

Abstract
Purpose:
The imidazopyridine moiety is important pharmacophore that has proven to be useful for a number of biologically relevant targets, also reported to display antibacterial, antifungal, antiviral properties. Riboflavin biosynthesis involving catalytic step of Lumazine synthase is absent in animals and human, but present in microorganism, one of marked advantage of this study. Still, this path is not exploited as antiinfective target. Here, we proposed different interactions between [1H,3H] imidazo[4,5-b] pyridine test ligands and target protein Lumazine synthase (protein Data Bank 2C92), one-step synthesis of title compounds and further evaluation of them for in vitro antimicrobial activity.
Materials and Methods:
Active pocket of the target protein involved in the interaction with the test ligands molecules was found using Biopredicta tools in VLifeMDS 4.3 Suite. In-silico docking suggests H-bonding, hydrophobic interaction, charge interaction, aromatic interaction, and Vanderwaal forces responsible for stabilizing enzyme-inhibitor complex. Disc diffusion assay method was used for in vitro antimicrobial screening.
Results and Discussion:
Investigation of possible interaction between test ligands and target lumazine synthase of Mycobacterium tuberculosis suggested 1i and 2f as best fit candidates showing hydrogen bonding, hydrophobic, aromatic and Vanderwaal's forces. Among all derivatives 1g, 1j, 1k, 1l, 2a, 2c, 2d, 2e, 2h, and 2j exhibited potent activities against bacteria and fungi compared to the standard Ciprofloxacin and Fluconazole, respectively. The superiority of 1H imidazo [4,5-b] pyridine compounds having R’ = Cl >No2 > NH2 at the phenyl/aliphatic moiety resident on the imidazopyridine, whereas leading 3H imidazo[4,5-b] pyridine compounds containing R/Ar = Cl > No2 > NH2> OCH3 substituents on the 2nd position of imidazole.
KEY WORDS: Antimicrobial, docking score, imidazopyridine, lumazine synthase, minimum inhibitory concentration
Vitamin B2, commonly called riboflavin, is one of eight water-soluble B vitamins. Like its close relative, vitamin B1 (thiamine), riboflavin plays a crucial role in certain metabolic reactions, for example, in the final metabolic conversion of monosaccharides, where reduction-equivalents and chemical energy in the form of adenosine triphosphate are produced through the Embden-Meyerhoff pathway.[1] Riboflavin is biosynthesized by plants and numerous microorganisms, but not by animals, whereas animals obtain riboflavin from dietary sources. A rational approach to therapeutically useful antibiotics would be to selectively inhibit an enzyme present in a parasite, but absent in the host. Inhibition of the bio-synthesis of riboflavin provides such a strategy, since pathogenic microorganisms synthesize their own riboflavin, whereas mammals obtain this vitamin through dietary sources. In particular, enterobacteria such as Salmonella and Escherichia species lack riboflavin due to the apparent absence of transport systems for riboflavin or flavocoenzymes, and are therefore absolutely dependent on endogenous riboflavin biosynthesis.[2,3,4,5] Riboflavin biosynthesis is therefore an attractive target for the design and synthesis of new antibiotics, which are urgently needed because pathogens are becoming drug resistant at an alarming rate.[6]
Lumazine synthase and riboflavin synthase catalyze the last two steps in the biosynthesis of riboflavin (4) [Figure 1]. The biosynthetic pathway starts off from one molecule of guanosine triphosphate,[5,6,7] which is converted to 5-amino-6-ribitylamino-2,4 (1H,3H)-pyrimidinedione (1) by a sequence of ring opening, deamination, reduction, and dephosphorylation.[8,9] Lumazine synthase catalyzes the condensation of 3,4-dihydroxy-2-butanone 4-phosphate (2) with 5-amino-6-ribitylamino-2,4-(1H,3H) pyrimidinedione (1) yielding 6,7-dimethyl-8-D-ribityllumazine (3).[10,11,12,13,14,15,16,17,18,19] The final process in the biosynthesis of riboflavin (4) involves a mechanistically unusual dismutation of two molecules of 6,7-dimethyl-8-D-ribityllumazine (3) that results in the formation of one molecule of riboflavin and one molecule of the pyrimidinedione derivative (1).[20,21,22,23,24] Although the precise details remain to be established, the lumazine synthase-catalyzed reaction most likely proceeds along a mechanistic pathway involving the formation of the Schiff base (5), phosphate elimination affording (6), tautomerization to (7), ring closure, and dehydration to yield the lumazine (3) [Figure 2].[9] Variations of this mechanism are possible depending on the Schiff base geometry and possible isomerization, conformational changes, and the timing of phosphate elimination.
Figure 1.
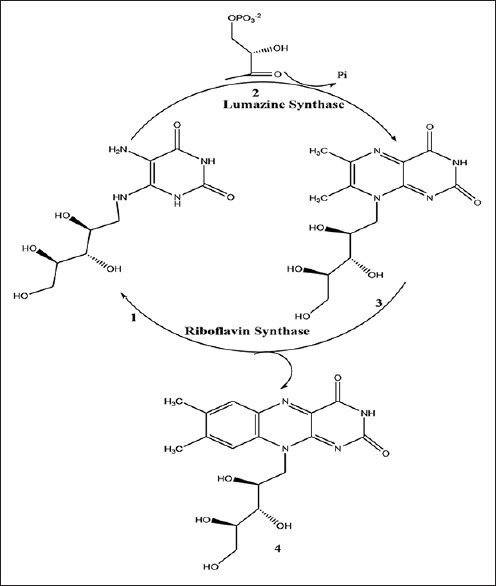
Riboflavin biosynthesis pathway
Figure 2.
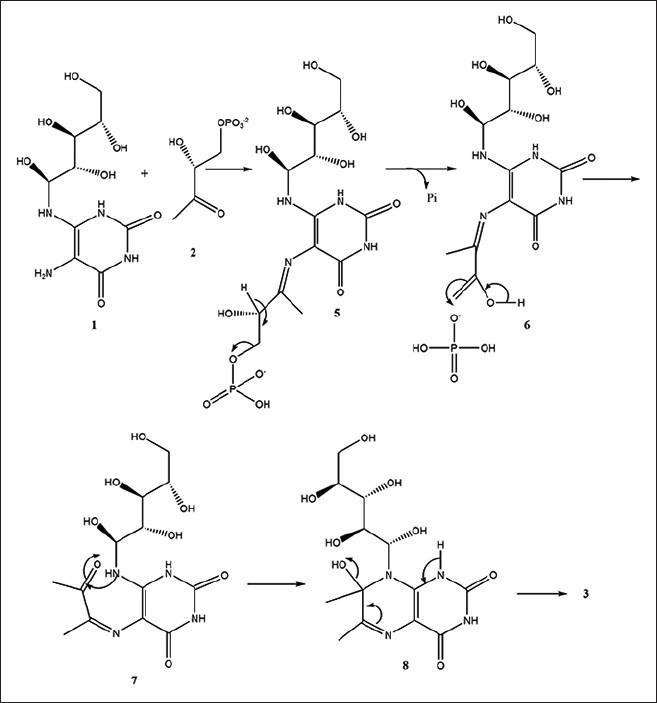
Catalytic mechanism of lumazine synthase
Until now, the riboflavin pathway has not been exploited as an antiinfective target. In the design and development of inhibitors, there would be no requirement for selective inhibition of the pathogen enzymes as opposed to (nonexistent) homologous enzymes of the human host. This is a marked advantage of biosynthesis pathway with regard to drug development.[7] We report herein an attempt for in-silico design of title compounds for inhibition of target lumazine synthase from Mycobacterium tuberculosis (Protein Data Bank [PDB] 2C92, Figure 3). Possible binding interactions were studied involving types of forces responsible for stabilizing the drug-receptor complex. Among all test ligands docked with receptor, we found 1f and 2j compounds as the best fit ligands. Study of docking is performed with the aim to design [1H,3H] Imidazo[4,5-b] pyridine analogues and to explore further as novel antibiotics against drug resistant pathogenic microbial strains. Recent studies from many laboratories, implicate the role of these imidazopyridine scaffolds in the treatment of many of the most common human diseases, including diabetes,[25] cancers,[26] infection by microorganisms,[27] and an array of neurological syndromes.[28] Furthermore, a literature search indicated that benzimidazoles,[29,30,31] oxadiazoles,[32,33,34] and phenyl imidazoles[35,36] with different substitution patterns possess a wide range of antimicrobial properties.
Figure 3.
