

Phosphonates comprise a class of compounds with a stable phosphorus-carbon bond, many of which have potent bioactivity. For example, fosfomycin and dehydrophos are both antimicrobial agents, and phosphinothricin is a commercially important herbicide (Figure 1).[1] Fosfomycin is produced by streptomycetes[2] and pseudomonads[3] and is used to treat urinary tract and gastrointestinal infections;[4] it has activity against both vancomycin- and methicillin-resistant strains of Staphylococcus aureus.[5] Fosfomycin covalently modifies an active site cysteine in UDP-N-acetyl-glucosamine-3-O-enolpyruvyltransferase (MurA),[6] thereby inhibiting cell wall biosynthesis. Dehydrophos functions as a Trojan horse antibiotic that upon uptake and proteolysis releases acetylphosphonate methyl ester that potently inactivates pyruvate oxidase and pyruvate dehydrogenase,[7] and phosphinothricin is a mimic of glutamate that inhibits glutamine synthase.[8] The biosynthetic gene clusters of all three compounds share a gene encoding a group III metal dependent alcohol dehydrogenase (AD).[9] FomC, DhpG, and PhpC use NAD(P)H to catalyze the reduction of phosphonoacetaldehyde (PnAA) to 2-hydroxyethylphosphonate (2-HEP).[9] We determined here the stereochemistry of the 2-HEP formed when deuterium labeled cofactor was utilized.
Figure 1.

Structures of fosfomycin, dehydrophos, and phosphinothricin.
In addition to providing the first determination of the face selectivity of substrate reduction of a group III metal dependent AD, these studies also provided additional insights into the fosfomycin biosynthetic pathway in Streptomyces fradiae (Scheme 1). The overall pathway[10] is still incomplete as a consequence of the inability to reconstitute the activity of a presumed methyltransferase, Fom3, as well as uncertainty over where in the biosynthetic pathway to place the reductase FomC (Scheme 1).[11]
Scheme 1.

Proposed pathways for the biosynthesis of fosfomycin.
Previous feeding studies have shown that 2-HEP is converted to fosfomycin by S. fradiae.[10] Two explanations have been offered for this observation. In one hypothesis, FomC acts on 2-HEP to generate phosphonoacetaldehyde (PnAA), which is subsequently methylated by Fom3 (Scheme 1, path A).[10, 12] Alternatively, genetic studies combined with feeding studies suggested FomC reduces PnAA to 2-HEP that is then methylated by Fom3 (Scheme 1, path B).[13] Because Fom3 activity has not been reconstituted to date, neither hypothesis can be ruled out unequivocally. Studies by Hammerschmidt and coworkers demonstrated that feeding (R)- or (S)-2-[2-2H1]-HEP to S. fradiae resulted in 32% incorporation of the deuterium label in fosfomycin from the S isomer and no deuterium incorporation from the R isomer.[14] This observation places a restriction on the stereochemistry of hydride transfer from 2-HEP by FomC for path A. Only if FomC removes the pro-R hydrogen from 2-HEP can pathway A explain the experimental results. Thus, experimental determination of the stereochemistry of hydride transfer by FomC or its close orthologs DhpG and PhpC could support or refute pathway A.
Of the three homologous enzymes, DhpG was used in this study as it proved much more stable under the conditions used (vide infra) and expresses better in Escherichia coli compared to FomC and PhpC. DhpG has 33% sequence identity with FomC with conservation of the predicted active site residues identified through a multiple sequence alignment with other group III alcohol dehydrogenases (see Figure 2).[9] The cofactor binding motif (Figure 2, orange box) and metal binding residues (Figure 2, blue boxes) are conserved in the three ADs involved in phosphonate biosynthesis. The residues responsible for binding of substrate are not conserved in this class of proteins, but they were identified for the three proteins of interest via homology models constructed based on the structures of 1,3-propanediol dehydrogenase from Thermotoga maritime (PDB code 1O2D) and lactaldehyde reductase from E. coli (PDB code 2BL4).[9] Two residues that are conserved only in the 2-HEP dehydrogenases, a Ser and a Tyr residue boxed in red in Figure 2, engage in hydrogen bonding to the phosphonate moiety in the homology model.[9] Their conservation across FomC, DhpG, and PhpC along with the overall sequence similarity indicates that hydride transfer from NAD(P)H to the aldehyde very likely occurs with the same facial selectivity for each protein.
Figure 2.

