, thus PRL-3 should be also considered as a therapeutic target.
Regarding the promotion of cell proliferation and the apoptosis evasion by PRL-3, the involved detailed questions that how PRL-3, as a non–transcription factor, upregulates the expression of cell cycle–related cyclin D1, CDK and suppresses P21 to enhance AML cell cycle needs to be further investigated. Similarly, the association analysis of PRL-3 mRNA expression to the clinical feature showed that PRL-3 expression is correlated to CD7 expression in FLT3-ITD–negative AML (Table 2), indicating that PRL-3 could synergize with CD7 to promote AML progression. However, the multivariant analysis of disease-free survival of FLT3-ITD negative AML showed that PRL-3 expression is significantly correlated to the disease-free survival, but not CD7 expression (Table 3), hinting that PRL-3 plays dominant role in the AML progression than CD7 does. Therefore, the CD7 expression could be somehow regulated by PRL-3 in FLT3-ITD–negative AML to certain extend, as PRL-3 is involved in NF-κB activation35 and other pathways,17 or in proteosome degradation36 or epigenetic chromatin modification23 to regulate a particular gene expression. Whether PRL-3 is interplaying with CD7 together or functioning independently is worth investigating clearly.
Given that PRL-3 is also upregulated in FLT3-ITD–negative AML, it seems that PRL-3 could be upregulated by several other factors apart from FLT3-ITD in AML. Results presented in this study show that PRL-3 overexpression can activate STAT5 (Fig. 4H) in FLT3-ITD–negative AML cells, which may in turn upregulate PRL-3 expression, as reported.28 Moreover, the well-known tumor suppressor p53, which is frequently mutated in various solid tumors37 and AML,2 has been shown to directly regulate PRL-3 at the transcriptional level.38 PRL-3 is a downstream effector of BCR-ABL signaling pathway in chronic myeloid leukemia,26 and PCBP1 also regulates PRL-3 at the translational level,39 all which could also be involved in PRL-3 upregulation in FLT3-ITD–negative AML. The exact underlying mechanism will be subsequently characterized in future studies.
FUNDING SUPPORT
This work was partially supported by National Science Foundation of China (grant 81171947) to Dr. Wang and Science and Technology Planning Project of Guangdong Province (no. 2012B031800388) to Dr. Tong. We thank Professor Vinay Tergaonkar for critical editing of this manuscript.
CONFLICT OF INTEREST DISCLOSURES
The authors made no disclosures.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- 1.Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Dohner K, Dohner H. Molecular characterization of acute myeloid leukemia. Haematologica. 2008;93:976–982. doi: 10.3324/haematol.13345. [DOI] [PubMed] [Google Scholar]
- 3.Fröhling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285–6295. doi: 10.1200/JCO.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Lerch E, Espeli V, Zucca E, et al. Prognosis of acute myeloid leukemia in the general population: data from southern Switzerland. Tumori. 2009;95:303–310. doi: 10.1177/030089160909500306. [DOI] [PubMed] [Google Scholar]
- 5.Juliusson G, Antunovic P, Derolf A, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood. 2009;113:4179–4187. doi: 10.1182/blood-2008-07-172007. [DOI] [PubMed] [Google Scholar]
- 6.Alibhai SM, Leach M, Minden MD, Brandwein J. Outcomes and quality of care in acute myeloid leukemia over 40 years. Cancer. 2009;115:2903–2911. doi: 10.1002/cncr.24373. [DOI] [PubMed] [Google Scholar]
