

Summary
Background
22q11·2 deletion syndrome (22q11·2DS) is a relatively common yet under-recognized genetic syndrome that may present with endocrine features. We aimed to address the factors that contribute to the high prevalence of hypocalcaemia.
Methods
We investigated hypocalcaemia in a well-characterized sample of 138 adults with 22q11·2DS (65 m, 73 F; mean age 34·2, SD 11·8, years) using laboratory studies and lifelong medical records. Logistic regression modelling was used to identify features associated with lifetime prevalence of hypocalcaemia.
Results
Of the total sample, 111 (80·4%) had a lifetime history of hypocalcaemia. Eleven (84·6%) of 13 subjects with neonatal hypocalcaemia had documented recurrence of hypocalcaemia. Lifetime history of hypocalcaemia was associated with lifetime prevalence of hypoparathyroidism (P < 0·0001) and hypothyroidism (P = 0·04), as statistically independent factors. Hypomagnesaemia was associated with concurrent hypocalcaemic measurements, especially in the presence of concurrent hypoparathyroidism (P = 0·02).
Conclusions
The results suggest that, in addition to the major effect of hypoparathyroidism, hypothyroidism may play a role in hypocalcaemia in 22q11·2DS and that there is a high recurrence rate of neonatal hypocalcaemia. Hypomagnesaemia may contribute to hypocalcaemia by further suppressing parathyroid hormone (PTH). Although further studies are needed, the findings support regular lifelong follow-up of calcium, magnesium, PTH and TSH levels in patients with 22q11·2DS. At any age, hypocalcaemia with hypoparathyroidism and/or hypothyroidism may suggest a diagnosis of 22q11·2DS.
Introduction
Hypocalcaemia is considered a classic feature of 22q11·2 deletion syndrome (22q11·2DS), a common yet still under-recognized genetic syndrome associated with hemizygous microdeletions of the long arm of chromosome 22.1 The deletion is present in over 1 in 4000 live births and usually occurs as a de novo mutation; only about 10% of individuals are found to have inherited the deletion from an affected parent.2–4 The clinical presentation of 22q11·2DS is highly variable with manifestations involving multiple organ systems and of wide-ranging severity, which differ from patient to patient.4 Thus far, there is no confirmed association between any two endocrine features. While some features are recognizable in infancy, many others, including hypocalcaemia, can occur later. The severe set of features associated with DiGeorge syndrome, one of the previous names for 22q11·2DS, is now known to be rare in the syndrome.5
Previous studies have reported that the prevalence of hypocalcaemia in 22q11·2DS ranges from 32·8% to 64·1%, but most have involved paediatric cohorts and/or relatively small samples of older individuals.1,6–9 Hypoparathyroidism is considered the primary cause of hypocalcaemia in the syndrome.10,11 The presentation of hypocalcaemia and hypoparathyroidism ranges from ‘transient’ neonatal hypocalcaemia to overt hypoparathyroidism.8,12 Patients can experience a wide range of symptoms, such as fatigue, ‘brain fog’ and paraesthesia, with more severe manifestations including a lowered seizure threshold and prolongation of the QT interval.13,14 The high prevalence of hypocalcaemia and limited knowledge about the long-term outcome of this treatable condition in 22q11·2DS warrant the systematic study of possible contributing factors. We therefore investigated the lifetime prevalence of hypocalcaemia and its associated clinical features in a large cohort of adults with 22q11·2DS.
Methods
Subjects
The sample comprised 138 adults with 22q11·2DS (65 males, 73 females; mean age at assessment 34·2, SD 11·8, years) followed at our clinic for adults with 22q11·2DS for an average of 7·72 years, where data on key endocrine variables were available. Written informed consent was provided for all subjects, and the study was approved by local research ethics boards. As previously described,15–17 subjects were ascertained because of congenital heart disease (CHD), psychiatric disorder or being first-degree relatives of other subjects; none were referred by an endocrinologist. The 22q11·2 microdeletion was confirmed by standard fluorescence in situ hybridization (FISH) or genome-wide microarray techniques using standard probes and methods,18,19 at a median age of 24·2 (range 3·8–66·5) years.
To assess the significance of the lowered ionized calcium levels in our 22q11·2DS sample, we used a comparison group of 100 adults with schizophrenia (69 males, 31 females; mean age 49·4, SD 13·8, years) where genome-wide microarray data had confirmed they did not have a 22q11·2 microdeletion15 and ionized calcium measurements were available.
