

Abstract
Knockout of genes with CRISPR/Cas9 is a newly emerged approach to investigate functions of genes in various organisms. We demonstrate that CRISPR/Cas9 can mutate endogenous genes of the ascidian Ciona intestinalis, a splendid model for elucidating molecular mechanisms for constructing the chordate body plan. Short guide RNA (sgRNA) and Cas9 mRNA, when they are expressed in Ciona embryos by means of microinjection or electroporation of their expression vectors, introduced mutations in the target genes. The specificity of target choice by sgRNA is relatively high compared to the reports from some other organisms, and a single nucleotide mutation at the sgRNA dramatically reduced mutation efficiency at the on-target site. CRISPR/Cas9-mediated mutagenesis will be a powerful method to study gene functions in Ciona along with another genome editing approach using TALE nucleases.
Keywords: ascidian, Ciona intestinalis, CRISPR/Cas9, knockout, mutagenesis
Introduction
Ciona intestinalis is a representative species of urochordate ascidians for elucidating genetic functions in the simplified chordate body (Satoh 2003). A draft genome sequence of Ciona intestinalis was reported in 2002 (Dehal et al. 2002), and deep annotations of genes encoded by the genome have been carried out since then (Satou et al. 2008). The basic technologies for examining gene functions have been established in Ciona, that include introduction of exogenous DNA and RNA by means of microinjection (Imai et al. 2000), efficient introduction of reporter constructs by electroporation (Corbo et al. 1997), knockdown of genes by antisense morpholino oligonucleotides (Satou et al. 2001), and transposon-mediated germ cell transformation and mutagenesis (Sasakura et al. 2003, 2005). These technologies have supported our detailed and thorough analyses to reveal molecular and cellular mechanisms that underlie development of Ciona.
Gene knockout is the ultimate way to investigate gene functions by introducing mutations directly and specifically into targeted genes in the genome. Gene knockout was limited to very few model organisms until recently; however, technological innovations brought by the artificial nucleases such as zinc finger nucleases (ZFNs) and TAL effector nucleases (TALENs) have enabled us to knockout genes in various organisms through quite simple approaches (Santiago et al. 2008; Ochiai et al. 2010, 2012; Miller et al. 2011; Watanabe et al. 2012; Ansai et al. 2013, 2014; Suzuki et al. 2013; Hayashi et al. 2014; Hosoi et al. 2014; Kondo et al. 2014; Sakane et al. 2014; Sakuma & Woltjen 2014; Sugi et al. 2014). In Ciona, our group have reported knockout of enhanced GFP (eGFP) gene inserted in the genome with ZFNs, and more recently, target mutagenesis of endogenous genes with TALENs (Kawai et al. 2012; Treen et al. 2014; Yoshida et al. 2014). In the latter two reports, we showed that TALENs have a high knockout efficiency in both somatic and germ cells of Ciona.
Although TALEN-mediated gene knockout is a powerful approach for addressing gene functions in Ciona, constructed TALENs do not always have sufficient mutation activity and therefore another approach that can compensate for inactive TALENs is necessary. Use of the clustered, regularly interspaced, short palindromic repeat (CRISPR) and Cas9-mediated genome editing is a relatively newer approach than TALENs for achieving targeted mutagenesis of genomes in various organisms (e. g. Hwang et al. 2013; Feng et al. 2013; Gratz et al. 2013; Nakayama et al. 2013; Friedland et al. 2013; Xie & Yang 2013; Daimon et al. 2014; Hisano et al. 2014; Mashiko et al. 2014; Mashimo 2014). CRISPR/Cas9 uses a short guide RNA (sgRNA) that contains 20-nucleotide stretch identical to the target DNA sequence, and an RNA-mediated nuclease Cas9 (Hwang et al. 2013; Mali et al. 2013). When sgRNA binds to its target site, Cas9 protein is recruited to the binding site, and then the nuclease induces a double-strand break at the target genomic region. The double-strand break is repaired by one of the two endogenous systems, homologous recombination or non-homologous end joining. The latter system is more dominant but more error-prone than the former, and therefore during the repair of the double-strand break insertions and/or deletions can accumulate at the target site. If a mutation is not introduced, the target site is again targeted by CRISPR/Cas9, and once mutations are introduced the region is no longer targeted because of the modification of the target DNA sequence. CRISPR/Cas9 could be a candidate that can compensate for TALENs, because their mechanisms that recognize target sites are quite different. In CRISPR/Cas9, the recognition of the target site is mediated by RNA, compared to the protein-DNA recognition in TALENs. Therefore, genomic regions that are resistant to one of the two methods could be sensitive to the other. An advantage of CRISPR/Cas9 compared to TALENs is easy construction of the components necessary for knockout. sgRNA is the only component that specifies target genes. As mentioned, sgRNA usually contains a 20-nt stretch that is specific to the target site. In addition, three nucleotides adjacent to the 3′ end of the target site should be NGG. This sequence is named the protospacer adjacent motif (PAM) (Wiedenheft et al. 2012). There is no critical restriction to select a target site other than the target length and the presence of a PAM in the CRISPR/Cas9 system. Because a 20-nt target site of sgRNA is quite short, the region can be easily substituted to another by conventional cloning techniques. In contrast, TALEN proteins are composed of about 20 repeats, each of which recognizes a single nucleotide, and the variable region of a TALEN is approximately 2000 bp long. Construction of highly repetitive TALENs requires special ligation methods (Cermak et al. 2011; Sakuma et al. 2013a,b). The easy construction of sgRNAs is an attractive advantage of CRISPR/Cas9 systems over TALENs.
In this study, we examined the efficiency of CRISPR/Cas9-mediated mutagenesis in Ciona. Our group is interested in the functions of Hox cluster genes in Ciona (Sasakura et al. 2012; Yoshida et al. 2014). With CRISPR/Cas9, we successfully introduced mutations in Hox3 and Hox5, suggesting that CRISPR/Cas9 can be a powerful approach for addressing functions of genes in Ciona.
Materials and methods
Animals
Wild-type C. intestinalis were collected from or cultivated at Maizuru (Kyoto), Mukaishima (Hiroshima), Misaki (Kanagawa) and Usa (Kochi). Eggs and sperm were surgically collected. After fertilization, the embryos and larvae were cultured at 18°C.