Sequence alignment of select group III alcohol dehydrogenases. The boxed residues are responsible for NAD(P)H binding (orange), metal cofactor binding (blue), or, in the case of FomC, DhpG, and PhpC, phosphonate binding (red), and were identified through homology modelling.
In vitro assays with DhpG did not produce sufficient quantities of product to determine the absolute configuration of 2-[2-2H1]- HEP formed upon reduction of PnAA with NADP2H by NMR spectroscopy.[15] Instead, we took advantage of the known stereochemistry of oxidation of 2-HEP to hydroxymethylphosphonate (HMP) and formate by 2-hydroxyethylphosphonate dioxygenase (HEPD) (Scheme 2). Previous studies have shown that oxidation of (R)-2-[2-2H1]-HEP with HEPD produced deuterium labeled formate, whereas oxidation of the (S) enantiomer resulted in unlabeled formate.[15] To eliminate the possibility of spurious formate interfering with analysis of the enzymatically produced formate,[16] a 13C label was introduced at the C2 position of PnAA such that either 13C,1H- or 13C,2H-formate would be generated, both of which can be readily distinguished from spurious formate by mass spectrometry. [2-13C1]-PnAA was prepared enzymatically from commercial [2-13C1]-phosphoenolpyruvate (PEP), using a recombinant N-terminally His6-tagged PEP mutase (RhiH) and a C-terminally His6-tagged PnPy decarboxylase from Bacteroides fragilis via a modification to a previously reported procedure.[17] Conversion of [2-13C1]-PEP of [2-13C1]-PnAA was confirmed by 31P NMR spectroscopy (Figure 3a).
Scheme 2.

Outline of the strategy used to determine the stereochemistry of DhpG-catalyzed reduction of PnAA.
Figure 3.
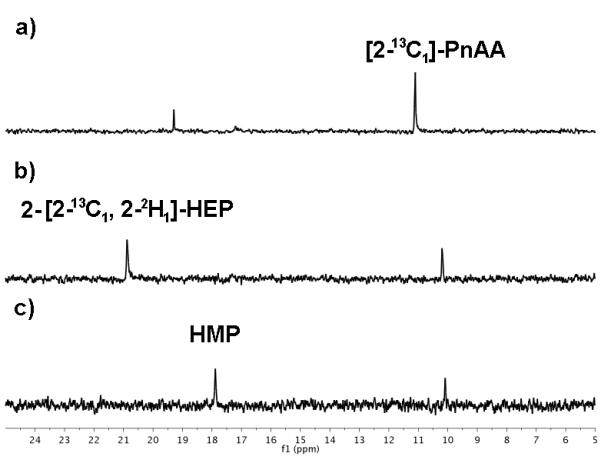
The progress of the enzymatic cascade outlined in Scheme 2 was followed by 31P NMR spectroscopy to confirm the generation of a) [2-13C1]-PnAA from [2-13C]-PEP by the successive actions of RhiH and PnPy decarboxylase, b) 2-[2-13C1, 2-2H1]-HEP via the reduction of [2-13C1]-PnAA by DhpG in the presence of (4R)-NADP2H, and c) HMP by the action of HEPD. 31P chemical shifts of phosphonates are very sensitive to small changes in pH. Therefore, the identity of 2-HEP and HMP was confirmed by spiking with an authentic synthetic standard. The additional resonance in b) and c) is residual [2-13C1]-PnAA. The additional resonance at ~19 ppm in a) is an unknown byproduct of PnAA generation.
Given that FucO, another group III metal-dependent AD, has been reported to transfer the pro-R hydride from C4 of the nicotinamide group of NADH to its substrate,[18] (4R)-NADP2H was prepared by incubating deuterated phosphite with an engineered phosphite dehydrogenase (PTDH) that can use either NAD+ or NADP+ as substrate.[19] After (4R)-NADP2H was generated, phosphite dehydrogenase was removed, and the (4R)-NADP2H was incubated with DhpG and excess [2-13C1]-PnAA. Once all (4R)-NADP2H was consumed as determined by UV-Vis spectroscopy, DhpG was removed, and the reaction was analyzed by 31P NMR spectroscopy to confirm the formation of 2-HEP (Figure 3b).