Clinical data
We systematically reviewed paediatric and adult medical records and recorded lifetime data available on endocrine and other relevant medical conditions1 and laboratory (serum) measurements, including ionized calcium, intact parathyroid hormone (PTH), thyroid-stimulating hormone (TSH), free thyroxine (FT4), magnesium and creatinine. Vitamin D and fasting phosphate levels were unavailable for sufficient numbers of subjects to be included in analyses. Renal failure was considered to have been present if documented in medical records or by an elevated creatinine level. We classified major CHD, such as tetralogy of Fallot, as previously described15–17 and confirmed DSM-IV lifetime diagnoses of schizophrenia and schizoaffective disorder, collectively termed schizophrenia, as before.20 Neonatal events were defined as those occurring within 2 weeks of birth.21 We recorded age at first documentation of endocrine conditions and laboratory values and treatments where available. Compliance with recommended treatment was uncertain however.
Subjects were considered to have a specific endocrinological condition, including hypocalcaemia, hypoparathyroidism and hypothyroidism, if it was documented in medical records and/or the laboratory measurements were outside of the reference ranges for the individual laboratory performing the respective test. Only for ease of graphic representation of intact PTH and ionized calcium levels did we use the single most common ranges: 1·3–7·6 pm and 1·2–1·35 mm, respectively (Figs1 and 2).
Figure 1.
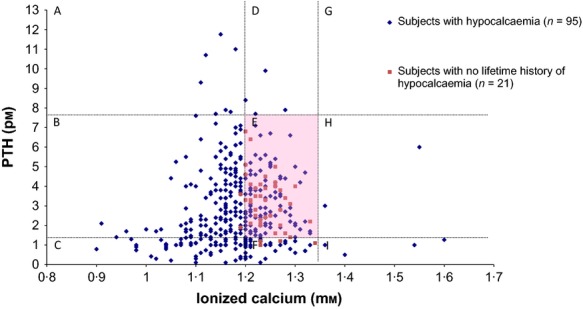
Scatterplot of 397 simultaneously obtained intact PTH and ionized calcium levels available for 116 subjects with 22q11·2DS. Most measurements (n = 364, 91·7%) were collected in adulthood. The dotted lines represent approximately normal ranges of intact PTH and ionized calcium levels. The pink shaded area E represents 145 values in the normal range for both ionized calcium and PTH from 66 subjects, of whom 32 (48·5%) subjects had previously documented episodes of hypocalcaemia and 46 (69·7%) subjects had a lifetime prevalence of hypocalcaemia. Area A (low calcium and high PTH levels) has 7 measurements from 7 subjects; area B (low calcium and normal PTH levels) 153 measurements from 77 subjects; area C (low calcium and low PTH levels) 47 measurements from 21 subjects; area D (normal calcium and high PTH levels) 4 measurements from 3 subjects; area F (normal calcium and low PTH levels) 35 measurements from 23 subjects (of whom 19 (82·6%) had previously documented episodes of hypocalcaemia). Areas H (high calcium and normal PTH levels) and I (high calcium and high PTH levels) together have 6 measurements from 6 subjects, all of whom had previously documented episodes of hypocalcaemia. Area B contains 99 measurements representing relative hypoparathyroidism.
Figure 2.

Scatterplot of 31 intact PTH and ionized calcium levels from six subjects with 22q11·2DS and elevated intact PTH (areas A and D from Fig.1 after excluding those with values just above the norm for PTH). The dotted lines represent approximately normal ranges of intact PTH and ionized calcium levels. Area labels A to I correspond to those in Figure1. At the time of elevated PTH level, subjects 1 and 2 had elevated creatinine (subject 2 also had worsening of Parkinson's disease); subject 3 had ruptured sinus of Valsalva; subjects 4 and 5 were apparently healthy, and subject 6 had a history of chronic renal insufficiency. Subject 4 was the only one of these six subjects for whom calcitriol was ever prescribed.