Constructs
The cDNA of Cas9 was amplified from pMLM3613 (purchased from Addgene; Hwang et al. 2013) with the primers 5′-AAATATCACCGGATCCGATAATGGATAAGAAATACTC-3′ and 5′-GTTAGATATCGAATTTCATCCTGCAGCTCCACC-3′, and PrimeSTAR thermostable DNA polymerase (Takara Bio). The polymerase chain reaction (PCR) fragment was inserted into the BamHI and EcoRI sites of pBS-HTB/N (Akanuma et al. 2002; Sasakura et al. 2010) by In-Fusion cloning technology (Clontech) to create pHTBCas9. The cDNA of Cas9 was amplified with the primers 5′-CGACTCTAGAGGATCGGATCCCCTTGCGGCCGCAATGGATAAGAAATACTCAATAG-3′ and 5′-CGCTCAGCTGGAATTGAATTCTCATCCTGCAGCTCCACCGCTC-3′, then the PCR fragment was inserted into the BamHI and EcoRI sites of pSPeGFP (Sasakura et al. 2003) to create pSPCas9. Five base pairs at the multicloning site (underlined in the above primer sequence) were deleted by inverse PCR in order to adjust the reading frame, and then the cis element of Ci-EF1α (Sasakura et al. 2010) was inserted into the SalI site of pSPCas9 to create pSPCiEF1α>Cas9. Primer sets for sgRNAs were designed with the ZiFiT program at the following URL (http://zifit.partners.org/ZiFiT/CSquare9Nuclease.aspx; Sander et al. 2007, 2010). The primer sequences are listed in TableS1. After annealing of the primer pairs, the DNA was inserted into the BsaI site of pDR274 (Hwang et al. 2013) to create vectors for in vitro transcription of sgRNAs. The DNA encoding Hox5-sg1 was amplified with the primers 5′-GGCGACGACGGGTTAGGTAAG-3′ and 5′-TTGAATTCAAAAAGCACCGACTCGGTGCCAC-3′ from pDR274Hox5-sg1, digested with EcoRI, and then inserted into the EcoRV and EcoRI sites of pSPU6EV (Nishiyama & Fujiwara 2008) to create pSPU6>Hox5-sg1. Other expression vectors for sgRNAs were created by identical procedures. Expression vectors of Hox3-sg3 that contain mismatched nucleotides were created by inverse PCR using pSPU6>Hox3-sg3 as the template and PrimeSTAR thermostable DNA polymerase (Takara Bio).
Microinjection and electroporation
pHTBCas9 was linearized with XhoI. mRNAs of Cas9 were synthesized using Megascript T3 kit (Ambion), poly (A) tailing kit (Ambion), and Cap structure analog (New England Biolabs). pDR274 vectors encoding sgRNAs were linearized with DraI. sgRNAs were synthesized using Megascript T7 kit (Ambion). RNA was microinjected into unfertilized eggs according to a previously described method (Hikosaka et al. 1992). The volume of the injected media in an egg was approximately 30 pl. Electroporation of plasmids into 1-cell embryos was performed according to the previous report (Corbo et al. 1997; Treen et al. 2014). Forty micrograms of the expression vector of Cas9 and 20 μg of sgRNA expression vectors were simultaneously electroporated for each electroporation. After electroporation, the embryos were washed in filtered seawater three times to remove excess plasmid DNA.
Genome analyses
For bulked analyses, 20–40 Cas9 and sgRNA-expressing G0 embryos were gathered and genomic DNA was extracted using Wizard Genomic DNA isolation kit (Promega) according to the manufacturer’s instructions. sgRNA targeted regions were amplified by PCR using Ex Taq thermostable DNA polymerase (Takara Bio). The primers used for PCR are shown in TableS1. After purifying PCR bands by electrophoresis, PCR products were subcloned into pGEM-T Easy vector (Promega) for sequencing analyses. Detection of mutations with Cel-I nuclease (Transgenomics) was done according to the previous report (Sakuma et al. 2013b; Treen et al. 2014) except that the amount of DNA was estimated by electrophoresis. The potential off-target sites of sgRNAs were searched for with the Blastn-based program (Altschul et al. 1990). The primers used for amplifying DNA fragments containing the potential off-target sites of Hox3-sg3 were 5′-AGAGTATACCAGCCCCATCTG-3′ and 5′-CCACAGCTTAGAAGAGTGCAG-3′ for off-target site 1, and 5′-GTAGCACCATAGATTGTAACGG-3′ and 5′-GATCTTGCAGCGTTGAACACC-3′ for off-target site 2.
Results
CRISPR/Cas9-mediated mutations of Hox genes
To knockout Hox genes of Ciona, we designed sgRNAs for Hox3,Hox5 and Hox12. Eight sgRNAs were created in total for these genes (Table1). They were microinjected into unfertilized Ciona eggs along with the mRNA encoding Cas9, then these eggs were fertilized with wild type sperm to start embryogenesis. After the embryos reached the larval stage, genomic DNA was isolated in bulk, and the DNA region including the target site of the injected sgRNA was amplified by PCR. The PCR fragments were treated with the surveyor nuclease Cel-I after denaturing and re-annealing processes. Cel-I nuclease cleaves mismatched sites of double stranded DNA. If the PCR product included nucleotide sequence variations, these variations could yield mismatched sites during the re-annealing. As a result the DNA fragments could be cleaved by Cel-I, and shorter bands could be detected by electrophoresis.
Table 1.
sgRNAs that target Hox genes in Ciona intestinalis
| Target gene | Name of sgRNA | Sequence of the target site (5′–3′)† | PAM | Activity‡ |
|---|---|---|---|---|
| Ci-Hox3 | Hox3-sg1 | GGCATACACAAACTCACAAT | TGG | Negative |
| Hox3-sg3 | GGCAGCCATAAGAGTCAACA | AGG | Active | |
| Hox3-sg4 | GGTCTGCCTACCCAACACAC | GGG | Negative | |
| Hox3-sg5 | GGTGTGGAACCGTAGTATAA | TGG | Negative | |
| Ci-Hox5 | Hox5-sg1 | GGCGACGACGGGTTAGGTAA | CGG | Active |
| Ci-Hox12 | Hox12-sg1 | GGAACCAGATTTTAACTTGC | GGG | Negative |
| Hox12-sg2 | GGAAGAGCCCCTTCAAGTTC | CGG | Negative | |
| Hox12-sg3 | GGCCGCTCGCTATCGACTTC | AGG | Negative |
†DNA sequence of the target sites is shown. ‡Activity tested by the Cel-I assay.
As shown in Figure1A, Cel-I cleaved Hox3 PCR product from larvae into which about 3.0 pg of a sgRNA for Hox3 (Hox3-sg3 in Table1) was introduced with 15 pg of Cas9 mRNA. The presence of Cel-I cleaved bands suggests that the PCR fragment contained nucleotide sequence variations, probably caused by mutations induced by the CRISPR/Cas9 system. Likewise, Cel-I cleaved Hox5 the PCR product derived from larvae into which 3.0 pg of the sgRNA for Hox5 (Hox5-sg1) was introduced with 15 pg of Cas9 mRNA (Fig.1B). The other six sgRNAs designed to target Hox3 or Hox12 were not able to yield a Cel-I sensitive band (Fig.1C), suggesting that appearance of Cel-I sensitive bands are dependent on the sequence of sgRNAs.
Figure 1.