Purified HEPD was then added to convert 2-[2-2H1]-HEP to formate and HMP. 31P NMR spectroscopy was used to confirm that all 2-HEP had been converted to HMP (Figure 3c). The formate produced in the last reaction was activated with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and reacted with 2-nitrophenylhydrazine to form the corresponding hydrazide that was analyzed by LC-MS.[15] As shown in Figure 4, the vast majority of the formate product contained both the 13C and 2H labels, indicating that reduction of PnAA with (4R)-NADP2H catalyzed by DhpG results in (R)-2-[2-2H1]-HEP. This finding establishes that DhpG transfers the pro-R hydride of the reduced nicotinamide to its substrate, in accord with the recent finding that the group III AD FucO transfers the pro-R hydride to its substrate.[18] Simultaneously, this result also establishes that reduction by this family of group III alcohol dehydrogenases takes place on the Re face of phosphonoacetaldehyde, and therefore that the enzymes remove the pro-R hydrogen during oxidation of 2-HEP.
Figure 4.

Extracted ion chromatograms of formyl 2-nitrophenylhydrazide. The major product observed is the 2-nitrophenylhydrazide of [13C, 2H]-formate (182 m/z, blue), whereas derivatized spurious formate (180 m/z, red) and formate containing either 13C or 2H (181 m/z, black) are minor constituents.
This study confirms that group III ADs transfer the pro-4R hydride to their substrate and establishes experimentally for the first time that hydride delivery occurs to the Re face of the phosphonacetaldehyde substrate. The experimental determination that DhpG, and by extension FomC, removes the pro-R hydrogen from 2-HEP combined with the demonstration by Hammerschmidt and coworkers that feeding of (R)-2-[2-2H1]-HEP results in production of unlabeled fosfomycin in S. fradiae does not rule out pathway A of fosfomycin biosynthesis. Ultimately, reconstitution of the putative methyltransferase, Fom3, will be be required to determine which pathway is operative during fosfomycin biosynthesis.
Experimental Section
Preparation of (4R)-NADP2H
NADP2H stereospecifically labeled with deuterium at C4 of the nicotinamide was synthesized by incubating NADP (2 mM) with deuterium labeled phosphite (3 mM) and phosphite dehydrogenase (PTDH, 7 μM) in 50 mM HEPES, pH 7.5.[19a] The mixture was allowed to incubate at 30° C for 90 min, and then PTDH was removed by centrifuge filtration (30 kDa MWCO). The presence of (4R)-NADP2H was confirmed by the generation of a peak at 340 nm in the UV-vis spectrum. Because of its instability, the cofactor was used without further purification.
Preparation of [2-13C1]-PnAA
Phosphonoacetaldehyde labeled with 13C at C2 was generated by incubating thiamine pyrophosphate (TPP, 1.4 mM) with [2-13C1]-phosphoenolpyruvate (PEP, 10 mM, Sigma Aldrich) in 50 mM HEPES pH 7.5 with 1 mM Mg2+.[17] The reaction was initiated by addition of RhiH (80 μM) and the PnPy decarboxylase (40 μM), causing turbidity. After incubating at 30° C for 45 min, proteins were removed by centrifuge filtration (30 kDa MWCO). An aliquot was taken from the eluent, and the presence of [2-13C1]-PnAA was confirmed by 31P NMR spectroscopy.
DhpG assay
(4R)-NADP2H (1 mM final concentration) was mixed with the prepared [2-13C1]-PnAA (4 mM final concentration) and DhpG (approximately 75 μM, expressed and purified as described previously[9]) was added in 50 mM HEPES, pH 7.5. The reaction was allowed to proceed at room temperature for 60 min, at which point the UV-Visible spectrum indicated the consumption of the 340 nm peak associated with the (4R)-NADP2H cofactor. Protein was removed via centrifuge filtration (30 kDa MWCO), and an aliquot was taken to confirm the presence of 2-[2-13C1, 2-2H1]-HEP via 31P NMR spectroscopy (Figure 3).
Determination of the stereochemistry of 2-[2-13C1, 2-2H1]-HEP
To the isotopically labeled 2-HEP (~0.8 mM) was added HEPD (final concentration of 20 μM) in 50 mM HEPES pH 7.5. The solution was incubated at room temperature to effect the conversion of 2-HEP to HMP and formate. Protein was removed via centrifuge filtration (30 kDa MWCO), and an aliquot of the flow through (10 μL) was derivatized to afford the 2-nitrophenylhydrazide adduct and analyzed by LC-MS using methodology developed previously.[16]
Acknowledgements
This work was supported by the National Institutes of Health (GM077596 to W.A.v.d.D). S.C.P. is a trainee in the Chemistry-Biology Interface Training Program (TM32 GM070421).