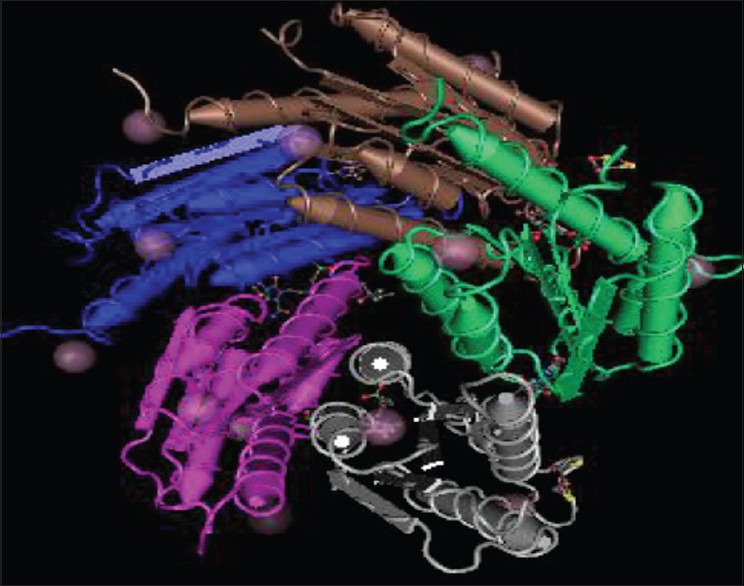
Lumazine synthase from Mycobacterium tuberculosis (PDB 2C92)
The imidazopyridine moiety is an important pharmacophore that has proven to be useful for a number of biologically relevant targets.[37] Imidazo[4,5-b] pyridine, known as 1-desasapurine, is a common structural motif found in numerous molecules that display antiviral, antifungal, antibacterial, and antiproliferative activities. The potent biological activity and the prevalence of 1-desazapurines in both natural products and pharmaceuticals has inspired significant interest in the synthesis of these heterocycles. Compounds that belong to the imidazo[4,5-b] pyridin-2-one class have been shown to be nonsteroidal antiinflammatory and analgesic agents,[38,39,40,41] and to possess antidepressant,[39,40,41,42] antiphlogistic,[41,42,43] cardiotonic,[41,42,43,44] hypotensive and antiarrhythmic activity.[42,43,44,45] In addition, certain members of this class had been reported to be a potent inhibitor of Aurora-A,[46] adenosine deaminase inhibitors,[47] potent inhibitors of inosine 5’-monophosphate dehydrogenase.[48]
Materials and Methods
Melting points (°C) were determined using a Fischer-Jones melting point apparatus and were uncorrected. Microanalyses (CHN) were performed at the microanalytical center, University of Pune using Rapid analyzer. Fourier Transform Infrared spectra (FT-IR, KBr cm−1) were run on JASCO 401 FT-IR spectrometer. 1H NMR and 13C NMR spectra were recorded on BRUKER AVANCE II FT-NMR (400 MHz) using TMS as an internal standard (chemical shifts in δ, ppm), s = singlet, d = doublet, m = multiplet, bs = broad singlet. The relative integrals of peak areas agreed with those expected for the assigned structures. Mass spectra were recorded on WATERS, Q-TOF MICROMASS (LC-MS), performed at SAIF, Punjab University, Chandigarh. Thin layer chromatography (TLC) analysis was carried out on silica gel precoated aluminum sheets (Type 60 F 254, Merck) and the spots were detected under ultraviolet-lamp at short wavelength λ 254 nm.
In-silico docking experiment
The crystal structures of the target protein was obtained from PDB and saved in standard three-dimensional coordinate format. The PDB is a repository for three-dimensional structural data of proteins and nucleic acids. This database provides the three-dimensional structure of all the proteins by NMR or by X-ray crystallography. After conducting adequate literature review lumazine synthase of M. tuberculosis (PDB entry code 2C92) was selected as the target for the present study. Preparation of ligands was done by drawing the structures using ChemSketch 12 (ACD) Advanced Chemistry Development (USA) and Chem Draw Ultra 7 Cambridge Soft Chem Draw Ultra in two-dimensional and saved as MDL Molfile format. Further conversion of ligands to three-dimensional format using VLife Engine tools of VlifeMDS 4.3 VLifesciences, A Division of NovaLead Pharma Pvt Ltd, Pune. Protein visualization was done by loading the structure in SWISS PDB viewer. Further the Energy Minimization was performed by VlifeMDS 4.3 docking suite. Docking simulations were performed with Biopredicta tool using grip docking mode. The number of docking runs was set to 10. Different types of binding interactions were studied between docked three-dimensional test ligands and three-dimensional macromolecule target [Figures 4-7].
Figure 4.
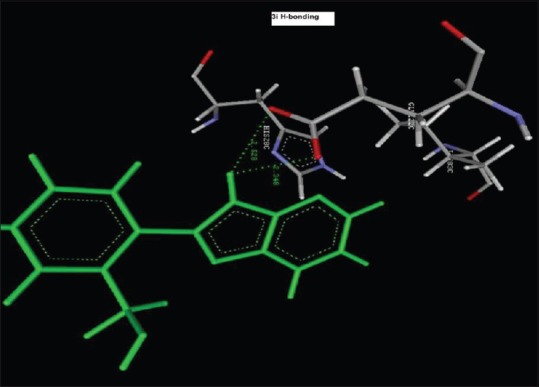
Hydrogen bond Interaction of 2-(2-nitrophenyl)-3H-imidazo[4,5-b]pyridine(1i) with GLU122C (2.348 A°, 2.528 A°)
Figure 7.
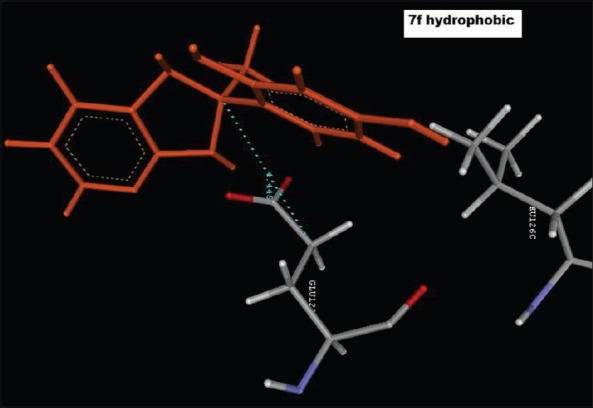
Hydrophobic interaction of 4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b]pyridin-2-yl)benzene-1,3-diol (2f), with GLU122C
Figure 5.
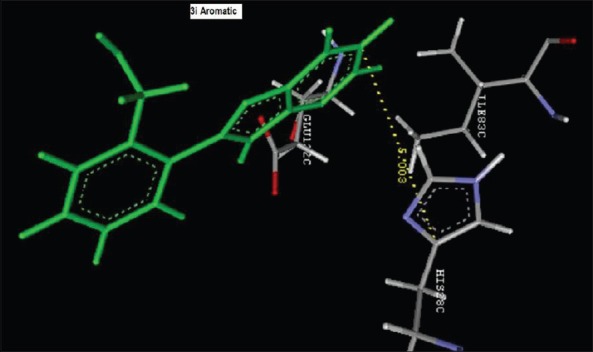
Aromatic Interaction of 2-(2-nitrophenyl)-3H-imidazo[4,5-b]pyridine (1i) with HIS28C
Figure 6.
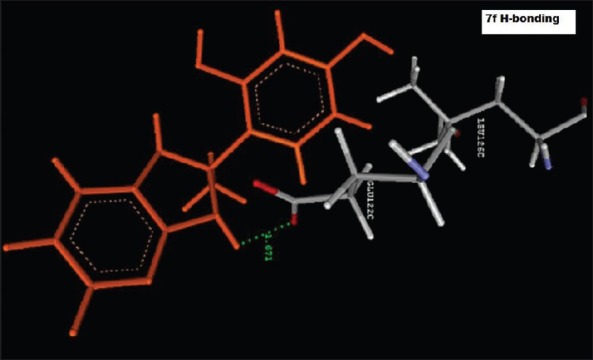
Hydrogen bond Interaction of 4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b]pyridin-2-yl)benzene-1,3-diol (2f) with GLU122C
Chemistry
General experimental procedure for the synthesis of 3H Imidazo[4,5-b] pyridine (1a-1l) (procedure A)
Solution of 2 nitro 3 aminopyridine (1.0 mmol) and substituted aldehyde (1.0 mmol) in DMF (4 mL) was treated with 1 M aqueous Na2S2O4 (3.0 mmol, 3 mL) [Figure 8, Scheme 1]. After heating the reaction mixture at 60°C for 24 h, reaction mixture was filtered to remove unreacted Na2S2O4. The clear filtrate was cooled to room tampere and excess solvent was removed by high vacuum distillation. The concentrated residue formed in the distillating flask was washed with water (2 ml × 15 ml) and dried under reduced pressure to afford the desired product in satisfactory purity. Further recrystallization was carried using ethanol. Purified compounds were subjected for melting point and reaction progress was monitored with TLC and respective chemical test.
Figure 8.
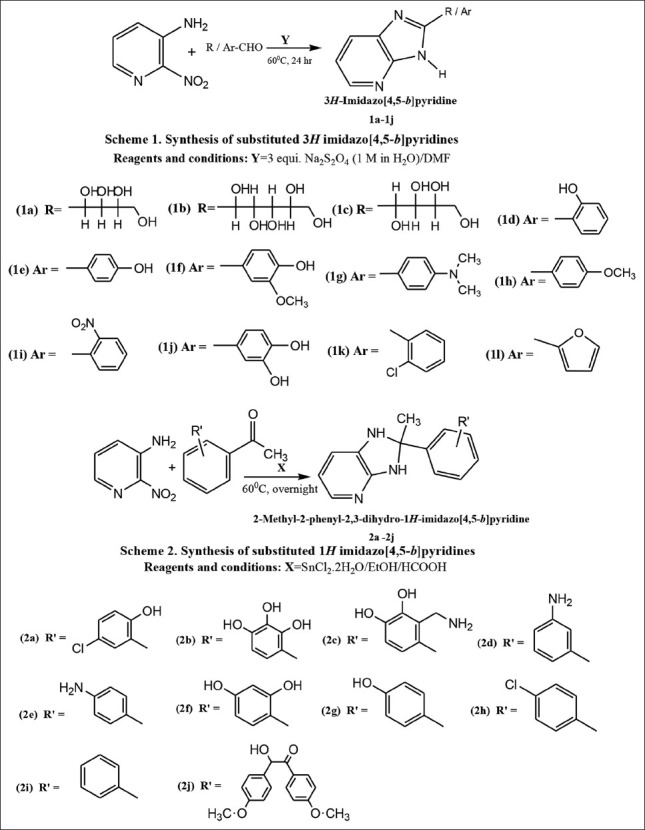
Scheme of synthesis of title compounds
1-(3H-Imidazo[4,5-b] pyridin-2-yl)-butane-1,2,3,4-tetraol (1a)
This derivative was synthesized according to the general procedure A. Yield 79.65%, as pale yellow solid, m.p. 160-165°C. IR (KBr, cm−1): 3600 (O-Haliph), 3350 (NH), 3250 (CHarom), 2900 (CHaliph), 1650 (C = Narom), 1500 (C = Carom), 1350 (C-Caliph), 1275 (C-Narom), 1100 (C-Oaliphalco), 800 (bend CHaliph), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.5 (s, 1H), 7.89 (t, 3H), 3.78 (s, 4H), 2.0 (s, 4H). 13C NMR (400 MHz, CDCl3, in ppm): 154, 149, 138, 128, 116, 33. HRMS (ESI): calcd. for C10H13N3O4 [M + H]+: 240.097; found 240.227. Analysis Calcd. for C10H13N3O4 (239.23); C, 49.06; H, 5.57; N, 15.59; O, 29.71%. Found: C, 49.06; H, 5.55; N, 15.59; O, 29.79%.