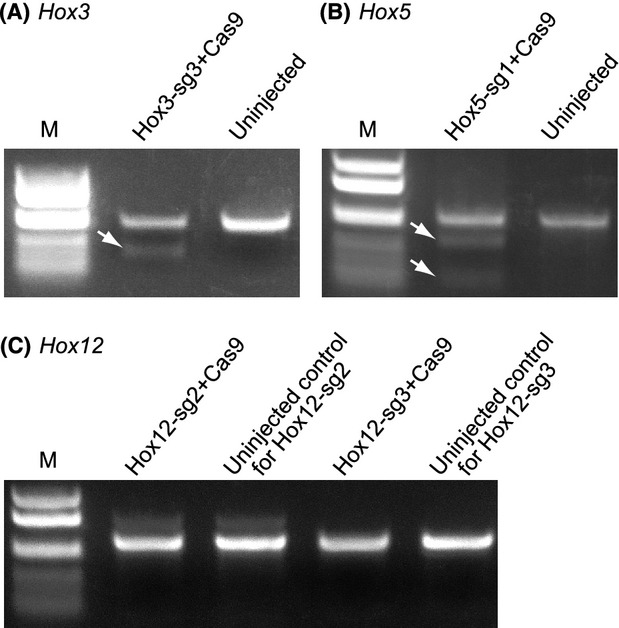
CRISPR/Cas9-mediated mutations of Hox genes in Ciona intestinalis. (A) Cel-I assay of polymerase chain reaction (PCR) amplifications that contained the target site of Hox3-sg3. PCR bands were treated with Cel-I prior to electrophoresis. The “Hox3-sg3+Cas9” lane indicates the PCR product derived from larvae into which 3.0 pg of Hox3-sg3 RNA and 15 pg of Cas9 mRNA were microinjected. “Uninjected” lane indicates the PCR product derived from uninjected control larvae. M, marker lane. The arrow indicates the position of Cel-I cleaved band. (B) Cel-I assay of PCR amplifications that contained the target site of Hox5-sg1. 3.0 pg of Hox5-sg1 RNA and 15 pg of Cas9 mRNA were microinjected. (C) Cel-I assays of PCR amplifications that contained the target sites of Hox12-sg2 and Hox12-sg3. 3.0 pg of a sgRNA and 15 pg of Cas9 mRNA were microinjected. In both cases, Cel-I sensitive bands were not detected.
To further investigate whether Hox3-sg3 and Hox5-sg1 introduced mutations at their corresponding target sites, we performed sequence analyses of the PCR fragments that showed Cel-I sensitivity. As a result, deletions and/or insertions of nucleotides were seen in the Hox3 target site derived from larvae into which Hox3-sg3 RNA and Cas9 mRNA were introduced (Fig.2A). The frequency of the mutated Hox3 clones among sequenced PCR clones was about 58% when 3.0 pg of Hox3-sg3 and 15 pg of Cas9 mRNA were injected. Likewise, Hox5 in Hox5-sg1 and Cas9 mRNA-introduced animals had mutations at the target site (Fig.2B), and the frequency of mutated PCR fragments was approximately 75.9%, when 3.0 pg of Hox5-sg1 and 15 pg of Cas9 mRNA were injected. These results suggest that CRISPR/Cas9 can induce insertion and/or deletion mutations at the target sites in the Ciona genome.
Figure 2.

Insertions and/or deletions induced by microinjection of CRISPR/Cas9 RNAs. (A) Examples of mutations induced by simultaneous injection of Hox3-sg3 RNA and Cas9 mRNA. The quantity of injected RNAs was 3.0 pg of Hox3-sg3 RNA and 15 pg of Cas9 mRNA. “Wild type” indicates the un-mutated sequence. The recognition site of Hox3-sg3 was shown in red. Deleted nucleotides were shown by “-”. Inserted nucleotides were shown in blue. (B) Examples of mutations induced by simultaneous injection of Hox5-sg1 RNA and Cas9 mRNA. The quantity of injected RNAs was 3.0 pg of Hox5-sg1 RNA and 15 pg of Cas9 mRNA.
Influence of the quantity of CRISPR/Cas9 RNAs on mutation frequency
We next examined different conditions of sgRNA/Cas9 mRNA introduction in order to achieve efficient knockout by CRISPR/Cas9. For this purpose, we compared the mutation frequencies among different amounts of sgRNA and Cas9 mRNA being introduced into embryos by changing their concentration in the injection media. We first investigated this with Hox5-sg1, which showed a higher mutation frequency than Hox3-sg3 in the above experiments. There was a tendency that the more RNAs being introduced, the higher the observed mutation frequency (Table2). When 0.3 pg of Hox5-sg1 was injected, the introduction of mutations was not detected by the Cel-I assay, suggesting that the mutation frequency was very low in this condition. Likewise, up to 3.0 pg of injected Cas9 mRNA was not able to introduce efficient mutations even though the quantity of injected sgRNA is high. When the amount of injected sgRNA and Cas9 mRNA was, respectively, adjusted to 1.5 and 15 pg, mutations were detected by Cel-I analysis, and sequencing of the PCR bands confirmed the occurrence of mutations at the target site (Table2). Mutation frequency increased when the quantity of injected Hox5-sg1 was increased from 1.5 to 3.0 pg. However, increasing the quantity of injected Cas9 mRNA from 15 to 30 pg slightly reduced the mutation frequency. A similar tendency was seen when Hox3-sg3 was examined (Table2). 0.3 pg of Hox3-sg3 or 3.0 pg of Cas9 mRNA did not cause efficient mutations that were detectable by Cel-I assay. We detected Cel-I sensitive bands when 1.5 pg of Hox3-sg3 and 15 pg of Cas9 mRNA were simultaneously microinjected. The difference of the mutation frequencies between 1.5 and 3.0 pg of Hox3-sg3 was not high, and increasing the injected Cas9 mRNA from 15 to 30 pg resulted in a 9% higher mutation frequency. Taking both results from Hox5-sg1 and Hox3-sg3 into consideration, we concluded that microinjection of 3.0 pg of sgRNA and 15 pg of Cas9 mRNA should be the standard condition due to its reproducible and high mutation activity, and there remains a possibility that the mutation frequency could be increased by further increasing the amount of sgRNA and Cas9 mRNA.
Table 2.
Mutation frequencies induced by Crispr/Cas9 system
| Name of sgRNA | Quantity of injected RNA | Mutation frequency (in %) | n† | |
|---|---|---|---|---|
| sgRNA (in pg) | Cas9 mRNA (in pg) | |||
| Hox5-sg1 | 0.3 | 3.0 | Cel-I negative | — |
| 0.3 | 15 | Cel-I negative | — | |
| 1.5 | 3.0 | Cel-I negative | — | |
| 3.0 | 3.0 | Cel-I negative | — | |
| 1.5 | 15 | 40.0 | 20 | |
| 3.0 | 15 | 75.9 | 31 | |
| 3.0 | 30 | 62.8 | 35 | |
| Hox3-sg3 | 0.3 | 15 | Cel-I negative | — |
| 3.0 | 3.0 | Cel-I negative | — | |
| 1.5 | 15 | 54.5 | 22 | |
| 3.0 | 15 | 58.0 | 31 | |
| 3.0 | 30 | 67.7 | 31 | |
†Number of sequenced polymerase chain reaction (PCR) clones. When the Cel-I assay did not yield cleaved band, sequenceing wan not performed.
Effect of Hox3 and Hox5 knockout on embryogenesis by CRISPR/Cas9
Expression patterns of Ci-Hox3 and Ci-Hox5 at early developmental stages have been described in previous reports (Gionti et al. 1998; Locascio et al. 1999; Ikuta et al. 2004). Ci-Hox3 starts its expression at the late tailbud stage in the anterior tail epidermis and a part of the central nervous system (CNS). At the larval stage, expression in the CNS is restricted to the motor ganglion. Ci-Hox5 starts its expression in the trunk lateral cells and tail nerve cord of early tailbud embryos, and the expression pattern persists until the late tailbud stage. At the larval stage, a weak expression of Ci-Hox5 remains at the nerve cord.