Data on the presence of a lifetime diagnosis of hypocalcaemia were available for all 138 subjects, based on history, serum total calcium or, most commonly, ionized calcium results. For ionized calcium, the pH-corrected level was used whenever available (261 of 397 values). Data on the presence of a lifetime diagnosis of hypoparathyroidism were available for 125 (90·6%) subjects. We considered hypoparathyroidism broadly to have been present if the intact PTH level was below the normal range8 or if the PTH level was in the lower half of the normal range, where the corresponding ionized calcium level was below normal, that is relative or partial hypoparathyroidism, hereafter also referred to as hypoparathyroidism.22
Data on the presence of a lifetime diagnosis of hypothyroidism were available for 134 (97·1%) subjects. Laboratory diagnosis was based on an elevated TSH (≥10 mIU/l) and/or decreased FT4 (≤11 pm) level.23 An elevated TSH level (5–10 mIU/l) was considered as subclinical hypothyroidism.23 The four (4·5%) subjects who had a history of thyroidectomy or radioactive iodine treatment for hyperthyroidism were not included in the hypothyroidism group.
Statistical analyses
We used a logistic regression model (n = 120 subjects with data on all variables) to investigate clinical factors that may predict hypocalcaemia. The dependent variable was documented hypocalcaemia. Variables considered in the model were the following: lifetime presence or absence of hypoparathyroidism, hypothyroidism and renal insufficiency,24,25 in addition to demographic variables (age and sex) and key clinical variables in our cohort (i.e. the diagnosis of schizophrenia or CHD). Hypothyroidism was included due to the importance of thyroid hormone on physiological functions26 and its high prevalence in 22q11·2DS.1 Post hoc χ2 tests were used to assess the individual regression coefficients. We used the Sobel27 test to evaluate whether hypoparathyroidism mediated the association observed between hypothyroidism and hypocalcaemia.
We calculated 95% confidence intervals (CI) for frequencies and used χ2 and Fisher's exact tests to compare categorical variables. We used nonparametric Wilcoxon z tests to compare continuous variables without normal distributions. All analyses were two-tailed and performed using SAS 9·2 (SAS Institute, Cary, NC); p values <0·05 were considered significant.
Results
The logistic regression model used to predict the lifetime diagnosis of hypocalcaemia in adults with 22q11·2DS was highly significant (likelihood ratio test: χ2 = 38·2, df = 7, P < 0·0001). Clinical and demographic variables used in this analysis are shown in Table1. Post hoc tests showed that hypoparathyroidism (χ2 = 16·5, df = 1, P < 0·0001) and hypothyroidism (χ2 = 4·4, df = 1, P = 0·04) were significantly associated with hypocalcaemia. None of the other clinical or demographic variables tested were significant (Table1). There was no significant interaction between any pair of variables (17 of 36 subjects with hypothyroidism had a lifetime history of hypoparathyroidism). A further test of the potential mediating effect of hypoparathyroidism on the relationship between hypothyroidism and hypocalcaemia was nonsignificant (P = 0·71).
Table 1.
Post hoc test results for the logistic regression analysis of the lifetime diagnosis of hypocalcaemia in 120 adults with 22q11·2 deletion syndrome
| Clinical features | N | % | OR | 95% Wald confidence limits | χ2 | df | P | |
|---|---|---|---|---|---|---|---|---|
| Hypoparathyroidism | 76 | 63·3 | 20·0 | 5·1 | 16·5 | 16·5 | 1 | <0·0001 |
| Hypothyroidism | 36 | 30·0 | 6·4 | 1·1 | 35·5 | 4·4 | 1 | 0·035 |
| Renal insufficiency | 14 | 11·7 | 0·4 | 0·04 | 3·6 | 0·7 | 1 | 0·42 |
| Congenital heart disease | 46 | 38·3 | 0·7 | 0·2 | 2·9 | 0·2 | 1 | 0·68 |
| Schizophrenia | 57 | 47·5 | 0·9 | 0·2 | 3·7 | 0·02 | 1 | 0·79 |
| Female sex | 64 | 53·3 | 0·9 | 0·3 | 3·0 | 0·03 | 1 | 0·85 |
These 120 subjects had medical records and/or available laboratory data available on calcium, PTH and TSH measurements; 99 (82·5%) subjects had a history of hypocalcaemia.
Serious congenital heart disease included tetralogy of Fallot and interrupted aortic arch type B.15
OR: odds ratio; df: degree of freedom.