- 7.Pemmaraju N, Kantarjian H, Ravandi F, Cortes J. FLT3 inhibitors in the treatment of acute myeloid leukemia: the start of an era? Cancer. 2011;117:3293–3304. doi: 10.1002/cncr.25908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stephens BJ, Han H, Gokhale V, Von Hoff DD. PRL phosphatases as potential molecular targets in cancer. Mol Cancer Ther. 2005;4:1653–1661. doi: 10.1158/1535-7163.MCT-05-0248. [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Li B. Expression of phosphatase of regenerating liver-3 in squamous cell carcinoma of the cervix. Med Oncol. 2011;28:775–780. doi: 10.1007/s12032-010-9514-3. [DOI] [PubMed] [Google Scholar]
- 10.Saha S, Bardelli A, Buckhaults P, et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–1346. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- 11.Miskad UA, Semba S, Kato H, et al. High PRL-3 expression in human gastric cancer is a marker of metastasis and grades of malignancies: an in situ hybridization study. Virchows Arch. 2007;450:303–310. doi: 10.1007/s00428-006-0361-8. [DOI] [PubMed] [Google Scholar]
- 12.Radke I, Gotte M, Kersting C, Mattsson B, Kiesel L, Wulfing P. Expression and prognostic impact of the protein tyrosine phosphatases PRL-1, PRL-2, and PRL-3 in breast cancer. Br J Cancer. 2006;95:347–354. doi: 10.1038/sj.bjc.6603261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren T, Jiang B, Xing X, et al. Prognostic significance of phosphatase of regenerating liver-3 expression in ovarian cancer. Pathol Oncol Res. 2009;15:555–560. doi: 10.1007/s12253-009-9153-1. [DOI] [PubMed] [Google Scholar]
- 14.Polato F, Codegoni A, Fruscio R, et al. PRL-3 phosphatase is implicated in ovarian cancer growth. Clin Cancer Res. 2005;11(19 pt 1):6835–6839. doi: 10.1158/1078-0432.CCR-04-2357. [DOI] [PubMed] [Google Scholar]
- 15.Guo K, Li J, Wang H, et al. PRL-3 initiates tumor angiogenesis by recruiting endothelial cells in vitro and in vivo. Cancer Res. 2006;66:9625–9635. doi: 10.1158/0008-5472.CAN-06-0726. [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Zeng H, Zhang X, et al. Phosphatase of regenerating liver-3 promotes motility and metastasis of mouse melanoma cells. Am J Pathol. 2004;164:2039–2054. doi: 10.1016/S0002-9440(10)63763-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Aidaroos AQ, Zeng Q. PRL-3 phosphatase and cancer metastasis. J Cell Biochem. 2010;111:1087–1098. doi: 10.1002/jcb.22913. [DOI] [PubMed] [Google Scholar]
- 18.Krndija D, Munzberg C, Maass U, et al. The phosphatase of regenerating liver 3 (PRL-3) promotes cell migration through Arf-activity-dependent stimulation of integrin alpha5 recycling. J Cell Sci. 2012;125(pt 16):3883–3892. doi: 10.1242/jcs.104885. [DOI] [PubMed] [Google Scholar]
- 19.Ustaalioglu BB, Bilici A, Barisik NO, et al. Clinical importance of phosphatase of regenerating liver-3 expression in breast cancer. Clin Transl Oncol. 2012;14:911–922. doi: 10.1007/s12094-012-0880-5. [DOI] [PubMed] [Google Scholar]
- 20.Bilici A, Ustaalioglu BB, Yavuzer D, Seker M, Mayadagli A, Gumus M. Prognostic significance of high phosphatase of regenerating liver-3 expression in patients with gastric cancer who underwent curative gastrectomy. Dig Dis Sci. 2012;57:1568–1575. doi: 10.1007/s10620-012-2076-9. [DOI] [PubMed] [Google Scholar]
- 21.Mayinuer A, Yasen M, Mogushi K, et al. Upregulation of protein tyrosine phosphatase type IVA member 3 (PTP4A3/PRL-3) is associated with tumor differentiation and a poor prognosis in human hepatocellular carcinoma. Ann Surg Oncol. 2013;20:305–317. doi: 10.1245/s10434-012-2395-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922–2926. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Zheng P, Ji T, et al. An epigenetic role for PRL-3 as a regulator of H3K9 methylation in colorectal cancer. Gut. 2013;62:571–581. doi: 10.1136/gutjnl-2011-301059. [DOI] [PubMed] [Google Scholar]