We observed the morphology of larvae injected with Hox3-sg3 or Hox5-sg1 together with Cas9 mRNA. We did not recognize any morphological defect associated with knockout of these genes (Fig.3A–C). This suggests that these genes may not be essential for forming the tadpole body of Ciona, as previous studies have pointed out (Ikuta et al. 2010; Yoshida et al. 2014).
Figure 3.
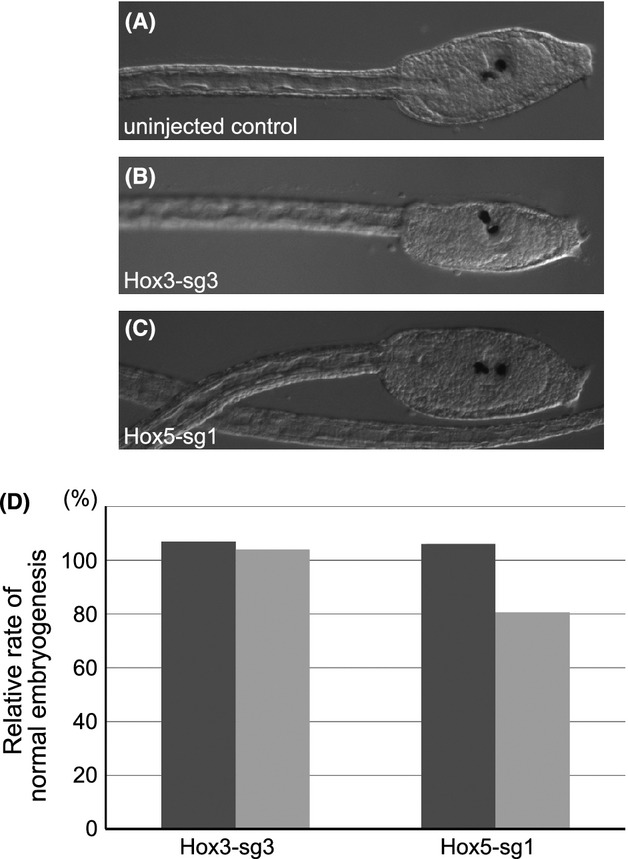
CRISPR/Cas9 system does not have a strong side-effect on embryogenesis. (A–C) Morphology of larvae. (A) An un-treated control larva, lateral view. Anterior is toward right. (B) A larvae into which 3.0 pg of Hox3-sg3 and 30 pg of Cas9 mRNA was microinjected. (C) A larvae into which 3.0 pg of Hox5-sg1 and 30 pg of Cas9 mRNA was microinjected. (D) Relative rate of normally developed larvae. In each experiment, the % of normally developed larvae in the population injected with 3.0 pg of sgRNA and 30 pg of Cas9 mRNA was normalized with the score of the uninjected control populations. Therefore, “100%” indicates the rates of normal larvae were identical between two populations.  , Experiment 1;
, Experiment 1;  , Experiment 2.
, Experiment 2.
By utilizing Hox3-sg3 and Hox5-sg1, we investigated the side-effect of expressing sgRNA and Cas9 mRNA on embryogenesis. For this purpose, we introduced 3.0 pg of sgRNAs and 30 pg of Cas9 mRNA, and compared the frequency of normally developed larvae with that of uninjected controls. As a result, the frequencies of larvae with normal morphology were almost identical between uninjected control larvae and larvae injected with sgRNA and Cas9 mRNA (Fig.3D), suggesting that the toxicity of sgRNA and Cas9 is not significant at the highest quantity of introduced sgRNA and Cas9 mRNA in this study.
Mutagenesis with expression vectors of sgRNA and Cas9
Although RNA-based knockout is a powerful method, this method has some disadvantages over using expression vectors of sgRNA and Cas9. First, RNA-mediated methods require in vitro transcription after preparation of DNA constructs. This step can be skipped in the expression vector-mediated method. Second, microinjection is necessary to introduce sgRNA and Cas9 mRNA into eggs and/or embryos in RNA-based method. In Ciona, plasmid DNAs can be introduced into hundreds of 1-cell embryos simultaneously by electroporation (Corbo et al. 1997). Electroporation is an easy and fast method compared to microinjection. Taking these advantages into consideration, we decided to construct expression vectors of sgRNA and Cas9. For Cas9, conventional expression vectors for reporter genes can be applied because Cas9 is a protein-coding gene. In this study we used a promoter of the ubiquitously expressed gene Ci-EF1α (Fig.4A; Sasakura et al. 2010; Treen et al. 2014). By contrast, sgRNA requires a special promoter because sgRNA functions as a short RNA. For expressing sgRNA, we used U6 promoter of Ciona intestinalis that was reported in a previous study for expressing short palindromic RNAs (Fig.4A; Nishiyama & Fujiwara 2008). We electroporated an sgRNA-expression vector and EF1α-driven Cas9 vector into 1-cell embryos. After the embryos were developed to the larval stage, genomic DNA was extracted in bulk and the occurrence of mutations were examined by Cel-I assay and sequencing analyses of the PCR products (Fig.4B). We found that both Ci-Hox3 and Ci-Hox5 were mutated by, respectively, introducing Hox3-sg3 and Hox5-sg1 expression vector with Cas9 vector (Fig.4C,D), although we were unable to obtain a Cel-I sensitive band for Hox3-sg3. The mutation frequencies were 36.3% and 46.2% for Hox3 and Hox5, respectively (Table3).
Figure 4.
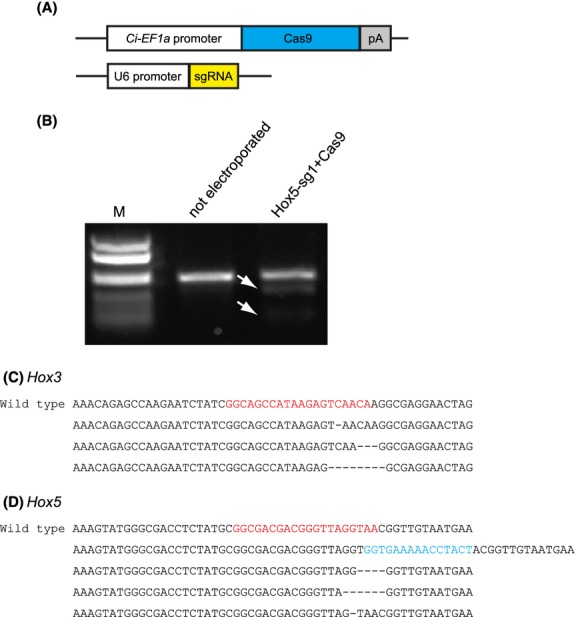
Mutations induced by electroporation of expression vectors of Cas9 and sgRNA. (A) Schematic illustrations of expression vectors for Cas9 and sgRNA. pA, poly adenylation sequence. (B) Cel-I assay of polymerase chain reaction (PCR) amplifications that contained the target site of Hox5-sg1. “Not electroporated” lane indicates the PCR band from untreated control larvae. “Hox5-sg1+Cas9” lane indicates the PCR band from larvae electroporated with pSPCiEF1α>Cas9 and pSPU6>Hox5-sg1 vectors. Cel-I cleaved bands were shown by arrows. M, marker lane. (C) Examples of mutations induced by expression vectors of Cas9 and Hox3-sg3. “Wild type” indicates the un-mutated sequence. The recognition site of Hox3-sg3 was shown in red. Deleted nucleotides were shown by “–”. (D) Examples of mutations induced by expression vectors of Cas9 and Hox5-sg1. Inserted nucleotides were shown in blue.