Bolded values represent statistically significant values.
| Mean | SD | OR | 95% Wald confidence limits | χ2 | df | P | ||
|---|---|---|---|---|---|---|---|---|
| Age (years) | 34·8 | 11·3 | 0·9 | 0·9 | 1·0 | 3·1 | 1 | 0·079 |
Hypocalcaemia and hypoparathyroidism
Hypocalcaemia was documented in 111 (80·4%; 95% CI, 73·8–87·1%) of 138 subjects at one or more points in their lives, comprising data available from low ionized calcium or serum total calcium values (n = 39), hypocalcaemia noted in medical records (n = 8), or both (n = 64). Among the 111 subjects, 72 (64·8%) were first diagnosed after the age of 17 years, and of these, 17 had had prior normal calcium measurements documented in adulthood. None of these 72 subjects had calcium tested as a child. Neonatal hypocalcaemia was reported in 13 (9·4%) of 138 subjects.21 Recurrence of hypocalcaemia in adulthood (>17 years) was documented in 11 (84·6%) of these 13 subjects, with no adult calcium results available for the other two subjects.
There were 125 subjects with data on PTH and/or hypoparathyroidism. For 78 of these, there was a lifetime documentation of hypoparathyroidism (62·4%; 95% CI, 53·3–70·9%). Among these 78 subjects, 11 (14·1%) were diagnosed by the age of 10 years, of whom 8 (72·7%) had recurrence of hypoparathyroidism in adulthood; 10 (12·8%) were diagnosed between age 10 and 17 years, and 57 (73·1%) were diagnosed in adulthood.
Calcium and vitamin D supplements were recommended to all 138 subjects with 22q11·2DS,28 regardless of whether they had been diagnosed with hypocalcaemia. Sixteen (14·4%) of the 111 hypocalcaemic subjects were prescribed calcitriol. It is important to note that many 22q11·2DS subjects were asymptomatic in spite of hypocalcaemic measurements.
Hypocalcaemia and hypothyroidism
The lifetime prevalence of hypothyroidism was documented in 39 (29·1%; 95% CI, 21·4%–36·8%) of 134 subjects with data available. Among these 39 subjects, 9 (23·1%) had subclinical hypothyroidism. Thirty-five (89·7%) of the 39 had hypocalcaemia. Where data on timing were available (n = 32), hypocalcaemia was documented concurrently in 3 subjects, after the diagnosis of hypothyroidism in 15 subjects and before the diagnosis of hypothyroidism in 14 subjects.
Of the 17 subjects where first documentation of hypocalcaemia was either prior to or concurrent with the first documentation of hypothyroidism, 10 were taking medications for hypothyroidism. There was no significant association between thyroid supplementation and the recurrence of hypocalcaemia thereafter (P = 0·77, two-tailed Fisher's exact test).
Ionized calcium levels and PTH response
The mean ionized calcium level in the 22q11·2DS group (1·19 ± 0·08 mm; 364 measurements for 116 subjects collected during adulthood) was significantly lower than that in the adult comparison group without 22q11·2DS (1·27 ± 0·12 mm;z = 8·88, P < 0·0001; 110 measurements for 100 subjects). Age and gender had no significant effects on calcium levels (data not shown).
Figure1 shows the distribution of 397 simultaneously measured ionized calcium and intact PTH levels in 116 subjects with 22q11·2DS. Of these, 184 (46·3%) of the ionized calcium values and 300 (75·6%) of the intact PTH values were within the normal ranges. We were particularly interested in evidence of any normal parathyroid gland response to hypocalcaemia. There were six subjects with values that involved elevated PTH and low to normal ionized calcium values (areas A and D of Fig.1), after excluding five measurements that were just slightly above the normal range. We therefore examined 31 ionized calcium-intact PTH level pairs obtained from these 6 subjects (Fig.2). Each subject had only one measurement above the normal intact PTH range with the exception of subject 4. Subjects 3, 4, 5 and 6 had elevated PTH upon first documented hypocalcaemia, suggesting an initially normal parathyroid response. Subjects 1, 2 and 6 had elevated creatinine levels accompanying the elevated PTH levels. Subject 4 presented with an elevated PTH level coupled with normal ionized calcium and creatinine levels, a condition that could be caused by vitamin D deficiency or early hyperparathyroidism; however, 25-hydroxy vitamin D3 values were not available.
The remaining areas of Fig.1 represent normal PTH measurements (areas B, E and H) or low PTH measurements (areas C, F and I). The six measurements in areas H and I reflect hypercalcaemia in six subjects; four had accompanying elevated creatinine values, and three subjects had elevated blood pH. One of these four subjects was hospitalized as a result of acute renal failure.
It is notable that the eight very low ionized calcium values (<1·0 mm) in areas B and C originated from six different subjects, all but one of whom had documented seizures associated with the hypocalcaemia.