1-(3H-imidazo[4,5-b] pyridin-2-yl) pentane-1,2,3,4,5-pentol (1b)
This derivative was synthesized according to the general procedure A. Yield 89.58%, as gray solid, m.p. 145-150°C. IR (KBr, cm−1): 3600 (O-Haliph), 3350 (N-H), 3250 (CHarom), 2900 (CHaliph), 1650 (C = Narom), 1500 (C = Carom), 1350 (C-Caliph), 1275 (C-Narom), 1100 (C-Oaliphalcoholic), 800(bend CHaliph), 770 (bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 13.5 (s, 1H), 7.87 (s, 3H), 3.79 (t, 5H), 2 (t, 5H). HRMS (ESI): calcd. for C11H15N3O5 [M + H]+: 270.108447; found 270.118349. Analysis Calcd. for C11H15N3O5 (269.253); C, 50.20; H, 5.43; N, 17.55; O, 26.75%. Found: C, 50.22; H, 5.33; N, 17.54; O, 26.65%.
1-(3H-imidazo[4,5-b] pyridin-2-yl) butane-1,2,3,4-tetrol (1c)
This derivative was synthesized according to the general procedure A. Yield 79.65%, as orange solid, m.p. 150-154°C. IR (KBr, cm−1): 3600 (O-Haliph), 3350 (NH), 3250 (CHarom), 2900 (CHaliph), 1650 (C = Narom), 1500 (C = Carom), 1350 (C-Caliph), 1275 (C-Narom), 1100 (C-Oaliphalcoholic), 800 (bend CHaliph), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.5 (s, 1H), 7.89 (t, 3H), 3.78 (s, 4H), 2.0 (s, 4H). 13C NMR (400 MHz, CDCl3, in ppm): 154, 149, 138, 128, 116, 33. HRMS (ESI): calcd. for C10H13N3O4 [M + H]+: 240.097; found 239.227. Analysis Calcd. for C10H13N3O4 (239.23); C, 49.06; H, 5.57; N, 15.59; O, 29.71%. Found: C, 49.06; H, 5.55; N, 15.59; O, 29.79%.
2-(3H-imidazo[4,5-b] pyridin-2-yl) phenol (1d)
This derivative was synthesized according to the general procedure A. Yield 68.00%, as faint yellow solid, m.p. 135-140°C. IR (KBr, cm−1): 3600 (OH), 3350 (NH), 3250 (CHarom), 1650 (C = Narom), 1500 (C = Carom), 1275 (C-Narom), 1250 (C-Ophenolic), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H), 7.8 (s, 3H), 7.3 (s, 4H), 5.1 (s, 1H). 13C NMR (400 MHz, CDCl3, in ppm): 154, 149, 138, 128, 116, 33. HRMS (ESI): calcd. for C12H9N3O [M + H]+: 212.08183; found 212.07346. Analysis Calcd. for C12H9N3O (211.21); C, 68.17; H, 4.26; N, 19.89; O, 5.57%. Found: C, 68.17; H, 4.24; N, 19.89; O, 5.56%.
4-(3H-imidazo[4,5-b] pyridin-2-yl) phenol (1e)
This derivative was synthesized according to the general procedure A. Yield 78.00%, as faint yellow solid, m. p. 150-155°C. IR (KBr, cm−1): 3600 (OH), 3350 (NH), 3250 (CHarom), 1650 (C = Narom), 1500 (C = Carom), 1275 (C-Narom), 1250 (C-Ophenolic), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H), 7.8 (s, 3H), 7.3 (s, 4H), 5.1 (s, 1H). 13C NMR (400 MHz, CDCl3, in ppm): 154, 149, 138, 128, 116, 33. HRMS (ESI): calcd. for C12H9N3O [M + H]+: 212.08183; found 212.07346. Analysis Calcd. for C12H9N3O (211.21); C, 68.17; H, 4.26; N, 19.89; O, 5.57%. Found: C, 68.17; H, 4.24; N, 19.89; O, 5.56%.
4-(3H-imidazo[4,5-b] pyridin-2-yl)-2-methoxyphenol (1f)
This derivative was synthesized according to the general procedure A. Yield 69.06%, as orange solid, m.p. 148-150°C. IR (KBr, cm−1): 3600 (OH), 3350 (NH), 3250 (CHarom), 1650 (C = Narom), 1500 (C = Carom), 1275 (C-Narom), 1250 (C-Ophenolic), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H), 7.8 (s, 3H), 7.3 (s, 4H), 5.1 (s, 1H). 13C NMR (400 MHz, CDCl3, in ppm): 154, 149, 138, 128, 116, 33. HRMS (ESI): Calcd. for C12H9N3O [M + H]+: 212.08183; found 212.07346. Analysis Calcd. for C12H9N3O (211.21); C, 68.17; H, 4.26; N, 19.89; O, 5.57%. Found: C, 68.17; H, 4.24; N, 19.89; O, 5.56%.
4-(3H-imidazo[4,5-b] pyridin-2-yl)-N, N-dimethylaniline (1g)
This derivative was synthesized according to the general procedure A. Yield 75.03%, as yellow solid, m.p. 150-153°C. IR (KBr, cm−1): 3600 (OH), 3350 (NH), 3250 (CHarom), 1650 (C = Narom), 1500 (C = Carom), 1275 (C-Narom), 1250 (C-Ophenolic), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H), 7.8 (s, 3H), 7.3 (s, 4H), 5.1 (s, 1H). 13C NMR (400 MHz, CDCl3, in ppm): 154, 149, 138, 128, 116, 33. HRMS (ESI): calcd. for C12H9N3O [M + H]+: 212.08183; found 212.07346. Analysis Calcd. for C12H9N3O (211.21); C, 68.17; H, 4.26; N, 19.89; O, 5.57%. Found: C, 68.17; H, 4.24; N, 19.89; O, 5.56%.
2-(4-methoxyphenyl)-3H-imidazo[4,5-b] pyridine (1h)
This derivative was synthesized according to the general procedure A. Yield 82.5%, as faint yellow-orange solid, m.p. 189-193°C. IR (KBr, cm−1): 3350 (NH), 3250 (CHarom), 2850 (CH3), 1500 (C = Carom), 1650 (C = Narom), 1375 (bend CH3), 1275 (C-Narom), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H). 7.8 (s, 3H), 7.5 (s, 4H). 13C NMR (400 MHz, CDCl3, in ppm): 149.8, 123.6, 122.3, 138, 128, 56. HRMS (ESI): Calcd. for C13H11N3O [M + H]+: 226.097488; found 226.096565. Analysis Calcd. for C13H11N3O (225.24); C, 69.20; H, 4.9; N, 18.64; O, 7.10%. Found: C, 69.20; H, 4.91; N, 18.59; O, 7.11%.
2-(2-nitrophenyl)-3H-imidazo[4,5-b] pyridine (1i)
This derivative was synthesized according to the general procedure A. Yield 63.02%, as dark yellow solid, m.p. 175-180°C. IR (KBr, cm−1): 3600 (OHarom), 3350 (NH), 3250 (CHarom), 2850 (CH3), 1650 (C = Narom), 1500 (C = Carom), 1475 (N = O), 1375 (bend CH3), 1275 (C-Narom), 1250 (C-Ophenolic), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H) 7.7(s, 3H), 7.3 (s, 4H). 13C NMR (400 MHz, CDCl3, in ppm): 149, 145, 135, 130, 117, 121.3. HRMS (ESI): Calcd. for C13H11N3O [M + H]+: 226.097488; found 226.096565. Analysis Calcd. for C13H11N3O (225.24); C, 69.20; H, 4.9; N, 18.64; O, 7.10%. Found: C, 69.20; H, 4.91; N, 18.59; O, 7.11%.
4-(3H-imidazo[4,5-b] pyridin-2-yl) benzene-1,2-diol (1j)
This derivative was synthesized according to the general procedure A. Yield 85.55%, as greenish brown solid, m.p. 180-185°C. IR (KBr, cm−1): 3600 (O-Hphenol), 3350 (N-H), 3250 (CHarom), 1650 (C = Narom), 1500 (C = Carom), 1350 (C-Caliph), 1275 (C-Narom), 1250 (C-Ophenol), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H), 7.5 (t, 3H), 6.5 (t, 3H), 5.0 (s, 2H). 13C NMR (400 MHz, CDCl3, in ppm): 149.8, 123.6, 122.3, 138, 128, 56. HRMS (ESI): Calcd. for C12H9N3O2 [M + H]+: 228.076753; found 228.108912. Analysis Calcd. for C12H9N3O2 (227.21); C, 63.37; H, 3.96; N, 18.47; O, 14.08%. Found: C, 63.37; H, 3.96; N, 18.47; O, 14.08%.