Table 3.
Mutation frequency by electroporating expression vectors of sgRNA and Cas9†
| Target gene | Name of sgRNA | Sequence of the recognition site in DNA‡ | Mutation frequency (in %) | n§ |
|---|---|---|---|---|
| Ci-Hox3 | wt Hox3-sg3¶ | GGCAGCCATAAGAGTCAACA | 36.3 | 22 |
| Ci-Hox5 | wt Hox5-sg1 | GGCGACGACGGGTTAGGTAA | 46.2 | 26 |
| Hox5-sg1/-G | GCGACGACGGGTTAGGTAA | 56.3 | 16 | |
| Hox5-sg1/-GG | CGACGACGGGTTAGGTAA | 0 | 15 | |
| Hox5-sg1/+A | AGGCGACGACGGGTTAGGTAA | 10.7 | 28 | |
| Hox5-sg1/+TA | TAGGCGACGACGGGTTAGGTAA | 3.1 | 32 | |
| Hox5-sg1/+ATC | ATCGGCGACGACGGGTTAGGTAA | 0 | 29 | |
| Hox5-sg1/+ATCA | ATCAGGCGACGACGGGTTAGGTAA | 0 | 14 |
†Twenty micrograms of sgRNA and 40 μg of Cas9 expression vectors were simultaneously electroporated. ‡Nucleotides added at the junction between U6 promoter were shown in red. §Number of sequenced polymerase chain reaction (PCR) clones. ¶wild type (wt) indicating the standard 20-nt recognition sites of the sgRNAs.
While we have much experience for expressing protein-coding genes from cis regulatory elements, we have limited experience expressing small RNAs from a U6 promoter. For this reason, we modified the junction sequence between U6 promoter and the DNA encoding sgRNA to see which variation could be acceptable. First, we deleted GG dinucletides at the 5′ end of the sgRNA coding region, (this was previously required for constructing the T7 promoter in pDR274 vector, see the materials and methods section; Hwang et al. 2013). When one G was deleted, the modification did not decrease the mutation frequency (Table3). However, when GG dinucleotides were deleted a significant reduction of mutation frequency was observed (Table3). Next, we added a few nucleotides at the junction between U6 promoter and sgRNA coding region. The addition of even one nucleotide strongly impaired mutation frequency. Our conclusion is that (i) one G can be omitted from the 5′ end of sgRNA in the expression vector system, and (ii) additional nucleotides should not be added at the junction region, when constructing sgRNA expression vectors.
Target-specificity of CRISPR/Cas9 in Ciona
Several reports in other organisms have suggested that CRISPR/Cas9 shows lesser target specificity than other genome editing methods such as TALENs (Fu et al. 2013; Shen et al. 2014). We addressed specificity of target recognition by CRISPR/Cas9 in Ciona to see how effective CRISPR/Cas9 is in this organism. We used two strategies to approach this issue. First, we searched for the similar sequences to Hox3-sg3 and Hox5-sg1 on-target sites in the Ciona genome. These sites could be potential off-target sites. We found three sites that contained four-base mismatches to the target sequence of Hox3-sg3. We examined whether mutations could be introduced in these potential off-target sites by introduction of Hox3-sg3 and Cas9 mRNAs by means of microinjection. As a result, we did not detect any mutation in two putative off-target sites (Table4), suggesting that a 4-bp mismatch is different from the on-target site enough for avoiding off-target mutations. We could not examine Hox5-sg1 because we were not able to find an off-target site of Hox5-sg1, which contains 4 bp or less mismatches.
Table 4.
Mutation frequency at the putative off-target sites of Hox3-sg3†
| Name of sites | Sequence of the target site in DNA‡ | Mutation frequency (in %) | n§ |
|---|---|---|---|
| On-target | GGCAGCCATAAGAGTCAACA | 58.0¶ | 31 |
| Off-target 1 | GGCAcCCATAtcAGTCAACt | 0 | 16 |
| Off-target 2 | GaCAaCCATAAGAGaCcACA | 0 | 16 |
| Off-target 3 | tcCAGCCATAAGAGTCtAtA | Not examined |
†3.0 pg of sgRNA and 15 pg of Cas9 mRNA were microinjected. ‡Mismatched nucleotides are shown in lower case. §Number of sequenced clones. ¶The score is the same as that shown in Table2.
In another approach, we created a series of Hox5-sg1 that contained single base mismatches at the recognition site, and examined their mutation capacity to the on-target site by PCR and sequencing analyses. We introduced these mismatched Hox5-sg1 series into Ciona embryos by microinjection of RNAs with Cas9 mRNA. As a result, these mismatched sgRNAs showed great reduction of the mutation frequency (Table5). Two out of six mismatched sgRNAs introduced mutations at the on-target sites; however, the frequency was more than ten times lower than that of wild type Hox5-sg1. We performed a similar experiment with Hox3-sg3 to see whether this tendency is also true of other sgRNAs. In the case of Hox3-sg3, we used expression vector-based knockout. As shown in Table5, we detected introduction of mutations with only one of the five mismatched Hox3-sg3, and the observed mutation frequency was about 40% of that of wild type Hox3-sg3. These results suggest that the recognition of the target site by sgRNAs exhibit high specificity in Ciona that could be significantly affected by even a single nucleotide difference, particularly when microinjections of sgRNA and Cas9 are performed.
Table 5.
Mutation frequency by sgRNAs that contain single base mismatches at the recognition site
| Target gene | Name of sgRNA | Method of introduction | Sequence of the recognition site in RNA† | Mutation frequency (in %) | n‡ |
|---|---|---|---|---|---|
| Ci-Hox5 | wt Hox5-sg1§ | Microinjection of 3.0 pg of sgRNA and 15 pg of Cas9 mRNA | GGCGACGACGGGUUAGGUAA | 75.9¶ | 31 |
| Mismatch1 | GGCGcCGACGGGUUAGGUAA | 3.3 | 30 | ||
| Mismatch2 | GGCGACGcCGGGUUAGGUAA | 0 | 31 | ||
| Mismatch3 | GGCGACGACGuGUUAGGUAA | 0 | 28 | ||
| Mismatch4 | GGCGACGACGGGUgAGGUAA | 6.7 | 29 | ||
| Mismatch5 | GGCGACGACGGGUUAGuUAA | 0 | 30 | ||
| Mismatch6 | GGCGACGACGGGUUAGGUAc | 0 | 31 | ||
| Ci-Hox3 | wt Hox3-sg3§ | Electroporation of 20 μg of sgRNA and 40 μg of Cas9 expression vectors | GGCAGCCAUAAGAGUCAACA | 36.3¶ | 22 |
| Mismatch1 | GGCAGCCcUAAGAGUCAACA | 0 | 23 | ||
| Mismatch2 | GGCAGCCAUAcGAGUCAACA | 13.6 | 22 | ||
| Mismatch3 | GGCAGCCAUAAGAuUCAACA | 0 | 24 | ||
| Mismatch4 | GGCAGCCAUAAGAGUCcACA | 0 | 24 | ||
| Mismatch5 | GGCAGCCAUAAGAGUCAACc | 0 | 24 |
Discussion
In this study, we demonstrated that CRISPR/Cas9 system could introduce mutations into endogenous genes in the Ciona genome. This suggests that CRISPR/Cas9 can be another powerful tool for investigating gene functions in this organism together with TALENs (Treen et al. 2014; Yoshida et al. 2014). In the following paragraphs we discuss the strengths and weaknesses of CRISPR/Cas9 in Ciona.