Hypomagnesaemia
We reviewed available serum magnesium values (n = 84) because hypomagnesaemia is known to inhibit PTH activity.24 There was no significant association between the lifetime prevalence of hypomagnesaemia (13 of 84 subjects) and lifetime hypocalcaemia. However, the cooccurrence of hypocalcaemia and hypomagnesaemia at measurement was significant (P = 0·046, two-tailed Fisher's exact test). Furthermore, among the 19 hypocalcaemic measurements with concurrent low intact PTH, six (31·6%) also had concurrent low serum magnesium documented. Conversely, only eight (10·8%) of 74 hypocalcaemic measurements with normal intact PTH were accompanied by hypomagnesaemia. As expected, in the presence of hypocalcaemia, the intact PTH level was significantly associated with hypomagnesaemia (χ2 = 2·91, df = 1, P = 0·02).
Discussion
The results of this study showed that the high lifetime prevalence of hypocalcaemia in adults with 22q11·2DS was not only associated with hypoparathyroidism, as expected, but also associated with hypothyroidism. In addition, hypomagnesaemia may contribute to hypocalcaemia in this population, perhaps by inh-ibiting the release of PTH. Furthermore, we found that early onset of hypocalcaemia and hypoparathyroidism had a high risk of later recurrence. This is in contrast to previous thinking about neonatal hypoparathyroidism as entirely a ‘transient’ phenomenon in 22q11·2DS.22 We further observed that subjects who had episodes of very low ionized calcium (<1·00 mm) were at greater risk of developing hypocalcaemic seizures, as expected.29,30 This is the first systematic study to test for potential features associated with lifetime prevalence of hypocalcaemia and to investigate its long-term manifestation in adults with 22q11·2DS. Results of this study contribute to our understanding of the long-term outcome of hypocalcaemia and possible interacting features in 22q11·2DS.
The overall lifetime prevalence of hypocalcaemia in our adult cohort (80·4%) is higher than that in two previous retrospective studies, reporting hypocalcaemia in 77 (49%) of 158 paediatric patients31 and 203 (60%) of 340 patients of mixed ages from across Europe.9 In our adult cohort, 64·8% of the first documented hypocalcaemic episodes were after age 17 years. This could be an overestimation due to the fact that routine calcium/PTH testing began in adulthood for most subjects, after the diagnosis of 22q11·2DS. We made every effort to review available paediatric records including laboratory values. However, subjects could have experienced asymptomatic or subclinical hypocalcaemic episodes as children that did not lead to follow-up by endocrinologists or a formal diagnosis entered into medical records. Nevertheless, we found that neonatal/paediatric onset of hypocalcaemia and hypoparathyroidism had high rates of recurrence in adulthood.
As originally described by DiGeorge,32 hypocalcaemia was attributed to aplasia or hypoplasia of the parathyroid glands due to a developmental defect. There are however few reported supporting autopsy findings to document structural parathyroid defects in 22q11·2DS.33,34 The analysis of the PTH response to ionized calcium in individuals with 22q11·2DS in our study suggests that the parathyroid glands were not completely dysfunctional but displayed variable (often poor) efficiency of response, likely related to reduced PTH reserve,22,28 in these subjects. The phenomenon of decreased parathyroid function has previously been described as relative or partial hypoparathyroidism.30 This is evident in Fig.1 where 33% of the ‘normal’ PTH levels would be considered inappropriately low relative to the low ionized calcium levels, representing 61·54% (48 of 78 subjects) at at least one point in time, suggesting a lack of adequate PTH response to falling serum total/ionized calcium. There would thus be an enhanced probability of detecting an intact PTH level within the normal range under these circumstances.10 Only a few subjects showed elevated PTH in response to low ionized calcium levels, and often, the elevated PTH levels were accompanied by elevated creatinine levels, suggesting underlying renal disease as a contributing factor.35
We observed that the few hypercalcaemic data points (areas H and I of Fig.1) documented had accompanying normal or suppressed PTH levels, suggesting non-PTH-related factors as the cause. Furthermore, the six subjects with these measurements all had had previously documented hypocalcaemic measurements. Some had elevated pH and/or elevated creatinine, suggesting a possible aetiology of milk–alkali syndrome associated with overuse of calcium supplements.36,37 However, this accounted for a small percentage of subjects, of whom none had experienced another documented hypercalcaemic episode. We conclude therefore that calcium and vitamin D supplements remain advisable for all individuals with 22q11·2DS28 while emphasizing the importance of adequate hydration.