2-(2-chlorophenyl)-3H-imidazo[4,5-b] pyridine (1k)
This derivative was synthesized according to the general procedure A. Yield 98.22%, as off white solid, m.p. 169-172°C. IR (KBr, cm−1): 3350 (N-H), 3250 (CHarom), 1650 (C = Narom), 1500 (C = Carom), C-N Ar 1275, 750 (C-Cl), 770 (bend CHarom). 1HNMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H), 7.7 (t, 3H), 7.2 (m, 4H). 13C NMR (400 MHz, CDCl3, in ppm): 149.8, 136.3, 135.7, 129.6, 128.3, 122.4. HRMS (ESI): Calcd. for C12H8 ClN3 [M + H]+: 230.66502; found 230.57092. Analysis Calcd. for C12H8 ClN3 (229.66); C, 62.88; H, 3.49; N, 18.34; O, 15.28%. Found: C, 62.88; H, 3.49; N, 18.34; O, 15.28%.
2-(furan-2-yl)-3H-imidazo[4,5-b] pyridine (1l)
This derivative was synthesized according to the general procedure A. Yield 89.34%, as purple solid, m.p. 89.6-94.8°C. IR (KBr, cm−1): 3350 (N-H), 3250(s CHarom), 1650 (C = Narom), 1500 (C = Carom), 1275 (C-Narom), 1100 (C-O), 770 (s bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 13.4 (s, 1H), 7.8 (s, 3H), 6.5 (s, 3H). 13C NMR (400 MHz, CDCl3, in ppm): 154, 135, 140, 123, 111. HRMS (ESI): calcd. for C10H7N3O [M + H]+: 186.066188; found 186.044543. Analysis Calcd. for C10H7N3O (185.18); C, 64.80; H, 3.78; N, 22.68; O, 8.64%. Found: C, 64.80; H, 3.78; N, 22.68; O, 8.64%.
General experimental procedure for the synthesis of 1H Imidazo[4,5-b] pyridine (2a-2j) (procedure B)
In the present synthesis, mixture of 2 nitro 3 aminopyridine and substituted acetophenones were refluxed in EtOH along with SnCl2 H2O and formic acid as a catalyst at 60°C for overnight time period. After the reaction is over, the reaction mixture was cooled at room temperature and filtered at normal temperature. Clear filtrate was subjected for distillation under reduced pressure (in vacuo). Concentrate in the distillating flask is kept open at room temperature for air oxidation and evaporation of trapped solvent. Crude solid mass was subjected further for washing with water (2 ml × 30 ml). Next recrystallization was performed with solvent benzene to obtain crystalline product of expected purity. Reaction progress was monitored with TLC on precoated plates. Dry crystals were subjected for m.p and respective chemical tests [Figure 8, Scheme 2]. This type of product has been reported previously by Scheuerman and Tumelty.[49] and is presumably formed through formylation of the aniline nitrogen, nitro reduction and cyclization.
Formylation of the aniline nitrogen is believed to assist nitro reduction. In summary, we have demonstrated that imidazopyridines can be efficiently prepared from a support-bound nitro aminopyridine using a “one-pot” reduction–cyclisation method. This approach has provided the shortest solid phase synthesis of imidazopyridines to date. Since a wide range of amines and aldehydes are commercially available, a large number of pyridoimidazoles and respective benzimidazoles can be easily prepared using this method.
4-chloro-2-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) phenol (2a)
This derivative was synthesized according to the general procedure B. Yield 78.11%, as faint white solid, m.p. 219-223°C. IR (KBr, cm−1): 3500 (O-Hphenolic), 3350 (N-H), 3250 (CHarom), 2850 (CH3), 1650 (C = Narom), 1456 (C = Carom), 1275 (C-Narom), 1250 (C-Ophenol), 800 (bend CHaliph), 770 (bend CHarom), 750 (C-Cl). 1H NMR (400 MHz, CDCl3, in ppm): 7.7 (m, 4H), 7.0 (m, 3H), 5 (s, 1H, OH), 4 (s, 2H, NH), 1.5 (s, 3H, CH3), 13C NMR (400 MHz, CDCl3, in ppm): 150, 135, 130, 123, 65, 35. HRMS (ESI): calcd. for C13H12 ClN3O [M + H]+: 262.074166; found 262.04697. Analysis Calcd. for C13H12 ClN3O (261.70); C, 59.61; H, 4.58; N, 16.04; O, 6.11; Cl, 13.37%. Found: C, 59.61; H, 4.58; N, 16.04; O, 6.11; Cl, 13.37%.
4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) benzene-1,2,3-triol (2b)
This derivative was synthesized according to the general procedure B. Yield 67.89%, as white solid, m.p. 167-170°C. IR (KBr, cm−1): 3600 (O-Haliph), 3350 (N-H), 3250 (CHarom), 2900 (CHaliph), 1650 (C = Narom), 1500 (C = Carom), 1350 (C-Caliph), 1275 (C-Narom), 1100 (C-Oaliphalcoholic), 800 (bend CHaliph), 770 (bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 13.5 (s, 1H), 7.87 (s, 3H), 3.79 (t, 5H), 2 (t, 5H). 13C NMR (400 MHz, CDCl3, in ppm): 149, 144, 139, 130, 123, 113, 65, 33. HRMS (ESI): calcd. for C13H13N3O3 [M + H]+: 260.102968; found 262.14397. Analysis Calcd. for C13H13N3O3 (259.26); C, 60.17; H, 6.17; N, 16.19; O, 18.51%. Found: C, 60.17; H, 6.18; N, 16.20; O, 18.52%.
3-(aminomethyl)-4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-l) benzene-1,2-diol (2c)
This derivative was synthesized according to the general procedure B. Yield 73.00%, as pale yellow solid, m.p. 211-214°C. IR (KBr, cm−1): 3500 (O-Hphenolic), 3350 (N-Harom), 3300 (N-Haliph), 3250 (CHarom), 2850 (CH3), 2700 (CH2), 1650 (C = Narom), 1456 (C = Carom), 1275 (C-Narom), 1250 (C-Ophenolic), 1100 (C-Naliph), 800 (bend CHaliph), 770 (bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 7.5 (m, 3H), 6.5 (m, 4H), 4.0 (s, 2H, NH), 1.5 (s, 3H, CH3). 13C NMR (400 MHz, CDsCl3, in ppm): 149, 142, 138, 135, 130, 123, 114, 33, 31. HRMS (ESI): calcd. for C14H16N4O2 [M + H]+: 273.134602; found 273.144312. Analysis Calcd. for C14H16N4O2 (272.30); C, 61.69; H, 5.87; N, 20.56; O, 11.75%. Found: C, 61.69; H, 5.87; N, 20.55; O, 11.72%.
3-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) aniline (2d)
This derivative was synthesized according to the general procedure B. Yield 77.00%, as yellow solid, m.p. 224-228°C. IR (KBr, cm−1): 3450 (N-H), 3350 (N-Harom), 3250 (CHarom), 2850 (CH3), 1650 C = Narom), 1456 (C = Carom), 1275 (C-Narom), 770 (bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 7.5 (m, 3H), 6.5 (m, 4H), 4.0 (s, 2H, NH), 1.5 (s, 3H, CH3). 13C NMR (400 MHz, CDCl3, in ppm): 149, 143, 138, 135, 123, 113, 117, 74, 32. HRMS (ESI): calcd. for C13H14N4 [M + H]+: 227.129123; found 227.11813. Analysis Calcd. for C13H14N42 (226.27); C, 68.4; H, 6.18; N, 24.74%. Found: C, 68.4; H, 6.18; N, 24.74%.
4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) aniline (2e)
This derivative was synthesized according to the general procedure B. Yield 80.11%, as pale yellow solid, m.p. 220-223°C. IR (KBr, cm−1): 3450 (N-H), 3350 (N-Harom), 3250 (CHarom), 2850 (CH3), 1650 C = Narom), 1456 (C = Carom), 1275 (C-Narom), 770 (bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 7.5 (m, 3H), 6.5 (m, 4H), 4.0 (s, 2H, NH), 1.5 (s, 3H, CH3). 13C NMR (400 MHz, CDCl3, in ppm): 149, 143, 138, 135, 123, 113, 117, 74, 32. HRMS (ESI): Calcd. for C13H14N4 [M + H]+: 227.129123; found 227.11813. Analysis Calcd. for C13H14N42 (226.27); C, 68.4; H, 6.18; N, 24.74%. Found: C, 68.4; H, 6.18; N, 24.74%.
4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) benzene-1,3-diol (2f)
This derivative was synthesized according to the general procedure B. Yield 76.89%, as pale yellow solid, m.p. 238-240°C. IR (KBr, cm−1): 3500 (OHphenolic), 3350 (N-H), 3250 (CHarom), 2850 (CH3), 1650 (C = Narom), 1456 (C = Carom), 1275 (C-Narom), 1250 (C-Ophenol), 800 (bend CHaliph), 770 (bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 7.5 (m, 3H), 6.9 (m, 3H), 5.0 (s, 1H, OH), 4.0 (s, 2H, NH), 1.5 (s, 3H, CH3). 13C NMR (400 MHz, CDCl3, in ppm): 157, 149, 138, 130, 122, 108, 102, 64, 31. HRMS (ESI): calcd. for C13H13N3O2 [M + H]+: 244.108053; found 244.117458. Analysis Calcd. for C13H13N3O2 (243.26); C, 64.12; H, 5.34; N, 17.26; O, 13.15%. Found: C, 64.12; H, 5.33; N, 17.24; O, 13.16%.