In this study we succeeded in the disruption of Hox3 and Hox5 with CRISPR/Cas9. For Hox3, we tested two pairs of TALENs but they failed to introduce mutations. This suggests that CRISPR/Cas9 can mutate genes that TALENs cannot, as suggested in the previous study (Hwang et al. 2013). The opposite situation also occurs, CRISPR/Cas9 failed to introduce mutations at the Hox12 locus, which we successfully mutated with TALENs (Treen et al. 2014). This difference between the two methods may be derived from their different mechanisms of mutagenesis. In CRISPR/Cas9, guide RNAs determine target sites through formation of heteroduplex of targeted DNA and sgRNAs, and then Cas9 cleaves the sites by a RNA-mediated mechanism (Wiedenheft et al. 2012). By contrast, TALENs recognize their target sites by forming protein-DNA complexes, and they cleave the target sites by dimerization of the nuclease domains from a restriction enzyme FokI (Christian et al. 2010). With this difference, the genomic loci that are resistant to one method could be sensitive to the other. Using CRISPR/Cas9 and TALENs to compensate for the weakness of each other, we will be able to knockout more genes than using a single method.
While CRISPR/Cas9 can be used as a substitute for TALENs in Ciona, our study has shown that designing sgRNAs that have high mutational activity is quite low compared to the cases reported in Xenopus and zebrafish (25% in this study versus >80% in Xenopus and zebrafish; Hwang et al. 2013; Guo et al. 2014). Therefore, we need extensive screening of sgRNA to destroy one genetic locus in Ciona. In the case of TALENs, our rough estimation of constructing a good TALEN pair exceeds 85%. Because of this disadvantage in CRISPR/Cas9, we currently use TALENs as the main tool for the knockout of Ciona genes. If several TALEN pairs fail to introduce mutations with good efficiency, then we consider CRISPR/Cas9. In future studies, the sequence information of good sgRNAs in Ciona will accumulate. By comparing their sequences we could deduce the rules to consistently design good sgRNAs for Ciona.
The recognition of the target sites by sgRNAs show high specificity in Ciona that could usually distinguish even a single nucleotide difference. This is in contrast to the reports in some other organisms like mammalian cell cultures and Xenopus (Fu et al. 2013; Guo et al. 2014). In Xenopus, mismatches at the core region of sgRNA named “seed” sequence significantly influenced the mutation frequency, but the other regions are basically resistant to single nucleotide mismatches. One possible explanation for the high specificity of CRISPR/Cas9 in Ciona is that the complex of sgRNA and Cas9 protein formed on the genome DNA may be less stable than those formed in other organisms, and is therefore less tolerant of individual mismatches. The high specificity of the target recognition is advantageous for analyzing gene functions, because this feature may reduce the occurrence of off-target effects, as we have shown that high quantities of sgRNA and Cas9 mRNA introduction do not have strong side effects on embryogenesis. The low off-target effect was also reported in zebrafish (Hruscha et al. 2013), suggesting that off-target effects of CRISPR/Cas9 systems are highly dependent on the conditions that differ among organisms.
Like other organisms (Feng et al. 2013; Kondo & Ueda 2013), sgRNAs as well as Cas9 mRNA can be supplied from expression vectors in Ciona. There are two applications of this method. One is examining the effectiveness of newly designed sgRNAs. As we mentioned above, the probability of designing an sgRNA with good mutational activity is not high in Ciona. Therefore, extensive screening of sgRNAs is necessary to mutate a gene. An electroporation-mediated method is an easier and faster way compared to microinjection of mRNA, and therefore this method is more suitable for screening of sgRNAs. The other application is thorough analysis of gene functions in the G0 generation, as we demonstrated with TALENs (Treen et al. 2014). In TALENs, we established the method of tissue-specific knockout of genes by expressing TALENs in tissue-specific cell lineages. We will be able to apply CRISPR/Cas9 to perform conditional gene knockouts by inducing expression of Cas9 with tissue-specific promoters.
In the present study, we have shown that the Ciona genome can be mutated by the CRISPR/Cas9 system. Although we were unable to address whether the mutations introduced by the system can be reflected by phenotypes, we think this is highly probable, based on the mutation frequency done by TALENs (Treen et al. 2014). Therefore, CRISPR/Cas9-mediated mutagenesis will be another powerful approach in addition to TALENs to investigate functions of genes in Ciona intestinalis, the excellent model for elucidating molecular mechanisms underlying formation of the chordate body plan and its evolution.
Acknowledgments
We would like to thank Drs Nobuo Yamaguchi, Kunifumi Tagawa and the National Bioresource Project for collecting the C. intestinalis adults. We are grateful to Drs Shigeki Fujiwara and Hiroki Nishida for their kind provision of wild type C. intestinalis, pSPU6EV, and pBS-HTB. We wish to thank Nicholas Treen for English editing of the manuscript and scientific discussions. This study was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) and Ministry of Education, Culture, Sports, Science and Technology (MEXT) to YS. YS was supported by the Toray Science and Technology Grant. Further support was provided by grants from the National Bioresource Project.
Author contributions
Y.S. designed research. H.S., K.Y., A.H. and Y.S. performed research. H.S., K.Y. and Y.S. analyzed data. Y.S. wrote the paper.
Supporting Information
Table S1. Primer list for construction and genomic analyses.