Some subjects who had mild hypocalcaemia recovered temporarily but had later recurrence. Hence, all of our subjects were recommended to take calcium and vitamin D supplements, prophylactically or to correct documented hypocalcaemia.28 Although the precise points in time that subjects received these recommendations and started to take the recommended supplements were not always well documented, it is reasonable to assume that a diagnosis of hypocalcaemia would warrant such a recommendation. About half of the subjects who had normal ionized calcium measurements (areas E and F of Fig.1) were known to have had previous episodes of hypocalcaemia. This is consistent with our observation of the positive effect of supplements, despite the problem of uncertain compliance, even though this was not the objective of the original study.
Hypomagnesaemia can cause hypoparathyroidism by suppressing the secretion of PTH and causing end-organ PTH resistance, subsequently leading to hypocalcaemia.24,38 The available data suggest that hypomagnesaemia may have contributed both to hypocalcaemic measurements with low intact PTH values and to a smaller proportion of subjects where PTH values were normal. These results indicate that serum magnesium should be closely followed in patients with 22q11·2DS who have hypocalcaemia. Treatment with magnesium supplements as needed could ameliorate this readily reversible cause of mild hypoparathyroidism.
Studies of thyroid function in 22q11·2DS have largely been limited to case reports.8 Compared to results we reported in 2005,1 the prevalence of hypothyroidism (29·1%) appears even higher in our cohort due in part to the inclusion of subclinical cases. Treatment with thyroxine had no apparent effect on decreasing the recurrence of hypocalcaemic episodes, suggesting that the association between hypothyroidism and hypocalcaemia is unlikely to be related to a direct functional effect of thyroid hormone levels on calcium homoeostasis. Also, there was no statistical support that hypoparathyroidism mediated the observed association between hypothyroidism and hypocalcaemia. An autoimmune mechanism could be proposed as a potential mechanism underlying both hypothyroidism and hypocalcaemia/hypoparathyroidism in 22q11·2DS. Such a possibility deserves further study.
The main limitation of this study is the retrospective design. Information bias due to missing medical information was likely present in the early childhood period of asymptomatic subjects whose calcium levels were untested. Recognizing this limitation, we referred to age of first documentation as opposed to age of onset. We excluded individuals with incomplete information on any of the key endocrine variables from the regression analyses to compensate for this limitation. We also recognized that some subjects in the 22q11·2DS group had calcium measurements annually, if not more often, while subjects in the comparison group would not have had annual calcium measurements.
In summary, while the results support primary hypoparathyroidism and decreased PTH reserve as the main mechanism involved in the high prevalence of hypocalcaemia in 22q11·2DS, this may not be the sole factor involved. Hypothyroidism and hypomagnesaemia may also be relevant to the manifestation of hypocalcaemia in 22q11·2DS. In addition, in individuals with neonatal/paediatric onset of hypocalcaemia and hypoparathyroidism, subsequent recurrence is common. Early recognition of hypocalcaemia and subsequent follow-up are important for the care of patients with 22q11·2DS at any age. In addition to the regular monitoring of pH-corrected ionized calcium, PTH and TSH recommended by clinical practice guidelines,28 monitoring of serum magnesium may be helpful. Magnesium supplementation for those with hypomagnesaemia could be considered to help minimize risks of hypocalcaemia. Our results may help health care providers in formulating long-term care plans for patients with 22q11·2DS, given that individuals with 22q11·2DS may not be good reporters of symptoms and that, as for any patient, endocrinologic symptoms may be confused with those of other conditions.26 Under-recognition of 22q11·2DS is broadly acknowledged.28 Endocrinologists and primary care physicians should consider the possibility of undiagnosed 22q11·2DS at any age in patients with hypocalcaemia, especially in the presence of relative/partial or complete hypoparathyroidism and/or hypothyroidism.
Acknowledgments
This study was supported by Canadian Institutes of Health Research grants (MOP-79518, MOP-89066 and MOP-97800), a Comprehensive Research Experience for Medical Students scholar grant (to E.N.M.C.) and a W. Garfield Weston Foundation grant (to A.S.B.). Anne S. Bassett holds the Canada Research Chair in Schizophrenia Genetics and Genomic Disorders. Susan R. George holds the Canada Research Chair in Molecular Neuroscience. The authors thank the patients and their families for their participation, research assistants and staff at the Toronto Congenital Cardiac Centre for Adults (TCCCA), staff at The Centre for Applied Genomics (TCAG), and fellows and students who assisted in the collection and analysis of data.