4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) phenol (2g)
This derivative was synthesized according to the general procedure B. Yield 82.56%, as yellow solid, m.p. 230-234°C. IR (KBr, cm−1): 3500 (O-Hphenolic), 3350 (N-H), 3250 (CHarom), 1650 (C = Narom), 1456 (C = Carom), 1275 (C-Narom), 1250 (C-Ophenol), 800 (bend CHaliph), 770 (bend CHarom), 1H NMR (400 MHz, CDCl3, in ppm): 7.5(m, 3H), 6.9 (m, 4H), 5.0 (s, 1H, OH), 4.0 (s, 2H, NH), 1.5(s, 3H, CH3). 13C NMR (400 MHz, CDCl3, in ppm): 155, 149, 138, 135, 130, 123, 115, 74, 32. HRMS (ESI): calcd. for C13H13N3O [M + H]+: 228.113138; found 228.113458. Analysis Calcd. for C13H13N3O (227.26); C, 68.64; H, 5.72; N, 18.48; O, 7.04%. Found: C, 68.64; H, 5.72; N, 18.48; O, 7.04%.
2-(4-chlorophenyl)-2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridine (2h)
This derivative was synthesized according to the general procedure B. Yield 65.23%, as yellow solid, m.p. 215-218°C. IR (KBr, cm−1): 3350 (N-H), 3250 (CHarom), 2850 (CH3), 1650 (C = Narom), 1456 (C = Carom), 1275 (C-N arom), 800 (bend, CHaliph), 770 (bend CHarom), 750 (C-Cl). 1H NMR (400 MHz, CDCl3, in ppm): 7.25 (m, 3H), 6.9 (m, 3H), 4.0 (s, 2H, NH), 1.5 (s, 3H, CH3). 13C NMR (400 MHz, CDCl3, in ppm): 149, 140, 138, 130, 123, 128, 113, 74, 32. HRMS (ESI): calcd. for C13H12 ClN3 [M + H]+: 246.079252; found 246.085552. Analysis Calcd. for C13H12 ClN3 (245.70); C, 63.49; H, 4.88; N, 17.09; Cl, 14.24%. Found: C, 63.49; H, 4.88; N, 17.09; Cl, 14.24%.
2-methyl-2-phenyl-2,3-dihydro-1H-imidazo[4,5-b] pyridine (2i)
This derivative was synthesized according to the general procedure B. Yield 75.99%, as yellow solid, m.p. 119-124°C. IR (KBr, cm−1): 3350 (N-H), 3250 (CHarom), 2850 (CH3), 1650 (C = Narom), 1456 (C = Carom), 1275 (C-Narom), 800 (bend CHaliph), 770 (bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 7.2 (m, 5H), 7.0 (m, 3H), 4.0 (s, 2H, NH), 1.5 (s, 3H, CH3). 13C NMR (400 MHz, CDCl3, in ppm): 149, 142, 138, 130, 128, 123, 74, 32. HRMS (ESI): calcd. for C13H13N3 [M + H]+: 212.118224; found 212.109232. Analysis Calcd. for C13H13N3 (211.26); C, 73.84; H, 6.15; N, 19.88%. Found: C, 73.84; H, 6.15; N, 19.88%.
4-methoxyphenyl)[2-(4-methoxyphenyl)-2,3-dihydro-1H-imidazo[4,5-b] pyridine-2-yl] methanol (2j)
This derivative was synthesized according to the general procedure B. Yield 62.11%, as white solid, m.p. 250-254°C. IR (KBr, cm−1): 3600 (O-H), 3350 (N-H), 3250 (CHarom), 2850 (CH3), 1650 (C = Narom), 1456 (C = Carom), 1290 (C-O), 1275 (C-Narom), 800 (bend CHaliph), 770 (bend CHarom). 1H NMR (400 MHz, CDCl3, in ppm): 7.2 (m, 3H), 6.9 (m, 4H), 5.3 (s, 1H, CHaliph), 4.0 (s, 2H, NH), 2.0 (s, 1H, OH), 3.5 (s, 6H). 13C NMR (400 MHz, CDCl3, in ppm): 160, 128, 123, 115, 90, 87, 57. HRMS (ESI): calcd. for C21H21N3O3 [M + H]+: 364.165568; found 364.155549. Analysis Calcd. for C21H21N3O3 (363.40); C, 69.34; H, 5.77; N, 11.55; O, 13.20%. Found: C, 69.34; H, 5.77; N, 11.55; O, 13.20%.
In vitro anti-bacterial procedure
In vitro antimicrobial activity was carried out using disc diffusion assay.[22,23] Whatman No. 1 filter paper discs of 5 mm diameter were sterilized by autoclaving for 15 min at 121°C. The sterile discs were impregnated with the test compounds (500 μg/disc). The agar plates were inoculated with standard inoculum (10 cells/ml broth) of the quality control test organism namely Gram-positive M. tuberculosis, Staphylococcus aureus (ATCC 25923) and Gram-negative Escherichia coli (ATCC 25922), Brucella abortus. The impregnated discs were placed on the agar plate medium, and the plates were incubated at 5-6°C for 1 h to permit good diffusion and then transferred to an incubator at 37°C for 24 h. The diameter of inhibition zone was measured using a caliber, to the nearest millimeter. Among the tested compounds, those exhibiting moderate activity (inhibition zone >15 mm) were subjected to a quantitative assay in order to determine their minimum inhibitory concentrations (MICs) using the 2-fold serial broth dilution assay.[24,7]
Standardized bacterial inoculum were prepared by touching the top of four or five colonies of a single type and inoculating them into a tube containing 5 mL of Mueller-Hinton broth (Difco) at pH 7.3. Incubations of these microorganism suspensions were carried out at 35°C until a visible turbidity was obtained. Finally, the culture was diluted so that, after inoculation, each microplate well had an inoculum size of 5 × 105 colony forming units (CFU)/ml. Antibacterial assays were performed in Mueller-Hinton broth (Difco) at pH 7.3. The dilutions in the test medium were prepared at the required concentration of 100–1 μg/ml, and for standard compound ciprofloxacin at 40-0.015 μg/ml.