- 24.Juric D, Lacayo NJ, Ramsey MC, et al. Differential gene expression patterns and interaction networks in BCR-ABL-positive and -negative adult acute lymphoblastic leukemias. J Clin Oncol. 2007;25:1341–1349. doi: 10.1200/JCO.2006.09.3534. [DOI] [PubMed] [Google Scholar]
- 25.Fagerli UM, Holt RU, Holien T, et al. Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood. 2008;111:806–815. doi: 10.1182/blood-2007-07-101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J, Cheong LL, Liu SC, et al. The pro-metastasis tyrosine phosphatase, PRL-3 (PTP4A3), is a novel mediator of oncogenic function of BCR-ABL in human chronic myeloid leukemia. Mol Cancer. 2012;11:72. doi: 10.1186/1476-4598-11-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J, Bi C, Chng WJ, et al. PRL-3, a metastasis associated tyrosine phosphatase, is involved in FLT3-ITD signaling and implicated in anti-AML therapy. PLoS One. 2011;6(5):e19798. doi: 10.1371/journal.pone.0019798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JE, Yuen HF, Zhou JB, et al. Oncogenic roles of PRL-3 in FLT3-ITD induced acute myeloid leukaemia. EMBO Mol Med. 2013;5:1351–1366. doi: 10.1002/emmm.201202183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Guo K, Koh VW, et al. Generation of PRL-3- and PRL-1-specific monoclonal antibodies as potential diagnostic markers for cancer metastases. Clin Cancer Res. 2005;11:2195–2204. doi: 10.1158/1078-0432.CCR-04-1984. [DOI] [PubMed] [Google Scholar]
- 31.Sun ZH, Bu P. Downregulation of phosphatase of regenerating liver-3 is involved in the inhibition of proliferation and apoptosis induced by emodin in the SGC-7901 human gastric carcinoma cell line. Exp Ther Med. 2012;3:1077–1081. doi: 10.3892/etm.2012.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003;17:1738–1752. doi: 10.1038/sj.leu.2403099. [DOI] [PubMed] [Google Scholar]
- 33.Knapper S. The clinical development of FLT3 inhibitors in acute myeloid leukemia. Expert Opin Investig Drugs. 2011;20:1377–1395. doi: 10.1517/13543784.2011.611802. [DOI] [PubMed] [Google Scholar]
- 34.Liang F, Liang J, Wang WQ, Sun JP, Udho E, Zhang ZY. PRL3 promotes cell invasion and proliferation by down-regulation of Csk leading to Src activation. J Biol Chem. 2007;282:5413–5419. doi: 10.1074/jbc.M608940200. [DOI] [PubMed] [Google Scholar]
- 35.Lian S, Meng L, Liu C, et al. PRL-3 activates NF-kappaB signaling pathway by interacting with RAP1. Biochem Biophys Res Commun. 2013;430:196–201. doi: 10.1016/j.bbrc.2012.11.036. [DOI] [PubMed] [Google Scholar]
- 36.Choi MS, Min SH, Jung H, et al. The essential role of FKBP38 in regulating phosphatase of regenerating liver 3 (PRL-3) protein stability. Biochem Biophys Res Commun. 2011;406:305–309. doi: 10.1016/j.bbrc.2011.02.037. [DOI] [PubMed] [Google Scholar]
- 37.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 38.Basak S, Jacobs SB, Krieg AJ, et al. The metastasis-associated gene Prl-3 is a p53 target involved in cell-cycle regulation. Mol Cell. 2008;30:303–314. doi: 10.1016/j.molcel.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Vardy LA, Tan CP, et al. PCBP1 suppresses the translation of metastasis-associated PRL-3 phosphatase. Cancer Cell. 2010;18:52–62. doi: 10.1016/j.ccr.2010.04.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.