References
- [1].Metcalf WW, van der Donk WA. Annu. Rev. Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB, Mata JM, Hernandez S, Mochales S. Science. 1969;166:122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- [3] a.Shoji J, Kato T, Hinoo H, Hattori T, Hirooka K, Matsumoto K, Tanimoto T, Kondo E. J. Antibiot. 1986;39:1011–1012. doi: 10.7164/antibiotics.39.1011. [DOI] [PubMed] [Google Scholar]; b Katayama N, Tsubotani S, Nozaki Y, Harada S, Ono H. J. Antibiot. 1990;43:238–246. doi: 10.7164/antibiotics.43.238. [DOI] [PubMed] [Google Scholar]
- [4].Lobel B. Int. J. Antimicrob. Agents. 2003;22(Supplement 2):85–87. doi: 10.1016/s0924-8579(03)00237-1. [DOI] [PubMed] [Google Scholar]
- [5] a.Cassone M, Campanile F, Pantosti A, Venditti M, Stefani S. Microb. Drug Resist. 2004;10:43–49. doi: 10.1089/107662904323047790. [DOI] [PubMed] [Google Scholar]; b Nakazawa H, Kikuchi Y, Honda T, Isago T, Nozaki M. J. Infect. Chemother. 2003;9:304–309. doi: 10.1007/s10156-003-0266-2. [DOI] [PubMed] [Google Scholar]
- [6].Marquardt JL, Brown ED, Lane WS, Haley TM, Ichikawa Y, Wong CH, Walsh CT. Biochemistry. 1994;33:10646–10651. doi: 10.1021/bi00201a011. [DOI] [PubMed] [Google Scholar]
- [7] a.Kuemin M, van der Donk WA. Chem. Commun. 2010;46:7694–7696. doi: 10.1039/c0cc02958k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Circello BT, Miller CG, Lee J-H, van der Donk WA, Metcalf WW. Antimicrob. Agents Chemother. 2011;55:3357–3362. doi: 10.1128/AAC.01483-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Abell LM, Villafranca JJ. Biochemistry. 1991;30:6135–6141. doi: 10.1021/bi00239a008. [DOI] [PubMed] [Google Scholar]
- [9].Shao Z, Blodgett JAV, Circello BT, Eliot AC, Woodyer R, Li G, van der Donk WA, Metcalf WW, Zhao H. J. Biol. Chem. 2008;283:23161–23168. doi: 10.1074/jbc.M801788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Seto H, Kuzuyama T. Nat. Prod. Rep. 1999;16:589–596. doi: 10.1039/a809398i. [DOI] [PubMed] [Google Scholar]
- [11].Woodyer RD, Shao Z, Metcalf WM, Thomas PM, Kelleher NL, van der Donk WA, Zhao H. Chem. Biol. 2006;13:1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- [12].Kuzuyama T, Hidaka T, Kamigiri K, Imai S, Seto H. J. Antibiot. 1992;45:1812–1814. doi: 10.7164/antibiotics.45.1812. [DOI] [PubMed] [Google Scholar]
- [13].Woodyer RD, Li G, Zhao H, van der Donk WA. Chem. Commun. 2007:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- [14].Hammerschmidt F. Liebigs Ann. Chem. 1992;1992:553–557. [Google Scholar]
- [15].Whitteck JT, Malova P, Peck SC, Cicchillo RM, Hammerschmidt F, van der Donk WA. J. Am. Chem. Soc. 2011;133:4236–4239. doi: 10.1021/ja1113326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Whitteck JT, Cicchillo RM, van der Donk WA. J. Am. Chem. Soc. 2009;131:16225–16232. doi: 10.1021/ja906238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Chem. Biol. 2010;17:28–37. doi: 10.1016/j.chembiol.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Blikstad C, Widersten M. J. Mol. Catal. B: Enzym. 2010;66:148–155. [Google Scholar]
- [19] a.Vrtis JM, White A, Metcalf WW, van der Donk WA. J. Am. Chem. Soc. 2001;123:2672–2673. doi: 10.1021/ja004301k. [DOI] [PubMed] [Google Scholar]; b Woodyer R, van der Donk WA, Zhao H. Biochemistry. 2003;42:11604–11614. doi: 10.1021/bi035018b. [DOI] [PubMed] [Google Scholar]