After inclusion of 100 μg/ml of the broth containing the standard drug or the test compound, 100 μg/ml of bacterial suspensions were inoculated into microplate wells. After incubation for 16-20 h at 35°C, the well-containing the lowest concentration of the standard drug or the test compound that inhibit microorganism growth as detected by the unaided eye, was recorded to represent the MIC expressed in μg/ml [Tables 1 and 2]. The MIC was defined as the lowest concentration of the antibiotic or test sample allowing no visible growth.
Table 1.
MIC (μg/ml)a for 3H Imidazo[4,5-b] pyridine (1a-1l)
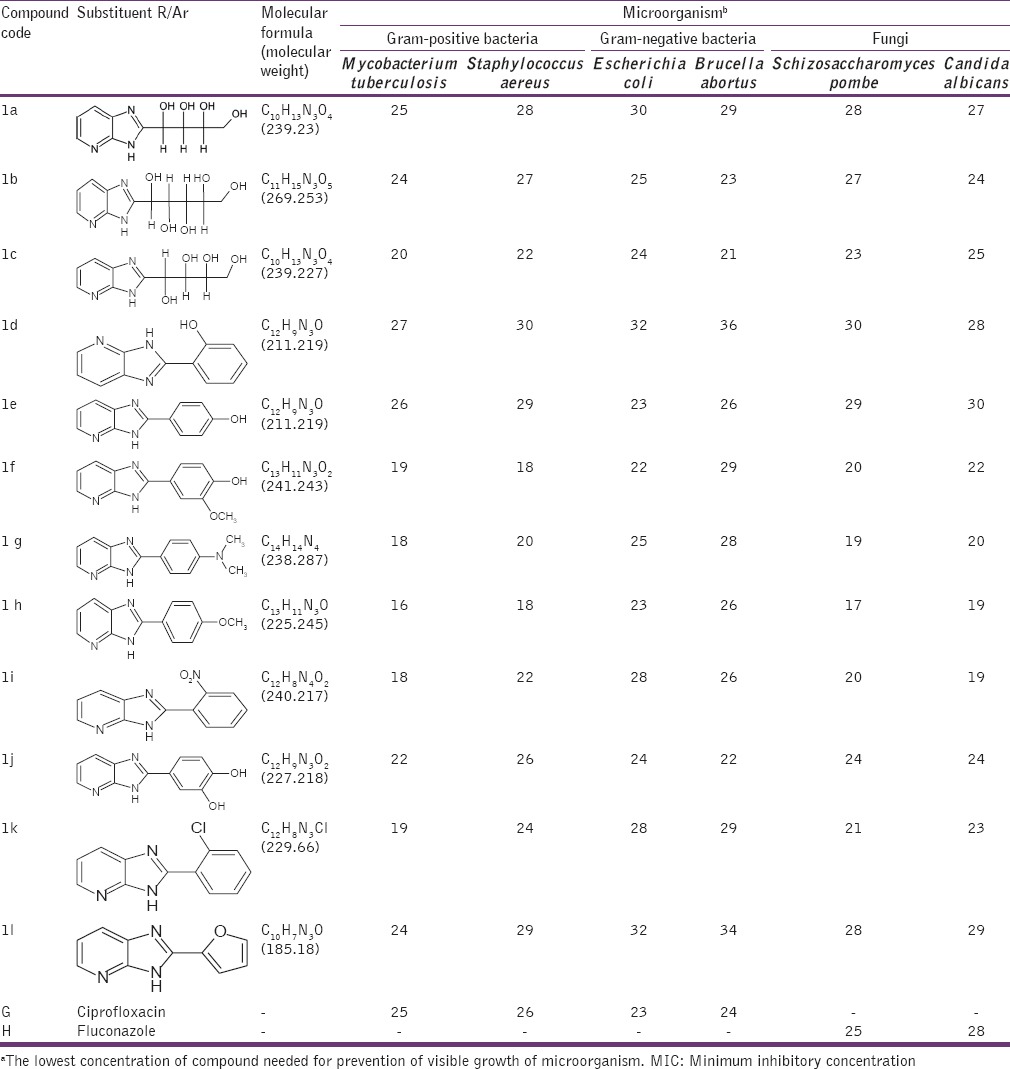
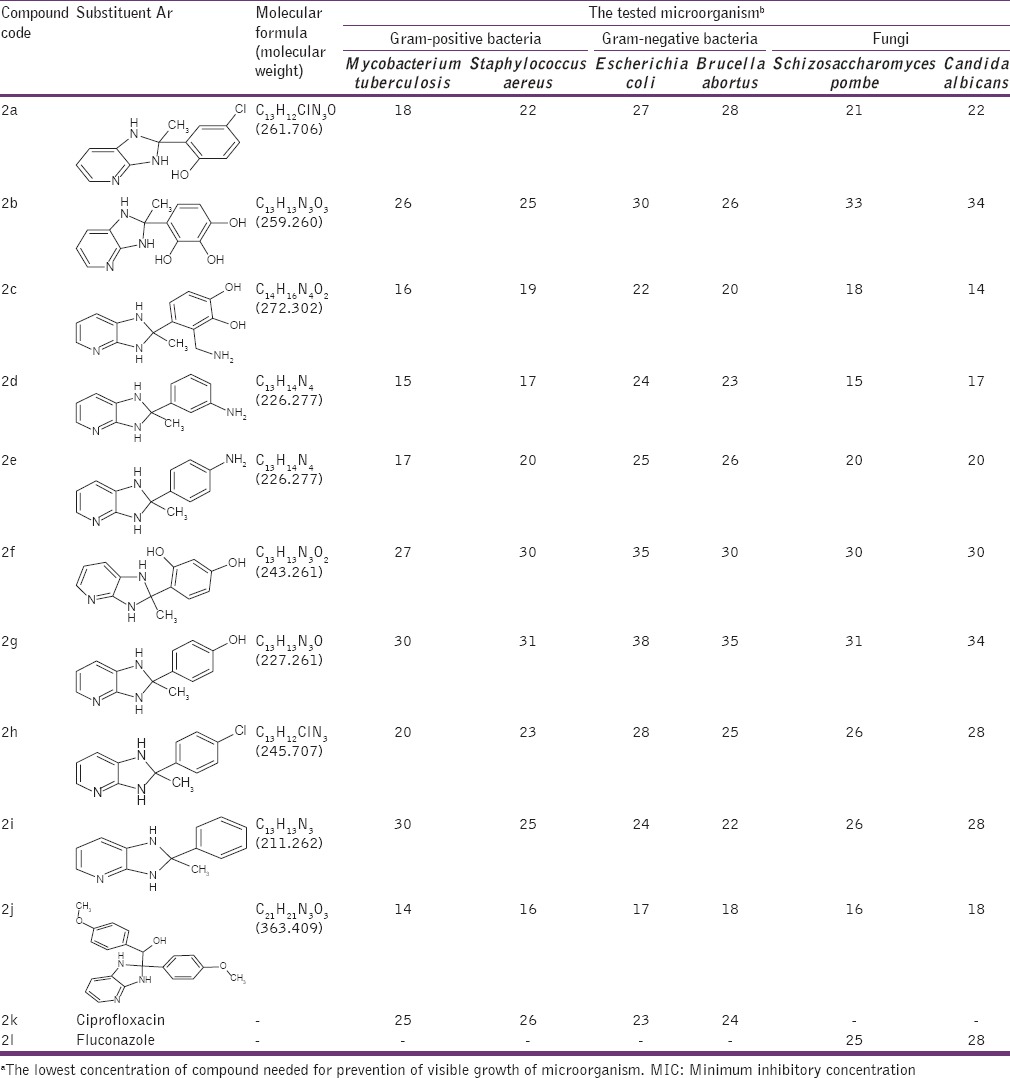
Table 2.
MIC (μg/ml)a of 1H imidazo[4,5-b] pyridine (2a-2j)
In vitro anti-fungal procedure
Fungal quality control strains used were Schizosaccharomyces pombe, Candida albicans (ATCC 90018). Disc diffusion assay[22,23] method was used as described above. All fungi used were cultivated in Sabouraud's Dextrose Agar medium (Merck) with L-glutamine, buffered with 3-(N-morpholino) propane-sulfonic acid at pH 7.4. The culture was further diluted after inoculation with each microplate well had final inoculum density as 0.5-2.5 × 103 CFU/ml. Standard drug used was fluconazole. The stock solutions of the test compounds and standard compound were prepared in dimethyl sulfoxide (DMSO). The microtiter plates were incubated at 35°C and evaluated visually after 48 h. It was established that dilution of DMSO lacked antimicrobial activity against any of the test microorganisms.
Results and Discussion
In-silico docking
An attempt tried for in-silico design of title compounds aiming to inhibit lumazine synthase. Binding interaction of test motifs studied using crystal protein lumazine synthase from M. tuberculosis (PDB 2C92).[1] With the aim of rationalizing the antimicrobial activity data obtained, docking study was performed for the imidazo [4,5-b] pyridine derivatives 1a-1l and 2a-2j in order to investigate the possible interactions with lumazine synthase. Minimum docking score for test ligands was showed in Table 3 in comparison with the reference ligand Trimethoprim.
Table 3.
MolDock scores of various (3H) imidazo[4,5-b] pyridines (1a-1l) and (1H) imidazo[4,5-b] pyridines (2a-2j)
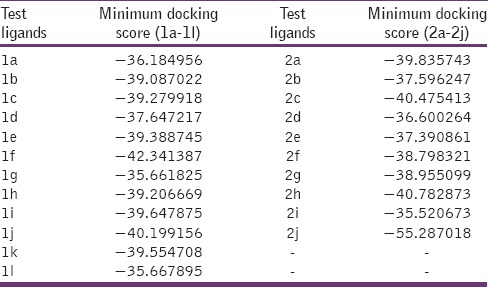
Reference ligand Trimethoprim binding to the active site of the enzyme showed in Figures 9 and 10 for validation of docking protocol and confirmation of the biological data. Complex between the enzyme's active site and compounds 2-(2-nitrophenyl)-3H-imidazo[4,5-b] pyridine (1i), 4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) benzene-1,3-diol (2f) showed in Figures 4-7. Different types of interactions studied provides some important SAR points supporting possible interaction.
Figure 9.
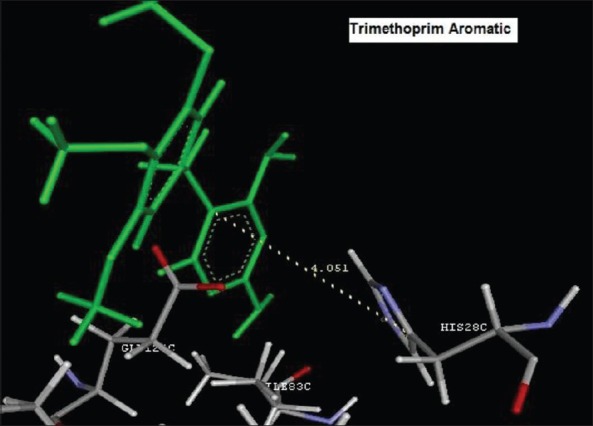
Aromatic binding of Trimethoprim to the active site HIS28C
Figure 10.
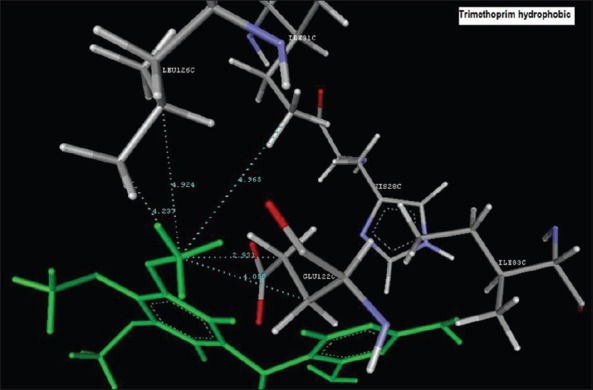
Hydrophobic binding of Trimethoprim to the active pocket ILE31C, GLU122C, GLU122C, LEU126C, LEU126C
Chemistry
Previously one-step method was reported for benzimidazole synthesis from o-nitro aniline and aldehydes.[50] In the present one-step synthesis of 3H imidazo [4,5-b] pyridines (1a-1j) successful attempt was made from aldehydes and 2-nitro-3-amino pyridine through reductive cyclisation using Na2S2O4. Aqueous paste of Na2S2O4 was prepared as 1M in H2O and added in 3 equivalent proportions to the reaction mixture [Figure 8, Scheme 1].
Second reaction for one-step synthesis of 1H imidazo [4,5-b] pyridine (2a-2l) was obtained from ketones and 2-nitro-3-amino pyridine through reductive cyclization using SnCl2.2H2O as reductive catalyst [Figure 8, Scheme 2]. This type of approach has been reported previously for obtaining benzimidazole motifs.[51] Imidazopyridine scaffolds were visited after treatment of the substituted acetophenones and 2-nitro-3-amino pyridine with the addition of SnCl2.H2O in the presence of formic acid. It is presumably formed through formylation of the aniline nitrogen, nitro reduction and cyclization. Formylation of the aniline nitrogen is believed to assist nitro reduction, In summary, we have demonstrated that imidazo pyridines can be efficiently prepared from a support-bound 2 nitro 3 amino pyridine as like the same way benzimidazoles reported using a ‘one-pot’ reduction-cyclisation method. Both reactions were clear and without any form of side products or byproducts as impurities.