References
- Akanuma T, Hori S, Darras S. Nishida H. Notch signaling is involved in neurogenesis in the ascidian embryos. Dev. Genes. Evol. 2002;212:459–472. doi: 10.1007/s00427-002-0264-x. &. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW. Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. &. [DOI] [PubMed] [Google Scholar]
- Ansai S, Sakuma T, Yamamoto T, Ariga H, Uemura N, Takahashi R. Kinoshita M. Efficient targeted mutagenesis in medaka using custom-designed transcription activator-like effector nucleases. Genetics. 2013;193:739–749. doi: 10.1534/genetics.112.147645. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansai S, Inohaya K, Yoshiura Y, Schartl M, Uemura N, Takahashi R. Kinoshita M. Design, evaluation, and screening methods for efficient targeted mutagenesis with transcription activator-like effector nucleases in medaka. Dev. Growth Differ. 2014;56:98–107. doi: 10.1111/dgd.12104. &. [DOI] [PubMed] [Google Scholar]
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ. Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ. Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo JC, Levine M. Zeller RW. Characterization of a notochord-specific enhancer from the Brachyury promoter region of the ascidian, Ciona intestinalis. Development. 1997;124:589–602. doi: 10.1242/dev.124.3.589. &. [DOI] [PubMed] [Google Scholar]
- Daimon T, Kiuchi T. Takasu Y. Recent progress in genome engineering techniques in the silkworm, Bombyx mori. Dev. Growth Differ. 2014;56:14–25. doi: 10.1111/dgd.12096. &. [DOI] [PubMed] [Google Scholar]
- Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, Harafuji N, Hastings KE, Ho I, Hotta K, Huang W, Kawashima T, Lemaire P, Martinez D, Meinertzhagen IA, Necula S, Nonaka M, Putnam N, Rash S, Saiga H, Satake M, Terry A, Yamada L, Wang HG, Awazu S, Azumi K, Boore J, Branno M, Chin-Bow S, DeSantis R, Doyle S, Francino P, Keys DN, Haga S, Hayashi H, Hino K, Imai KS, Inaba K, Kano S, Kobayashi K, Kobayashi M, Lee BI, Makabe KW, Manohar C, Matassi G, Medina M, Mochizuki Y, Mount S, Morishita T, Miura S, Nakayama A, Nishizaka S, Nomoto H, Ohta F, Oishi K, Rigoutsos I, Sano M, Sasaki A, Sasakura Y, Shoguchi E, Shin-I T, Spagnuolo A, Stainier D, Suzuki MM, Tassy O, Takatori N, Tokuoka M, Yagi K, Yoshizaki F, Wada S, Zhang C, Hyatt PD, Larimer F, Detter C, Doggett N, Glavina T, Hawkins T, Richardson P, Lucas S, Kohara Y, Levine M, Satoh N. Rokhsar DS. The draft genome of Ciona intestinalis: insight into chordate and vertebrate origins. Science. 2002;298:2157–2167. doi: 10.1126/science.1080049. &. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang B, Ding W, Liu X, Yang DL, Wei P, Cao F, Zhu S, Zhang F, Mao Y. Zhu JK. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 2013;23:1229–1232. doi: 10.1038/cr.2013.114. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM. Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods. 2013;10:741–743. doi: 10.1038/nmeth.2532. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK. Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gionti M, Ristoratore F, Di Gregorio A, Aniello F, Branno M. Di Lauro R. Cihox5, a new Ciona intestinalis Hox-related gene, is involved in regionalization of the spinal cord. Dev. Genes. Evol. 1998;207:515–523. doi: 10.1007/s004270050142. &. [DOI] [PubMed] [Google Scholar]
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J. O’Connor-Giles KM. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–1035. doi: 10.1534/genetics.113.152710. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang T, Hu Z, Zhang Y, Shi Z, Wang Q, Cui Y, Wang F, Zhao H. Chen Y. Efficient RNA/Cas9-mediated genome editing in Xenopus tropicalis. Development. 2014;141:707–714. doi: 10.1242/dev.099853. &. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Sakamoto K, Sakuma T, Yokotani N, Inoue T, Kawaguchi E, Agata K, Yamamoto T. Takeuchi T. Transcription activator-like effector nucleases efficiently disrupt the target gene in Iberian ribbed newts (Pleurodeles waltl), an experimental model animal for regeneration. Dev. Growth Differ. 2014;56:115–121. doi: 10.1111/dgd.12103. &. [DOI] [PubMed] [Google Scholar]
- Hikosaka A, Kusakabe T, Satoh N. Makabe KW. Introduction and expression of recombinant genes in ascidian embryos. Dev. Growth Differ. 1992;34:627–634. doi: 10.1111/j.1440-169X.1992.tb00031.x. &. [DOI] [PubMed] [Google Scholar]
- Hisano Y, Ota S. Kawahara A. Genome editing using artificial site-specific nucleases in zebrafish. Dev. Growth Differ. 2014;56:26–33. doi: 10.1111/dgd.12094. &. [DOI] [PubMed] [Google Scholar]
- Hosoi S, Sakuma T, Sakamoto N. Yamamoto T. Targeted mutagenesis in sea urchin embryos using TALENs. Dev. Growth Differ. 2014;56:92–97. doi: 10.1111/dgd.12099. &. [DOI] [PubMed] [Google Scholar]
- Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C. Schmid B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development. 2013;140:4982–4987. doi: 10.1242/dev.099085. &. [DOI] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR. Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta T, Satoh N. Saiga H. Limited functions of Hox genes in the larval development of the ascidian. Ciona intestinalis. Development. 2010;137:1505–1513. doi: 10.1242/dev.046938. &. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Yoshida N, Satoh N. Saiga H. Ciona intestinalis Hox gene cluster: Its dispersed structure and residual colinear expression in development. Proc. Natl. Acad. Sci. USA. 2004;101:15118–15123. doi: 10.1073/pnas.0401389101. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Takada N, Satoh N. Satou Y. β-catenin mediates the specification of endoderm cells in ascidian embryos. Development. 2000;127:3009–3020. doi: 10.1242/dev.127.14.3009. &. [DOI] [PubMed] [Google Scholar]
- Kawai N, Ochiai H, Sakuma T, Yamada L, Sawada H, Yamamoto T. Sasakura Y. Efficient targeted mutagenesis of the chordate Ciona intestinalis genome with zinc-finger nucleases. Dev. Growth Differ. 2012;54:535–545. doi: 10.1111/j.1440-169X.2012.01355.x. &. [DOI] [PubMed] [Google Scholar]
- Kondo S. Ueda R. Highly improved gene targeting by germline-specific Cas9 expression in Drosophila. Genetics. 2013;195:715–721. doi: 10.1534/genetics.113.156737. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Sakuma T, Wada H, Akimoto-Kato A, Yamamoto T. Hayashi S. TALEN-induced gene knock out in Drosophila. Dev. Growth Differ. 2014;56:86–91. doi: 10.1111/dgd.12097. &. [DOI] [PubMed] [Google Scholar]
- Locascio A, Aniello F, Amoroso A, Manzanares M, Krumlauf R. Branno M. Patterning the ascidian nervous system: structure, expression and transgenic analysis of the CiHox3 gene. Development. 1999;126:4737–4738. doi: 10.1242/dev.126.21.4737. &. [DOI] [PubMed] [Google Scholar]
- Mali P, Esvelt KM. Church GM. Cas9 as a versatile tool for engineering biology. Nat. Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiko D, Young SA, Muto M, Kato H, Nozawa K, Ogawa M, Noda T, Kim YJ, Satouh Y, Fujihara Y. Ikawa M. Feasibility for a large scale mouse mutagenesis by injecting CRISPR/Cas plasmid into zygotes. Dev. Growth Differ. 2014;56:122–129. doi: 10.1111/dgd.12113. &. [DOI] [PubMed] [Google Scholar]