Disclosure statement
Nothing to declare.
References
- Bassett AS, Chow EWC, Husted J, et al. Clinical features of 78 adults with 22q11 deletion syndrome. American Journal of Medical Genetics. Part A. 2005;138A:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scambler P. The 22q11 deletion syndromes. Human Molecular Genetics. 2000;9:2421–2426. doi: 10.1093/hmg/9.16.2421. [DOI] [PubMed] [Google Scholar]
- Swillen A, Vogels A, Devriendt K, et al. Chromosome 22q11 deletion syndrome: update and review of the clinical features, cognitive-behavioral spectrum, and psychiatric complications. American Journal of Medical Genetics. 2000;97:128–135. doi: 10.1002/1096-8628(200022)97:2<128::aid-ajmg4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Bales AM, Zaleski CA. McPherson EW. Newborn screening programs: should 22q11 deletion syndrome be added? Genetics in Medicine. 2010;12:135–144. doi: 10.1097/GIM.0b013e3181cdeb9a. [DOI] [PubMed] [Google Scholar]
- Kobrynski LJ. Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- Cuneo BF, Driscoll DA, Gidding SS, et al. Evolution of latent hypoparathyroidism in familial 22q11 deletion syndrome. American Journal of Medical Genetics. 1997;69:50–55. [PubMed] [Google Scholar]
- Scir G, Dallapiccola B, Iannetti P, et al. Hypoparathyroidism as the major manifestation in two patients with 22q11 deletions. American Journal of Medical Genetics. 1994;52:478–482. doi: 10.1002/ajmg.1320520415. [DOI] [PubMed] [Google Scholar]
- Choi JH, Shin YL, Kim GH, et al. Endocrine manifestations of chromosome 22q11.2 microdeletion syndrome. Hormone Research. 2005;63:294–299. doi: 10.1159/000086745. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. Journal of Medical Genetics. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner R, De Gonneville AL, Kindermans C, et al. Parathyroid function and growth in 22q11.2 deletion syndrome. Journal of Pediatrics. 2003;142:504–508. doi: 10.1067/mpd.2003.156. [DOI] [PubMed] [Google Scholar]
- Taylor SC, Morris G, Wilson D, et al. Hypoparathyroidism and 22q11 deletion syndrome. Archives of Disease in Childhood. 2003;88:520–522. doi: 10.1136/adc.88.6.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinzimer SA. Endocrine aspects of the 22q11.2 deletion syndrome. Genetics in Medicine. 2001;3:19–22. doi: 10.1097/00125817-200101000-00005. [DOI] [PubMed] [Google Scholar]
- Bilezikian JP, Khan A, Potts JT, Jr, et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. Journal of Bone and Mineral Research. 2011;26:2317–2337. doi: 10.1002/jbmr.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eryol N, Colak R, Ozdogru I, et al. Effects of calcium treatment on QT interval and QT dispersion in hypocalcemia. American Journal of Cardiology. 2003;91:750–752. doi: 10.1016/s0002-9149(02)03423-9. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Costain G, Fung WLA, et al. Clinically detectable copy number variations in a Canadian catchment population of schizophrenia. Journal of Psychiatric Research. 2010;44:1005–1009. doi: 10.1016/j.jpsychires.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung WLA, Chow EWC, Webb GD, et al. Extracardiac features predicting 22q11.2 Deletion Syndrome in adult congenital heart disease. International Journal of Cardiology. 2008;131:51–58. doi: 10.1016/j.ijcard.2007.08.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EWC, Husted J, et al. Premature death in adults with 22q11.2 deletion syndrome. Journal of Medical Genetics. 2009;46:324–330. doi: 10.1136/jmg.2008.063800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, et al. Prevalence of 22q11 microdeletions in digeorge and velocardiofacial syndromes - implications for genetic-counseling and prenatal-diagnosis. Journal of Medical Genetics. 1993;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Marshall CR, Lionel AC, et al. Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Human Molecular Genetics. 