In vitro antimicrobial screening
The antimicrobial activities of the compounds 1a-1j and 2a-2l were tested against Gram-positive bacteria M. tuberculosis, S. aureus, Gram-negative bacteria E. coli, B. abortus and fungi S. pombe, C. albicans. All selected isolates were reported to express protein lumazine synthase involved in Riboflavin path. MIC was measured as described in the experimental section. Ciprofloxacin and Fluconazole were used as positive controls for antibacterial and antifungal activity, respectively. Solutions of different concentrations of Ciprofloxacin, Fluconazole and test compounds (1a-1l, 2a-2j) were prepared by dissolving them in DMSO. Bacterial suspensions at 5 × 105 CFU and fungal suspension 2.5 × 103 (CFU/mL) were inoculated into the wells. Antibacterial plates were incubated at 37°C for 16-20 h and antifungal plates were evaluated after incubation at 35°C for 48 h. The MIC (μg/ml) values were determined using a disc diffusion method. The results of in vitro antimicrobial activities of the synthesized compounds are listed in Tables 1 and 2.
The results showed a wide range of antimicrobial activities among the different derivatives tested with MIC values, defined as the lowest concentration of drug at which no visible growth is observed from 100 to 1.0 μg/ml [Tables 1 and 2] with the most active compounds belonging to those having chloro- and nitro-substituents. Derivatives 2-(2-chlorophenyl)-3H-imidazo[4,5-b] pyridine (1k), 2-(2-nitrophenyl)-3H-imidazo[4,5-b] pyridine (1i), 4-chloro-2-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) phenol (2a), 3-(aminomethyl)-4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-l) benzene-1,2-diol (2c), 3-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) aniline (2d), 4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) aniline (2e), 2-(4-chlorophenyl)-2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridine (2h) exhibited potent activities. Compounds 4-(3H-imidazo[4,5-b] pyridin-2-yl)-2-methoxyphenol (1f), 4-(3H-imidazo[4,5-b] pyridin-2-yl)-N, N-dimethylaniline (1g), 2-(4-methoxyphenyl)-3H-imidazo[4,5-b] pyridine (1h), 2-(furan-2-yl)-3H-imidazo[4,5-b] pyridine (1l), 4-methoxyphenyl)[2-(4-methoxyphenyl)-2,3-dihydro-1H-imidazo[4,5-b] pyridine-2-yl] methanol (2j) showed moderate anti-microbial activity. Whereas compounds 4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) benzene-1,2,3-triol (2b), 4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) benzene-1,3-diol (2f), 4-(2-methyl-2,3-dihydro-1H-imidazo[4,5-b] pyridin-2-yl) phenol (2g), 2-methyl-2-phenyl-2,3-dihydro-1H-imidazo[4,5-b] pyridine (2i) behave with poor antimicrobial activity against bacteria and fungi compared to standard Ciprofloxacin and Fluconazole respectively. Some qualitative structure activity relationships could be concluded from Tables 1 to 2. The superiority of the compounds having R’ = Cl ˃ No2 ˃ NH2 at the phenyl moiety resident on the imidazopyridine ring (1H imidazo 4,5-b pyridine) and R/Ar = Cl ˃ No2 ˃ NH2 containing substituent on the 2nd position of the imidazole ring (3H imidazo 4,5-b pyridine) is obviously evident from Tables 1 and 2. Those compounds having to possess alkoxy (OCH3), alkylamine (dimethyl amine) and furan heterocycle were observed with moderate activity, whereas compounds with more polar substituents (polyhydroxy) were observed with poor antimicrobial activity. We next strove to arrive at the title motifs and study their growth inhibitory activity (MIC) against Gram-positive bacteria M. tuberculosis, S. aureus, Gram-negative bacteria E. coli, B. abortus and fungi S. pombe, C. albicans.
Conclusion
In summary, we have performed in-silico docking with the aim to study possible interactions between title compounds [1H,3H] 4,5-b imidazopyridine and macromolecule lumazine synthase. Further comparison of types of interaction with the reference ligand trimethoprim. On the basis of docking experiment next we synthesized and evaluated new imidazo [4,5-b] pyridines against various Gram-positive and Gram-negative bacteria and fungi expressing lumazine synthase. Many of the synthesized motifs showed potent antimicrobial activity compared to the control antibiotics ciprofloxacin and fluconazole. Characterization of the antimicrobial spectrum of the synthesized compounds as shown in Tables 1 and 2, indicating a broad spectrum of antimicrobial activity against the tested strains. Docking interaction study of lumazine synthase revealed perfect binding of test ligands in comparison with reported ligands. Minimum docking score of best fit ligands confirm their usefulness as Lumazine synthase inhibitors for in vitro tested pathogenic microorganism causing deadliest infection and further to arrest spread of infectious disease. These results form the foundation for further investigations in our laboratories.
Acknowledgments
Authors are grateful toward SAIF, Punjab University, Chandigarh for performing spectral studies of synthesized compounds. Also sincere thanks to Microanalytical Department, University of Pune for performing CHNO analysis.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Morgunova E, Illarionov B, Sambaiah T, Haase I, Bacher A, Cushman M, et al. Structural and thermodynamic insights into the binding mode of five novel inhibitors of lumazine synthase from Mycobacterium tuberculosis. FEBS J. 2006;273:4790–804. doi: 10.1111/j.1742-4658.2006.05481.x. [DOI] [PubMed] [Google Scholar]
- 2.Nakajima H. Tuberculosis: A global emergency. World Health. 1993;46:3. [Google Scholar]
- 3.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 4.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 5.Cushman M, Mavandadi F, Kugelbrey K, Bacher A. Synthesis of 2,6-dioxo-(1H,3H)-9-N-ribitylpurine and 2,6-dioxo-(1H,3H)-8-aza-9-N-ribitylpurine as inhibitors of lumazine synthase and riboflavin synthase. Bioorg Med Chem. 1998;6:409–15. doi: 10.1016/s0968-0896(98)00013-3. [DOI] [PubMed] [Google Scholar]
- 6.Talukdar A, Morgunova E, Duan J, Meining W, Foloppe N, Nilsson L, et al. Virtual screening, selection and development of a benzindolone structural scaffold for inhibition of lumazine synthase. Bioorg Med Chem. 2010;18:3518–34. doi: 10.1016/j.bmc.2010.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaiser J, Illarionov B, Rohdich F, Eisenreich W, Saller S, den Brulle JV, et al. A high-throughput screening platform for inhibitors of the riboflavin biosynthesis pathway. Anal Biochem. 2007;365:52–61. doi: 10.1016/j.ab.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Jin G, Illarionov B, Bacher A, Fischer M, Cushman M. Structure-based model of the reaction catalyzed by Lumazine Synthase from Aquifex aeolicus. J Mol Biol. 2003;328:167–82. doi: 10.1016/s0022-2836(03)00186-4. [DOI] [PubMed] [Google Scholar]
- 9.Volk R, Bacher A. Biosynthesis of riboflavin. The structure of the four-carbon precursor. J Am Chem Soc. 1988;110:3651–3. [Google Scholar]
- 10.Steinbacher S, Schiffmann S, Richter G, Huber R, Bacher A, Fischer M. Structure of 3,4-dihydroxy-2-butanone 4-phosphate synthase from Methanococcus jannaschii in complex with divalent metal ions and the substrate ribulose 5-phosphate: Implications for the catalytic mechanism. J Biol Chem. 2003;278:42256–65. doi: 10.1074/jbc.M307301200. [DOI] [PubMed] [Google Scholar]
- 11.Fischer M, Haase I, Kis K, Meining W, Ladenstein R, Cushman M, et al. Enzyme catalysis via control of activation entropy: Site-directed mutagenesis of 6,7-dimethyl-8-ribityllumazine synthase. J Mol Biol. 2003;326:783–93. doi: 10.1016/s0022-2836(02)01473-0. [DOI] [PubMed] [Google Scholar]
- 12.Fischer M, Haase I, Feicht R, Richter G, Gerhardt S, Changeux JP, et al. Biosynthesis of riboflavin: 6,7-dimethyl-8-ribityllumazine synthase of Schizosaccharomyces pombe. Eur J Biochem. 2002;269:519–26. doi: 10.1046/j.0014-2956.2001.02674.x. [DOI] [PubMed] [Google Scholar]
- 13.Bacher A, Eberhardt S, Fischer M, Mörtl S, Kis K, Kugelbrey K, et al. Biosynthesis of riboflavin: Lumazine synthase and riboflavin synthase. Methods Enzymol. 1997;280:389–99. doi: 10.1016/s0076-6879(97)80130-9. [DOI] [PubMed] [Google Scholar]
- 14.Haase I, Mörtl S, Köhler P, Bacher A, Fischer M. Biosynthesis of riboflavin in archaea.6,7-dimethyl-8-ribityllumazine synthase of Methanococcus jannaschii. Eur J Biochem. 2003;270:1025–32. doi: 10.1046/j.1432-1033.2003.03478.x. [DOI] [PubMed] [Google Scholar]
- 15.Haase I, Fischer M, Bacher A, Schramek N. Temperature-dependent presteady state kinetics of lumazine synthase from the hyperthermophilic eubacterium Aquifex aeolicus. J Biol Chem. 2003;278:37909–15. doi: 10.1074/jbc.M303090200. [DOI] [PubMed] [Google Scholar]