- Mashimo T. Gene targeting technologies in rats: zinc finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats. Dev. Growth Differ. 2014;56:46–52. doi: 10.1111/dgd.12110. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD. Rebar EJ. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. &. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Fish MB, Fisher M, Oomen-Hajagos J, Thomsen GH. Grainger RM. Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. Genesis. 2013;51:835–843. doi: 10.1002/dvg.22720. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A. Fujiwara S. RNA interference by expressing short hairpin RNA in the Ciona intestinalis embryo. Dev. Growth Differ. 2008;50:521–529. doi: 10.1111/j.1440-169X.2008.01039.x. &. [DOI] [PubMed] [Google Scholar]
- Ochiai H, Fujita K, Suzuki K, Nishikawa M, Shibata T, Sakamoto N. Yamamoto T. Targeted mutagenesis in the sea urchin embryo using zinc-finger nucleases. Genes Cells. 2010;15:875–885. doi: 10.1111/j.1365-2443.2010.01425.x. &. [DOI] [PubMed] [Google Scholar]
- Ochiai H, Sakamoto N, Fujita K, Nishikawa M, Suzuki K, Matsuura S, Miyamoto T, Sakuma T, Shibata T. Yamamoto T. Zinc-finger nuclease-mediated targeted insertion of reporter genes for quantitative imaging of gene expression in sea urchin embryos. Proc. Natl. Acad. Sci. USA. 2012;109:10915–10920. doi: 10.1073/pnas.1202768109. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane Y, Sakuma T, Kashiwagi K, Kashiwagi A, Yamamoto T. Suzuki KT. Targeted mutagenesis of multiple and paralogous genes in Xenopus laevis using two pairs of transcription activator-like effector nucleases. Dev. Growth Differ. 2014;56:108–114. doi: 10.1111/dgd.12105. &. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Hosoi S, Woltjen K, Suzuki K, Kashiwagi K, Wada H, Ochiai H, Miyamoto T, Kawai N, Sasakura Y, Matsuura S, Okada Y, Kawahara A, Hayashi S. Yamamoto T. Efficient TALEN construction and evaluation methods for human cell and animal applications. Genes Cells. 2013a;18:315–326. doi: 10.1111/gtc.12037. &. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Ochiai H, Kaneko T, Mashimo T, Tokumasu D, Sakane Y, Suzuki K, Miyamoto T, Sakamoto N, Matsuura S. Yamamoto T. Repeating pattern of non-RVD variations in DNA-binding modules enhances TALEN activity. Sci. Rep. 2013b;3:3379. doi: 10.1038/srep03379. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma T. Woltjen K. Nuclease-mediated genome editing: at the front-line of functional genomics technology. Dev. Growth Differ. 2014;56:2–13. doi: 10.1111/dgd.12111. &. [DOI] [PubMed] [Google Scholar]
- Sander JD, Zaback PZ, Joung JK, Voytas DF. Dobbs D. Zinc Finger Targeter (ZiFiT): an engineered zinc finger/target site design tool. Nucleic Acids Res. 2007;35:W599–W605. doi: 10.1093/nar/gkm349. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK. Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res. 2010;38:W462–W468. doi: 10.1093/nar/gkq319. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago Y, Chan E, Liu PQ, Orlando S, Zhang L, Urnov FD, Holmes MC, Guschin D, Waite A, Miller JC, Rebar EJ, Gregory PD, Klug A. Collingwood T. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc. Natl. Acad. Sci. USA. 2008;105:5809–5814. doi: 10.1073/pnas.0800940105. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasakura Y, Awazu S, Chiba S. Satoh N. Germ-line transgenesis of the Tc1/mariner superfamily transposon Minos in Ciona intestinalis. Proc. Natl. Acad. Sci. USA. 2003;100:7726–7730. doi: 10.1073/pnas.1230736100. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasakura Y, Nakashima K, Awazu S, Matsuoka T, Nakayama A, Azuma J. Satoh N. Transposon-mediated insertional mutagenesis revealed the functions of animal cellulose synthase in the ascidian Ciona intestinalis. Proc. Natl. Acad. Sci. USA. 2005;102:15134–15139. doi: 10.1073/pnas.0503640102. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasakura Y, Suzuki MM, Hozumi A, Inaba K. Satoh N. Maternal factor-mediated epigenetic gene silencing in the ascidian Ciona intestinalis. Mol. Genet. Genomics. 2010;283:99–110. doi: 10.1007/s00438-009-0500-4. &. [DOI] [PubMed] [Google Scholar]
- Sasakura Y, Kanda M, Ikeda T, Horie T, Kawai N, Ogura Y, Yoshida R, Hozumi A, Satoh N. Fujiwara S. Retinoic acid-driven Hox1 is required in the epidermis for forming the otic/atrial placodes during ascidian metamorphosis. Development. 2012;139:2156–2160. doi: 10.1242/dev.080234. &. [DOI] [PubMed] [Google Scholar]
- Satoh N. The ascidian tadpole larva: comparative molecular development and genomics. Nat. Rev. Genet. 2003;4:285–295. doi: 10.1038/nrg1042. [DOI] [PubMed] [Google Scholar]
- Satou Y, Imai KS. Satoh N. Action of morpholinos in Ciona embryos. Genesis. 2001;30:103–106. doi: 10.1002/gene.1040. &. [DOI] [PubMed] [Google Scholar]
- Satou Y, Mineta K, Ogasawara M, Sasakura Y, Shoguchi E, Ueno K, Yamada L, Matsumoto J, Wasserscheid J, Dewar K, Wiley GB, Macmil SL, Roe BA, Zeller RW, Hastings KE, Lemaire P, Lindquist E, Endo T, Hotta K. Inaba K. Improved genome assembly and evidence-based global gene model set for the chordate Ciona intestinalis: new insight into intron and operon populations. Genome Biol. 2008;9:R152. doi: 10.1186/gb-2008-9-10-r152. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X. Skarnes WC. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. &. [DOI] [PubMed] [Google Scholar]
- Sugi T, Sakuma T, Ohtani Y. Yamamoto T. Versatile strategy for isolating transcription activator-like effector nuclease-mediated knockout mutants in Caenorhabditis elegans. Dev. Growth Differ. 2014;56:78–85. doi: 10.1111/dgd.12108. &. [DOI] [PubMed] [Google Scholar]
- Suzuki KT, Isoyama Y, Kashiwagi K, Sakuma T, Ochiai H, Sakamoto N, Furuno N, Kashiwagi A. Yamamoto T. High efficiency TALENs enable F0 functional analysis by targeted gene disruption in Xenopus laevis embryos. Biol. Open. 2013;2:448–452. doi: 10.1242/bio.20133855. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treen N, Yoshida K, Sakuma T, Sasaki H, Kawai N, Yamamoto T. Sasakura Y. Tissue-specific and ubiquitous gene knockouts by TALEN electroporation provide new approaches to investigating gene function in Ciona. Development. 2014;141:481–487. doi: 10.1242/dev.099572. &. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Ochiai H, Sakuma T, Horch HW, Hamaguchi N, Nakamura T, Bando T, Ohuchi H, Yamamoto T, Noji S. Mito T. Non-transgenic genome modifications in a hemimetabolous insect using zinc-finger and TAL effector nucleases. Nat. Commun. 2012;3:1017. doi: 10.1038/ncomms2020. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg SH. Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. &. [DOI] [PubMed] [Google Scholar]
- Xie K. Yang Y. RNA-guided genome editing in plants using CRISPR-Cas system. Mol. Plant. 2013;6:1975–1983. doi: 10.1093/mp/sst119. &. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Treen N, Hozumi A, Sakuma T, Yamamoto T. Sasakura Y. Germ cell mutations of the ascidian Ciona intestinalis with TALE nucleases. Genesis. 2014;52:431–439. doi: 10.1002/dvg.22770. &. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer list for construction and genomic analyses.