2008;17:4045–4053. doi: 10.1093/hmg/ddn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow EWC, Watson M, Young DA, et al. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophrenia Research. 2006;87:270–278. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ENM, George SR, Andrade DM, et al. Neonatal hypocalcemia, neonatal seizures, and intellectual disability in 22q11.2 deletion syndrome. Genetics in Medicine. 2014;16:40–44. doi: 10.1038/gim.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima K, Abrahamsen TG, Wolff AB, et al. Hypoparathyroidism and autoimmunity in the 22q11.2 deletion syndrome. European Journal of Endocrinology. 2011;165:345–352. doi: 10.1530/EJE-10-1206. [DOI] [PubMed] [Google Scholar]
- Stagi S, Lapi E, Gambineri E, et al. Thyroid function and morphology in subjects with microdeletion of chromosome 22q11 (del(22)(q11)) Clinical Endocrinology (Oxf) 2010;72:839–844. doi: 10.1111/j.1365-2265.2009.03736.x. [DOI] [PubMed] [Google Scholar]
- Agus Z, Wasserstein A. Goldfarb S. Disorders of calcium and magnesium homeostasis. American Journal of Medicine. 1982;72:473–488. doi: 10.1016/0002-9343(82)90519-8. [DOI] [PubMed] [Google Scholar]
- Jain A, Agarwal R, Sankar MJ, et al. Hypocalcemia in the newborn. Indian Journal of Pediatrics. 2010;77:1123–1128. doi: 10.1007/s12098-010-0176-0. [DOI] [PubMed] [Google Scholar]
- McGraw-Hill Medical. Harrison's Endocrinology. 2nd edn. New York: McGraw-Hill Medical; 2010. [Google Scholar]
- Baron R. Kenny D. The moderator mediator variable distinction in social psychological-research - conceptual, strategic, and statistical considerations. Journal of Personality and Social Psychology. 1986;51:1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- Bassett AS, McDonald-McGinn DM, Devriendt K, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. Journal of Pediatrics. 2011;159:332–339.e1. doi: 10.1016/j.jpeds.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maalouf N, Sakhaee K. Odvina C. A case of chromosome 22q11 deletion syndrome diagnosed in a 32-year-old man with hypoparathyroidism. Journal of Clinical Endocrinology & Metabolism. 2004;89:4817–4820. doi: 10.1210/jc.2004-0442. [DOI] [PubMed] [Google Scholar]
- Al-Jenaidi F, Makitie O, Grunebaum E, et al. Parathyroid gland dysfunction in 22q11.2 deletion syndrome. Hormone Research. 2007;67:117–122. doi: 10.1159/000096421. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, et al. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genetic Counseling. 1999;10:11–24. [PubMed] [Google Scholar]
- DiGeorge AM. Discussion on a new concept of the cellular basis of immunity. Journal of Pediatrics. 1965;67:907–908. [Google Scholar]
- Conley ME, Beckwith JB, Mancer JFK, et al. Spectrum of the DiGeorge syndrome. Journal of Pediatrics. 1979;94:883–890. doi: 10.1016/s0022-3476(79)80207-3. [DOI] [PubMed] [Google Scholar]
- Kapadia CR, Kim YE, McDonald-McGinn DM, et al. Parathyroid hormone reserve in 22q11.2 deletion syndrome. Genetics in Medicine. 2008;10:224–228. doi: 10.1097/GIM.0b013e3181634edf. [DOI] [PubMed] [Google Scholar]
- Khan S. Vitamin D deficiency and secondary hyperparathyroidism among patients with chronic kidney disease. American Journal of the Medical Sciences. 2007;333:201–207. doi: 10.1097/MAJ.0b013e31803bb129. [DOI] [PubMed] [Google Scholar]
- Medarov BI. Milk-alkali syndrome. Mayo Clinic Proceedings. 2009;84:261–267. doi: 10.4065/84.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picolos M, Lavis V. Orlander P. Milk-alkali syndrome is a major cause of hypercalcaemia among non-end-stage renal disease (non-ESRD) inpatients. Clinical Endocrinology (Oxf) 2005;63:566–576. doi: 10.1111/j.1365-2265.2005.02383.x. [DOI] [PubMed] [Google Scholar]
- Rude R, Oldham S. Singer F. Functional hypoparathyroidism and parathyroid-hormone end-organ resistance in human magnesium-deficiency. Clinical Endocrinology (Oxf) 1976;5:209–224. doi: 10.1111/j.1365-2265.1976.tb01947.x. [DOI] [PubMed] [Google Scholar]