- 16.Schramek N, Haase I, Fischer M, Bacher A. Biosynthesis of riboflavin. Single turnover kinetic analysis of 6,7-dimethyl-8-ribityllumazine synthase. J Am Chem Soc. 2003;125:4460–6. doi: 10.1021/ja028226k. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Meining W, Fischer M, Bacher A, Ladenstein R. X-ray structure analysis and crystallographic refinement of lumazine synthase from the hyperthermophile Aquifex aeolicus at 1.6 A resolution: Determinants of thermostability revealed from structuralcomparisons. J Mol Biol. 2001;306:1099–114. doi: 10.1006/jmbi.2000.4435. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Meining W, Cushman M, Haase I, Fischer M, Bacher A, et al. A structure-based model of the reaction catalyzed by lumazine synthase from Aquifex aeolicus. J Mol Biol. 2003;328:167–82. doi: 10.1016/s0022-2836(03)00186-4. [DOI] [PubMed] [Google Scholar]
- 19.Scheuring J, Kugelbrey K, Weinkauf S, Cushman M, Bacher A, Fischer M. 19F NMR ligand perturbation studies on 6,7-bis (trifluoromethyl)-8-ribityllumazine-7-hydrates and the lumazine synthase complex of Bacillus subtilis Site-directed mutagenesis changes the mechanism and the stereoselectivity of the catalyzedhaloform-type reaction. J Org Chem. 2001;66:3811–9. doi: 10.1021/jo001739u. [DOI] [PubMed] [Google Scholar]
- 20.Mörtl S, Fischer M, Richter G, Tack J, Weinkauf S, Bacher A. Biosynthesis of riboflavin. Lumazine synthase of Escherichia coli. J Biol Chem. 1996;27(271):33201–7. doi: 10.1074/jbc.271.52.33201. [DOI] [PubMed] [Google Scholar]
- 21.Meining W, Mörtl S, Fischer M, Cushman M, Bacher A, Ladenstein R. The atomic structure of pentameric lumazine synthase from Saccharomyces cerevisiae at 1.85 A resolution reveals the binding mode of a phosphonate intermediate analogue. J Mol Biol. 2000;299:181–97. doi: 10.1006/jmbi.2000.3742. [DOI] [PubMed] [Google Scholar]
- 22.Illarionov B, Eisenreich W, Bacher A. A pentacyclic reaction intermediate of riboflavin synthase. Proc Natl Acad Sci U S A. 2001;98:7224–9. doi: 10.1073/pnas.131610698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Illarionov B, Kemter K, Eberhardt S, Richter G, Cushman M, Bacher A. Riboflavin synthase of Escherichia coli. Effect of single amino acid substitutions on reaction rate and ligand binding properties. J Biol Chem. 2001;276:11524–30. doi: 10.1074/jbc.M008931200. [DOI] [PubMed] [Google Scholar]
- 24.Eberhardt S, Richter G, Gimbel W, Werner T, Bacher A. Cloning, sequencing, mapping and hyperexpression of the ribC gene coding for riboflavin synthase of Escherichia coli. Eur J Biochem. 1996;242:712–9. doi: 10.1111/j.1432-1033.1996.0712r.x. [DOI] [PubMed] [Google Scholar]
- 25.Kuehnert S, Oberboersch S, Sundermann C, Haurand M, Jostock R, Schiene K, et al. Gruenenthal GmbH, Germany. Preparation of 1-(1-oxo-2-propynyl) piperazines as mGluR5 receptor modulators for the treatment of pain. WO2006002981. 2006 WO 2006029980. [Google Scholar]
- 26.Berset C, Audetat S, Tietz J, Gunde T, Barberis A, Schumacher A, et al. Protein Kinase Inhibitors. PCT Int Appl. 2005 WO/2005/120513, 2005 (Oncalis AG) [Google Scholar]
- 27.Goldfarb DS. U.S: Patent Appl Publ; 2009. Applications of N, N’-bis (2-pyridyl) aryldiamines. US 2008-341615. [Google Scholar]
- 28.Van Niel MB, Miah A. British UK: Patent Appl; 2008. 3-hydroxy-pyridine-4-ones useful for treating parasitic infections. [Google Scholar]
- 29.Kumar BV, Vaidya SD, Kumar RV, Bhirud sSB, Mane RB. Synthesis and anti-bacterial activity of some novel 2-(6-fluorochroman-2-yl)-1-alkyl/acyl/aroyl-1H-benzimidazoles. Eur J Med Chem. 2006;41:599–604. doi: 10.1016/j.ejmech.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Ozkay Y, Tunali Y, Karaca H, Isikdag I. Antimicrobial activity and a SAR study of some novel benzimidazole derivatives bearing hydrazone moiety. Eur J Med Chem. 2010;45:3293–8. doi: 10.1016/j.ejmech.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Coban G, Zencir S, Zupkó I, Réthy B, Gunes HS, Topcu Z. Synthesis and biological activity evaluation of 1H-benzimidazoles via mammalian DNA topoisomerase I and cytostaticity assays. Eur J Med Chem. 2009;44:2280–5. doi: 10.1016/j.ejmech.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 32.Rai NP, Narayanaswamy VK, Govender T, Manuprasad BK, Shashikanth S, Arunachalam PN. Design, synthesis, characterization, and antibacterial activity of {5-chloro-2-[(3-substitutedphenyl-1,2,4 -oxadiazol-5-yl)-methoxy]-phenyl}-(phenyl)-methanones. Eur J Med Chem. 2010;45:2677–82. doi: 10.1016/j.ejmech.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 33.Chandrakantha B, Shetty P, Nambiyar V, Isloor N, Isloor AM. Synthesis, characterization and biological activity of some new 1,3,4-oxadiazole bearing 2-flouro-4-methoxy phenyl moiety. Eur J Med Chem. 2010;45:1206–10. doi: 10.1016/j.ejmech.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 34.Gaonkar SL, Rai KM, Prabhuswamy B. Synthesis and antimicrobial studies of a new series of 2-[4-[2-(5-ethylpyridin-2-yl) ethoxy] phenyl]-5-substituted-1,3,4-oxadiazoles. Eur J Med Chem. 2006;41:841–6. doi: 10.1016/j.ejmech.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Dawane BS, Konda SG, Mandawad GG, Shaikh BM. Poly (ethylene glycol) (PEG-400) as an alternative reaction solvent for the synthesis of some new 1-(4-(4’-chlorophenyl)-2-thiazolyl)- 3-aryl-5-(2-butyl-4-chloro-1H-imidazol-5yl)-2-pyrazolines and their in vitro antimicrobial evaluation. Eur J Med Chem. 2010;45:387–92. doi: 10.1016/j.ejmech.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 36.Rohini R, Shanker K, Reddy PM, Ho YP, Ravinder V. Mono and bis-6-arylbenzimidazo[1,2-c] quinazolines: A new class of antimicrobial agents. Eur J Med Chem. 2009;44:3330–9. doi: 10.1016/j.ejmech.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 37.Dubey PK, Kumar RV, Naidu A, Kulkarni SM. A review on the biological activity of imidazo (4,5 b) pyridines and related compounds. Asian J Chem. 2002;14:1129–38. [Google Scholar]
- 38.Abstract Clark RL, Pessolano AA, Shen TY, Jacobus DP, Jones H, Lotti VJ, Flataker LM. Synthesis and analgesic activity of 1,3-dihydro-3-(substituted phenyl) imidazo[4,5-b] pyridin-2-onesand 3-(substituted phenyl)-1,2,3-triazolo[4,5-b] pyridines. J Med Chem. 1978;21:965–78. doi: 10.1021/jm00207a023. [DOI] [PubMed] [Google Scholar]
- 39.Robinson MM, Finch N. Imidazo 4,5 b pyridines U.S. Patent. 1973 3719683. [Google Scholar]
- 40.Von Bebenberg W. Azabenzimidazole derivatives which are thromboxane receptor antagonists. U.S.Patent. 1974 [Google Scholar]
- 41.Lesher GY, Brundage RP, Opalka CJ, Page DF. French Patent 1981, 2, 478 637. Chem Abstr. 1982;96:85551k. [Google Scholar]
- 42.Kuczynski L, Mrozikiewicz A, Poreba K. Synthesis and biological properties of imidazo-[4,5-b]-pyridine derivatives. Pol J Pharmacol Pharm. 1982;34:229–38. [PubMed] [Google Scholar]
- 43.Bianchi M, Butti A, Rossi S, Barzaghi F, Marcaria V. Compounds with antiulcer and antisecretory activity. III. N-Substituted imidazolones condensed with nitrogen-containing heteroaromatic rings. Eur J Med Chem. 1983;18:501. [Google Scholar]
- 44.Vaughn JR., Jr Organo phosphorous compounds Final technical reports Section A,B. U.S. Patent. 1953;263:7731. [Google Scholar]
- 45.Schmidt B, Schieffer B. Angiotensin II AT1 receptor antagonists. Clinical implications of active metabolites. J Med Chem. 2003;46:2261–70. doi: 10.1021/jm0204237. [DOI] [PubMed] [Google Scholar]
- 46.Bavetsias V, Sun C, Bouloc N, Reynisson J, Workman P, Linardopoulos S, et al. Hit generation and exploration: Imidazo[4,5-b] pyridine derivatives as inhibitors of Aurora kinases. Bioorg Med Chem Lett. 2007;17:6567–71. doi: 10.1016/j.bmcl.2007.09.076. [DOI] [PubMed] [Google Scholar]
- 47.Shu Q, Nair V. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med Res Rev. 2008;28:219–32. doi: 10.1002/med.20104. [DOI] [PubMed] [Google Scholar]
- 48.Hedstrom L. IMP dehydrogenase: Structure, mechanism, and inhibition. Chem Rev. 2009;109:2903–28. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheuerman R, Tumelty AD. The reduction of aromatic nitro groups on solid supports using sodium hydrosulfite Tetrahedron Lett. 2000;41:6531–5. [Google Scholar]
- 50.Yang D, Fokas D, Li L, Yu C, Baldino M. A versatile method for the synthesis of benzimidazoles from o-nitroanilines and aldehydes in one step via a reductive cyclization. Synthesis. 2005;1:47–56. [Google Scholar]
- 51.Hongfeng C, Nilsen NC, Choudhury A, Sorgi KL. Process for preparing substituted diaminopyrimidine oximes. Justia Patents Database. 2012 Patent number 8278446. [Google Scholar]